

Graphical abstract
Keywords: Noninvasive prenatal diagnosis, Variant calling, NIPT, NIPD, cell-free DNA, cfDNA
Abstract
The technology of noninvasive prenatal testing (NIPT) enables risk-free detection of genetic conditions in the fetus, by analysis of cell-free DNA (cfDNA) in maternal blood. For chromosomal abnormalities, NIPT often effectively replaces invasive tests (e.g. amniocentesis), although it is considered as screening rather than diagnostics. Most recently, the NIPT has been applied to genome-wide, comprehensive genotyping of the fetus using cfDNA, i.e. identifying all its genetic variants and mutations. Previously, we suggested that NIPD should be treated as a special case of variant calling, and presented Hoobari, the first software tool for noninvasive fetal variant calling. Using a unique pipeline, we were able to comprehensively decipher the inheritance of SNPs and indels. A few caveats still exist in this pipeline. Performance was lower for indels and biparental loci (i.e. where both parents carry the same mutation), and performance was not uniform across the genome. Here we utilized standardized methods for benchmarking of variant calling pipelines and applied them to noninvasive fetal variant calling. By using the best performing pipeline and by focusing on coding regions, we showed that noninvasive fetal genotyping greatly improves performance, particularly in indels and biparental loci. These results emphasize the importance of using widely accepted concepts to describe the challenge of genome-wide NIPT of point mutations; and demonstrate a benchmarking process for the first time in this field. This study brings genome-wide and complete NIPD closer to the clinic; while potentially alleviating uncertainty and anxiety during pregnancy, and promoting informed choices among families and physicians.
1. Introduction
Since the discovery of fetal DNA in maternal plasma [1], interest in noninvasive prenatal testing (NIPT) of genetic disorders in the fetus has been steadily growing. Such cell-free DNA (cfDNA) is a mixture of both maternal and fetal DNA; both the amount of cfDNA and the fraction of fetal DNA within it increases throughout pregnancy. The most prominent success of NIPT is the screening of chromosomal abnormalities, especially Down syndrome, but also trisomies 13 and 18, and sex chromosome abnormalities [2], [3], [4]. Due to this success, the current reason that many women undergo an invasive procedure, e.g. amniocentesis or chorionic villus sampling (CVS) is to test for large sub-chromosomal deletions and duplications. Therefore, these types of genetic abnormalities are becoming available through NIPT as well [5], [6], [7], [8]. In the last few years, NIPT has also become available for monogenic disorders caused by point mutations. Initially, bespoke tests were suggested for up to one mutation or one gene simultaneously [9]. Nowadays, commercially available NGS panels consist of up to 30 genes [10]. However, false negative results in tailored tests and panels [11], together with the growing interest in prenatal whole exome/genome sequencing (WES/WGS) from amniocentesis and CVS [12], [13], [14], [15], [16], [17], [18], have prompted a demand for noninvasive prenatal WES/WGS.
In several studies, genome-wide noninvasive sequencing of the cfDNA in maternal plasma was shown to reveal the entire fetal genome [19], [20], [21]. In these studies, fetal positions where only the father was heterozygous were genotyped in a straightforward manner, based on the presence or absence of the paternal alternate allele in the plasma. However, for maternal-only heterozygous positions, these methods required the maternal haplotype information, since both maternal alleles are present in the plasma. Genome-wide haplotyping relies on costly technologies that require expertise and that are less available; their resolution is lower than in site-by-site methods. Moreover, some regions cannot be phased due to low density of markers; and recombination events near mutations can yield incorrect genotype classifications [22], [23], [24], [25]. Other attempts that did not require parental haplotype information have also been performed [20], [24]. However, these approaches were not applicable to positions in which both parents are heterozygous (biparental loci). Such loci pose a greater algorithmic challenge, since the fetus can be homozygous to either allele, or heterozygous. Algorithmic challenges have also precluded the inclusion of indels in these attempts. Moreover, some unique characteristics of the fetal cfDNA have not been utilized, although they might improve the genotyping process.
We recently suggested a different approach for genome-wide NIPT of monogenic disorders [26]. We defined this issue as a unique case of variant calling, termed noninvasive prenatal variant calling. We subsequently followed the well-established principles of standard variant calling. Accordingly, a Bayesian genotyping algorithm utilizes the information of each read covering each candidate variant, and a machine learning-based fine-tuning step subsequently incorporates information from previously verified results. By accounting for each read, we were able to utilize characteristics that separate fetal and maternal DNA, such as fragment length. Our algorithm was implemented as Hoobari, the first noninvasive fetal variant caller, which was able to genotype all fetal positions, including biparental loci and indels. These results were achieved in first trimester pregnancies, for the first time. However, performance in biparental loci and indels was lower than in positions in which only one parent is heterozygous.
Standard variant calling pipelines have been available for about one decade, and they have undergone continuous improvement during this time. Such pipelines have enabled us and other teams to detect countless deleterious mutations [27], [28], [29], [30], [31]. A number of studies have compared pipelines according to sequencing technologies, alignment software, post-alignment processing, and variant calling software [32], [33], [34]. Several teams of experts have published best practice guidelines for variant calling pipelines. The Global Alliance for Genomics and Health (GA4GH) Benchmarking Team publishes and updates best practice guidelines for benchmarking of variant calling pipelines [35]. The Genome Analysis Toolkit team publishes its own best practices [36]. Other attempts have been performed as well [37]. In noninvasive fetal variant calling, however, guidelines have not been established for benchmarking. New methods often suggest an end-to-end solution that relies on various samples, sets of variants, and statistical methods; and a pipeline that is based on a specific set of off-the-shelf software. Thus, comparing methods and pipelines is effectively impossible, and it is unclear whether achievements should be attributed to the methods or to other factors.
Here we suggest initial guidelines for benchmarking of noninvasive prenatal variant calling and demonstrate their use. We tested Hoobari’s pipeline with various alignment software, post-alignment processing methods, and variant callers. We also re-implemented large parts of Hoobari’s code to enable its compatibility with more software tools. The results showed great improvement in Hoobari’s performance, demonstrating that NIPD can benefit from a standardized benchmarking process. Moreover, the most prominent improvement was achieved in biparental loci and in indels. When assessing only the coding regions, rather than the whole genome, the results achieved in these loci demonstrated the feasibility of performing a genome-wide NIPT of monogenic diseases. Thus, our results bring this test closer to the clinic, while promoting informed choices among physicians and families, and potentially alleviating uncertainty and anxiety during pregnancy.
2. Results
2.1. Datasets and pipelines
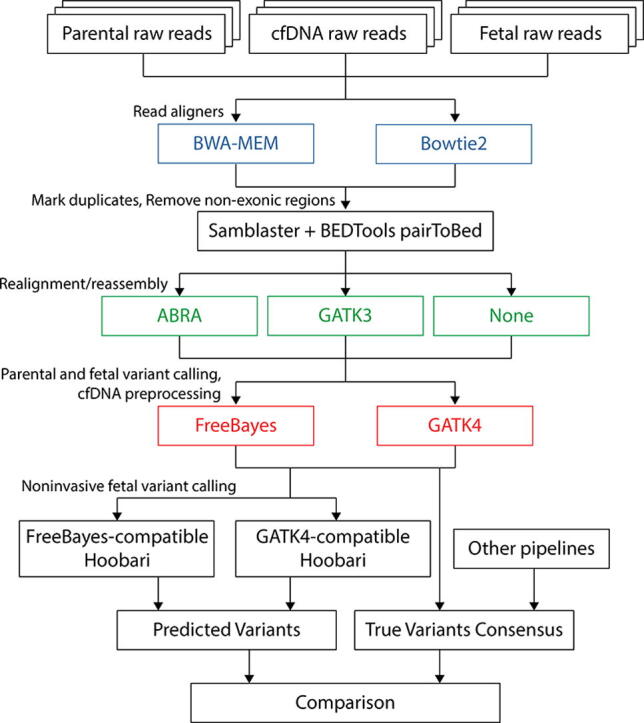
In this analysis, twelve pipelines were assembled and compared (Fig. 1). Sequence reads were aligned to the reference human genome (GRCh38/hg38), using either BWA-MEM [38] or Bowtie2 [39]. We examined only the coding regions of the genome, since outliers and variants of unknown significance are found in the non-coding areas, and since the coding regions contain most of the clinically relevant variants. Next we marked duplicate reads; we performed this step uniformly through Samblaster [40], in all pipelines. We tested post-alignment processing, such as indel realignment and reassembly, against avoiding such processing. One approach included realignment using the former Genome Analysis Toolkit version, i.e. GATK3 [41], through RealignerTargetCreator, IndelRealigner, and BaseRecalibrator. The second approach included reassembly-based realignment, using Assembly Based ReAligner (ABRA) [42]. Variant calling of the parents and the fetal sample, and pre-processing of the cfDNA reads, were performed using either FreeBayes or GATK4. Eventually, Hoobari was run on the pre-processed data.
Fig. 1.

Experimental workflow for comparing fetal genotyping pipelines. This is the workflow for comparing pipelines for noninvasive fetal variant callers. Twelve pipelines were tested, based on possible combinations of two read aligners, three post-alignment approaches, and two variant calling programs. Each pipeline begins with raw sequence FASTQ files of the parents, the true fetal sample, and cfDNA. Alignment is performed using either BWA-MEM or Bowtie2 (Blue). Duplicate reads are removed using Samblaster. Non-exonic regions are sliced out using BEDTools pairToBed, by keeping only read pairs that have at least one read covering an exonic region. ABRA and GATK3 IndelRealign are compared against avoidance of any realignment or reassembly (Green). FreeBayes and GATK4 are used for variant calling of the parents and the true fetal samples, and for preprocessing of the cfDNA sample (Red). Eventually, FreeBayes- or GATK4-comparitble Hoobari is run to call fetal variants using the cfDNA reads and the parental genotypes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Benchmarking requires an agreed and verified true-set to serve as a reference. To this end, we used family G1, which was sequenced in a previous study [24]. This family has preferable technical and biological settings; its cfDNA sample contains 30% fetal-derived DNA and was sequenced using PCR-free WGS to a depth of 300×. This ensures that our results are attributed to computational differences, rather than to the quality of the data, and thus enables isolation of variables. Although we suggest family G1 as the benchmark dataset for noninvasive fetal variant calling, this dataset is not as verified as the benchmark dataset of standard variant calling (see Discussion). Hence, we demonstrated that the same results were achieved for two additional families: family G2, from the same study as family G1 [24]; and family G3, which we sequenced in our previous study [26], where it was named family G5 (see Materials and Methods). Information about the three families is summarized in Table 1.
Table 1.
A summary of the samples used in this study.
| Family | Individual | Sample | Depth of coverage1 | Fetal fraction |
|---|---|---|---|---|
| G1 | Mother | White blood cells | 40 | |
| Plasma (38 weeks2) | 270 | 30.2% | ||
| Father | White blood cells | 45 | ||
| Offspring | Umbilical cord blood | 50 | ||
| G2 | Mother | White blood cells | 40 | |
| Plasma (18 weeks) | 195 | 23.2% | ||
| Father | White blood cells | 60 | ||
| Offspring | Placental tissues | 60 | ||
| G3 | Mother | White blood cells | 38 | |
| Plasma (11 weeks) | 310 | 18.5% | ||
| Father | White blood cells | 41 | ||
| Offspring | Chorionic villus sampling | 38 | ||
Median, on target; 2 Gestational age.
2.2. Comparing pipelines in exonic SNPs
When using the F1-score as the main metric, we noticed several phenomena. First, no single pipeline was optimal across all families (Table 2; Supplemental Tables S3 and S4). However, some gross-scale phenomena were apparent. First, aligning using BWA-MEM resulted in better F1-scores, both on average and when each BWA-MEM-based pipeline was compared with its Bowtie2-based analogue. This finding is very important, since alignment of deep WGS data from the cfDNA is computationally intensive, and should therefore not be performed by more than one tool (e.g. for different types of mutations). Second, inconclusive results and only subtle differences were found when the same aligner and variant caller were used, i.e. the realignment or reassembly steps did not have a major effect. Moreover, the post-alignment effect was inconsistent across families; in each family, a different realignment tool provided better results. Finally, FreeBayes provided slightly better results than GATK4 for SNPs, but the differences were minute, and the F1-scores were very high in all pipelines across all families, in all forms of inheritance.
Table 2.
Comparison of noninvasive fetal variant calling pipelines in family G1 – SNPs.
| Pipeline | F1_Score | Recall | Precision | TOTAL | TP | FN | FP | FP.gt | FP.al |
|---|---|---|---|---|---|---|---|---|---|
| bwa-none-freebayes | 0.991090 | 0.986208 | 0.996020 | 22,332 | 22,024 | 308 | 88 | 68 | 0 |
| bwa-abra-freebayes | 0.990910 | 0.986117 | 0.995749 | 22,330 | 22,020 | 310 | 94 | 75 | 0 |
| bwa-gatk-freebayes | 0.991157 | 0.986208 | 0.996155 | 22,332 | 22,024 | 308 | 85 | 67 | 0 |
| bwa-none-gatk | 0.988222 | 0.981314 | 0.995228 | 22,316 | 21,899 | 417 | 105 | 71 | 0 |
| bwa-abra-gatk | 0.988245 | 0.981447 | 0.995138 | 22,315 | 21,901 | 414 | 107 | 72 | 0 |
| bwa-gatk-gatk | 0.988063 | 0.981397 | 0.994820 | 22,308 | 21,893 | 415 | 114 | 72 | 0 |
| bowtie-none-freebayes | 0.943192 | 0.898413 | 0.992669 | 22,306 | 20,040 | 2266 | 148 | 105 | 0 |
| bowtie-abra-freebayes | 0.989173 | 0.985115 | 0.993265 | 22,305 | 21,973 | 332 | 149 | 106 | 0 |
| bowtie-gatk-freebayes | 0.942454 | 0.897206 | 0.992508 | 22,297 | 20,005 | 2292 | 151 | 101 | 0 |
| bowtie-none-gatk | 0.974657 | 0.956364 | 0.993663 | 22,298 | 21,325 | 973 | 136 | 88 | 0 |
| bowtie-abra-gatk | 0.974633 | 0.956319 | 0.993663 | 22,298 | 21,324 | 974 | 136 | 88 | 0 |
| bowtie-gatk-gatk | 0.974679 | 0.956407 | 0.993663 | 22,297 | 21,325 | 972 | 136 | 87 | 0 |
The results attained for coding regions demonstrated very high precision, >0.996, and recall of >0.986 in family G1 (Table 2). Both the recall and precision metrics, and the absolute number of false positives (FPs) and false negatives (FNs), suggest that the algorithm tends to overlook existing variants more than it calls non-existing variants. In a clinical setting, FNs are more problematic than FPs, since FNs can result in the birth of an offspring with a severe genetic condition. FPs, on the other hand, are validated by an invasive procedure, so in the worst-case scenario, an unnecessary procedure is performed.
2.3. Performance in different inheritance modes
As shown throughout this study, the performance of the noninvasive fetal variant calling algorithm depends on the parental genotypes. Paternal-only heterozygous loci are the easiest to predict, followed by maternal-only heterozygous loci. The most challenging positions are biparental-heterozygous loci, in which both parents are heterozygous. We stratified the pipelines we compared according to these three categories of positions. Here as well, paternal loci consistently received the highest F1-score, followed by maternal and then biparental loci. These patterns were consistent in all pipelines and families (Fig. 2, A; Supplemental Fig. S1, A; Supplemental Fig. S2, A). No single pipeline was shown to have the highest F1-score over all the categories. However, BWA-MEM was always preferred, and the question as to whether to apply a realignment/reassembly method or not is still inconclusive (Supplemental Table S1). A key finding is that when testing over the exome (as performed here), and by using BWA-MEM followed by post-alignment steps, and by GATK4 for variant calling, genotyping of biparental loci becomes highly accurate. The recall value was >0.981 and the precision value was 0.989 in family G1. This result was achieved even without using the ML-based variant recalibration process that was described in our previous study [26].
Fig. 2.

Comparison of fetal genotyping pipelines stratified by inheritance in family G1.
2.4. Comparing performance over indels
We compared the execution of twelve pipelines in indel positions (Table 3). Our initial analysis of Hoobari used BWA-MEM as the aligner, with no realignment, and with FreeBayes as the variant calling software. As such, genotyping of indels was found to be more challenging and less accurate than SNP genotyping. Accordingly, the bwa-none-freebayes pipeline showed the lowest performance (Table 3; Fig. 2, B). Unlike the SNP results, the F1-score achieved in indels was notably higher when GATK4 was used for parental variant calling and cfDNA pre-processing. This is a key finding in our study, which, for the first time, brought the accuracy of noninvasive fetal indel detection to levels that are similar to those of SNP detection.
Table 3.
Comparison of noninvasive fetal variant calling pipelines in family G1 – Indels.
| Pipeline | F1_Score | Recall | Precision | TOTAL | TP | FN | FP | FP.gt | FP.al |
|---|---|---|---|---|---|---|---|---|---|
| bwa-none-freebayes | 0.722148 | 0.656098 | 0.802985 | 410 | 269 | 141 | 66 | 62 | 0 |
| bwa-abra-freebayes | 0.843710 | 0.805825 | 0.885333 | 412 | 332 | 80 | 43 | 41 | 0 |
| bwa-gatk-freebayes | 0.749669 | 0.695332 | 0.813218 | 407 | 283 | 124 | 65 | 58 | 0 |
| bwa-none-gatk | 0.961586 | 0.934940 | 0.989796 | 415 | 388 | 27 | 4 | 4 | 0 |
| bwa-abra-gatk | 0.964064 | 0.937349 | 0.992347 | 415 | 389 | 26 | 3 | 3 | 0 |
| bwa-gatk-gatk | 0.962779 | 0.934940 | 0.992327 | 415 | 388 | 27 | 3 | 3 | 0 |
| bowtie-none-freebayes | 0.736698 | 0.658537 | 0.835913 | 410 | 270 | 140 | 53 | 49 | 0 |
| bowtie-abra-freebayes | 0.864385 | 0.827670 | 0.904509 | 412 | 341 | 71 | 36 | 34 | 0 |
| bowtie-gatk-freebayes | 0.805851 | 0.737226 | 0.888563 | 411 | 303 | 108 | 38 | 35 | 0 |
| bowtie-none-gatk | 0.940881 | 0.901205 | 0.984211 | 415 | 374 | 41 | 6 | 6 | 0 |
| bowtie-abra-gatk | 0.940881 | 0.901205 | 0.984211 | 415 | 374 | 41 | 6 | 6 | 0 |
| bowtie-gatk-gatk | 0.944862 | 0.908434 | 0.984334 | 415 | 377 | 38 | 6 | 6 | 0 |
Similar to the SNP results, the pipelines that included BWA-MEM for alignment rather than Bowtie2 reached higher F1-scores. Indel genotyping was also shown to improve following additional steps of careful reassembly and realignment. First, the three BWA-MEM- and GATK4-based pipelines had similar results overall, but a slight improvement was achieved by using ABRA for reassembly or GATK for realignment. Second, the use of ABRA before FreeBayes increased the F1-scores substantially. Indel results were consistent also in families G2 and G3 (Supplemental Tables S3 and S4). Notably, recall for indels was lower than for SNPs, thus indicating a higher relative number of FNs.
When indel results were stratified based on parental inheritance, GATK4 still resulted in a notable effect on the F1-score (Fig. 2, B; Supplemental Table S2). This was also consistent in families G2 and G3 (Supplemental Fig. S1, B; Supplemental Fig. S2, B). Choosing different approaches for realignment and reassembly did not achieve conclusive results. BWA-MEM usually showed better results than Bowtie2, for all three categories.
2.5. Advanced quality control measurements
Two additional quality control measurements that are often used in genetic studies are the transition/transversion (Ti/Tv) ratio and the heterozygous/nonreference-homozygous (het/hom) ratio [43]. The Ti/Tv ratio is expected to be approximately 2 across the genome, and 2.8–3 in exonic regions [43]. In the fetal samples of family G1, Ti/Tv ratios initially reached values of 2.4, suggesting contamination of non-exonic regions. After filtering out variants that occurred in the 100 bp padding regions of the exonic coordinates, Ti/Tv ratios reached values of ~3 (Fig. 3, A). More interesting are the differences between the examined pipelines in this ratio; differences were smaller for FreeBayes- and BWA-MEM-based pipelines, in accordance with their better F1-scores. The expected het/hom ratio for family G1 is ~1.3, based on ethnicity. This is not expected to vary within genomic regions [43]. Again, of greater interest are the values in the fetal sample compared across pipelines (Fig. 3, B). Here, when using BWA-MEM, smaller differences were shown for FreeBayes-based pipelines; this suggests that the GATK4-based pipelines tend to call heterozygosity and miss nonreference-heterozygosity too often. This analysis was only demonstrated using family G1.
Fig. 3.

Comparison of fetal genotyping pipelines – transitions/transversions and heterozygous/alternate-homozygous ratios in family G1.
3. Discussion
In our previous study, we developed and demonstrated a novel method for genome-wide NIPD of monogenic diseases. When we attempted to compare it to existing methods, we noticed that the field of NIPD is limited by the lack of standardization, compared to other common analyses in bioinformatics. We noted that novel methods often suggest a fixed solution that relies on specific samples, sets of variants, statistical methods, etc. For these reasons, we suggested that genome-wide NIPD of monogenic diseases should be considered a special case of variant calling. This enabled our following the widely accepted and studied concepts of variant calling, to make NIPD more accessible.
The goal of our current study was to demonstrate the great benefits to NIPD achieved from the application of a standardized benchmark of variant calling. To this end, we propose that family G1, which was sequenced in a previous study, is the optimal candidate for a benchmark dataset. We showed how guidelines and software tools that were originally designed for standard variant calling can be used in the context of NIPD. Hopefully, this study will enable researchers in the field of NIPD to speak the same language, better demonstrate the capabilities of their methods, and advance the field towards the clinic.
As demonstration of the benchmarking process, we aimed to identify a preferred pipeline for noninvasive prenatal variant calling. The issue is complex and depends on the specific analysis carried out. Consistent results were achieved in several conditions, such as choosing the preferred aligner and variant caller. Effects of post-alignment steps such as reassembly, realignment, and base quality recalibration were usually negligible. Other key results and conclusions are the great improvement over biparental loci, and over indels, when post-alignment steps and GATK4 are used. Such overarching ideas and directions can help bioinformaticians in future NIPT analyses.
The first key results of this study relate to the preferred aligner. Alignment is the most computationally expensive step of the analysis, and thus entails the least flexibility. Other steps, such as variant calling, are known to be faster than the alignment step. Thus, different tools can be run for different types of loci. In other words, the consensus of several variant callers can be used. However, since performing the same activity with different aligners might be too complex, defining one preferable aligner is important. Two popular aligners were compared in this study. As seen, BWA-MEM was superior and is preferred over Bowtie2. Other aligners are available that we did not test here, some were too slow for the extremely deep cfDNA sample. However, when a stronger architecture will be achieved, they may have an advantage in accuracy.
An important question that arises in the context of noninvasive fetal variant calling is whether it can be improved using realignment and reassembly. In both SNPs and indels, when realignment and reassembly were not used, the F1-score was usually lower, unless GATK4 was the variant caller. As explained, this happened because reassembly of candidate variants was embedded within the GATK4 algorithm. For this reason, the use of RealignerTargetCreator, IndelRealigner, and BaseRecalibrator before applying GATK4 was usually redundant and not beneficial. The use of ABRA, however, did improve the results in some cases in which GATK4 was applied, even though both methods implemented similar algorithms. This should be further explored with more data. The most prominent advantages of the realignment and reassembly methods were apparent in indel calling, effectively bringing the F1-score near the range of the F1-score achieved in SNP calling. Here as well, GATK4 presented a central advantage.
When noninvasive fetal variant calling was first introduced, cfDNA preprocessing was performed using FreeBayes, as read-level information could not be obtained from GATK. Recent versions of GATK included accessibility to this information, and the great advantage of such is evident. Although FreeBayes provided slightly better results for SNPs, GATK4 was prominently better for indels, as mentioned. Even for SNPs, the results were close to those of the FreeBayes-based pipelines, indicating that the GATK4 pipeline can be used as a single solution. Eventually, since no single pipeline presented superiority in both SNPs and indels, and in all three inheritance categories, different pipelines could be used in different settings. For example, FreeBayes can be used for SNPs and GATK4 for indels, as these steps are not computationally intensive. Other variant callers, e.g. Google’s DeepVariant, can be tested here as well. However, since this variant caller shows similar results to those of GATK4, we do not expect it would yield significant improvement.
The last step in the pipeline is noninvasive fetal variant calling. Although several algorithms are available for NIPT of monogenic diseases, none of them is a variant calling algorithm, and none was implemented as a variant calling software tool. Each algorithm requires distinct implementation for each pipeline. Since Hoobari is currently the only noninvasive fetal variant caller, it was the only one that was included in this comparison. We welcome other researchers to release new and improved noninvasive fetal variant callers that can be used for comparison.
Our study has several limitations. First, although family G1 is our benchmark dataset, it is not as verified as the benchmark dataset of standard variant calling. Therefore, we wanted to demonstrate our results on more families, but we could reach only a total of three families that were similarly sequenced, i.e. the cfDNA was sequenced using very deep WGS. Second, we compared only the computational pipeline, though various biological methods might also affect the results. For instance, families G1-2 were sequenced by the same team, and family G3 was sequenced by another. Moreover, the team that sequenced G1-2 is one of the most experienced teams in the field, and their methods are presumably well calibrated. Read lengths differed between the families, and were larger in family G3. Third, our focus on coding regions could introduce bias to our comparison, for instance if a certain pipeline can deal better with challenging positions in non-coding regions. Finally, we demonstrated that different aligners lead to different results in the context of noninvasive fetal genotyping. Small differences in the percent of aligned reads become important in noninvasive fetal variant calling, which relies heavily on subtle differences in the quantification of reads supporting each allele. Unfortunately, a wider comparison of aligners could not be performed in the scope of this study. Thus, we compared the most popular aligners for genomes. Even when only two aligners were chosen, this was shown to be an important factor.
An important limitation of any comparison of pipelines in the field of NIPT is the lack of a benchmark sample. When comparing standard variant calling pipelines, the benchmark is typically the NA12878 sample, a publicly available genome sequenced in the 1000 Genome Project of the Genome in a Bottle (GIAB) Consortium [32], [44]. The variant calls for this individual were previously validated by sequencing, mapping, and genotyping its genome using various NGS technologies, read aligners, and variant callers [44]. This resulted in several variant datasets, which were manually integrated to filter discordant variants. To add confidence to this database, pedigree information was also used, together with calls from other projects, such as previous versions of GIAB, and the Illumina Platinum Project. Such a validated dataset does not exist for noninvasive variant calling, thus limiting the ability to perform a reliable benchmarking of NIPT methods. The use of methods similar to the aforementioned NA12878 sample to create a benchmark dataset requires a distinct collaboration and was impractical within the scope of this study. Notably, although a consensus VCF file was eventually used in our comparison, the total size of the true-set still varied by several variants between the various pipelines. This might be related to candidate variants that are homozygous to the reference allele and that are therefore excluded by hap.py. The possible effect of such could be mitigated in a comparison that would include more families, thus reducing the variance. Alternatively, this matter should be further explored.
In conclusion, our study presents an example of a suggested benchmarking process of noninvasive fetal variant calling. It illuminates the advantages of such standardization, as well as various considerations and limitations. Our comparison of pipelines for noninvasive fetal variant calling identified BWA-MEM as the preferred aligner for this task. Realignment and reassembly can be introduced using ABRA or GATK4; these steps might improve the genotyping of both biparental and indel positions. GATK4 was shown to enable highly accurate indel prediction, but to perform similarly to FreeBayes with ABRA in SNPs. In choosing the best pipelines, while focusing solely on the coding regions, we showed a major improvement compared with the original noninvasive fetal variant calling, which was FreeBayes-based and did not include post-alignment steps.
This study highlighted the advantages of considering NIPT of monogenic diseases as a unique case of standard variant calling. This enables the use of protocols, standards, tools, and other knowledge that were acquired over the course of 10–15 years of standard variant calling in this exciting new field, as well as in adjacent fields related to cfDNA.
4. Materials and methods
Our comparison of noninvasive fetal variant calling pipelines was similar to systematic comparisons of standard variant calling pipelines. However, some biological, technical, and computational considerations required adjustments to the unique case of fetal genotyping from the mixed fetal-maternal cfDNA samples.
4.1. Datasets
Families G1 and G2 were sequenced as part of a previous NIPT study [45]. Family G3 corresponds to family G5 in our previous study and was sequenced as described there [26]. Prior to the comparison, the VCF files were filtered such that the depth of the fetal sample and each parent was > 10, and the QUAL was > 20; the depth of the cfDNA positions was in the range of 100–1000.
4.2. Software used throughout each pipeline
Raw sequence FASTQ files of each sample were mapped to the human genome using BWA-MEM v0.7.8-r455 [38] or Bowtie2 v2.3.4.3 [39], with default parameters. Aligned reads were streamed directly from the alignment tool through the BEDTools v2.18.1′s pairToBed [46] option, which maintains only reads that overlap the given coordinates, or that their mates overlap them. Keeping both mates of each pair enabled accurately marking duplicate reads. Agilent SureSelect Exome V7 was used as the genomic coordinates of the coding regions, extended by 100 bp padding on either side. Coding region coordinates were downloaded as a browser extensible data (BED) file from Agilent’s website. Samblaster v0.1.24 [40] was used for marking duplicates. GATK v3.1-1-g07a4bf8 was used for realignment and base recalibration according to the recommendations of GATK Best Practices [36], [47]. ABRA v0.97 was used for reassembly. FreeBayes v1.1.0-3-g961e5f3 and GATK v4.0.11.0 were used for variant calling of the parents and the fetus, and for pre-processing of reads for Hoobari. All VCF files were filtered again using BEDTools intersect tools, this time without the 100 bp padding, to reassure that only variants within the exonic regions would be included in the downstream analysis.
4.3. The gold standard genotyped data
To evaluate the performance of each pipeline, a gold standard set of verified variant calls is required. Two strategies to assemble a true set were explored. The first included comparing between each noninvasive fetal pipeline and its invasive equivalent, which consists of the same aligner, post-alignment methods, and variant caller. In the second strategy, an attempt was made to create a validated dataset of variants. Several approaches are possible for creating such a dataset. One option is to use a majority vote, which can be executed in a naïve manner, without accounting for similarities between pipelines (which cause dependence); or alternatively, in a more sophisticated manner. A more straightforward but stringent option, which was used here, is to intersect the twelve invasively-achieved variant sets. Intersecting VCF files requires standardizing the representation of the variants they contain. Differences can occur due to various reasons, such as relating to the aligners, post-alignment methods, and variant callers. To standardize the VCF files, hap.py was first run over each pipeline’s VCF file and its corresponding true set VCF, i.e. the fetal sample. The output VCF files of the hap.py run contained normalized position representations; these files were then intersected to create the consensus true set. Eventually, when each pipeline’s VCF file was compared again to the corresponding fetal sample’s VCF file, the comparison was restricted to positions included in the consensus true set.
4.4. Performance measure of variant calling pipelines
Since genotyping results are not binary, defining positive and negative results is important, as is subsequently defining FP, FN, and true positive (TP) results; and the metrics to use. For genotyping, a FP can result from either a mismatch in the allele that was found by the variant caller, or a mismatch in the genotype (heterozygous or homozygous). These two challenges, as well as many others, are well described in the literature in the context of variant calling benchmarking. To address this issue, best practices were published by the GA4GH Benchmarking Team [35], after they convened representatives from sequencing technology developers, government agencies, academic bioinformatics researchers, clinical laboratories, and commercial technology and bioinformatics developers for whom benchmarking variant calls is essential to their work. The aforementioned comparison methods and conventions were implemented by the GA4GH Benchmarking Team into Illumina’s benchmarking tool, named hap.py, which was used in this study to compare pipelines.
Formerly, we assessed Hoobari’s performance against other noninvasive fetal genotyping algorithms, and also demonstrated the advantage of accounting for the cfDNA fragment lengths, compared with a version of Hoobari that ignores this information [26]. The metrics that were used are different from those used by hap.py. The former includes metrics that are similar to those used in previous attempts to perform genome-wide genotyping of a fetus. Accordingly, positive and negative results were defined by whether the fetus was homozygous or heterozygous, with regard to the parental genotypes at the tested position [20], [21]. The GA4GH Benchmarking Team stated that due to the inherent complexity of the human genome, TP, FP, and FN can be defined in different ways. In their best practices, which were implemented here, a genotype match is defined as a position where the same allele and the same genotype appear in both the placental (truth) and the cfDNA (query) VCF files, regardless of the parental genotype (Table 4). Positions are considered TP only if the genotype matches. If only an allele match exists, but not a genotype match, the position is considered as either FN or FP. FP is further classified as a genotype mismatch (FP.gt), in which the alleles match but the genotypes do not match, and an allele mismatch (FP.al), in which even the alleles do not match. Finally, the metrics used for the comparison are precision (aka positive predictive value = TP/(TP + FP)) and recall (aka sensitivity = TP/(TP + FN)), as well as F1-score, which is their harmonic mean.
Table 4.
Definitions for TPs, FPs, and FNs by the GA4GH Benchmarking Team.
| Truth (fetal sample) |
||||
|---|---|---|---|---|
| Genotype | homref1 | het2 | homalt3 | |
| Query (cfDNA) | homref | n/a | FN | FN |
| het | FP | TP | FP.GT | |
| homalt | FP | FP.GT | TP | |
homozygous to the reference allele; 2 heterozygous; 3homozygous to the alternate allele TPs: true positives; FP: false positives; FN: false positives.
4.5. Data access
Hoobari is accessible via GitHub (https://github.com/nshomron/hoobari). The sequencing data for family G3 was submitted to the database of Genotype and Phenotype (dbGaP) under accession number phs001659.v1.p1.
Funding
The Shomron Laboratory is supported by the Israel Science Foundation (ISF; 1852/16); Israeli Ministry of Defense, Office of Assistant Minister of Defense for Chemical, Biological, Radiological and Nuclear (CBRN) Defense; Foundation Fighting Blindness; The Edmond J. Safra Center for Bioinformatics at Tel Aviv University; Zimin Institute for Engineering Solutions Advancing Better Lives; Eric and Wendy Schmidt Breakthrough Innovative Research Award; Tel Aviv University Richard Eimert Research Fund on Solid Tumors; Djerassi-Elias Institute of Oncology; Canada-Montreal Friends of Tel Aviv University; Donations from Harold H. Marcus, Amy Friedkin, Natalio Garber, Tal Zohar; Kirschman Dvora Eleonora Fund for Parkinson's Disease; Joint funding between Tel Aviv University and Yonsei University; Israeli Ministry of Science and Technology, Israeli–Russia; Aufzien Family Center for the Prevention and Treatment of Parkinson’s Disease; and a generous donation from the Adelis Foundation.
CRediT authorship contribution statement
Tom Rabinowitz: Conceptualization, Data curation, Formal analysis, Investigation, Software, Visualization, Validation, Writing - original draft. Shira Deri-Rozov: Formal analysis, Validation. Noam Shomron: Conceptualization, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Samples G1-2 used in this paper were generated by The Chinese University of Hong Kong (CUHK) Circulating Nucleic Acids Research Group, as reported by Chan et al in Proc Natl Acad Sci USA (doi: 10.1073/pnas.1615800113).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.12.032.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lo Y.M.D., Corbetta N., Chamberlain P.F., Rai V., Sargent I.L., Redman C.WG., Wainscoat J.S. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350(9076):485–487. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann B., Hill M., Gemelos G., Demko Z., Banjevic M., Baner J., Ryan A., Sigurjonsson S., Chopra N., Dodd M., Levy B., Rabinowitz M. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci: Noninvasive prenatal aneuploidy testing at chromosomes 13, 18, 21, X, and Y. Prenat Diagn. 2012;32(13):1233–1241. doi: 10.1002/pd.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan H.C., Blumenfeld Y.J., Chitkara U., Hudgins L., Quake S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci. 2008;105(42):16266–16271. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo Y.M.D., Lun F.M.F., Chan K.C.A., Tsui N.B.Y., Chong K.C., Lau T.K., Leung T.Y., Zee B.C.Y., Cantor C.R., Chiu R.W.K. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc Natl Acad Sci. 2007;104(32):13116–13121. doi: 10.1073/pnas.0705765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen TJ, Dzakula Z, Deciu C, van den Boom D, Ehrich M. Detection of microdeletion 22q11.2 in a fetus by next-generation sequencing of maternal plasma. Clin Chem. 2012 Jul;58(7):1148–51. [DOI] [PubMed]
- 6.Peters D., Chu T., Yatsenko S.A., Hendrix N., Hogge W.A., Surti U., Bunce K., Dunkel M., Shaw P., Rajkovic A. Noninvasive prenatal diagnosis of a fetal microdeletion syndrome. N Engl J Med. 2011;365(19):1847–1848. doi: 10.1056/NEJMc1106975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srinivasan A., Bianchi D., Huang H., Sehnert A., Rava R. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. The American Journal of Human Genetics. 2013;92(2):167–176. doi: 10.1016/j.ajhg.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neofytou MC, Tsangaras K, Kypri E, Loizides C, Ioannides M, Achilleos A, et al. Targeted capture enrichment assay for non-invasive prenatal testing of large and small size sub-chromosomal deletions and duplications. PLOS ONE. 2017 Feb 3;12(2):e0171319. [DOI] [PMC free article] [PubMed]
- 9.Lun F.M.F., Tsui N.B.Y., Chan K.C.A., Leung T.Y., Lau T.K., Charoenkwan P., Chow K.C.K., Lo W.Y.W., Wanapirak C., Sanguansermsri T., Cantor C.R., Chiu R.W.K., Lo Y.M.D. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci. 2008;105(50):19920–19925. doi: 10.1073/pnas.0810373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J., Li J., Saucier J.B., Feng Y., Jiang Y., Sinson J. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med. 2019;25(3):439. doi: 10.1038/s41591-018-0334-x. [DOI] [PubMed] [Google Scholar]
- 11.Scotchman E, Chandler NJ, Mellis R, Chitty LS. Noninvasive Prenatal Diagnosis of Single-Gene Diseases: The Next Frontier. Clin Chem. 2020 Jan 1;66(1):53–60. [DOI] [PubMed]
- 12.Best S., Wou K., Vora N., Van der Veyver I.B., Wapner R., Chitty L.S. Promises, pitfalls and practicalities of prenatal whole exome sequencing: Promises and pitfalls of prenatal whole exome sequencing. Prenat Diagn. 2018;38(1):10–19. doi: 10.1002/pd.5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drury S., Williams H., Trump N., Boustred C., Lench N., Scott R.H. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn. 2015;35(10):1010–1017. doi: 10.1002/pd.4675. [DOI] [PubMed] [Google Scholar]
- 14.Lord J., McMullan D.J., Eberhardt R.Y., Rinck G., Hamilton S.J., Quinlan-Jones E. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019 Feb 23;393(10173):747–757. doi: 10.1016/S0140-6736(18)31940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackie FL, Carss KJ, Hillman SC, Hurles ME, Kilby MD. Exome Sequencing in Fetuses with Structural Malformations. J Clin Med. 2014 Jul 8;3(3):747–62. [DOI] [PMC free article] [PubMed]
- 16.Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017 Dec 1;171(12):e173438–e173438. [DOI] [PMC free article] [PubMed]
- 17.Vora N.L., Powell B., Brandt A., Strande N., Hardisty E., Gilmore K., Foreman A.K.M., Wilhelmsen K., Bizon C., Reilly J., Owen P., Powell C.M., Skinner D., Rini C., Lyerly A.D., Boggess K.A., Weck K., Berg J.S., Evans J.P. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med. 2017;19(11):1207–1216. doi: 10.1038/gim.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayward J., Chitty L.S. Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Seminars in Fetal and Neonatal Medicine. 2018;23(2):94–101. doi: 10.1016/j.siny.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Lo YMD, Chan KCA, Sun H, Chen EZ, Jiang P, Lun FMF, et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Science Translational Medicine. 2010 Dec 8;2(61):61ra91-61ra91. [DOI] [PubMed]
- 20.Fan H.C., Gu W., Wang J., Blumenfeld Y.J., El-Sayed Y.Y., Quake S.R. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487(7407):320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitzman JO, Snyder MW, Ventura M, Lewis AP, Qiu R, Simmons LE, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012 Jun 6;4(137):137ra76. [DOI] [PMC free article] [PubMed]
- 22.Hui WWI, Jiang P, Tong YK, Lee W-S, Cheng YKY, New MI, et al. Universal Haplotype-Based Noninvasive Prenatal Testing for Single Gene Diseases. Clin Chem. 2017 Feb;63(2):513–24. [DOI] [PubMed]
- 23.Wei X., Lv W., Tan H.u., Liang D., Wu L. Development and validation of a haplotype‐free technique for non‐invasive prenatal diagnosis of spinal muscular atrophy. J Clin Lab Anal. 2020;34(2) doi: 10.1002/jcla.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan K.C.A., Jiang P., Sun K., Cheng Y.K.Y., Tong Y.K., Cheng S.H., Wong A.I.C., Hudecova I., Leung T.Y., Chiu R.W.K., Lo Y.M.D. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci USA. 2016;113(50):E8159–E8168. doi: 10.1073/pnas.1615800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabinowitz T., Shomron N. Genome-wide noninvasive prenatal diagnosis of monogenic disorders: current and future trends. Comput Struct Biotechnol J. 2020;18:2463–2470. doi: 10.1016/j.csbj.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinowitz T., Polsky A., Golan D., Danilevsky A., Shapira G., Raff C., Basel-Salmon L., Matar R.T., Shomron N. Bayesian-based noninvasive prenatal diagnosis of single-gene disorders. Genome Res. 2019;29(3):428–438. doi: 10.1101/gr.235796.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malki L., Sarig O., Romano M.-T., Méchin M.-C., Peled A., Pavlovsky M., Warshauer E., Samuelov L., Uwakwe L., Briskin V., Mohamad J., Gat A., Isakov O., Rabinowitz T., Shomron N., Adir N., Simon M., McMichael A., Dlova N.C., Betz R.C., Sprecher E. Variant PADI3 in central centrifugal cicatricial alopecia. N Engl J Med. 2019;380(9):833–841. doi: 10.1056/NEJMoa1816614. [DOI] [PubMed] [Google Scholar]
- 28.Tatour Y., Tamaiev J., Shamaly S., Colombo R., Bril E., Rabinowitz T. A novel intronic mutation of PDE6B is a major cause of autosomal recessive retinitis pigmentosa among Caucasus Jews. Mol Vis. 2019;25:155–164. [PMC free article] [PubMed] [Google Scholar]
- 29.Mohamad J., Sarig O., Godsel L.M., Peled A., Malchin N., Bochner R., Vodo D., Rabinowitz T., Pavlovsky M., Taiber S., Fried M., Eskin-Schwartz M., Assi S., Shomron N., Uitto J., Koetsier J.L., Bergman R., Green K.J., Sprecher E. Filaggrin 2 deficiency results in abnormal cell-cell adhesion in the cornified cell layers and causes peeling skin syndrome type A. J Invest Dermatol. 2018;138(8):1736–1743. doi: 10.1016/j.jid.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohamad J., Sarig O., Malki L., Rabinowitz T., Assaf S., Malovitski K. Loss-of-function variants in SERPINA12 underlie autosomal recessive palmoplantar keratoderma. J Investig Dermatol [Internet] 2020 doi: 10.1016/j.jid.2020.02.030. http://www.sciencedirect.com/science/article/pii/S0022202X20312549 Apr 2 [cited 2020 Apr 7]; Available from: [DOI] [PubMed] [Google Scholar]
- 31.Vodo D., Sarig O., Jeddah D., Malchin N., Eskin-Schwarz M., Mohamad J., Rabinowitz T., Goldberg I., Shomron N., Khamaysi Z., Bergman R., Sprecher E. Punctate palmoplantar keratoderma: an unusual mutation causing an unusual phenotype. Br J Dermatol. 2018;178(6):1455–1457. doi: 10.1111/bjd.16502. [DOI] [PubMed] [Google Scholar]
- 32.Hwang S., Kim E., Lee I., Marcotte E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015;5(1) doi: 10.1038/srep17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman B. Updated comparison of variant detection methods: Ensemble, FreeBayes and minimal BAM preparation pipelines [Internet]. Blue Collar Bioinformatics. 2013. Available from: http://bcb.io/2013/10/21/updated-comparison-of-variant-detection-methods-ensemble-freebayes-and-minimal-bam-preparation-pipelines/.
- 34.Cornish A., Guda C. A comparison of variant calling pipelines using genome in a bottle as a reference. Biomed Res Int. 2015;2015:1–11. doi: 10.1155/2015/456479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krusche P., Trigg L., Boutros P.C., Mason C.E., De La Vega F.M., Moore B.L., Gonzalez-Porta M., Eberle M.A., Tezak Z., Lababidi S., Truty R., Asimenos G., Funke B., Fleharty M., Chapman B.A., Salit M., Zook J.M. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol. 2019;37(5):555–560. doi: 10.1038/s41587-019-0054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013 Oct 15;11(1110):11.10.1-11.10.33. [DOI] [PMC free article] [PubMed]
- 37.Brownstein C.A., Beggs A.H., Homer N., Merriman B., Yu T.W., Flannery K.C. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014;15(3):R53. doi: 10.1186/gb-2014-15-3-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faust G.G., Hall I.M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30(17):2503–2505. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014 Oct 1;30(19):2813–5. [DOI] [PMC free article] [PubMed]
- 43.Wang J, Raskin L, Samuels DC, Shyr Y, Guo Y. Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics. 2015 Feb 1;31(3):318–23. [DOI] [PMC free article] [PubMed]
- 44.Zook J.M., Chapman B., Wang J., Mittelman D., Hofmann O., Hide W., Salit M. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32(3):246–251. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
- 45.Chan K.C.A., Jiang P., Sun K., Cheng Y.K.Y., Tong Y.K., Cheng S.H. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. PNAS. 2016 Oct;31:201615800. doi: 10.1073/pnas.1615800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010 Mar 15;26(6):841–2. [DOI] [PMC free article] [PubMed]
- 47.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., McKenna A., Fennell T.J., Kernytsky A.M., Sivachenko A.Y., Cibulskis K., Gabriel S.B., Altshuler D., Daly M.J. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.