

Abstract
Background
Recent publications from a single research group have suggested that aldehyde-based high-level disinfectants (HLDs), such as ortho-phthalaldehyde (OPA), are not effective at inactivating HPVs and that therefore, patients may be at risk of HPV infection from medical devices. These results could have significant public health consequences and therefore necessitated evaluation of their reproducibility and clinical relevance.
Methods
We developed methods and used standardised controls to: (1) quantify the infectious levels of clinically-sourced HPVs from patient lesions and compare them to laboratory-derived HPVs, (2) evaluate experimental factors that should be controlled to ensure consistent and reproducible infectivity measurements of different HPV genotypes, and (3) determine the efficacy of select HLDs.
Findings
A novel focus forming unit (FFU) infectivity assay demonstrated that exfoliates from patient anogenital lesions and respiratory papillomas yielded infectious HPV burdens up to 2.7 × 103 FFU; therefore, using 2.2 × 102 to 1.0 × 104 FFU of laboratory-derived HPVs in disinfection assays provides a relevant range for clinical exposures. RNase and neutralising antibody sensitivities were used to ensure valid infectivity measures of tissue-derived and recombinant HPV preparations. HPV infectivity was demonstrated over a dynamic range of 4–5 log10; and disinfection with OPA and hypochlorite was achieved over 3 to >4 log10 with multiple genotypes of tissue-derived and recombinant HPV isolates.
Interpretation
This work, along with a companion publication from an independent lab in this issue, address a major public health question by showing that HPVs are susceptible to HLDs.
Funding
Advanced Sterilization Products; US NIH (R01CA207368, U19AI084081, P30CA118100).
Keywords: Sexually transmitted disease, Nosocomial infection, Virus disinfection, Medical devices, Oncogenic virus, High-level disinfectant
Research In Context.
Evidence before this study
Questions have been raised about the effectiveness of high-level disinfectants (HLD) such as ortho-phthalaldehyde (OPA) based on data from in vitro infectivity assays using HPV16 and HPV18 isolated from organotypic (raft) epithelial tissue cultures. The work has not been replicated by any other laboratory and there are multiple methodological and translational gaps that need to be addressed to evaluate the reproducibility and clinical relevance of these data. The gaps can be addressed by:
-
•
Developing novel assays to quantify the levels of infectious HPV in clinically-derived, human lesions to ascertain whether the viral doses tested from model systems (e.g. tissue-derived HPV and recombinant quasivirions) are relevant to human exposures.
-
•
Developing approaches and suggesting guidelines for consistent and objective evaluation of HPV infection by RT-qPCR.
-
•
Evaluating the impact of virus isolation techniques on the infectivity readout.
-
•
Evaluating the impact of timing for readouts of infectivity in vitro.
-
•
Evaluating the impact of neutralising agents on viral infectivity.
-
•
Evaluating differences in infectivity of tissue-derived, recombinant, and clinically sourced viruses
-
•
Assessing the similarities and differences in clinically sourced virus titres across different HPV high-risk (e.g. anogenital) and low-risk (e.g. recurrent respiratory) lesions
Added value of this study
The data presented in this publication together with work described in the companion manuscript by another laboratory have addressed the gaps listed above. They have advanced our understanding of the important methodological considerations for how to design infectivity and disinfection testing studies that have greater clinical relevance. The results have also demonstrated that high-level disinfectants such as ortho-phthalaldehyde and hypochlorite are effective at inactivating HPV. In our view, the reports that have called the efficacy of these disinfectants into question have methodologic limitations with respect to the virus isolation process, assay fidelity, and infection controls that likely confounded their results and interpretations.
Implication of all the available evidence
The data generated allow the healthcare and research communities to have confidence in their use of disinfectants such as ortho-phthalaldehyde and hypochlorite to protect patients against HPV fomite transmission. The methods that have been developed can also be used to evaluate new prophylactic and disinfectant agents.
Alt-text: Unlabelled box
1. Introduction
Human papillomaviruses (HPVs) are highly transmissible nonenveloped viruses that cause significant health and economic burden worldwide [1]. HPV infections are spread by close contact and cause epithelial warts and papillomas with some infections progressing to malignancies [2]. HPV DNA and/or RNA can be detected on medical devices [3], [4], [5], equipment, and surfaces in clinical settings [6], and is commonly described as “viral load”. However, the relationship between viral nucleic acid load and HPV infectious potential has not been previously evaluated.
Questions about the effectiveness of high-level disinfectants such as ortho-phthalaldehyde (OPA) against laboratory-produced HPVs have arisen following publications from a single research group [7], [8], [9]. The prospect that certain commonly-used disinfection approaches may not be effective at inactivating HPV infectivity contradicts the accepted hierarchy of microbial resistance to biocides [10] and merits thorough examination. To investigate the relationship between HPV DNA load in lesions and infectious potential and to comprehensively assess the efficacy of different disinfectants against HPV, infection assays were carefully controlled and novel methods were developed. To our knowledge, these approaches facilitated, for the first time, evidence for the levels of infectious HPV in clinical lesions to which medical instruments might be exposed. We quantified and compared infectious levels of clinically-sourced HPVs from patient lesions to laboratory-derived HPV levels used in in vitro assays, and evaluated the factors that must be controlled to ensure consistency and reproducibility for in vitro HPV infectivity assessments. We then utilised these well-controlled methods to determine the efficacy of disinfectants such as OPA and hypochlorite in inactivating multiple HPV genotypes isolated by common experimental approaches.
2. Methods
2.1. Ethics
The collection of clinical material used in this study complied with the Helsinki Declaration of 1975, as revised in 1983 and described previously and was approved by the University of New Mexico Health Sciences Center's Human Research Protections Office (UNM HRPO #17-202 IRB Approved Protocol).
2.2. Papilloma sampling and HPV typing
Between August 2017 and April 2018, subjects with papillomas consistent with HPV infection were approached to participate in the study. After receiving written, informed consent, physicians rubbed the lower half of a small (1 × 4-cm), sterile piece of emery paper (600A-grit Wetordry Tri-M-ite; 3M) 5 times across the surface of the papilloma, similar to a study wherein HPV DNA prevalence was tested [11]. Exfoliates were eluted with sterile PBS. Briefly, each emery paper swab was held at the upper half with sterile tweezers, with the lower half in a 1.7 mL microcentrifuge tube. PBS (250 µl) was used to rinse the exfoliate from the lower portion of the paper, rinsing 2–3 times into the microcentrifuge tube. The rinse was repeated with a second 250-µl aliquot of PBS for a total recovery volume of 0.5 ml. The PBS clinical virion stocks were stored at 4 °C. HPV DNA typing was performed according to manufacturer's instructions using the SEEGENE HPV28 assay (Anyplex II HPV28 detection kit, Seegene). This multiplexed genotyping system is semiquantitative and facilitates simultaneous detection and identification of 19 high-risk HPVs (16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 69, 73, 82) and 9 low-risk HPVs (6,11, 40, 42, 43, 44, 54, 61, 70). For infectivity comparison to laboratory-derived HPV stocks, clinical samples positive for HPV11, HPV16 and/or HPV31 were subject to quantification of viral genome equivalents (VGE) by quantitative PCR (qPCR).
2.3. Viral genome equivalent (VGE) quantification of virus stocks
All HPV virion preparations were quantified for encapsidated viral genomes using TaqMan qPCR specific to the HPV long control region (LCR), a noncoding segment of the viral genome, as previously described [12,13]. HPV stocks were diluted (e.g., 1:50 and 1:500) in carrier DNA (sheared salmon sperm DNA [Invitrogen] or human placental DNA [Millipore Sigma]) at a carrier concentration of 50 ng/ul. qPCR reactions were performed using 5 µL of the 1:50 and 1:500 virus stocks using primers targeted to the HPV LCR (Bio-Rad SsoFast EvaGreen Supermix) (see Supplemental Table 2). For each assay a calibration curve spanning 108–100 genome copies was run in triplicate and was generated by serial dilutions of cloned HPV genomes. Data were collected on a Bio-Rad CFX96 and analysed using Bio-Rad CFX Manager (version1.6.541.1068). Only experiments with calibration curve R2 values of ≥0.995 and amplification efficiencies between 90 and 100% were included.
2.4. Cell lines
The cell lines W12 (RRID:CVCL_T290) and CIN-612 were established from human cervical intraepithelial neoplasia grade I (CIN1) biopsies [14,15]. The clonal CIN-612 9E cell line (RRID:CVCL_ER27; a gift of L. Laimins, Northwestern Univ.) maintains episomal HPV31 (subtype 31b) genomes at ≈50–100 copies per cell [16]. W12 clone 20863 (W12-E) cells (gifted by P. Lambert, Univ. of Wisconsin-Madison and M. Stanley, Univ. of Cambridge) harbour episomal HPV16 genomes at ≈1000 copes per cell [17]. CIN-612 9E and W12-E cells were grown in monolayer culture in the presence of mitomycin C-treated J2 3T3 mouse fibroblast feeder cells (RRID:CVCL_W667) with E medium containing 5% fetal bovine serum (FBS) and 100 U/ml Nystatin (Sigma-Aldrich) as previously described [12]. HEK-293T cells (RRID:CVCL_0063) obtained from C. Buck (U.S. National Cancer Institute) were maintained in DMEM (Gibco Cat. # 11965-084). The HaCaT cell line (RRID:CVCL_0038), a gift of N. Fuesnig (DKFZ), are a spontaneously-immortalized, HPV-negative epithelial line derived from normal adult skin [18]. HaCaT cells were maintained in DMEM/F12-Ham's Nutrient Mixture containing 4X amino acids, 2 mM L-glutamine, 100 U/ml penicillin, and 1 ug/ml streptomycin (each from Sigma) and supplemented with 10% FBS (Atlanta Biologicals). Cell lines were authenticated by short tandem repeat analysis (IDEXX) and used within 15 passages of verification; mycoplasma screening was performed once per month.
2.5. Human papillomavirus production and isolation
Infectious HPV virion stocks were obtained from three laboratory model systems, including virion biosynthesis in mouse xenograft tissues and organotypic (raft) epithelial tissue cultures, and the recombinant assembly of quasivirions (QV). HPV11 virions from infected human keratinocyte xenografts grown in athymic mice as previously described [19] were a gift of Neil Christensen (Penn State College of Medicine). In short, the xenograft-derived HPV11 stock was obtained as a supernatant following mincing and disruption of infected tissue by Virtis homogenization in PBS at 4 °C at 25,000 rpm. for 30 min. Supernatants and pellets were separated by sedimentation at 11,000 x g. HPV16 and HPV31 virions were acquired from organotypic (raft) epithelial cultures [12]. HPV16 and HPV31 virions were acquired from organotypic (raft) epithelial cultures grown from W12-E and CIN-612 9E cells, respectively, as previously described [12,20]. For the raft-derived HPV31 stock described here, 80 raft tissues were cultivated to obtain the virus titres reported. Two methods were used to isolate virus particles from the raft tissues. First, crude virus preparations (CVPs) were obtained as described by Conway et al., wherein 5–10 raft tissues were homogenized in sodium phosphate solution (1M NaCl/0.05 M Na-Phosphate buffer, pH 8.0) using a Dounce apparatus and large cellular debris were pelleted [21]. The CVP supernatant was collected and stored at −20 °C for short-term storage. Second, a more refined raft virus preparation was obtained as previously detailed [12,22]. Briefly, raft tissues were homogenized using a BeadBeater™ tissue grinder. After a series of low- and high- speed centrifugation steps to remove cellular debris, DNA-containing virus particles were pelleted based on a sedimentation coefficient of 296S–300S for viral DNA-containing particles [23]. Recombinant “quasivirion” (QV) production was modified from that previously published [24]. Briefly, cloned viral genomes were released from plasmids by restriction endonuclease digestion. Intramolecular ligations were performed, and DNA was concentrated by isopropanol precipitation. HEK-293T cells were co-transfected with the re-circularized HPV genome, and a genotype-matched, codon optimized HPV L1- and L2- expressing plasmid (see Table 1). At 72 h post-transfection, the cells were lysed and treated with 99% pure Benzonase (EMD Millipore) and Plasmid-Safe ATP Dependent DNase (Epicentre). Virions were incubated at 37 °C for 24 h to permit capsid maturation [25]. Cellular debris were pelleted, and the supernatants were subjected to a 1.25–1.4 g/mL CsCl step gradient and centrifugation at 20,000xg for 18 h. The opalescent viral band was extracted by side puncture, virions were washed 2–3X against Virus Stabilization Buffer (VSB) (25 mM HEPES pH 7.5, 500 mM NaCl, 1 mM MgCl2) and concentrated using an Amicon Ultra-4 centrifugal filter 100 K (Millipore).
Table 1.
HPV genotypes, genome loads and titres from clinical lesions.
| Samplea | HPV genotype(s) (semiquantitative DNA load)b | Total HPV genome load (VGE)c | Physical titre (VGE/mL) | Infectious by RT-qPCR | VGE per FFUd | Infectious units (FFU) per samplee |
|---|---|---|---|---|---|---|
| RRP 1 | LR: 11 (+++) | HPV11–1.4 × 107 | 2.7 × 107 | Yes | 5.6 × 103 | 2.5 × 103 |
| RRP 2 |
LR: 11 (+++) HR: 31 (+) |
HPV11–1.2 × 105 HPV31–3.0 × 104 |
2.3 × 105 6.0 × 104 |
N.D.f N.D. |
N.D. | N.D. N.D. |
| RRP 3 | LR: 11 (++) | HPV11–2.2 × 105 | 4.5 × 105 | N.D. | N.D. | N.D. |
| RRP 4 | LR: 11 (+++) | HPV11–2.9 × 105 | 5.8 × 105 | N.D. | N.D. | N.D. |
| RRP 5 |
LR: 11 (++) HR: 31 (+) |
HPV11–5.7 × 105 HPV31–2.0 × 104 |
1.1 × 106 4.0 × 104 |
N.D. N.D. |
7.6 × 103 N.D. |
7.5 × 101 N.D. |
| RRP 6 | LR: 11 (++) | HPV11–2.6 × 105 | 5.3 × 105 | N.D. | N.D. | N.D. |
| RRP 7 |
LR: 11 (+++), 43 (+) HR: 31 (+), |
HPV11–2.5 × 105 HPV31–4.0 × 104 |
4.9 × 105 2.0 × 104 |
N.D. N.D. |
N.D. N.D. |
N.D. N.D. |
| AW 1 |
LR: 11 (+++), 54 (++), 70 (+), 44 (++) HR: 59 (+++), 16 (++), 56 (+), 68 (++) |
HPV11–3.3 × 106 HPV16–2.5 × 105 |
6.6 × 106 5.0 × 105 |
Yes N.D. |
N.D.g N.D. |
N.D. N.D. |
| AW 2 |
LR: 61 (+) HR: 16 (+++), 35 (++) |
HPV16–2.1 × 107 | 4.2 × 107 | No | 2.9 × 104 | 7.2 × 102 |
| AW 3 |
LR: 54 (++), 6 (+++) HR: 16 (++) |
HPV16–2.5 × 105 | 4.9 × 105 | N.D. | N.D. | N.D. |
| AW 4 |
LR: 54 (++) HR: 16 (+++), 33 (++), 39 (+++) |
HPV16–3.9 × 108 | 7.9 × 108 | Yes | 1.4 × 105 | 2.7 × 103 |
| AW 5 | HR: 16 (++), 31 (++), 53 (++) | HPV16–5.0 × 105 HPV31–3.3 × 105 |
1.0 × 106 6.6 × 105 |
N.D. N.D. |
N.D. N.D. |
N.D. N.D. |
| AW 6 |
LR: 61 (++) HR:16 (+++), 35 (++) |
HPV16–3.0 × 106 | 6.0 × 106 | N.D. | N.D. | N.D. |
| AW 7 |
LR: 61 (++) HR: 16 (+++), 35 (+++) |
HPV16–1.6 × 106 | 3.3 × 106 | N.D. | N.D. | N.D. |
Recurrent respiratory papilloma (RRP), Anal wart (AW).
Determined by Anyplex II HPV28 detection (Seegene), LR (low risk), HR (high risk).
Based on total recovery volume of 0.5 mL.
Based on RNA-ISH.
Based on total VGE load/focus-forming units (FFU) measured by RNA ISH data (Fig. 4 panels i, j, m, n and data not shown).
Not determined (N.D.).
Stock was depleted with the RT-qPCR assay.
All virion stocks were quantified by qPCR based upon VGE content as noted above [12,13]. When virus stock yields allowed, SDS-PAGE and Coomassie Brilliant Blue staining were used to assess purity and presence of appropriately sized L1 (≈55 kDa) and L2 (≈72 kDa) capsid proteins (Suppl. Fig. 1). The virus stock concentrations of L1 and L2 proteins were determined by Chemidoc analytic software (Bio-Rad) and comparison to a BSA standard curve.
2.6. Infectivity assays
HaCaT cells were seeded at 1.5 × 105 cells per well into 12-well tissue culture plates and incubated overnight at 37 °C, 5% CO2 prior to infections. The clinically sourced HPV11- and HPV16-positive virus isolates with the highest physical viral genome titres were freeze-thawed three times and sonicated prior to exposure to cells. HaCaT cells at ≈70% confluency were exposed to virions in the absence or presence of genotype-specific neutralising antibodies [12] for up to 48 h at 37 °C with 5% CO2. Cells were lysed in Trizol (Sigma) and RNA was extracted per the manufacturer's instructions. Reverse transcription (RT) of a constant amount of total RNA, typically 30 µg, with random hexamer primers (Invitrogen), dNTPs (Invitrogen), RNase inhibitor (Applied Biosystems) and M-MLV reverse transcriptase (Invitrogen) in 20-µl reactions was performed at 37 °C for 60 min. Quantitative polymerase chain reaction (qPCR) was performed using 10X PCR buffer II and AmpliTaq Gold (Applied Biosystems) in triplicate with 5.0 µl of cDNA using primers and probes in Suppl. Table 3; ß-actin was amplified as a reference control for RT efficiency as previously reported [26]. A calibration curve with copy number controls spanning 108–100 were prepared by serial 10-fold dilution from genotype specific, cloned HPV E1^E4 cDNA. Amplifications were in 96-well plate format on a on a Bio-Rad CFX96 and analysed using Bio-Rad CFX Manager (version1.6.541.1068). To be considered a valid qPCR reaction, the R2 values were ≥0.995 and amplification efficiencies were 90–100% for each assay.
RNAscope in situ hybridization (RNA-ISH; Advanced Cell Diagnostics [ACD]) of viral E6/E7 mRNAs[27] was adapted as a focus forming assay to detect HPV infected cells. HaCaT cells were seeded into 8-well chamber slides at 5 × 104 cells per well and exposed to virus stocks. NIKS-SG3 cells persistently infected with HPV16 [28] were used as a positive control and were treated with 20 mg/ml RNase A to verify the specificity of E6/E7 mRNA detection by RNA-ISH. RNAscope was performed with HPV-HR7 E6/E7 probes (ACD Cat. #312351), specific for HPV types 16, 18, 31, 33, 35, 52 and 58, or HPV-LR10 E6/E7 probes (ACD Cat. #314551), recognizing HPV types 6,11, 40, 43, 44, 54, 69, 70, 71 and 74. Stained slides were subject to high-definition digital imaging using Aperio Image Scope microscopy (Leica). Cells were considered to be positive when there were ≥2 punctate signals indicative of HPV E6/E7 mRNA detection. Infrequently, a single punctate signal was observed in a cell, which was considered to be non-specific staining. Positive cells were counted by eye from digitized images.
2.7. Virus neutralization procedures
Genotype specific anti-HPV monoclonal antibodies [29], anti-HPV11 clone 11F.G1, anti-HPV16 clone H16.V5, anti-HPV31 clone H31.A6, (a gift of N. Christensen, Penn State College of Medicine) were incubated with HPV virion stocks at 37 °C for 1 h prior to exposure to cells. Chemical disinfection was performed using a modified suspension test method[30] (Suppl. Fig. 2). Herein, one volume of virions (5 µL) was incubated with nine volumes of either 80% Cidex (final 0.44% OPA) or 10% bleach (final 0.825% hypochlorite [ClO-]) diluted in complete HaCaT medium for 12 min at RT. This 10−1 dilution of treated virus was combined with 450 µL of the appropriate neutralizer (1% or 7% v/v glycine for OPA; 2% v/v sodium thiosulphate [Na2S2O3] for hypochlorite), yielding a 10−2 virus dilution. Untreated controls included virus incubated with 1X VSB; HaCaT medium alone was considered the neutralizer for the virus/buffer positive control. The virus/disinfectant was subject to four 10-fold serial dilutions (10−3–10−6) in the appropriate neutralizer and incubated 15 min, RT. HaCaT cells were exposed to the virus-antibody or virus-disinfectant solutions and allowed to incubate for up to 48 h at 37 °C with 5% CO2. Cells were harvested for RNA; for experimental consistency, 3.0 µg of DNase I-treated RNA from each infection was subject to RT (total 20 µl reaction) and three 5.0-µl technical cDNA replicates were assayed for HPV E1^E4 mRNA levels by qPCR as described above. ß-actin cDNA was amplified as an internal RT control.
2.8. Statistical analyses
Data were analysed using GraphPad Prism 8 software, including simple linear regression and two-tailed Student's t test, which was used to compare differences between two groups. All data are presented as means ± standard deviation (SD). A p value of <0.05 was considered statistically significant and p values are shown in the figures.
2.9. Role of the funders
The study sponsors had no role in the collection, analysis, and interpretation of data or in the decision to submit the paper for publication. The U.S. NIH had no role in the study design or the writing of the report. ASP had a limited role in consulting on disinfection experimental design and in the writing the report.
3. Results
3.1. HPV genome loads in patient papillomas
Exfoliates from the surfaces of seven RRP and seven anogenital papillomas positive for HPV11, HPV16 and HPV31 were quantitatively assessed for encapsidated VGE (Fig. 1, Table 1). Total HPV11 recovered from the RRPs and one anal wart ranged from 3.3 × 104 to 1.4 × 107 VGE. Total HPV31 from the RRP samples ranged from 2.0 to 4.0 × 104 VGE. Samples from three HPV16-positive anal warts (AW) were assayed to have HPV16 DNA loads ranging from 2.5 × 105 to 3.9 × 108 VGE; one also had HPV31 at 3.3 × 105 VGE. Lacking definitive means to verify complete endonuclease digestion of free viral genomes, these VGE load measures may be over-estimates of that encapsidated within viral particles.
Fig. 1.

Clinical lesion sampling and quantification of HPV genome load from samples. (a) Exfoliating cells and squames were collected from the surface of clinically-diagnosed HPV lesions using five passes of a sterile emery paper (see Supplemental Information). Image made in collaboration with BioRender. (b) DNA was isolated from exfoliated cells from atop clinically-diagnosed papillomas and subject to genotyping. Samples positive for HPV11, HPV16 and/or HPV31 were assessed for genotype specific viral DNA load in triplicate by qPCR. Also shown are the estimated sum levels of genome copies per sample for non-HPV11 low risk types (types 6, 43, 44, 54, 61, 70) or non-HPV16/31 high-risk types, (types 33, 35, 39, 53, 56, 59, 68). Bars represent the mean values of total VGE from each sample; mean values and 95% CI are shown. Details of cases are shown in Table 1.
3.2. Impact of viral preparation on HPV infectivity across different virus types
In a prior report, vast differences in disinfection susceptibility were observed when comparing crude HPV16 preparations (CVP) derived from raft epithelial tissues with recombinant HPV16 QV refined by gradient centrifugation [7]. Yet, the differing virus isolation methods could introduce confounding variables, such as free viral nucleic acids, if not properly controlled. Predominantly, HPV virion isolation methods have utilised whole epithelial tissue homogenisation followed by a series of low and high-speed centrifugations to separate virions from unpackaged nucleic acids and cellular debris [22] (see Supplemental Methods). Previous disinfection studies employed a CVP of raft-derived HPV wherein homogenised tissues were simply clarified using low-speed centrifugation [21]. Therefore, we directly compared physical and infectivity characteristics amongst HPV virions obtained from the common laboratory models: xenograft tissues, organotypic (raft) epithelial tissues, and the recombinant QV assembly system. Salient characteristics of laboratory-derived virus stocks are summarised in Table 2 and are highlighted below.
Table 2.
Characteristics of HPV virus preparations.
| Virus Genotype (stock ID) | Source | Final Virion Isolationa | Physical titre (VGE/mL) | Antibody neutralisation | Dynamic range of infection | VGE per FFUh |
|---|---|---|---|---|---|---|
| HPV11 (10.10) | 293T QV | CsCl density gradient | 1.2 × 1010 | 99% | 3-4 log10 | 3.8 × 104 |
| HPV11 | Xenografts b | Debris Sedimentation at 11,000 x g c | 2.9 × 109 | 98% | 3 log10 | 5.0 × 104 |
| HPV16 (12.35) | 293T QV | CsCl density gradient | 3.6 × 1011 | 99% | 4-5 log10 | 3.8 × 105 |
| HPV16 (12.40) | 293T QV | CsCl density gradient | 1.5 × 1011 | 100% | 4-5 log10 | N.D. |
| HPV16 | HCK 16-8 Rafts | Crude Virus Preparation (CVP) d,e | 1.0 × 109 | 53% | N.D.g | no FFU |
| HPV16 (19.17) | W12-E Rafts | CVP | 2.4 × 107 | 55% | N.D. | no FFU |
| HPV16 (19.40) | W12-E Rafts | CVP | 8.4 × 106 | 40% | N.D. | no FFU |
| HPV31 (18.81) | 293T QV | CsCl density gradient | 3.4 × 1010 | 100% | 3-4 log10 | 1.6 × 104 |
| HPV31 (13.02) | CIN-612 9E Rafts | Virion Sedimentation Ultracentrifugation f | 2.0 × 108 | 97% | ≥5 log10 | 3.0 × 103 |
Details of isolation in Methods.
Gift from N. Christensen (PennState College of Medicine).
VirTis homogenisation, supernatant following debris sedimentation at 11,000 x g.
Gift from C. Meyers (PennState College of Medicine).
Dounce homogenisation, low speed debris pelleting per Conway et al. 2009.
BeadBeater homogenisation, high-speed centrifugation, ultracentrifugation at 130,000 x g; based on sedimentation coefficient of 296S–300S for viral DNA-containing particles.
Not determined.
Based on RNA-ISH with FFU related to the total VGE exposed to cells.
Experiments were undertaken to verify bona fide infection from each laboratory-derived virus stock using the established criteria of time-dependent infection measures and neutralisation of infection by well-characterised L1 genotype-specific monoclonal antibodies [29]. HPV infections fail to produce cytopathic effects in cultured cells and thus cannot be measured by quantitative viral plaques or quantal assays. HPV infections are measured via detection of spliced E1^E4 mRNAs, which are the most abundant HPV transcripts detected post infection (p.i.) [12,31,32]. E1^E4 mRNAs are undetectable in the first hours p.i., their levels rise over 48h p.i. [13,33], and detection is abrogated by antibody-mediated neutralisation of virions [12,[34], [35], [36]]. Therefore, HaCaT cells were inoculated with viral stocks and E1^E4 transcripts were quantified after 5 min (T = 0 h), 24 h, or 48 h. In parallel, virus stocks were incubated with genotype-specific neutralising antibodies prior to inoculation and infection for 48 h. Infection for 48 h allows maximal viral transcription without permitting cells to become overly confluent, which we show suppresses early HPV transcription [37]. Each QV stock and the xenograft HPV11 virus exposures were devoid of detectable E1^E4 transcripts at 0 h p.i., and as expected, HPV mRNA levels increased substantially after 48 h p.i. (Fig. 2). Mock infected controls were consistently negative for E1^E4 targets. The refined, raft-derived HPV31 stock had low levels of E1^E4 mRNAs at 0 h; however, the E1^E4 mRNA levels increased significantly with an average of 10-fold at 48 h p.i., consistent with considerable viral transcription following nuclear viral genome delivery (Fig. 2f). Additionally, each of these virus stocks was neutralised >98% with genotype-specific monoclonal antibodies (p ≤ 0.0001). Conversely, the HPV16 CVPs yielded anomalous results, inconsistent with all other virion stocks. Not only were relatively high E1^E4 mRNA levels detected immediately after inoculation, but the E1^E4 levels decreased significantly over 24–48 h p.i. (p = 0.0004; Fig. 2d). Additionally, the CVP stocks were predominantly resistant to antibody neutralisation (Fig. 2d).
Fig. 2.

Infection time course and antibody neutralisation characteristics of HPV virion stocks. Replicate cultures of subconfluent HaCaT cells were incubated with recombinant HPV quasivirions (a, c, e) or tissue-derived HPV preparations (b, d, f). Inocula were exposed to cells for 5 min and were either harvested for total RNA (0h infection), or fresh media were added for infection for 24h or 48 h. A replicate set for each virus stock was incubated with a genotype-specific anti-virus-like particle, monoclonal antibody prior to cell exposure for 48h. The antibodies included: anti-HPV11 clone 11F.G1, anti-HPV16 clone H16.V5, anti-HPV31 clone H31.A6. (g, h) Raft tissue-derived virus stocks were untreated or RNase A treated prior to cell exposure for 0 or 48 h. RNAs were subjected to RT and triplicate qPCR for spliced E1^E4 mRNA quantification (circles); values were normalised to 100% infection at 48 h p.i. Bars represent the mean values of triplicate qPCR reactions from two experimental replicate infections; error bars indicate SD [with paired, two-tailed t-tests performed on indicated values].
RNase treatment of the crude and refined raft-derived HPV stocks prior to inoculation confirmed that both contained contaminating HPV E1^E4 mRNAs which could be misjudged as infection (Fig. 2g–h). However, RNase treatment of the refined, raft-derived HPV31 stock did not significantly reduce the overall levels of E1^E4 mRNAs detected at 48 h p.i. (Fig. 2h), verifying that E1^E4 detection at 48 h p.i. was a valid measure of infection for this virus isolate. Conversely, RNase treatment of the HPV16 CVP resulted in little to no detection of E1^E4 mRNAs at 48 h p.i. (Fig. 2g), indicating that contaminating viral RNA in the CVP confounded the infection assay. Taken together, the infection time course studies and the low antibody-mediated neutralisation of HPV infections revealed that the CVPs contained E1^E4 mRNAs that could be inaccurately attributed to infection at 24 h and 48 h p.i. This could explain CVP resistance to the protein crosslinking actions of OPA and glutaraldehyde in the previously reported disinfection studies.
In assessing virus preparation purity by SDS-PAGE, when adequate stocks permitted, virus purity was not an absolute predictor of infectious capacity or susceptibility to neutralising monoclonal antibodies (Suppl. Fig. 1). Infection-validated, xenograft-derived HPV11 was composed of proteins with sizes corresponding to HPV capsid proteins, L1 (≈55kD) and L2 (≈72kD), and cellular histones (≈10–15kD); but numerous other protein species were also present (Suppl. Fig. 1a, lane 6). High-efficiency HPV QV assembly yielded relatively pure preparations with protein sizes consistent with L1, L2 and cellular histones (Suppl. Fig. 1b–d). Limited recovery of HPVs from the raft tissues precluded our ability to both perform infectivity studies and SDS-PAGE characterisation. Yet, we previously reported that neither sedimentation- nor density gradient-ultracentrifugation refinements yielded infection-validated raft-tissue virus preparations with obvious L1 or L2 species amongst the many proteins detected after SDS-PAGE [22].
3.3. Comparison of infectious titres and dose-responsiveness across HPV genotypes and preparations
Studies involving microbial titrations and neutralisation efficacy (e.g., biocides, antibodies) rely on assays that have a broad detection range, ideally over many log10 dilutions [10]. Whereas qPCR approaches are well suited to detect and quantify viral nucleic acid products resulting from infection over a broad range, accurate data interpretation relies on four critical assay performance characteristics: qPCR efficiency, linear dynamic range, limit of detection (LoD) and precision [38]. Studies evaluating HPV disinfection purportedly assayed over a 5 log10-range [7], [8], [9]. However, the four essential qPCR characteristics were unreported and there was no evidence that the assays were capable of detecting infection over 5 log10. Therefore, as a prerequisite for systematic evaluation of HPV disinfection, we first determined the qPCR performance characteristics. qPCR calibration curves spanning 108–100 copies of cloned, E1^E4 cDNA for HPV types 11, 16 and 31 each achieved efficiencies of ≥95%, revealed 7–8 log10 linear dynamic ranges, and LoD were 101 or 10° copies per reaction (Fig. 3).
Fig. 3.

Linear dose response dynamic range of RT-qPCR quantification and infectivity titration of infectious HPV stocks. HaCaT cells were exposed to serial dilutions of HPV virus stocks that were validated as in Fig. 2. At 48 h p.i., total RNA was extracted and subjected to RT-qPCR with triplicate copy number controls from 108 to 101 for quantification of HPV E1^E4; ß-actin was a reference target. The y-axis represents the total number of E1^E4 cDNA copies present in triplicate amplifications relative to internal calibration curves attaining ≥95% efficiency. The limit of detection (LoD) for each calibration curve is indicated for each qPCR series. Bars represent the mean of qPCR triplicates (symbols) for each infection replicate; SD error bars are shown. Hashtags (#) specify samples with no detectable E1^E4 targets. Simple linear regression of the mean triplicate values (R2) is shown for each replicate dilution series.
Using internal calibration curves for every assay, we assessed the linear dynamic range of detecting HPV infections over as wide a range of inocula as our virus yields permitted. CVPs were not analysed due to their failure to meet the infection criteria detailed above. Cells were exposed to serial virus dilutions (doses reported in VGE/cell) for 48 h. Between two and six replicate infections were performed for each viral dose, depending upon the amount of virus stock available. Following RT-qPCR, absolute quantities of E1^E4 mRNAs were determined relative to the E1^E4 cDNA qPCR calibration curves (Fig. 3) and revealed a number of important findings. The replicate infections demonstrated consistency (precision) among levels of E1^E4 detected, which decreased as each assay LoD was approached. Whether using tissue-derived virions or QV, simple linear regression analyses indicated a strong association between the virus dose and the quantity of E1^E4 mRNA detected. Parallel dose-response infections with HPV11 xenograft and QV 10.10 stocks yielded R2 values ≥ 0.97 (Fig. 3a). The highest dynamic ranges achieved for HPV11 were 3–4 log10 due to limited stocks, with similar infectivity when directly comparing xenograft-sourced and QV HPV11 in a single assay (Fig. 3a; Table 2). Dose-response infections with HPV16 and HPV31 stocks showed strong linearity with R2 values ≥ 0.98; a linear dynamic infection range of ≥ 4–5 log10 was achieved for HPV16 and HPV31 infections (Fig. 3b, c, e; Table 2). The reproducibly low qPCR LoD between 1 and 10 E1^E4 copies per reaction provides a platform to compare HPV virus stocks for relative infectivity based on physical doses of VGE per cell. Most virus stocks yielded reproducible infection at doses ≥1.0 VGE/cell. Refined, raft-derived HPV31 reproducibly demonstrated detectable infection at doses of ≥0.001 VGE/cell (Fig. 3e) for reasons not immediately apparent.
Four clinically-derived HPV-positive samples with the highest physical VGE titres were evaluated for infectivity alongside characterised QV stocks (Fig. 4). RT-qPCR analyses showed HPV11-positive RRP1 and AW1 were infectious (Fig. 4a). HPV16 infection was detected with AW4, but not the AW2 sample (Fig. 4b). As the clinical isolates were limited and had lower VGE/mL titres than lab-sourced HPV virions (Table 1), we developed a novel, quantitative cell-based infection assay using RNA-ISH that requires one-third fewer cells and lower inoculum volumes. Uninfected HaCaT cells were negative for RNA-ISH signal whereas a persistently HPV16-infected cell line (NIKS-SG3), showed predominantly cytoplasmic detection of HPV mRNAs (Fig. 4c and d). RNase A treatment of NIKS-SG3 cells eliminated the positive ISH signals demonstrating RNA target specificity (Fig. 4e). Infections were evaluated with doses ranging from 1.0 to 500 VGE/cell revealing isolated cells with positive RNA-ISH signals expressed as an infectious centre, or “focus forming unit” (FFU), relative to the total VGE dose (Fig. 4f–n). Enumeration of ISH-positive cells revealed that HPV11 QV and xenograft stocks had similar RNA-ISH titres at 3.8 × 104 and 5. VGE/cell revealing isolated cells with positive RNA-ISH signals expressed as an infectious centre, or \215focus forming unit\216 (FFU), relative to the total VGE dose (nullf\055-n). Enumeration of ISH-positive cells revealed that HPV11 QV and xenograft stocks had similar RNA-ISH titres at 3.8 \327 104 and 5.<span class=``error-correction''><delete>.<delete></span>0 \327 104 VGE/FFU, respectively (nullf\055-h), consistent with their similar HPV11 RT-qPCR infection profiles (nulla). HPV11-positive clinical RRP1 was assayed at 5.6 \327 103 VGE/FFU (nulli- and j). The AW1 sample was depleted in the RT-qPCR assay, but HPV11-positive RRP5 scored as 73 \327 103 VGE/FFU (null). Quantification of HPV16 QV infection revealed 3.8 \327 105 VGE/FFU (nullk-l); AW2 and AW4 isolates scored as 2.9 \327 104 and 16 \327 105 VGE/FFU, respectively (nullm- and n). CVP yielded no positive signals in this assay, consistent with their failure in the infection studies noted above. The lower VGE/FFU ratio for clinical isolates compared to HPV16 QV may be attributed to the fact that ISH probes can recognize, in addition to HPV16, other high-risk HPV genotypes that were detected in the clinical samples by DNA typing (null). This novel infectious centre assay revealed that total infectious units isolated from these clinical lesions ranged from 71 \327 101 to 27 \327 103 FFU/sample (null). To our knowledge, this is the first assessment of infectious potential of HPVs from clinical lesions."?>0 × 104 VGE/FFU, respectively (Fig. 4f–h), consistent with their similar HPV11 RT-qPCR infection profiles (Fig. 3a). HPV11-positive clinical RRP1 was assayed at 5.6 × 103 VGE/FFU (Fig. 4i and j). The AW1 sample was depleted in the RT-qPCR assay, but HPV11-positive RRP5 scored as 7.3 × 103 VGE/FFU (Table 1). Quantification of HPV16 QV infection revealed 3.8 × 105 VGE/FFU (Fig. 4k-l); AW2 and AW4 isolates scored as 2.9 × 104 and 1.6 × 105 VGE/FFU, respectively (Fig. 4m and n). CVP yielded no positive signals in this assay, consistent with their failure in the infection studies noted above. The lower VGE/FFU ratio for clinical isolates compared to HPV16 QV may be attributed to the fact that ISH probes can recognize, in addition to HPV16, other high-risk HPV genotypes that were detected in the clinical samples by DNA typing (Table 1). This novel infectious centre assay revealed that total infectious units isolated from these clinical lesions ranged from 7.1 × 101 to 2.7 × 103 FFU/sample (Table 1). To our knowledge, this is the first assessment of infectious potential of HPVs from clinical lesions.
Fig. 4.

Infectivity of HPV-positive clinical samples compared with laboratory-sourced HPVs. Genotyped clinical samples from recurrent respiratory papillomas (RRP) and anogenital warts (AW) were freeze-thawed 3x prior to exposure to subconfluent HaCaT cells for 48 h. Xenograft and QV HPV stocks and mock-infected samples were included as positive and negative controls, respectively. (a-b). RT-qPCR with copy number controls from 107 to 101 for quantification of HPV E1^E4 as in Fig. 2. (c-n) Cells were seeded on chamber slides and exposed to HPV stocks. RNA in situ hybridisation (RNA-ISH) was performed for high-risk (HR) and low-risk (LR) HPV genotypes (see Supplemental Information). (c-e) Controls showing specificity of RNA-ISH as labeled. RNA-ISH was performed for LR HPV11 in lab and clinical HPV stocks (f-j) and HR RNA-ISH for HPV16 in lab and clinical HPV stocks (k-n) with focus forming unit (FFU) comparisons to the inocula dose (VGE) for each assay. Arrows point to positive cells containing ≥2 positive puncta indicative of HPV E6/E7 mRNA. Bars = 100 µm unless otherwise noted.
3.4. Efficacy of disinfectants in vitroacross HPV genotypes and isolates
One research group reported aldehyde-based disinfectants to be ineffective in inactivating HPV QV and raft-derived CVP infections in vitro [8,9]. However, as detailed above, we noted several limitations in these studies. Here we provide an independent and critical evaluation using our high-fidelity RT-qPCR assay, which shows that both OPA and hypochlorite effectively disinfect tissue-derived and recombinant HPV preparations. Based on ASTM International guidelines [30], we challenged our infection-validated and well-characterised HPV stocks (Suppl. Fig. 2). Each virus preparation was incubated with either buffer or disinfectants for 12 min at room temperature followed by 10-fold serial dilutions in the appropriate neutraliser for 15 min. Following cell exposure for 48 h, infection levels were quantified (Fig. 5). For each tissue-derived or QV HPV stock, hypochlorite and OPA were each effective disinfectants, consistent with the results of Egawa et al. in a companion manuscript [39]. Hypochlorite completely prevented infection for all HPV stocks tested. Infectivity was undetected after OPA treatment of both xenograft-derived and QV HPV11 virus stocks, although limited virion stock concentrations permitted assessment only over a 2–3 log10 range (Fig. 5a and b). OPA led to a ≥4 log10 titer reduction in HPV16 and HPV31 QV infectivity (Fig. 5c and d). Although OPA showed lower effectiveness against refined raft-derived HPV31 infectivity (≥3 log10 titer reduction), its efficacy was similar to or slightly better than that of neutralising antibody in direct comparisons (Fig. 5e and f). We cannot rule out the possibility that residual contaminating viral RNA in the raft HPV31 stock is responsible for the OPA- and antibody-resistant E1^E4 signals remaining after treatment. Nevertheless, our data strongly indicate that HPVs are sensitive to disinfection by OPA and hypochlorite.
Fig. 5.

Disinfection efficiency of OPA and hypochlorite against validated HPV stocks. According to the schematic in Suppl. Fig. 2, 5 µL of virus stocks (infection validated as in Fig. 2) were incubated with buffer as a control or disinfectants (OPA or hypochlorite [ClO-]) for 12min. Virus-disinfectant and virus control (buffer) solutions were neutralised and subject to 10-fold serial dilutions in neutraliser and incubated 15 min (final virus dilutions included 10-2–10−6). Virus was also incubated with neutralised disinfectants (light pink bars) or in cell medium (control; dark pink bars). (a-c) 1% glycine was the OPA neutraliser; (d-f) 7% glycine was the OPA neutraliser. As indicated, virus was incubated with dilutions of a neutralising monoclonal antibody (NAb; striped pink bars). HaCaT cells were exposed to each virus stock for 48 h; the physical load (VGE) and infectious load (FFU) of each virus in the 10−2 dilution is indicated. Infectivity levels were determined by RT-qPCR for HPV E1^E4 mRNAs compared to intra-assay, E1^E4 cDNA internal calibration curves; bars represent the mean values of triplicate qPCR reactions. Hashtags (#) specify samples with no detectable E1^E4 targets and the limit of detection (LoD) for each qPCR assays is indicated. Log reduction (yellow boxes) was determined by comparing disinfectant-treated virus to virus treated with buffer (black lines). Neutralised disinfectant and antibody effects on virus infection were compared to untreated control virus (red lines).
Control experiments evaluated viral inhibitory effects of neutraliser alone and of neutralised disinfectant compared to untreated virions. OPA inactivated with 1% glycine had a limited inhibitory effect on HPV infection, with the largest inhibition (1.21-1.99 log10) in xenograft and QV HPV11 virus stocks (Fig. 5a and b). Increasing from 1% to 7% glycine appeared to neutralise OPA more effectively (Fig. 5d–f). Inactivated hypochlorite had minimal effects (<0.6 log10) on HPV infectivity, with the exception of HPV16 QV (Fig. 5c). HPV16 QV were similarly inhibited by 2–3 log10 when exposed to hypochlorite neutralised with 2% sodium-thiosulfate (not shown). Simply, this could not be explained by obvious differences in physical characteristics (Suppl. Fig. 1c) or infection phenotypes (Fig. 3b) compared with other virus stocks.
4. Discussion
We determined for the first time to our knowledge the infectious quantities of HPVs from human papillomas and warts that could contaminate medical devices or fomites, and, which lacking adequate decontamination, pose a public health threat for nosocomial transmission. Additionally, using laboratory-produced HPVs at infectious levels that met or exceeded the infectious HPV levels detected from sampling to model clinical exposure, we determined that both OPA and hypochlorite are effective disinfectants in contrast to published reports from another laboratory [7], [8], [9].
To quantify the infectious potential of HPV virions shed from clinical lesions, we collected cell exfoliates from the uppermost layers of HPV-induced lesions (Fig. 1a), which are directly relevant to those that may contaminate medical equipment. HPV11 was the predominant genotype in RRPs, whereas HPV16 was the most frequent and high-level genotype in anal warts. Using a novel, cell-based RNA-ISH assay to quantify the infectious potential of clinical HPV isolates, we determined select clinical samples to contain between 75 and 2.7 × 103 infectious units (FFU). HPV genome loads (VGE) outnumbered infectious virions by a factor of 103–105 (Tables 1 and 2), consistent with reported viral particle-to-infectivity ratios [40]. This emphasises that genome loads should not be assumed to be accurate predictors for infectious virions. Whereas this work was not intended to be a comprehensive evaluation of the viral loads across a population, it does provide an initial assessment of the range of infectious viral loads on the surface of lesions in a small population.
Although aldehyde disinfectants display potent and broad antimicrobial activity and are used in clinical settings worldwide [41], recent reports from a single group indicated that laboratory-produced HPVs are resistant to aldehyde-based disinfection [7], [8], [9]. Furthermore, this group found stark differences between raft-derived and recombinant HPVs in their susceptibilities to other disinfectants. Therefore, we evaluated quantitative in vitro HPV infection characteristics, including disinfection susceptibility. Our disinfection experiments employed infectious levels of HPV comparable to those we recovered from clinical lesions. Contrary to previous reports, we show that both tissue-derived and recombinant QV are susceptible to disinfection by both OPA and hypochlorite. These findings are in harmony with the established hierarchy of microbicidal activity [10] based on the non-enveloped nature of HPV virions. Our methodical studies of the infection characteristics of laboratory-produced HPV virions reveal two predominant explanations as to why our OPA disinfection results are in opposition to the previously published reports. First, the prior studies employed crude HPV preparations (CVP) derived from raft tissues [7], [8], [9], which we show did not meet common infection criteria of time dependent viral gene expression and strong antibody neutralisation. We demonstrated CVPs were sensitive to RNase treatment, despite being nuclease treated during isolation; this suggests prior studies using CVPs likely measured residual viral RNA contaminating the preparations, rather than authentic infection. Nucleic acids are unlikely to be sensitive to protein crosslinking by aldehydes or antibody-mediated neutralisation. Thus, it is unsurprising that aldehyde-based disinfectants failed to reduce the observed measure of viral mRNAs in aforementioned studies [7], [8], [9], or that neutralising antibodies were ineffective on CVPs investigated herein. Conversely, hypochlorite solutions are effective neutralisers of RNA contamination [42] and many viruses [41]. Second, the above-mentioned studies showed no evidence that their RT-qPCR assay had a linear dynamic range capable of detecting infection across the ≥4-log10 range they claimed to evaluate. Rather, the data suggested they could only detect infection reduction of ≤ 2-log10 from their untreated control. Lacking essential information, it is impossible to differentiate between their assay LoD and any potential for reduced infectivity. Herein, we employed qPCR calibration curves with a 7-log10 linear dynamic range and a LoD of 101 or 100 copies per reaction, providing confidence that our qPCR-based infection assay detects infection over the 4- to 5-log10 doses of viral inocula we tested. Since prior reports lacked important controls, any conclusions, particularly those drawn from the testing of disinfectants, should be viewed cautiously.
Our comparison of HPV virus preparations that were isolated by different protocols revealed that purity was not an absolute predictor of infectious potential. Yet, the presence of proteins corresponding in size to L1, L2 and histones correlated with infectivity of xenograft and QV stocks. We have found that QV stocks that lack either L1 or L2 protein species of the appropriate size by SDS-PAGE analyses are infection compromised (data not shown). Although nuclease treatment of virion stocks can reduce contaminating viral nucleic acids, it could not guarantee removal of HPV mRNAs that might confound infection assays. For future studies of HPV infection and disinfection, we recommend that HPV virion stocks meet bona fide infection criteria, including ≥98% antibody neutralization and time dependent infection detection. Additionally, viral titration assay performance characteristics should be established and reported.
The novel “infectious centre” FFU assay developed herein facilitated titration of clinical virus samples alongside laboratory-derived HPV stocks. While technically more cumbersome than RT-qPCR, advantages of the FFU assay include the need for lower inoculum and the direct enumeration of infected cells. Comparatively, RT-qPCR cannot differentiate between many infected cells expressing low viral RNA levels versus few infected cells expressing higher RNA levels. Due to the low virus yields from the clinical samples, we cannot make definitive conclusions about infectious loads obtained therefrom. However, our physical and infectious virus comparisons strongly suggest that the infectious titres of lab-derived virions used in the disinfection experiments represent similar doses to those from infectious virions that may contact medical devices. It should be emphasized that with any disinfection method, cleaning is of utmost importance; physical removal of microbes and the organic soil that carry them improves the margin of safety contributed by any disinfection method [41]. Therefore, our results, in concert with the companion study by Egawa et al. [39], lead us to conclude that with proper washing of contaminated instruments, OPA and hypochlorite disinfection will minimise potential risk of HPV transmission in associated medical settings.
Contributors
M.A.O., J.Y., M.R., G.E. conceived and designed the study. M.A.O. obtained funding and IRB approvals, and coordinated clinical sampling. V.B., N.A.P., R.T.S. collected the data. M.A.O., V.B., N.A.P., R.T.S. analysed and interpreted the data. M.A.O., V.B., N.A.P. made the figures. A.G.W., E.C.B., R.M. recruited patients, obtained written informed consent, and collected samples. M.A.O., V.B., N.A.P., A.S., J.Y., M.R., G.E. wrote the original manuscript draft. All authors read, revised and approved the manuscript in its final form.
Data sharing statement
The data that support the findings of this study are available from the corresponding author, MAO, upon reasonable request.
Declaration of Competing Interest
M.A.O. reports grants, personal fees, and non-financial support from Advanced Sterilization Products, grants from U.S. National Institutes of Health, during the conduct of the study; A.G.W. reports personal fees and non-financial support from ASCCP, non-financial support from ACOG, non-financial support from Loktal Medical Electronics, personal fees and non-financial support from CSCCP (Chinese Society for colposcopy and cervical pathology, personal fees and non-financial support from Henan People's Regional Hospital, personal fees from Southcentral Foundation (Alaska Native Corp), non-financial support from FECOLSOG (Colombian colposcopy society), non-financial support from COMEGIC (Mexican colposcopy society), non-financial support from Doctors Hospital at Renaissance, non-financial support from MD Anderson, non-financial support from Univ Texas Rio Grande Valley, non-financial support from Global Coalition against Cervical Cancer, non-financial support from AIDS Malignancy Consortium, personal fees and non-financial support from Deaconess Beth Israel Hospital, personal fees and non-financial support from Texas Tech University, personal fees and non-financial support from ABPTGIC (Brazilian colposcopy society), outside the submitted work. V.B., N.A.P, R.T.S., E.C.B. and R.M. have nothing to disclose. A.S., J.Y. and M.R. are employees of Advanced Sterilization Products. G.E. is an employee and shareholder of Johnson & Johnson, which was the parent company for Advanced Sterilization Products and Janssen Pharmaceutica NV at the time the research was conducted.
Acknowledgments
We are extremely grateful for the participation of the women and men who provided apical HPV samples. We thank Fred Schultz for digital scanning of the RNA ISH slides. Funding for this work was primarily provided by Advanced Sterilization Products, with additional contributions from the U.S. NIH (R01CA207368, U19AI084081) and via the UNM Comprehensive Cancer Center Human Tissue Repository Shared Resource (P30CA118100). The contents are solely the responsibility of the authors and the funders played no role in data collection or analyses.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ebiom.2020.103165.
Appendix. Supplementary materials
Supplementary Figure 1. Characterization of HPV quasivirions (QV) and xenograft-derived (XG) virus isolates by SDS-PAGE and Coomassie Brilliant Blue staining. HPV stocks (neat or diluted 1:10) were loaded on SDS-polyacrylamide gels and electrophoresed with molecular size standards and dilutions of BSA as protein loading standards. (a) 4-20% SDS-PAGE; (b-d) 10% SDS-PAGE. Predominant species of L2 (≈72 kD), L1 (≈55 kD), and cellular histones (10-15 kD) are noted. Panel A shows annotation marks used to verify the band sizes compared to the MW standards (lane 1) and to estimate band densities in viral stocks compared to BSA standards (lanes 2-4 or 2-5). In panels a and b, the vertical line indicates gel images that were cropped and spliced together to remove irrelevant data.
{kind=link}
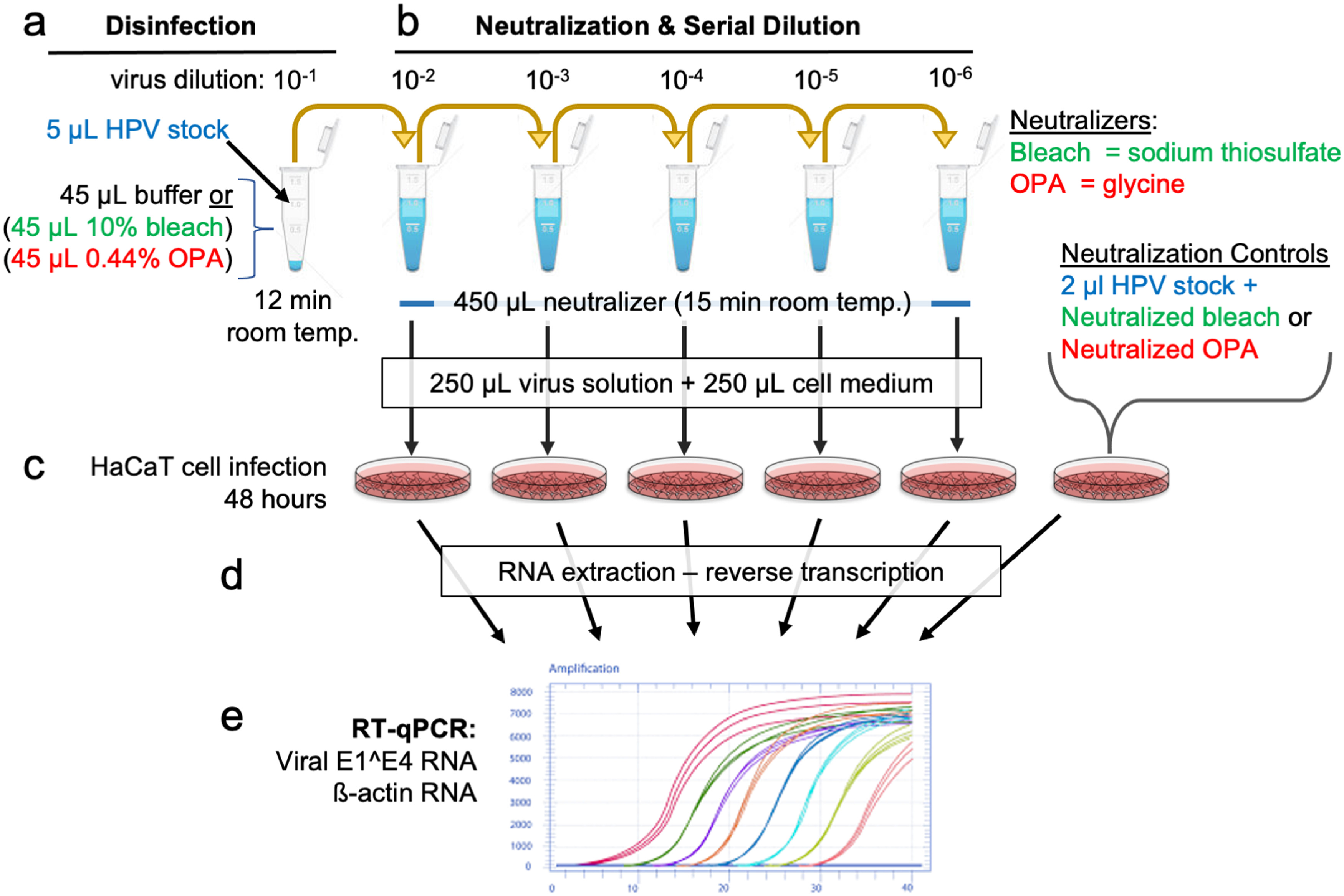
Supplementary Figure 2. Schematic of disinfection protocol. Virus preparations validated for infectious virus based on time-dependent infection measures and antibody-mediated neutralizations (Fig. 2) were subjected to disinfection testing based on the ASTM International’s standard test method to assess the activity of microbicides against viruses in suspension [30]. (a) one part of undiluted virus stock in 1X virus stabilization buffer (VSB; 5 µl) was mixed with nine parts of either 80% Cidex (final 0.44% OPA), or 10% bleach (final 0•825% hypochlorite) for 12 min at room temperature. Virus in buffer was the untreated reference control. (b) Virus was combined with appropriate neutralizers (450 µl) diluted in HaCaT medium containing 10% FBS included 1% v/v glycine for OPA or 2% v/v sodium thiosulphate for hypochlorite. Virus treated with buffer was “neutralized” with HaCaT medium containing 10% FBS an untreated control. The virus/disinfectant was subject to four 10-fold serial dilutions in the appropriate neutralizer and incubated for 15 min at RT. Controls included 2 µL virus incubated with pre-neutralized OPA or bleach. (c) 250 µL of each virus dilution was combined with complete medium and exposed to HaCaT cells for 48 h. No cytotoxicity was observed in controls including cells incubated with pre-neutralized OPA or hypochlorite or the neutralizers alone. (d-e) Cells were harvested for RNA and assayed for HPV E1^E4 mRNA levels via RT-qPCR with an HPV E1^E4 cDNA calibration curve.
{kind=link}
References
- 1.Serrano B, Brotons M, Bosch FX, Bruni L. Epidemiology and burden of HPV-related disease. Best Pract Res Clin Obstet Gynaecol. 2018;47:14–26. doi: 10.1016/j.bpobgyn.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Doorbar J. Host control of human papillomavirus infection and disease. Best Pract Res Clin Obstetr Gynaecol. 2018;47:27–41. doi: 10.1016/j.bpobgyn.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Ma STC, Yeung AC, Chan PKS, Graham CA. Transvaginal ultrasound probe contamination by the human papillomavirus in the emergency department. Emerg Med J. 2013;30:472–475. doi: 10.1136/emermed-2012-201407. [DOI] [PubMed] [Google Scholar]
- 4.Casalegno J-S, Le Bail Carval K, Eibach D, Valdeyron M-L, Lamblin G, Jacquemoud H. High risk HPV contamination of endocavity vaginal ultrasound probes: an underestimated route of nosocomial infection? PLoS One. 2012;7:e48137. doi: 10.1371/journal.pone.0048137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.M'Zali F, Bounizra C, Leroy S, Mekki Y, Quentin-Noury C, Kann M. Persistence of microbial contamination on transvaginal ultrasound probes despite low-level disinfection procedure. PLoS One. 2014;9:e93368. doi: 10.1371/journal.pone.0093368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallay C, Miranda E, Schaefer S, Catarino R, Jacot-Guillarmod M, Menoud P-A. Human papillomavirus (HPV) contamination of gynaecological equipment. Sex Transm Infect. 2016;92:19–23. doi: 10.1136/sextrans-2014-051977. [DOI] [PubMed] [Google Scholar]
- 7.Meyers J, Ryndock E, Conway MJ, Meyers C, Robison R. Susceptibility of high-risk human papillomavirus type 16 to clinical disinfectants. J Antimicrob Chemother. 2014;69:1546–1550. doi: 10.1093/jac/dku006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryndock E, Robison R, Meyers C. Susceptibility of HPV16 and 18 to high level disinfectants indicated for semi-critical ultrasound probes. J Med Virol. 2016;88:1076–1080. doi: 10.1002/jmv.24421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyers C, Milici J, Robison R. UVC radiation as an effective disinfectant method to inactivate human papillomaviruses. PLoS One. 2017;12 doi: 10.1371/journal.pone.0187377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rutala W, Weber D. Registration of disinfectants based on relative microbicidal activity. Infect Control Hosp Epidemiol. 2004;25:333–341. doi: 10.1086/502401. [DOI] [PubMed] [Google Scholar]
- 11.Weaver BA, Feng Q, Holmes KK, Kiviat N, Lee S-K, Meyer C. Evaluation of genital sites and sampling techniques for detection of human papillomavirus DNA in men. J Infect Dis. 2004;189:677–685. doi: 10.1086/381395. [DOI] [PubMed] [Google Scholar]
- 12.Ozbun MA. Infectious human papillomavirus type 31b: purification and infection of an immortalized human keratinocyte cell line. J Gen Virol. 2002;83:2753–2763. doi: 10.1099/0022-1317-83-11-2753. [DOI] [PubMed] [Google Scholar]
- 13.Ozbun MA. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J Virol. 2002;76:11291–11300. doi: 10.1128/JVI.76.22.11291-11300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley MA, Browne HM, Appleby M, Minson AC. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer. 1989;43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- 15.Rader JS, Golub TR, Hudson JB, Patel D, Bedell MA, Laimins LA. In vitro differentiation of epithelial cells from cervical neoplasias resembles in vivo lesions. Oncogene. 1990;5:571–576. [PubMed] [Google Scholar]
- 16.Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J Virol. 1991;65:2254–2260. doi: 10.1128/jvi.65.5.2254-2260.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–2997. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreider JW, Howett MK, Leure-Dupree AE, Zaino RJ, Weber JA. Laboratory production in vivo of infectious human papillomavirus type 11. J Virol. 1987;61:590–593. doi: 10.1128/jvi.61.2.590-593.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song H, Moseley P, Lowe SL, Ozbun MA. Inducible heat shock protein 70 enhances HPV31 viral genome replication and virion production during the differentiation-dependent life cycle in human keratinocytes. Virus Res. 2010;147:113–122. doi: 10.1016/j.virusres.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Conway MJ, Alam S, Ryndock EJ, Cruz L, Christensen ND, Roden RBS. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J Virol. 2009;83:10515–10526. doi: 10.1128/JVI.00731-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozbun MA, Kivitz MP. The art and science of obtaining virion stocks for experimental human papillomavirus infections. In: Gaston K, editor. Small DNA tumor viruses. Caister Academic Press; Norfolk. U.K.: 2012. pp. 19–35. editor. [Google Scholar]
- 23.Crawford LV, Crawford EM. A comparative study of polyoma and papilloma viruses. Virology. 1963;21:258–263. doi: 10.1016/0042-6822(63)90265-4. [DOI] [PubMed] [Google Scholar]
- 24.Pyeon D, Lambert PF, Ahlquist P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc Natl Acad Sci. 2005;102:9311–9316. doi: 10.1073/pnas.0504020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buck CB, Thompson CD, Pang Y-YS, Lowy DR, Schiller JT. Maturation of papillomavirus capsids. J Virol. 2005;79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei L, Gravitt PE, Song H, Maldonado A, Ozbun MA. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus infected cells. Cancer Res. 2009;69:4878–4884. doi: 10.1158/0008-5472.CAN-08-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ukpo OC, Flanagan JJ, Ma X-J, Luo Y, Thorstad WL, Lewis JS. High-risk human papillomavirus E6/E7 mRNA detection by a novel in situ hybridization assay strongly correlates with p16 expression and patient outcomes in oropharyngeal squamous cell carcinoma. Am J Surg Pathol. 2011;35:1343–1350. doi: 10.1097/PAS.0b013e318220e59d. [DOI] [PubMed] [Google Scholar]
- 28.Genther SM, Sterling S, Duensing S, Münger K, Sattler C, Lambert PF. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J Virol. 2003;77:2832–2842. doi: 10.1128/JVI.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen ND, Dillner J, Eklund C, Carter JJ, Wipf GC, Reed CA. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology. 1996;223:174–184. doi: 10.1006/viro.1996.0466. [DOI] [PubMed] [Google Scholar]
- 30.ASTM International . ASTM International; West Conshohocken, PA: 2011. Standard test method to assess the activity of microbicides against viruses in suspension. ASTM Standard E1052-11. [Google Scholar]
- 31.Stoler MH, Broker TR. In situ hybridization detection of human papillomavirus DNA and messenger RNAs in genital condylomas and a cervical carcinoma. Hum Pathol. 1986;17:1250–1258. doi: 10.1016/s0046-8177(86)80569-x. [DOI] [PubMed] [Google Scholar]
- 32.Smith LH, Foster C, Hitchcock ME, Isseroff R. In vitro HPV-11 infection of human foreskin. J Investig Dermatol. 1993;101:292–295. doi: 10.1111/1523-1747.ep12365409. [DOI] [PubMed] [Google Scholar]
- 33.Culp TD, Christensen ND. Kinetics of in vitro adsorption and entry of papillomavirus virions. Virology. 2004;319:152–161. doi: 10.1016/j.virol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Christensen ND, Kreider JW, Cladel NM, Patrick SD, Welsh PA. Monoclonal antibody-mediated neutralization of infectious human papillomavirus type 11. J Virol. 1990;64:5678–5681. doi: 10.1128/jvi.64.11.5678-5681.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith LH, Foster C, Hitchcock ME, Leiserowitz GS, Hall K, Isseroff R. Titration of HPV-11 infectivity and antibody neutralization can be measured in vitro. J Invest Dermatol. 1995;105:438–444. doi: 10.1111/1523-1747.ep12321173. [DOI] [PubMed] [Google Scholar]
- 36.White WI, Wilson SD, Palmer-Hill FJ, Woods RM, Ghim SJ, Hewitt LA. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. J Virol. 1999;73:4882–4889. doi: 10.1128/jvi.73.6.4882-4889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luna AJ, Sterk RT, Griego-Fisher AM, Chung J-Y, Berggren KL, Bondu V. MEK/ERK signaling is a critical regulator of high-risk human papillomavirus oncogene expression revealing therapeutic targets for HPV-induced tumors. PLoS Pathog. 2020 doi: 10.1371/journal.ppat.1009216. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 39.Egawa N, Shiraz A, Crawford R, Sanders-Wood T, Yarwood J, Rogers M. Dynamics of papillomavirus in vivo disease formation & susceptibility to high-level disinfection. Implications for transmission in clinical settings. EBioMedicine. 2020 doi: 10.1016/j.ebiom.2020.103177. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Condit RC. Principles of virology. In: Knipe DM, Howley PM, editors. Fields Virology. 6th ed. Vol. 1. Lippincott Williams & Wilkins; Philadelphia, PA: 2013. pp. 21–51. editors. [Google Scholar]
- 41.Rutala WA, Weber DJ. Disinfection and sterilization in health care facilities: an overview and current issues. Infect Dis Clin N Am. 2016;3:609–637. doi: 10.1016/j.idc.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer M, Renevey N, Thür B, Hoffmann D, Beer M, Hoffmann B. Efficacy assessment of nucleic acid decontamination reagents used in molecular diagnostic laboratories. PLoS One. 2016;11 doi: 10.1371/journal.pone.0159274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Characterization of HPV quasivirions (QV) and xenograft-derived (XG) virus isolates by SDS-PAGE and Coomassie Brilliant Blue staining. HPV stocks (neat or diluted 1:10) were loaded on SDS-polyacrylamide gels and electrophoresed with molecular size standards and dilutions of BSA as protein loading standards. (a) 4-20% SDS-PAGE; (b-d) 10% SDS-PAGE. Predominant species of L2 (≈72 kD), L1 (≈55 kD), and cellular histones (10-15 kD) are noted. Panel A shows annotation marks used to verify the band sizes compared to the MW standards (lane 1) and to estimate band densities in viral stocks compared to BSA standards (lanes 2-4 or 2-5). In panels a and b, the vertical line indicates gel images that were cropped and spliced together to remove irrelevant data.
Supplementary Figure 2. Schematic of disinfection protocol. Virus preparations validated for infectious virus based on time-dependent infection measures and antibody-mediated neutralizations (Fig. 2) were subjected to disinfection testing based on the ASTM International’s standard test method to assess the activity of microbicides against viruses in suspension [30]. (a) one part of undiluted virus stock in 1X virus stabilization buffer (VSB; 5 µl) was mixed with nine parts of either 80% Cidex (final 0.44% OPA), or 10% bleach (final 0•825% hypochlorite) for 12 min at room temperature. Virus in buffer was the untreated reference control. (b) Virus was combined with appropriate neutralizers (450 µl) diluted in HaCaT medium containing 10% FBS included 1% v/v glycine for OPA or 2% v/v sodium thiosulphate for hypochlorite. Virus treated with buffer was “neutralized” with HaCaT medium containing 10% FBS an untreated control. The virus/disinfectant was subject to four 10-fold serial dilutions in the appropriate neutralizer and incubated for 15 min at RT. Controls included 2 µL virus incubated with pre-neutralized OPA or bleach. (c) 250 µL of each virus dilution was combined with complete medium and exposed to HaCaT cells for 48 h. No cytotoxicity was observed in controls including cells incubated with pre-neutralized OPA or hypochlorite or the neutralizers alone. (d-e) Cells were harvested for RNA and assayed for HPV E1^E4 mRNA levels via RT-qPCR with an HPV E1^E4 cDNA calibration curve.