

Abstract
Background and Objective
Ampreloxetine is a novel norepinephrine reuptake inhibitor in development for the treatment of symptomatic neurogenic orthostatic hypotension. The objectives of this analysis were to define the pharmacokinetics of once-daily oral ampreloxetine and provide dose recommendations for clinical development.
Methods
We fitted a population pharmacokinetic model to ampreloxetine plasma concentrations from single- and multiple-ascending dose trials in healthy subjects and two phase II studies in adult subjects with attention-deficit/hyperactive disorder or fibromyalgia at doses of 2–50 mg.
Results
Ampreloxetine pharmacokinetics was best described by a two-compartment model with first-order absorption and elimination. The terminal half-life was 30–40 h, resulting in sustained drug concentrations for the entire 24-h dosing interval at steady state. Covariates of age, weight, or renal impairment did not impact ampreloxetine exposure. Cytochrome P450 2D6 phenotype had no influence on ampreloxetine exposure. Sex and smoking status were identified as statistically significant covariates, suggesting a role for cytochrome P450 1A2 in the elimination of ampreloxetine. Despite statistical significance, differences in ampreloxetine exposure in male vs female subjects and smokers vs non-smokers were not clinically meaningful at the recommended dose. At the 10-mg dose, > 75% norepinephrine transporter inhibition and < 50% serotonin transporter inhibition are anticipated for adult subjects.
Conclusions
The population pharmacokinetic model effectively described the plasma concentration–time profile of ampreloxetine after single and multiple doses. Population pharmacokinetic/pharmacodynamic analysis justified using a fixed dosing regimen with no dose adjustments across a broad population and can be used to inform dosing strategies in future clinical studies.
Clinical Trial Registration
ClinicalTrials.gov identifier numbers NCT01693692 (fibromyalgia); NCT01458340 (attention-deficit/hyperactive disorder).
Electronic supplementary material
The online version of this article (10.1007/s40262-020-00918-7) contains supplementary material, which is available to authorized users.
Key Points
| Pharmacokinetic analysis of plasma ampreloxetine exposure indicated that the terminal half-life was 30–40 h, reached steady state after 6 days, and accumulated three- to four-fold with once-daily dosing. |
| Population pharmacokinetic modeling identified sex and smoking status as statistically significant covariates indicating that cytochrome P450 1A2-based metabolism is the key mechanism for ampreloxetine elimination. |
| Based on the pharmacokinetic/pharmacodynamic relationship, a once-daily ampreloxetine dose of 10 mg is predicted to achieve sustained norepinephrine transporter engagement during the entire dosing interval. |
Introduction
Ampreloxetine (previously known as TD-9855) is a novel small-molecule inhibitor of both norepinephrine (NE) and serotonin (5-HT) uptake, with selectivity for NE [1]. Based on the results of both non-clinical [2] and human [3] studies, ampreloxetine is anticipated to offer benefits for the management of symptomatic neurogenic orthostatic hypotension (nOH). Symptomatic nOH remains largely unaddressed by current therapies [4–7]. The pressor agents, fludrocortisone [5], a synthetic mineralocorticoid, midodrine, an α1-adrenergic receptor agonist approved for the treatment of orthostatic hypotension [6], and droxidopa, an NE precursor approved in the USA for the treatment of nOH [7], are effective in acutely treating declines in blood pressure but lack durability of effect in treating the debilitating symptoms of nOH [8–12]. Moreover, midodrine and droxidopa have short half-lives and require multiple doses per day [6, 7].
Pharmacological inhibitors of NE transporters (NETs) afford an opportunity to treat the core symptoms of nOH that are attributable to the variety of underlying neurological disorders (e.g., synucleinopathies, such as multiple system atrophy, Parkinson’s disease, or pure autonomic failure) [4]. Norepinephrine transporter inhibitors are anticipated to be beneficial in the treatment of nOH through potentiation of residual sympathetic tone that support positional changes from supine to standing [2, 13]. Atomoxetine, an NET inhibitor approved in the USA for the treatment of attention-deficit/hyperactivity disorder (ADHD) [13], has been evaluated in a proof-of-concept clinical study for the treatment of nOH, with results suggestive of symptomatic benefits in patients with nOH [14].
The pharmacological and pharmacokinetic behavior of ampreloxetine has been studied in previous clinical trials. An absorption, metabolism, and excretion study of [14C]-ampreloxetine was conducted in six healthy male volunteers after a single oral administration of 20 mg (100 μCi) of ampreloxetine [15]. Ampreloxetine elimination is primarily driven by metabolism, as less than 5% of the dose was recovered in urine and feces as unchanged drug. Furthermore, ampreloxetine has been evaluated in healthy male subjects to assess NET and serotonin transporter occupancy in the human brain using positron emission tomography after administration of oral doses from 4 to 20 mg [1]. The results of the positron emission tomography study demonstrated NET occupancy to be predominant (NET > serotonin transporter) and a concentration-dependent increase in NET occupancy with increasing ampreloxetine plasma concentrations over the tested dose range. Limited serotonin transporter occupancy was observed at the highest evaluated dose (20 mg), 25% (± 8%) at a plasma concentration of 6.8 ng/mL [1]. The relationship between plasma ampreloxetine concentration and NET occupancy data was well described by an Emax model, with an ampreloxetine plasma concentration of 1.2 ng/mL (95% confidence interval 0.76–1.7 ng/mL) predicted to yield 50% occupancy of NET within the brain.
The objectives of this analysis were to define the pharmacokinetics of ampreloxetine in healthy volunteers and subjects with ADHD and fibromyalgia (FM) and identify any covariates or special populations to inform the dosing strategy for further clinical development.
Materials and Methods
Ethical Conduct of the Study
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee, the 1964 Helsinki Declaration and its later amendments, and Good Clinical Practice [16]. The trial protocols were approved by independent ethics committees and/or institutional review boards (single- and multiple-ascending dose studies, Independent Investigational Review Board, Inc., 6738 West Sunrise Blvd, Suite 102, Plantation, FL 33313, USA; ADHD and FM studies, Western Institutional Review Board®, 3535 7th Avenue, SW Olympia, WA 98502-5010, USA, and IntegReview 3001 S. Lamar Blvd, Suite 210, Austin, TX 78704, USA). All subjects provided written informed consent before initiation of any trial-related activities.
Study Designs
Single-Ascending Dose
A phase I, double-blind, randomized, placebo-controlled, single-ascending dose (SAD) study of ampreloxetine was conducted to evaluate the pharmacokinetic (PK) profile in healthy male and female subjects. A total of 40 subjects were enrolled in this study into five sequential cohorts (n = 8/cohort) to receive a single oral dose of either ampreloxetine (2, 4, 8, 20, or 50 mg) or placebo after 8 h of fasting, in a ratio of 6:2 active drug to placebo. No concomitant medications were allowed during the course of this study.
In all cohorts, blood samples for measurement of ampreloxetine concentration were collected pre-dose and at 0.5, 1, 1.5, 2, 3, 3.5, 4, 6, 8, 10, 12, 24, 36, 48, and 72 h post-dose. Based on plasma half-life and time to maximum concentration values observed in cohort 1, additional blood samples were collected at 14 and 96 h post-dose in cohort 2, and at 14, 96, and 144 h post-dose in cohorts 3, 4, and 5.
Multiple-Ascending Dose
A phase I, double-blind, randomized, placebo-controlled, multiple-ascending dose (MAD) study was conducted to evaluate the safety and pharmacokinetics of multiple doses of ampreloxetine in healthy subjects aged 18–55 years and in an additional cohort of healthy subjects aged 56–65 years. Thirty-two subjects aged 18–55 years and eight subjects aged 56–65 years were enrolled in this study; thus, a total of 40 subjects were randomized into five sequential cohorts (n = 8/cohort). Within each cohort, subjects were randomized to receive either ampreloxetine or placebo, in a ratio of 6:2 active drug to placebo. In cohorts 1, 2, and 3, ampreloxetine doses of 4 mg, 10 mg, and 20 mg were administered orally once daily for 14 days, respectively, in subjects aged 18–55 years. Cohort 4 received ampreloxetine 20 mg once daily for 14 days in subjects aged 56–65 years. In cohort 5, dose titration was explored, starting with 20 mg of ampreloxetine administered once daily for 3 days followed by 40 mg administered once daily for 14 days, in subjects aged 18–55 years. No concomitant medications were allowed during the course of this study.
In cohorts 1–4, PK blood samples for assays of ampreloxetine were collected on days 1 and 14 at pre-dose and at 0.5, 1, 2, 4, 6, 8, 10, 12, and 14 h post-dose. Additional samples were collected after the day 14 dose at 24, 48, 72, and 96 h post-dose. Trough samples were collected pre-dose on days 2, 4, 6, 8, 10, and 12. In cohort 5, during the 3 days at the 20-mg dose level, PK blood samples were collected at pre-dose and 10 h post-dose on day 1, and at pre-dose on day 2. Following escalation to the 40-mg dose level, serial, trough, and post-dose samples were collected on the same schedule as in cohorts 1–4.
To evaluate the effect of food on the pharmacokinetics of ampreloxetine, following the 14 days of ampreloxetine administration and an additional 14-day washout period, subjects in cohort 1 were administered a single oral dose of ampreloxetine 30 min after eating a high-fat meal [17]. Pharmacokinetic sample times mirrored the sampling following the day 14 dose.
Attention-Deficit/Hyperactivity Disorder Study
A phase II, double-blind, placebo-controlled, parallel-group, three-arm, randomized study (NCT01458340) was conducted to compare 5 mg and 20 mg of ampreloxetine with placebo in 285 adult subjects with clinically confirmed ADHD according to the Adult Attention-Deficit/Hyperactivity Disorder Clinical Diagnostic Scale (ACDS Version 1.2) [18]. Eligible subjects were randomized equally to receive either active treatment or placebo daily for 6 weeks. Subjects in the ampreloxetine 5-mg or placebo arm received ampreloxetine 5 mg or placebo once daily for 6 weeks. Subjects in the ampreloxetine 20-mg arm received an initial dose of 10 mg once daily for 1 week, followed by 20 mg once daily for 5 weeks. Each subject’s dose was increased to 20 mg unless a safety or tolerability concern was identified during the 10-mg once-daily dosing week, in which case the subjects continued to receive 10 mg once daily for a total of 6 weeks. Blood samples for the measurement of ampreloxetine plasma concentration were obtained on study days 8, 15, 29, and 43. On study days 15 and 43, the samples were obtained between 2 and 10 h post-dose. Cytochrome P450 (CYP) 2D6 genotype and phenotype were assigned using the AmpliChip CYP450 test (Roche Molecular Diagnostics, Branchburg, NJ, USA) based on the manufacturer’s algorithm. Polymorphisms in CYP2D6 have been shown to be associated with significant differences in the PK profile for CYP2D6 substrate drugs [19].
Fibromyalgia Study
A phase II multicenter, randomized, double-blind, parallel-group, placebo-controlled study (NCT01693692) was conducted to compare a lower-dose regimen (2.5 mg once daily for 1 week, followed by 5 mg once daily for 5 weeks) and a higher-dose regimen (10 mg once daily for 1 week, followed by 20 mg once daily for 5 weeks) of ampreloxetine vs placebo in approximately 375 adult subjects with FM according to the American College of Rheumatology diagnostic criteria for FM [20]. Blood samples for measurement of ampreloxetine plasma concentrations were obtained on study days 8, 15, 29, and 43 (end of treatment). Blood samples were taken before dosing on days 8, 29, and 43 and at any time between 2 and 10 h post-dose on day 15.
Pharmacokinetic Analyses
Bioanalysis of Pharmacokinetic Samples
Plasma samples were assayed using a validated liquid chromatography with tandem mass spectrometry method at Advion/Quintiles (Ithaca, NY, USA). The ampreloxetine assay quantification range was 0.05–100 ng/mL in all studies. Overall precision for the calibration standards and quality-control samples, as measured by coefficient of variation (%), was always within 10%.
Non-Compartmental Analysis
Non-compartmental analysis of the SAD and MAD studies was conducted using WinNonLin Version 5.2 (Pharsight Corporation, Sunnyvale, CA, USA). Reported PK parameters include maximum concentration (Cmax), time to maximum concentration, lag time, terminal elimination half-life, oral volume of distribution during terminal phase, oral clearance (CL/F), area under the concentration–time curve (AUC) from time 0 to 24 h (AUC0–24), and 0 h to time t (AUC0–t). The effect of food was assessed descriptively by examining the mean Cmax and AUC0–24 in cohort 1 on day 29 (fed state) relative to the mean Cmax and AUC0–24 values in cohort 1 on day 1 (fasted state).
Population Pharmacokinetic Analysis
Data from the phase I SAD/MAD and phase II studies were combined into a single dataset containing individual demographics and time course of plasma ampreloxetine concentrations. A total of 2978 ampreloxetine concentrations from 499 subjects were used to conduct a non-linear mixed-effects modeling analysis and identify sources of intra- and inter-individual variability. A two-compartment model best described the ampreloxetine PK profile collected from the phase I SAD/MAD studies. The models assumed a log-normal distribution of the individual PK parameters and a mixed residual error model. Models were evaluated using the objective function value, computed as -2 times the log-likelihood, diagnostic plots, and the precision of parameter estimates. All analysis was conducted using NONMEM 7.2 (Icon plc, Dublin, Ireland) using the first-order conditional estimation method.
A covariate analysis was conducted to identify the effects of age, weight, creatinine clearance (as continuous covariates using exponential models of inter-individual variation), sex, and smoking status (as categorical covariates using an additive model) on the apparent CL/F of ampreloxetine and volume of the central compartment (V1/F). Identification of statistically significant covariates was conducted using a stepwise-forward selection with an α = 0.01 (a decrease of 6.63 points in the objective function) and a stepwise-backward elimination process with an α = 0.001 (an increase of 10.83 points in the objective function). Model fit was evaluated using goodness-of-fit plots and visual predictive checks.
The exposure to ampreloxetine was simulated for each subject using the individual post hoc PK parameters from the final population PK model to determine the clinical significance of the statistically significant covariates. The post hoc exposures were also used to determine whether ampreloxetine exposure remained consistent across the healthy, ADHD, and FM patient populations. Differences in ampreloxetine plasma exposure between poor, intermediate, extensive, and ultra-rapid CYP2D6 metabolizers was evaluated using a one-way analysis of variance test of the log-transformed AUC values across each group.
Population Pharmacokinetic-Pharmacodynamic Simulation
The individually estimated PK parameters determined by the population PK analysis were used to simulate the steady-state concentration–time profiles for each subject in the dataset at two doses of ampreloxetine (5 mg once daily and 10 mg once daily). The steady state AUC0–24 was calculated for each simulated subject using the linear trapezoidal rule.
Simulated individual plasma concentration–time profiles and the pharmacodynamic model established from the positron emission tomography study (half maximal inhibitory concentration = 1.2 ng/mL) were subsequently used to predict the time course of NET occupancy. The predicted NET occupancy after multiple doses of ampreloxetine was averaged over the 24-h dosing interval to assess the steady-state level of NET inhibition.
Results
Ampreloxetine Pharmacokinetic Profile in Healthy Volunteers
Ampreloxetine PK assessments following single oral-dose administration based on non-compartmental analysis are presented in Table 1 and mean plasma concentrations of ampreloxetine over time are plotted in Fig. 1a, b. Plasma concentration–time profiles were characterized by an initial lag in the absorption phase with a median lag time value range of 0.5–1.0 h, followed by a prolonged absorption phase with a median time to maximum concentration value range of 8–12 h post-dose (Fig. 1a, Table 1). Mean ampreloxetine terminal half-life values had a range of 29.5–41.0 h. Exposure to ampreloxetine in plasma increased in a dose-proportional manner across a dose range of 2–50 mg.
Table 1.
Summary of mean (standard deviation [SD]) plasma pharmacokinetic parameters for ampreloxetine: single-ascending dose study
| Dose level (mg) | Cmax (ng/mL) | Tmaxa (h) | Tlaga (h) | AUC0–24 (ng*h/mL) | AUC0–t (ng*h/mL) | t1/2 (h) | CL/F (L/h) | VZ/F (L) | |
|---|---|---|---|---|---|---|---|---|---|
| 2 | Mean | 0.925 | 12 | 1 | 15.8 | 36.3 | 29.5 | 56.4 | 2397 |
| SD | 0.335 | 8.00, 12.0 | 0.5, 1.5 | 5.6 | 11.1 | 4.6 | 22.3 | 916 | |
| n | 6 | 6 | 6 | 6 | 6 | 3 | 3 | 3 | |
| 4 | Mean | 2.09 | 8 | 1 | 32.8 | 87.6 | 36.0 | 38.7 | 1983 |
| SD | 0.34 | 8.00, 10.0 | 0.5, 1.0 | 4.3 | 11.4 | 6.4 | 6.4 | 353 | |
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 8 | Mean | 5.02 | 12 | 0.50 | 85.6 | 231 | 34.5 | 38.2 | 1659 |
| SD | 1.4 | 8.05, 14.0 | 0, 1.0 | 22.5 | 98 | 12.8 | 17.4 | 386 | |
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 20 | Mean | 8.59 | 12 | 0.5 | 142 | 445 | 41.0 | 52.2 | 2745 |
| SD | 2.45 | 8.00, 14.0 | 0, 2.0 | 43 | 195 | 12.2 | 33.8 | 1034 | |
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 50 | Mean | 26.4 | 12 | 0.5 | 461 | 1430 | 40.9 | 39.9 | 2337 |
| SD | 5.7 | 8.00, 14.0 | 0, 1.0 | 105 | 530 | 8.8 | 15 | 1046 | |
| n | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
In each cohort, 6 subjects were dosed with the active drug
AUC0–24 area under the concentration–time curve from time 0 to 24 h, AUC0–t area under the concentration–time curve from time 0 to the time of last quantifiable concentration, CL/F oral clearance, Cmax maximum concentration, t1/2 terminal elimination half-life, Tlag lag time, Tmax time to maximum concentration, VZ/F oral volume of distribution during terminal phase
aMedian (minimum, maximum)
Fig. 1.

Mean plasma concentrations of ampreloxetine: a day 1 of single-ascending dose (SAD) study, b day 14 of multiple-ascending dose (MAD) study, and c attention-deficit/hyperactivity disorder study grouped by cytochrome P450 (CYP) 2D6 phenotype
Ampreloxetine PK assessments, following 14 days of once-daily dosing based on a non-compartmental analysis, are presented in Table 2 and mean plasma concentrations of ampreloxetine after once-daily administration for 14 days are presented in Fig. 1b. Steady-state plasma concentrations of ampreloxetine were achieved by day 6 in most subjects after once-daily doses of 4–40 mg (Table 2). Accordingly, the ampreloxetine PK profile on day 14 represents steady-state behavior. Oral clearance estimates following multiple dosing are more robust than those determined after a single dose owing to a large percentage (> 20%) of the AUC0–inf that was extrapolated in some subjects in the single-dose study. After reaching Cmax, plasma concentrations of ampreloxetine declined with the mean half-life range of 34.3–43.1 h at steady state. Consistent with the half-life, mean accumulation ratios were based on the AUC0–24 range of 3.18- to 4.38-fold at steady state. Consistent with the single-dose PK profile, plasma exposure at steady state also increased in an approximate dose-proportional manner at a dose range of 4–40 mg (Table 2, Table 1 of the Electronic Supplementary Material [ESM], and Fig. 1b). Mean Cmax and AUC0–24 were 9.4% and 6.5% lower, respectively, in the fed state compared with the fasted state. These differences are considerably lower than the magnitude of inter-individual variability in Cmax and AUC0–24 and are therefore not anticipated to be clinically meaningful (Table 1 of the ESM).
Table 2.
Summary of plasma pharmacokinetic parameters for ampreloxetine: multiple-ascending dose study (day 14a)
| Dose level (mg) | Cmax (ng/mL) | Tmaxb (h) | AUC0–4 (ng*h/mL) | t1/2 (h) | CL/F (L/h) | VZ/F (L) | AR | Ctrough (ng/mL) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 4 | Day 6 | Day 8 | Day 10 | Day 12 | |||||||||
| 4 | Mean | 7.88 | 8 | 150 | 43.1 | 29.9 | 1833 | 4.98 | 3.4 | 4.03 | 4.16 | 4.67 | 4.47 |
| SD | 2.39 | 8.00, 8.00 | 47 | 4.1 | 13.2 | 737 | 0.84 | 1.09 | 1.46 | 1.39 | 1.61 | 1.47 | |
| n | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 6 | 5 | 5 | 5 | |
| 10 | Mean | 18.2 | 6.01 | 336 | 37.7 | 36.4 | 1880 | 3.18 | 8.36 | 12.3 | 13.1 | 12.1 | 11.5 |
| SD | 8.7 | 6.00, 8.00 | 156 | 6.2 | 17.7 | 726 | 0.45 | 3.56 | 6.8 | 6.4 | 5.7 | 5.6 | |
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 20 | Mean | 30.5 | 8 | 600 | 37.6 | 35.7 | 2068 | 3.74 | 14.6 | 18.5 | 20.2 | 21.9 | 20.8 |
| SD | 9.6 | 5.50, 12.0 | 169 | 3.3 | 10.1 | 614 | 0.78 | 4.9 | 7.1 | 6.6 | 5.4 | 5 | |
| n | 6 | 6 | 6 | 5 | 6 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 20 | Mean | 52.5 | 7.03 | 1019 | 37.1 | 23.7 | 1528 | 4.38 | 21.9 | 28.5 | 29.2 | 29.5 | 30.8 |
| (older)c | SD | 22.8 | 6.00, 8.00 | 469 | 4.4 | 11.4 | 532 | 1.41 | 7.2 | 10.1 | 12.2 | 15.3 | 15.5 |
| n | 6 | 6 | 6 | 4 | 6 | 4 | 6 | 6 | 6 | 6 | 6 | 6 | |
| 40 | Mean | 74.5 | 8 | 1346 | 34.3 | 34.7 | 1611 | NA | 36.9 | 49.9 | 44.1 | 46.3 | 45 |
| SD | 36.2 | 6.00, 10.0 | 570 | 7.3 | 15 | 496 | NA | 12 | 19.7 | 18.9 | 21.3 | 15.3 | |
| n | 6 | 6 | 6 | 6 | 6 | 6 | NA | 6 | 6 | 6 | 6 | 6 | |
AR accumulation ratio (calculated using AUC0–24 on day 14 vs day 1), AUC0–24 area under the concentration–time curve from time 0 to 24 h, CL/F oral clearance, Cmax maximum concentration, Ctrough trough concentration (obtained prior to dosing on indicated days), NA not applicable, SD standard deviation, t1/2 terminal elimination half-life, Tmax time to maximum concentration, VZ/F oral volume of distribution during terminal phase
aDay 14 of dosing for the 40-mg treatment arm corresponds to study day 17
bMedian (minimum, maximum)
cHealthy subjects aged 56–65 years
At the 20-mg dose level, ampreloxetine exposure in subjects aged 56–65 years was approximately 35% and 70% higher on days 1 and 14, respectively, when compared with adult subjects aged 18–55 years who were administered the same 20-mg dose (Table 2 and Table 1 of the ESM). However, the ampreloxetine 20-mg older group consisted entirely of female subjects (n = 6), compared with three male subjects and three female subjects in the ampreloxetine 20-mg adult reference group. Accordingly, the impact of age or sex on ampreloxetine pharmacokinetics could not be differentiated in this study.
Ampreloxetine Pharmacokinetics in Subjects with Attention-Deficit/Hyperactivity Disorder and Fibromyalgia
After continuous once-daily dosing of ampreloxetine in the phase II study in subjects with ADHD, mean ampreloxetine plasma concentrations were generally consistent with exposures observed in the SAD and MAD studies. Mean plasma concentrations at steady state had a range from 4.5 ng/mL (day 8) to 5.1 ng/mL (day 15) in the ampreloxetine 5-mg treatment arm (Table 3). Subjects in the 20-mg treatment arm received a once-daily dose of 10 mg from day 1 to day 7. Subjects who were not escalated to 20 mg and continued to receive 10 mg up to day 42 had mean plasma concentrations at steady state from 12.0 ng/mL (day 29) to 13.4 ng/mL (day 8). Subjects who were escalated to 20 mg had a mean plasma concentration at the steady-state range from 18.2 ng/mL (day 43) to 20.3 ng/mL (day 29) (Table 3). No statistical difference in ampreloxetine plasma exposure was observed between poor, intermediate, extensive, and ultra-rapid CYP2D6 metabolizers within each dose level at each timepoint based on a one-way analysis of variance test (p > 0.05) (Fig. 1c).
Table 3.
Summary of mean plasma concentrations (ng/mL) of ampreloxetine (attention-deficit/hyperactive disorder study)
| Treatment arm | Dose level (mg) | PK collection dayc | ||||
|---|---|---|---|---|---|---|
| 8 | 15 | 29 | 43 | |||
| Ampreloxetine 5 mg | 5 | Mean | 4.53 | 5.11 | 4.85 | 4.91 |
| SD | 2.18 | 2.86 | 3.43 | 3.91 | ||
| %CV | 48 | 56 | 71 | 80 | ||
| Min | 1.33 | 0.190 | 0 | 0 | ||
| Max | 10.2 | 13.5 | 17.4 | 17.5 | ||
| n | 93 | 91 | 85 | 81 | ||
| Ampreloxetine 20 mg | 10a | Mean | 13.4 | 13.2 | 12.0 | 12.4 |
| SD | 6.50 | 6.20 | 7.76 | 10.0 | ||
| %CV | 48 | 47 | 65 | 81 | ||
| Min | 4.72 | 4.74 | 1.27 | 3.55 | ||
| Max | 27.5 | 24.1 | 27.9 | 41.0 | ||
| n | 14 | 13 | 13 | 13 | ||
| 20b | Mean | 9.29 | 20.1 | 20.3 | 18.2 | |
| SD | 4.50 | 11.2 | 13.4 | 14.9 | ||
| %CV | 48 | 56 | 66 | 82 | ||
| Min | 1.37 | 0.208 | 0 | 0 | ||
| Max | 21.1 | 56.4 | 63.4 | 81.2 | ||
| n | 79 | 75 | 66 | 66 | ||
CV coefficient of variability, Max maximum, Min minimum, PK pharmacokinetic, SD standard deviation
aSubjects remained on ampreloxetine 10 mg/day for the entire treatment period and were not escalated to 20 mg
bSubjects received 10 mg/day on days 1 through 7 and then on day 8 were escalated to ampreloxetine 20 mg/day for the remainder of the treatment period
cCollection time was random prior to dosing, between 2 and 10 h
After continuous once-daily dosing of ampreloxetine in the phase II study in subjects with FM, ampreloxetine concentrations were consistent with observed concentrations in the SAD, MAD, and phase II ADHD studies (Table 3). In the 5-mg and 20-mg treatment arms, mean ampreloxetine plasma concentrations were 5.3 ng/mL (day 43) to 6.3 ng/mL (day 15) and 21.0 ng/mL (day 43) to 28.0 ng/mL (day 15), respectively, at steady state (Table 4).
Table 4.
Summary of mean plasma concentrations (ng/mL) of ampreloxetine (fibromyalgia study)
| Treatment arm | PK collection day | |||
|---|---|---|---|---|
| 8 | 15 | 29 | 43 | |
| Ampreloxetine 5 mg | ||||
| Mean | 2.42 | 6.32 | 5.54 | 5.31 |
| SD | 1.43 | 3.39 | 3.67 | 3.63 |
| %CV | 59 | 54 | 66 | 68 |
| Min | 0 | 0 | 0 | 0 |
| Median | 2.45 | 5.99 | 5.07 | 5.15 |
| Max | 7.10 | 15.4 | 15.2 | 13.7 |
| n | 112 | 107 | 100 | 98 |
| Ampreloxetine 20 mg | ||||
| Mean | 9.93 | 28.0 | 23.4 | 21.0 |
| SD | 6.06 | 16.1 | 16.1 | 15.5 |
| %CV | 61 | 58 | 69 | 74 |
| Min | 0 | 0 | 0 | 0 |
| Median | 9.33 | 26.6 | 22.6 | 18.0 |
| Max | 24.8 | 79.0 | 87.2 | 76.3 |
| n | 113 | 107 | 93 | 90 |
CV coefficient of variability, Min minimum, Max maximum, PK pharmacokinetic, SD standard deviation
Population Pharmacokinetic Analysis
The pooled PK dataset consisted of demographics and plasma concentrations from 499 subjects (Table 2 of the ESM). A two-compartment model with absorption lag, first-order absorption, and elimination provided the best structural fit to the data, based on the minimum value of the objective function, diagnostic plots (Fig. 1 of the ESM), and the precision of parameter estimates. The model was parameterized with CL/F, V1/F, volume of distribution of the peripheral compartment, inter-compartmental clearance, and an absorption rate constant. The effects of covariates on the absorption lag, absorption rate constant, inter-compartmental clearance, and volume of distribution of the peripheral compartment were not characterized because of limited data from the sparsely sampled phase II studies.
Covariates of age (18–65 years), weight (43.1–157 kg), and creatinine clearance (45.2–201 mL/min) did not have statistically significant associations with either CL/F or V1/F (Table 5). In contrast, both sex and smoking status had statistically significant associations with CL/F, and sex had a statistically significant association with V1/F (Table 5). Male subjects had an increased CL/F and V1/F, resulting in approximately 33% lower ampreloxetine exposure compared with female subjects (Fig. 2a). Similarly, subjects who were smokers had increased CL/F compared with non-smokers, resulting in approximately 32% lower ampreloxetine exposure in smokers compared with non-smokers for male as well as female subjects (Fig. 2a). No differences in ampreloxetine exposure were observed between the healthy volunteers vs subjects with ADHD or FM (Fig. 2b). Despite the statistical significance of sex and smoking covariates, the differences in ampreloxetine exposure between male smokers, male non-smokers, female smokers, and female non-smokers demonstrated a significant overlap due to the inter-individual variability of ampreloxetine exposure within each subgroup (Fig. 3a).
Table 5.
Population pharmacokinetic analysis
| Description | Population estimate (%RSE) | Inter-subject variability %CV (%RSE) |
|---|---|---|
| Dose lag (h) | 1.74 (1.52) | – |
| CL/F (L/h) | 47.2 (3.72) | 50.4 (10.9) |
| V1/F (L) | 2040 (6.25) | 38.4 (31.9) |
| Q/F (L/h) | 15.4 (20.1) | – |
| V2/F (L) | 357 (16.6) | – |
| Ka (L/h) | 0.2 (7.17) | 34.1 (48.1) |
| Sex effect on CL [if female] (L/h) | − 16.9 (11.6) | – |
| Smoking effect on CL (L/h) | 16.0 (16.1) | – |
| Sex effect on V1 [if female] (L) | − 720.0 (18.1) | – |
| Proportional residual error (%) | 24.7 (8.7) | |
| Additive residual error (ng/mL) | 0.199 (24.0) |
CL clearance, CL/F oral clearance, CV coefficient of variability, Ka absorption rate constant, PK pharmacokinetics, Q/F inter-compartmental clearance, RSE relative standard error, V1 volume central compartment, V1/F volume of distribution of the central compartment, V2/F volume of distribution of the peripheral compartment
Fig. 2.
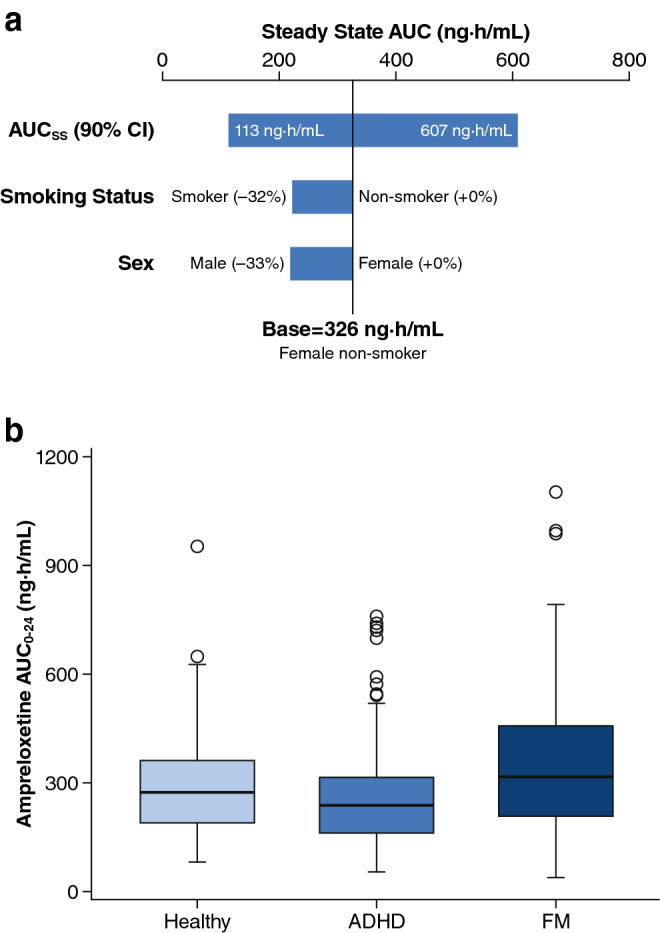
Effect of covariates on the model-predicted ampreloxetine exposure following repeated 10-mg once-daily dosing: a sensitivity plot comparing the effect of significant covariates, b grouped by disease state. Base, as represented by the black vertical line, refers to the predicted typical area under the concentration–time curve from time 0 to 24 h (AUC0–24) in a female non-smoker from the population pharmacokinetic model. The top horizontal bar with values at each end shows the 5th to 95th percentile AUC0–24 range across the entire population. The middle and lower horizontal bars represent the influence of a single categorical covariate on AUC0–24, with one subject created from each category with other covariates fixed at the typical values for the base subject. The solid black line denotes the median; the top and the bottom of the box denote the first and third quartiles; the whiskers denote 1.5 times the interquartile range. ADHD attention-deficit/hyperactivity disorder, AUCss AUC at steady state, CI confidence interval, FM fibromyalgia
Fig. 3.
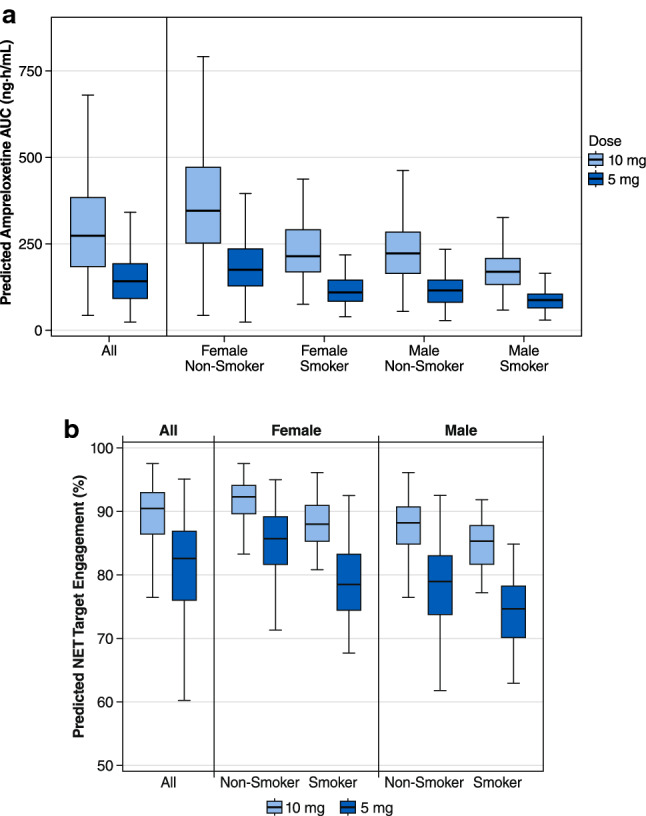
Predicted ampreloxetine steady-state pharmacokinetics and pharmacodynamics following repeated 10-mg and 5-mg doses: a area under the concentration–time curve (AUC). b Predicted brain norepinephrine transporter (NET) occupancy at an average plasma concentration of ampreloxetine. The solid black line denotes the median; the top and the bottom of the box denote the first and third quartiles; the whiskers denote 1.5 times the interquartile range
Population Pharmacokinetic-Pharmacodynamic Predictions
The individually predicted steady-state ampreloxetine exposures following a 5-mg once-daily regimen and a 10-mg once-daily dosing regimen were simulated for 499 subjects (Fig. 3a). Predicted exposures were highest in the female non-smoker group, followed by comparable exposures between female smokers and male non-smokers, with the lowest exposures in male smokers. The predicted pharmacodynamic effect (steady-state NET occupancy) for the ampreloxetine 5-mg group has a range of 71–95% in the female non-smoker group, 62–93% in female smokers and male non-smokers, and 63–85% in male smokers. However, at the 10-mg dose level, the predicted population pharmacodynamic effect was > 75% NET inhibition in nearly 100% of the patient population (Fig. 3b).
Discussion
Dose selection for phase II and phase III studies is influenced by many factors, and drug pharmacokinetics plays a central role in the dose-selection rationale and concomitant medication strategy. The objectives of this population PK analysis were to define the pharmacokinetics of ampreloxetine and identify covariates to inform dose selection for clinical development in subjects with symptomatic nOH.
Following oral administration, the pharmacokinetics of ampreloxetine was best characterized by a two-compartment PK model with first-order absorption and elimination. Dose proportionality observed in the population PK model was consistent with the results from the non-compartmental analysis of the SAD and MAD studies. Pharmacokinetic profiles were similar in healthy subjects and subjects with ADHD and FM, allowing for direct translation of clinical pharmacology study results in healthy volunteers to patient populations. Following once-daily dosing, ampreloxetine exposure reached steady state by day 6 and accumulated three- to four-fold, consistent with an ampreloxetine elimination half-life of 30–40 h. Accordingly, ampreloxetine exposure maintained a relatively flat drug concentration profile throughout the 24-h dosing interval at steady state. The low peak-to-trough ratio provides the potential for more sustained maintenance of stable orthostatic blood pressure throughout the entire 24-h dosing interval unlike the short-acting standard-of-care agents such as midrodrine [6] and droxidopa [7] used for treating symptomatic nOH. Furthermore, the once-daily dosing regimen for ampreloxetine is also anticipated to increase patient compliance [21] relative to midrodine [6] and droxidopa [7], which need to be administered three times a day owing to their short half-lives.
The population PK analysis of all the phase I and phase II clinical data indicates that the covariates of age, weight, or renal impairment (creatinine clearance in the range of mild-to-moderate renal impairment) did not impact ampreloxetine exposure. No differences in ampreloxetine exposure were observed based on the CYP2D6 phenotype and thus, the CYP2D6 enzyme is expected to have a negligible contribution towards ampreloxetine metabolism. Accordingly, the same dose is recommended for subjects across all age groups, CYP2D6 phenotype, irrespective of baseline weight or body mass index and inclusive of subjects with mild and moderate renal impairment.
Sex and smoking status were statistically significant covariates shown to impact CL/F and V1/F. Male subjects demonstrated an ~ 33% lower ampreloxetine exposure compared with female subjects, while smokers demonstrated an ~ 32% lower ampreloxetine exposure compared with non-smokers for both male and female subjects. These covariates indicate that CYP1A2-based metabolism is the key mechanism for ampreloxetine elimination because smoking is known to induce CYP1A2 activity [22]. Accordingly, a clinical drug–drug interaction study with a CYP1A2 inhibitor and inducer will be conducted with ampreloxetine.
Despite the statistical significance of the covariate effect, ampreloxetine exposure in each of the subgroups including male smokers, male non-smokers, female smokers, and female non-smokers overlaps significantly, and predicted NET brain occupancy is still anticipated to be greater than 75% at the ampreloxetine 10-mg dose for all subjects. Thus, no difference in the safety and efficacy profile is expected across the subgroups, and no dose adjustment based on sex or smoking status is recommended for clinical development.
Conclusions
The proposed population PK model effectively described the plasma concentration–time profile of ampreloxetine after single and multiple doses in both healthy subjects and subjects with ADHD and FM. While the covariates included in the model were useful for interpreting inter-individual variability in the data, and providing a rationale for dose selection, dose adjustment in future clinical studies is not warranted. Based on the PK/pharmacodynamic profile of ampreloxetine, a once-daily 10-mg dose is recommended as the optimal dosage to inhibit NET significantly in a majority of the intended patient population.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
AL, RMB, and JK designed the research, performed the research, analyzed the data, and wrote the manuscript. DB and RG designed the research, performed the research, and wrote the manuscript.
Compliance with Ethical Standards
Funding
This study was funded by Theravance Biopharma, Inc. Kirsty Nahm MD of The Curry Rockefeller Group, LLC provided assistance with writing/formatting/proofreading/collation of the author comments and this assistance was funded by Theravance Biopharma, Inc. Open access publication of this manuscript was sponsored by Theravance Biopharma, Inc.
Conflict of interest
Jitendra Kanodia, Arthur Lo, R. Michael Baldwin, Richard A. Graham, and David L. Bourdet are current or former employees of Theravance Biopharma US, Inc.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants in the study.
Data sharing
Theravance Biopharma, Inc. (and its affiliates) will not be sharing individual de-identified participant data or other relevant study documents.
Footnotes
R. Michael Baldwin: Former employee of Theravance Biopharma US, Inc.
Jitendra Kanodia and Arthur Lo contributed equally to this article.
References
- 1.Smith JA, Bourdet DL, Daniels OT, Ding YS, Gallezot JD, Henry S, et al. Preclinical to clinical translation of CNS transporter occupancy of TD-9855, a novel norepinephrine and serotonin reuptake inhibitor. Int J Neuropsychopharmacol. 2015;18(2):pyu027. doi: 10.1093/ijnp/pyu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegde S, Pulido-Rios M, McNamara A, Smith J, Smith W, Kanodia J, et al. Preclinical cardiovascular sympathoexcitatory effects of TD-9855, a novel norepinephrine transporter (NET) inhibitor in development for the treatment of symptomatic neurogenic orthostatic hypotension (nOH) in subjects with primary autonomic failure [abstract] Mov Disord. 2018;33(Suppl. 2):S748. [Google Scholar]
- 3.Kaufmann H, Biaggioni I, Chatamra K, Panneerselvam P, Haumann B, Vickery R. A Phase 2 study of the efficacy, durability, and safety of ampreloxetine (TD-9855), a norepinephrine reuptake inhibitor, given once-daily to treat neurogenic orthostatic hypotension (nOH) in subjects with synucleinopathies. Poster presented at the International Parkinson and Movement Disorder Society; 22–26 September 2019; Nice.
- 4.Metzler M, Duerr S, Granata R, Krismer F, Robertson D, Wenning GK. Neurogenic orthostatic hypotension: pathophysiology, evaluation, and management. J Neurol. 2013;260(9):2212–2219. doi: 10.1007/s00415-012-6736-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Florinef (fludrocortisone acetate) [package insert]. Bristol: Monarch Pharmaceuticals, Inc,; 2003.
- 6.ProAmatine (midodrine) [package insert]. Lexington: Shire US Inc., 2017.
- 7.Northera (droxidopa) [package insert]. Deerfield: Lundbeck NA Ltd., 2017.
- 8.Eschlböck S, Wenning G, Fanciulli A. Evidence-based treatment of neurogenic orthostatic hypotension and related symptoms. J Neural Transm (Vienna) 2017;124(12):1567–1605. doi: 10.1007/s00702-017-1791-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palma JA, Kaufman H. Epidemiology, diagnosis, and management of neurogenic orthostatic hypotension. Mov Disord Clin Pract. 2017;4(3):298–308. doi: 10.1002/mdc3.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufmann H, Freeman R, Biaggioni I, Low P, Pedder S, Hewitt LA, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–335. doi: 10.1212/WNL.0000000000000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rouse G. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson's disease (nOH306B) Mov Disord. 2015;30(5):646–654. doi: 10.1002/mds.26086. [DOI] [PubMed] [Google Scholar]
- 12.Biaggioni I, Freeman R, Mathias CP, Low P, Hewitt LA, Kaufmann H, et al. Randomized withdrawal study of subjects with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension. 2015;65(1):101–107. doi: 10.1161/HYPERTENSIONAHA.114.04035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibao C, Raj SR, Gamoa A, Diedrich A, Choi L, Black BK, et al. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension. 2007;50(1):47–53. doi: 10.1161/HYPERTENSIONAHA.107.089961. [DOI] [PubMed] [Google Scholar]
- 14.Strattera (atomoxetine hydrochloride) [package insert]. Indianapolis: Eli Lilly and Company; 2017.
- 15.Yeola S, Bolleddula J, Brassil P, Worboys P, Bourdet D. Absorption, metabolism, and excretion of [14C]TD-9855 after single oral administration to healthy human subjects. Poster presented at the American Association of Pharmaceutical Scientists; 13–17 November 2016; Denver (CO).
- 16.World Medical Association . Declaration of Helsinki: recommendations guiding doctors in clinical research. Ferney-Voltaire: World Medical Association; 1964. [Google Scholar]
- 17.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Assessing the effect of food and drugs in INDs and NDAs: clinical pharamacology considerations. Guidance for industry. February 2019. Available from: https://www.fda.gov/media/121313/download. Accessed 15 June 2020.
- 18.Adler L, Spencer T. The adult ADHD Clinical Diagnostic Scale (ACDS) New York: New York University School of Medicine; 2004. [Google Scholar]
- 19.Crews KR, Gaedigk A, Dunnenberger HM, Leeder JS, Klein TE, Caudle KE, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther. 2014;95(4):376–382. doi: 10.1038/clpt.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia: report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160–172. doi: 10.1002/art.1780330203. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava K, Arora A, Kataria A, Cappelleri JC, Sadosky A, Peterson AM. Impact of reducing dosing frequency on adherence to oral therapies: a literature review and meta-analysis. Patient Prefer Adher. 2013;7:419–434. doi: 10.2147/PPA.S44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why, how, when? Basic Clin Pharmacol Toxicol. 2005;97(3):125–134. doi: 10.1111/j.1742-7843.2005.pto_973160.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.