

Summary
Synovitis is common in patients with osteoarthritis (OA) and is associated with pain and disease progression. We have previously demonstrated that the chemokine C-C motif chemokine 22 (CCL22) induces chondrocyte apoptosis in vitro; however, the effects of CCL22 on the synovium remain unknown. Therefore, our goal was to investigate the effect of CCL22 on fibroblast-like synoviocytes (FLS). CCL22 treatment suppressed expression of IL-4 and IL-10 and promoted expression of S100A12 in FLS. The response of FLS to CCL22 was not dependent on the disease state of the joint (e.g., normal versus OA), but was instead correlated with the individuals' synovial fluid level of CCL22. CCL22 induction of S100A12 in FLS was attenuated after knockdown of CCR3, yet ligands of CCR3 (CCL7, CCL11) did not induce S100A12 expression. In the presence of CCL22, CCR3-positive FLS upregulate CCL22 and S100A12 driving a potential feedforward pro-inflammatory mechanism distinct from canonical CCL22 and CCR3 pathways.
Subject areas: Molecular Biology, Immunology
Graphical Abstract
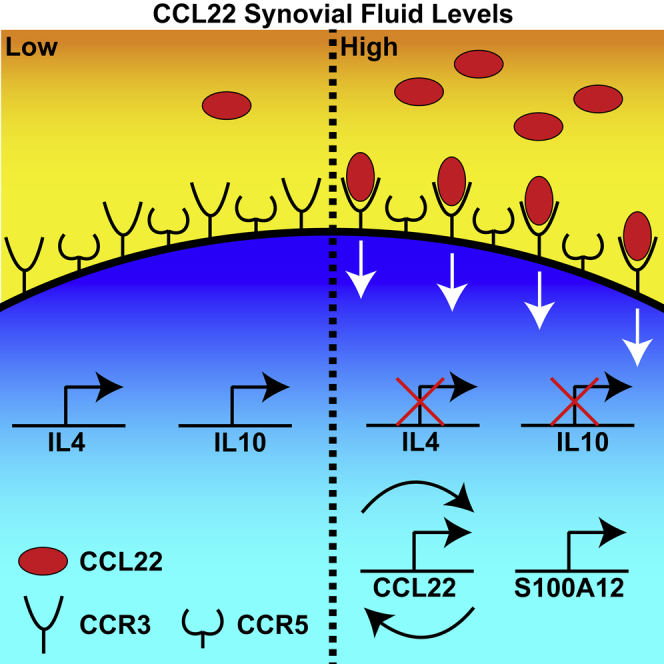
Highlights
-
•
CCL22 increases the expression of the pro-inflammatory mediator S100A12
-
•
CCL22 decreases the expression of anti-inflammatory mediators IL-4 and IL-10
-
•
Fibroblast-like synoviocytes from normal and OA joints do not express CCR4
-
•
CCL22 induces S100A12 expression through CCR3 but not CCR5
Molecular Biology; Immunology
Introduction
Osteoarthritis (OA) is among the most common chronic diseases that can lead to disability (Neogi, 2013). Although OA is characterized by progressive degeneration of the articular cartilage, it is widely considered as a whole joint disease that involves pathological changes of many joint tissues, such as inflammation of the synovium (synovitis), the inner surface of joint capsule that seals the joint cavity (Glyn-Jones et al., 2015; Robinson et al., 2016). The cells within the synovium are responsible for producing synovial fluid (SF) lubricants (e.g., lubricin/PRG4 and hyaluronic acid, Das et al., 2019) as well as filtering plasma as a source of nutrients for chondrocytes (Mathiessen and Conaghan, 2017). Synovitis is often associated with histological changes (e.g., synovial lining hyperplasia) and leukocytic infiltration of the synovial lining (Mathiessen and Conaghan, 2017). Furthermore, synovitis is also associated with the onset and progression of OA, and it is often observed in OA joints from the earliest to advanced stages of the disease (Atukorala et al., 2016). Synovitis is also associated with OA pain, with the synovium being a highly innervated tissue compared with the non-innervated cartilage (Attur et al., 2010; Neogi, 2017).
In a previous study, we described an association between OA pain, cartilage degeneration, and serum levels of C-C motif chemokine 22 (CCL22) (Ren et al., 2019). CCL22 is a chemokine that acts on CCR4+ cells including T cells and dendritic cells (among others) (Yoshie and Matsushima, 2015), and application of CCL22 to human chondrocytes induced apoptosis in a dose-dependent manner in vitro (Ren et al., 2019). We further demonstrated that chondrocytes present within OA cartilage (human and rat models) co-expressed CCL22 and cleaved caspase-3 (a marker of apoptosis) (Ren et al., 2019). These results suggested that, besides regulating chemotaxis in part through calcium signaling, CCL22 may also regulate pathways that lead to the degeneration of cartilage. Yet, it remains unknown if CCL22 can influence changes in cell behavior in additional joint tissues such as the synovium. Although CCL22 is present in human SF and does act directly on chondrocytes, we have previously found no evidence of CCL22 staining in synovium from patients with OA (Ren et al., 2018), and this result agrees with a previous study demonstrating minimal CCL22 expression in OA or normal synovium (Flytlie et al., 2010). This is quite interesting as we previously observed (but did not report) CCL22 staining in the synovium of rats that underwent joint injury (DMM) to induce an OA-like phenotype.
Therefore, to address these potentially conflicting results, and to determine if CCL22 acts upon fibroblast-like synoviocytes (FLS), we have evaluated the effects of CCL22 on FLS inflammatory cytokine production and gene expression in vitro. We also sought to determine if there was any difference in these outcome measures between FLS isolated from normal knee joints and those from patients with clinically diagnosed OA.
Results
CCL22 treatment regulates cytokine expression in FLS
As we previously observed that CCL22 expression was increased in human OA SF (Heard et al., 2013) and articular chondrocytes upregulate CCL22 in areas of cartilage damage (Ren et al., 2019), we wanted to determine if CCL22 treatment could modify cytokine expression in FLS. Normal (n = 10) and OA (n = 13) FLS were included for cytokine expression analysis. CCL22 treatment did not elicit a dramatic change in pro-inflammatory cytokine expression in normal (Figure 1A) or OA (Figure 1B) FLS. However, a decrease in the anti-inflammatory cytokines IL-4 and IL-10 was observed with CCL22 treatment in both normal (Figure 1A) and OA FLS (Figure 1B).
Figure 1.

Cytokine expression response to CCL22 treatment
(A–D) CCL22 treatment of normal (n = 10) (A) and OA (n = 13) (B) FLS decreased the expression of IL-4 and IL-10. When the OA cohort was sub-divided into patients with low SF levels of CCL22 (<0.5 ng/mL) (n = 5) or high SF levels of CCL22 (>0.5 ng/mL) (n = 8), it was observed that low SF CCL22 FLS demonstrated a decrease in IL-4 and IL-10 with CCL22 treatment (C), whereas high SF CCL22 FLS only demonstrated a decrease in IL-4 at the highest CCL22 concentration tested (3 ng/mL) (D). Furthermore, only high SF CCL22 FLS treated with CCL22 demonstrated an increase in GM-CSF (D). ∗p < 0.05; n.d. = no difference; data are represented as mean ± SD.
As we previously observed a difference in SF concentration of CCL22 between the normal cohort and patients with OA (Heard et al., 2013), we assayed for CCL22 concentration in the SF in the cohorts used in the current study (Table 1). In the normal cohort, SF CCL22 levels were found to range from 0.04 to 0.41 ng/mL (0.17 ± 0.11 ng/mL), whereas the CCL22 levels ranged from 0.12 to 4.31 ng/mL (1.63 ± 1.33 ng/mL) in the OA cohort (p = 0.0034). Furthermore, a positive relationship was observed between synovitis and CCL22 SF concentration (Figure S1). As endogenous SF CCL22 levels may have affected the response of FLS to exogenous CCL22 treatment, we re-analyzed the FLS from OA cohort after placing them into two sub-cohorts: patients with OA with low SF CCL22 levels (n = 5, <0.5 ng/mL) or high SF CCL22 levels (n = 8, >0.5 ng/mL). The boundary line of 0.5 ng/mL was selected because none of the normal SF samples displayed a CCL22 level greater than 0.5 ng/mL (Table 1). When cytokine expression in response to exogenous CCL22 treatment was examined in OA FLS separated by SF CCL22 expression, it was observed that FLS derived from patients with low CCL22 SF demonstrated a reduction in interleukin (IL)-4 and IL-10 in response to CCL22 treatment similar to normal FLS (Figure 1C). However, in FLS derived from patients with high CCL22 SF levels there was a reduction in IL-4 with only the highest concentration of exogenous CCL22, and no decrease in IL-10, as IL-10 was practically absent in FLS from patients with high CCL22 SF (Figure 1D). Furthermore, only FLS derived from patients with high CCL22 SF levels demonstrated an increase in granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor alpha (TNF-α) in response to CCL22 treatment (Figure 1D).
Table 1.
Cohort demographics including age, sex, and SF concentration of CCL22
| Cohort | Age | Sex | Synovial fluid CCL22 conc. (ng/mL) | Synovial fluid CCL22 conc. (ng/mL) mean +SD |
|---|---|---|---|---|
| Normal | 45 | Female | 0.15 | 0.17 ± 0.11 |
| Normal | 55 | Female | 0.16 | |
| Normal | 57 | Female | 0.13 | |
| Normal | 62 | Female | 0.41 | |
| Normal | 49 | Male | 0.28 | |
| Normal | 50 | Male | 0.04 | |
| Normal | 52 | Male | 0.09 | |
| Normal | 52 | Male | 0.10 | |
| Normal | 57 | Male | 0.08 | |
| Normal | 65 | Male | 0.23 | |
| OA | 43 | Female | 1.38 | 1.63 ± 1.33 |
| OA | 51 | Female | 2.16 | |
| OA | 52 | Female | 1.18 | |
| OA | 54 | Female | 0.16 | |
| OA | 55 | Female | 2.14 | |
| OA | 55 | Female | 0.23 | |
| OA | 61 | Female | 0.28 | |
| OA | 64 | Female | 0.12 | |
| OA | 50 | Male | 2.47 | |
| OA | 52 | Male | 0.44 | |
| OA | 52 | Male | 3.44 | |
| OA | 58 | Male | 2.82 | |
| OA | 64 | Male | 4.31 |
CCL22 levels were different between normal versus OA cohorts (p = 0.0034). Age was not different between the cohorts (p = 0.9086).
CCL22 induces the expression of S100A12 in vitro
As CCL22 downregulated the expression of IL-4 and IL-10 in FLS derived from normal individuals and patients with OA (with CCL22 SF levels lower than 0.5 ng/mL), RT-qPCR array analysis was employed to explore the response to CCL22 treatment in terms of expression related to chemotaxis, inflammation, and signal transduction. Only six genes found to be differentially expressed after normal FLS (n = 3: 2F, 1 M SF CCL22 concentration = 0.16, 0.41, 0.23 ng/mL, respectively) were treated with 3 ng/mL CCL22 (Figure 2A) (Table S1).
Figure 2.

S100A12 expression in CCL22-treated FLS
(A–D) Normal (n = 3) FLS were treated with the high concentration of CCL22 (3 ng/mL) and differential gene expression was examined by RT-qPCR array (A). The expression of S100A12 was validated using RT-qPCR (B). CCL22 treatment significantly increased the expression of S100A12 in normal FLS (n = 10) and FLS derived from patients with OA with low SF CCL22 levels (n = 5) versus respective PBS controls, whereas no effect was observed in FLS derived from patients with OA with high SF CCL22 levels (n = 8) versus the respective PBS control (B). Furthermore, no difference in S100A12 expression was observed between normal cohort and patients with OA with low SF CCL22 levels when treated with PBS (B). A similar result was observed for CCL22 mRNA expression (C). S100A12 expression was confirmed at the protein level by dot blot analysis using Histone H3 as a loading control (D). Treatment of all FLS with CCL22 (3 ng/mL) resulted in the upregulation of S100A12 versus the respective PBS-treated control (E). ∗p < 0.05; ∗∗∗p < 0.001; n.d. no difference. Data are represented as mean ± SD.
The decreased expression of IL-4 and IL-10 was confirmed at the transcript level, and an increase in CCL17, CCL22, Grb2, and S100A12 was observed with CCL22 treatment. We were particularly interested in S100A12, which is a protein that may be associated with the pathogenesis of OA (Han et al., 2012; Nakashima et al., 2012; Wang et al., 2013), and used RT-qPCR to validate the effect of CCL22 on S100A12 expression in all FLS lines. In accordance with the array results, S100A12 was elevated in normal FLS after CCL22 treatment (Figure 2B). The OA cohort was again sub-divided into FLS from individuals with low (n = 5, <0.5 ng/mL) versus high (n = 8, >0.5 ng/mL) CCL22 SF. It was observed that S100A12 was upregulated in response to CCL22 in FLS derived from patients with low SF CCL22 levels, but not in patients with high SF levels of CCL22 (Figure 2B). It should also be noted that FLS from patients with high SF levels of CCL22 displayed higher basal levels of S100A12 compared with normal FLS or FLS derived from patients with OA with low SF CCL22 levels (Figure 2B).
To validate the array finding that exogenous CCL22 induced endogenous CCL22, CCL22 mRNA levels were quantified in all FLS lines. Similar to S100A12 levels, normal FLS expressed minimal levels of CCL22 that increased upon treatment with exogenous CCL22 (Figure 2C). This effect was also observed in FLS derived from patients with OA with low SF CCL22 levels (Figure 2C), but no increase was observed in FLS derived from patients with OA with high SF CCL22 levels (Figure 2C). Similar to S100A12, FLS from patients with OA with high SF CCL22 levels expressed the highest baseline levels of CCL22 (Figure 2C).
To confirm that CCL22 induced expression of S100A12 at the protein level, lysates from the control/treated cells were assayed for S100A12 levels using Histone H3 as a loading control (Figure 2D). The results at the protein level were consistent with the mRNA levels for the most part, with the exception that FLS from patients with high SF CCL22 levels still demonstrated an increase in S100A12 expression when exposed to CCL22 (Figure 2E).
CCL22 expression levels increase once FLS are expanded in vitro
Although CCL22 mRNA expression was detected in normal and OA FLS after establishing the cells in vitro, it remained unknown if FLS expressed CCL22 in vivo/ex vivo. Therefore synovial membrane samples from all the normal individuals and patients with OA (with low and high SF CCL22) were digested and gated on the CDH-11-positive (Lee et al., 2007) FLS population (Figures 3A, 3D, and 3G). Two populations of FLS, both expressing CCL22 (low positive and high positive), were detected in patients with OA with high SF CCL22 (Figure 3H), whereas CCL22-positive FLS were not detected in normal individuals (Figure 3B) or patients with OA with low SF CCL22 levels (Figure 3E).
Figure 3.

Expression of CCL22 in FLS ex vivo and in vitro
(A–K) Freshly digested synovial samples were analyzed by flow cytometry and representative data are shown (A–I). CDH-11-positive FLS were gated on, and expression of CCL22 was analyzed. Normal FLS (n = 10) (B) and FLS derived from patients with OA with low SF CCL22 levels (n = 5) (E) did not express CCL22 ex vivo, but did express CCL22 once passaged in vitro (C and F). FLS derived from patients with OA with high SF CCL22 levels (n = 8) expressed CCL22 ex vivo (H) and in vitro (I), with two distinct populations of CCL22-positive cells observed in both conditions, and this was also the case in low SF CCL22 FLS in vitro (F). The percentage of FLS cells positive was quantified in each cohort (J). The amount of CCL22 protein was detected by western blot analysis and quantified based on the relative expression compared with the loading control (Histone H3) (K). The raw data for the western blots are presented (L and M). ∗p < 0.05; n.d. no difference. Data are represented as mean ± SD.
Interestingly, CCL22 expression was detected in FLS from normal individuals (Figure 3C) and patients with OA with low SF CCL22 (Figure 3F) once the cells had been expanded in culture, with patient with low SF CCL22 demonstrating low and high positive populations. Cell culture expansion of FLS from patients with OA with high SF CCL22 still presented with two populations of CCL22-positive cells (low positive and high positive) (Figure 3I). The percentage of CCL22-positive FLS was quantified in normal (n = 10) and OA (low [n = 5] and high [n = 8] SF CCL22) cohorts. It was observed that whereas CCL22 was not expressed in FLS from normal cohort or patients with OA with low SF OA CCL22 ex vivo, culture expansion resulted in the FLS becoming CCL22 positive (Figure 3J). FLS from patients with OA with high SF CCL22 expressed CCL22 both ex vivo and in vitro (Figure 3J).
As CCL22 expression was observed in normal FLS in vitro, it was decided to examine how much CCL22 protein was expressed as minimal CCL22 mRNA expression was observed (Figure 2B). Western blot analysis was undertaken on normal FLS directly isolated from synovial membrane and the same cells after 1 passage in vitro (Figures 3K–3M). In agreement with the flow cytometry data, little to no CCL22 protein was detected in normal FLS or FLS from patients with OA with low SF CCL22 ex vivo, whereas FLS from patients with high SF CCL22 expressed higher levels of CCL22 (Figures 3K and 3L). In vitro, higher levels of CCL22 were found in normal FLS and FLS from patients with OA with low SF CCL22 (compared with ex vivo), yet both presented with less CCL22 protein expression than FLS from patients with OA with high SF CCL22 (Figures 3K and 3M). When normal FLS were treated with exogenous CCL22 (3 ng/mL), and then washed to remove soluble CCL22, it was observed that normal FLS expressed similar CCL22 protein levels compared with FLS derived from patients with OA with high SF CCL22 (Figures 3K and 3M).
CCL22 and S100A12 co-localizes in situ at the protein level
As we observed a relationship between CCL22 and S100A12 in FLS in vitro, we investigated if the expression of these proteins was related in situ. Synovial biopsies from all individuals in the study were examined. In normal synovium, CCL22 staining was absent and minimal S100A12 staining was observed (Figure 4A). In contrast, a wide range of CCL22 and S100A12 staining (patterns and intensity) was observed in OA synovium (Figures 4B–4E). In some OA synovium, little to no staining for either CCL22 or S100A12 was observed, whereas other samples demonstrated robust staining for both. When the mean fluorescent intensity of CCL22 and S100A12 staining was quantified (then normalized to the sample with the greatest intensity of staining for each marker) and examined in the context of CCL22 concentration within the SF (e.g., low versus high SF CCL22), a positive correlation between CCL22 (R2 = 0.92) and S100A12 (R2 = 0.83) staining in the synovium was observed with SF CCL22 levels (Figure 4F). Isotype controls demonstrated minimal reactivity (Figure 4G).
Figure 4.

Expression of CCL22 and S100A12 protein in vivo
(A–G) Synovium from normal individuals (n = 10) was negative for CCL22 staining and only demonstrated sporadic S100A12 staining (representative data from n = 2, shown in A). Synovium from patients with OA (n = 13) demonstrated a range of CCL22 and S100A12 staining that co-localized (arrows) and appeared to increase with the SF concentration of CCL22 (representative data from n = 4 shown in B–E). A linear regression analysis was performed that included all samples (normal and OA) and a significant correlation between CCL22 staining (R2 = 0.92 p < 0.001), S100A12 staining (R2 = 0.83 p < 0.001), and SF levels of CCL22 (F). Isotype controls demonstrate limited reactivity (G). Scale bars, 50 μm.
CCL22 and S100A12 mRNA are co-localized in situ
We next decided to examine if CCL22 was being produced by the FLS in situ or if the CCL22 detected in the synovium (Figure 4) originated from the SF and accumulated within the synovium. In situ hybridization was used to detect CCL22 and S100A12 mRNA using β-actin as a positive control. All normal synovial samples assayed (n = 10) were negative for CCL22 and S100A12 (Figures 5A and 5B representative images presented). In the OA synovium samples examined (n = 13), two distinct staining patterns were observed. The first pattern was observed in patients with lower (but not exclusively under 0.5 ng/mL) CCL22 SF concentrations, wherein little to no CCL22 mRNA signal was detected (Figure 5C representative images presented), yet S100A12 mRNA was detected at the surface of the synovium (Figure 5D representative images presented), suggesting that this S100A12 mRNA and protein expression is either driven by CCL22 derived from the SF and/or regulated by a CCL22-independent mechanism. The other staining pattern observed was restricted to patients with OA with higher levels of CCL22 present in the SF. In these patients, CCL22 mRNA was detected in the synovium (Figure 5E representative images presented) with robust S100A12 mRNA expression observed throughout the synovium (Figure 5F representative images presented). When the mean fluorescent intensity of CCL22 and S100A12 staining was quantified (then normalized to the sample with the greatest intensity of staining for each marker and β-actin staining) and examined in the context of CCL22 concentration within the SF (e.g., low versus high SF CCL22), a positive correlation between CCL22 (R2 = 0.92) and S100A12 (R2 = 0.89) mRNA staining in the synovium was observed with SF CCL22 levels (Figure 5G). Yet, a clear demarcation was observed above versus below an SF CCL22 concentration of 2 ng/mL. SF samples below this level showed minimal to no CCL22 or S100A12 staining, whereas samples above 2 ng/mL demonstrated robust staining for both markers (Figure 5G). This is in contrast to the protein levels of each marker, which showed an intermediate level of expression in joints with 1–2 ng/mL CCL22 (Figure 4F). It is also important to note that the experimental design employed does not discriminate by cell type within the synovium and we cannot specifically state that the CCL22 and/or S100A12 mRNA is solely being expressed by FLS and not by macrophage-like synoviocytes and/or additional cell types.
Figure 5.

Expression of CCL22 and S100A12 mRNA in vivo
(A–G) Synovium from normal individuals (n = 10) was negative for CCL22 and S100A12 mRNA (representative data from n = 1 shown in A and B). In synovium from patients with OA with low SF CCL22 levels (n = 5), minimal CCL22 mRNA expression was observed (representative data from n = 1 shown in C), and S100A12 mRNA was observed at the surface (arrow, D). In synovium from patients with OA with high SF CCL22 levels (n = 8), CCL22 mRNA expression was observed throughout the synovium (representative data from n = 1 shown in E) and co-localized with S100A12 (arrows, E and F) mRNA, which was observed throughout the synovium (arrow, F). A linear regression analysis was performed that included all samples (normal and OA), and a significant correlation between CCL22 staining (R2 = 0.92 p < 0.0001), S100A12 staining (R2 = 0.89 p < 0.0001), and SF levels of CCL22 was observed (G). β-Actin was utilized as a positive control (green). Scale bars, 50 μm.
FLS lack expression of CCR4
As CCR4 is the only known receptor for CCL22 and previous studies have demonstrated the absence of CCR4 in normal human synovium (Flytlie et al., 2010), flow cytometry and immunofluorescence analysis was used to determine if CCR4 was expressed on the cell surface of normal (n = 10) or OA (n = 13) FLS or synovial biopsies employed in this study. None of synovial membrane samples examined expressed CCR4, but all expressed CCR3 and CCR5 (Figure 6A). To confirm these results, synovium was digested and primary FLS cells (CDH-11 positive, Lee et al., 2007) were examined by flow cytometry (Figure 6B). All primary FLS were negative for CCR4 (Figure 6C), and these findings were validated with a second CCR4 antibody (data not shown). However, all primary FLS used in this study were positive for CCR3 (Figure 6D) and CCR5 (Figure 6E). Interestingly, two distinct CCR5-positive FLS populations were observed in all samples, which was reminiscent of the CCL22-positive FLS populations observed (Figure 3E). The percentage of positive CCR3, CCR4, or CCR5 primary FLS were quantified, and no differences were observed between normal and OA samples (Figure 6F).
Figure 6.

Expression of CCR3, CCR4, and CCR5 in synovium and on FLS
(A–F) In all synovium membrane biopsies examined (n = 23), no expression of CCR4 (blue, A′) was detected; however, in all samples both CCR3 (green, A″) and CCR5 (red, A‴) were present. This result was validated on freshly derived normal (n = 10, representative data from n = 2 shown) and OA (n = 13, representative data from n = 5 shown) FLS using flow cytometry (B–E). The flow cytometry results were quantified, and no difference was observed between normal and OA FLS in terms of CCR4, CCR3, or CCR5 expression (F). Scale bars, 50 μm. n.d. = no difference; Data are represented as mean ± SD.
CCL22 induces SA100A12 expression through CCR3
Although it has been previously shown that CCL22 does not signal through CCR3 or CCR5 (Bochner et al., 1999; Imai et al., 1998) in terms of inducing calcium flux and/or chemotaxis, it remains unknown if CCL22 can induce other signaling pathways (e.g., aside from calcium flux and/or chemotaxis) through either of these receptors. Therefore, we used a short hairpin RNA (shRNA) knockdown approach to determine if CCL22 was able to induce S100A12 mRNA expression through CCR3 or CCR5. Normal (n = 3) and OA (n = 6; n = 3 from patients with low CCL22 SF, n = 3 from patients with high CCL22 SF) FLS were transfected with the shRNA plasmids for CCR3, CCR5, or a nonsense control shRNA and analyzed for receptor expression using flow cytometry (Figures 7A–7H). The nonsense construct had no effect of CCR3 or CCR5 expression, the CCR3 shRNA decreased the expression of CCR3 (but not CCR5), whereas the CCR5 shRNA decreased the expression of CCR5 (but not CCR3) (Figures 7A–7H). This result was validated at the mRNA level using RT-qPCR (Figure 7I).
Figure 7.

CCL22 induces expression of S100A12 through CCR3
(A–I) FLS were gated on CDH-11-positive cells and examined for CCR3 and CCR5 expression (A–H). FLS (normal n = 3; OA low CCL22 n = 3; OA high CCL22 n = 3) transfected with a nonsense control shRNA demonstrated expression of CCR3 and CCR5 (C and F). FLS transfected with a CCR3 shRNA demonstrated reduced expression of CCR3 only (D and G). FLS transfected with a CCR5 shRNA demonstrated reduced expression of CCR5 only (E and H). These results were validated at the mRNA level with RT-qPCR (I). Control and transfected FLS treated with CCL22 induced S100A12 at similar levels except in FLS transfected with CCR3 shRNA (J). ∗p < 0.05. n.d. = no difference. Data are represented as mean ± SD.
Transfected FLS cells were treated with CCL22 (3 ng/mL) and assayed for S100A12 expression by RT-qPCR (Figure 7J). In normal FLS, CCL22 induced S100A12 mRNA expression in untransfected, nonsense-transfected, and CCR5 shRNA-transfected cells. However, S100A12 mRNA expression was reduced in CCR3 shRNA-transfected cells (Figure 7J). The same trend was observed in FLS derived from patients with low SF levels of CCL22, whereas no effect on S100A12 mRNA expression in CCR3 shRNA transfected cells was observed in FLS derived from patients with high SF levels of CCL22 (Figure 7J). To further investigate the role of CCR3 in this mechanism, the CCR3 inhibitor SB 297006 (White et al., 2000) was added to normal FLS with and without CCR3 shRNA. The CCR4 inhibitor AZD 2098 (Kindon et al., 2017) was employed to examine any potential signaling/cross talk through CCR4 (Figure S2). The addition of AZD 2098 or SB 297006 had no effect on S100A12 expression in normal FLS in the absence of CCL22 regardless of CCR3 knockdown. Furthermore, inhibition of CCR4 through AZD 2098 also had no effect on S100A12 expression in the presence of CCL22. Inhibition of CCR3 by SB 297006 significantly reduced S100A12 expression in the presence of CCL22 with the nearly complete inhibition of S100A12 expression observed in normal FLS with CCR3 knockout and SB 297006 treatment (Figure S2A). A similar effect was observed in FLS from patients with low CCL22 SF (Figure S2B), yet no effect of either inhibitor was observed in FLS from patients with high CCL22 SF (Figure S2C). These results strongly suggest that this CCL22-S100A12 signaling mechanism is dependent on CCR3 and independent of CCR4, and that once this pathway has researched a certain threshold neither CCR3 nor CCR4 is required for perpetuation of this signaling mechanism.
To further investigate the potential mechanism of CCL22 signaling through CCR3, FLS were exposed to known ligands of CCR3 (CCL11/eotaxin-1, CCL13/MCP-4) and the expression of S100A12 mRNA was quantified. In normal and OA (both low and high SF levels of CCL22) FLS that were untransfected, nonsense transfected, CCR3 shRNA transfected, or CCR5 shRNA transfected, neither CCL11 nor CCL13 induced the expression of S100A12 (Figure S3). As previous studies examining CCL22-CCR3 signaling primarily focused on calcium flux, we investigated if CCL22 could induce calcium flux in normal or OA FLS with/without CCR3 shRNA knockdown (Figure S4). Known CCR3 ligands CCL11 and CCL14 were able to induce calcium flux in normal and OA FLS, whereas exposure to CCL22 did not result in an increase in calcium signaling. When FLS with reduced CCR3 levels were examined, the CCL11 response was noticeably attenuated, no response was observed with CCL22, and the CCL13 response was not impacted. This result was not unexpected as CCL13 is known to signal through CCR2, CCR3, and CCR5 (Blanpain et al., 1999). Importantly, these results confirm previous studies demonstrating that CCL22 cannot induce calcium signaling through CCR3, but yet suggest that CCL22 can induce S100A12 expression through a CCR3-dependent mechanism.
Discussion
The role of cytokines in the pathogenesis of OA has become increasingly recognized (Wojdasiewicz et al., 2014). Pro-inflammatory cytokines such as TNF-α and IL-1β have a direct and destructive impact on articular cartilage not only by promoting the expression of proteinases that degrade the extracellular matrix but also through induction of chondrocyte apoptosis (Dayer et al., 2017; Goda et al., 2015; Sun et al., 2017). In addition to these highly studied pro-inflammatory mediators, a variety of new cytokines and chemokines have been implicated with tissue degeneration and pain in OA (Kapoor et al., 2011). A number of studies have implicated CCL2 (MCP-1) in the onset and progression of OA, including roles in cartilage degeneration and pain (Appleton et al., 2015; Harris et al., 2013; Jablonski et al., 2019; Miller et al., 2012; Miotla Zarebska et al., 2017). Expression of CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) have also been associated with OA in clinical and pre-clinical studies (Beekhuizen et al., 2013; Raghu et al., 2017; Zhao et al., 2015). CCL19 and CCL21 have been implicated in synovitis, but a direct relationship with the onset/progression of OA remains unclear (Scanzello, 2017). Another example is CCL22, which is elevated in the SF and serum of patients with OA and has been correlated with pain (Flytlie et al., 2010; Ren et al, 2018, 2019). CCL22 has a complex role in inflammation and is recognized as a potent chemotactic molecule, recruiting both anti- and pro-inflammatory immune cells (Curiel et al., 2004; Imai et al., 1999). It has been recently reported by our laboratory that CCL22 can induce apoptosis in human chondrocytes in vitro and that it is expressed in chondrocytes throughout the progression of OA in vivo (rat DMM model). These results suggest that CCL22 may play a role in the initiation of cartilage degeneration in OA. Therefore, the aim of the present study was to investigate if CCL22 could also regulate inflammation in FLS from healthy donors and patients with OA as FLS are known to be a main driver of inflammation within the joint (Huh et al., 2015; Sokolove and Lepus, 2013). We found that CCL22 can inhibit the expression of anti-inflammatory cytokines (IL-4 and IL-10) and induce the expression of S100A12. Interestingly, whereas this effect was observed in FLS derived from all normal individuals, it was not observed in FLS derived from all patients with OA. When we further sub-divided the OA cohort based on endogenous SF CCL22 levels, we observed that FLS from patients with low SF levels of CCL22 acted similar to normal FLS, whereas FLS from patients with high SF levels of CCL22 did not show a robust response to CCL22. This lack of response from FLS derived from patients with high CCL22 SF is most likely due to these FLS already expressing high levels of CCL22 and S100A12 and therefore not susceptible to further regulation by exogenous CCL22.
Synovitis is often observed in the earliest stages of OA (Sellam and Berenbaum, 2010); however, it is not clear whether synovitis is primarily caused by systemic immune responses or occurs secondarily to joint tissue damage. One common hypothesis is that once a given threshold of inflammation is reached within the joint, a variety of cytokines including IL-6 and TNF-α in the inflamed SF could stimulate synovial cells to also produce inflammatory cytokines, resulting in a positive feedback pro-inflammatory cycle within the joint (Liu-Bryan, 2013). In agreement with this hypothesis, we observed that CCL22 treatment downregulated IL-4 and IL-10 while upregulating S100A12. IL-4 and IL-10 are considered anti-inflammatory cytokines, capable of suppressing the immune response through a variety of mechanisms, including inhibiting the synthesis of pro-inflammatory cytokines such as interferon-γ and TNF-α and GM-CSF (Iyer and Cheng, 2012). S100A12 is a pro-inflammatory cytokine-like protein. It is has previously been implicated in the development of OA (Han et al., 2012; Wang et al., 2013), potentially playing a role by upregulating MMPs and activating the NF-κB pathway (Nakashima et al., 2012). The downregulation of IL-4 and IL-10 and the upregulation of S100A12 in normal FLS indicate that CCL22 might play in important role in initiating/promoting a pro-inflammatory response in synovium.
As CCR4 is the only known receptor for CCL22 (Yoshie and Matsushima, 2015) and we observed changes to IL-4, IL-10, and S100A12 expression after CCL22 treatment, we were surprised to find that neither normal nor OA FLS expressed CCR4 and that CCL22 expression was not co-localized with CCR4 expression in situ. These results are difficult to reconcile in the current paradigm, therefore we suggest that there are additional receptors/co-receptors for CCL22. Although it has been previously demonstrated that CCL22 does not activate calcium signaling or chemotaxis through CCR3 or CCR5 (Bochner et al., 1999; Imai et al., 1998), it is important to note that one of these studies also observed CCL22-induced eosinophil chemotaxis, although the cells lacked expression of CCR4 (Bochner et al., 1999), which corroborates our result and strengthens the hypothesis that CCL22 has an additional receptor(s) aside from CCR4. As FLS express CCR3 and CCR5, we decided to test if CCL22 could signal through these receptors. We observed reduced S100A12 upregulation post-CCL22 treatment in FLS receiving CCR3 but not CCR5 shRNA suggesting that CCL22 can signal through CCR3, yet also demonstrated that CCL22 could not induce calcium flux through CCR3. We further demonstrated that known CCR3 ligands (CCL11, CCL13) could not induce S100A12 expression. Although these results do not demonstrate that CCR3 is a canonical receptor for CCL22 signaling, it does show that CCL22 is capable of signaling through CCR3 in the context of S100A12 and that this pathway is independent of canonical CCR3 signaling. Further investigation should be undertaken to identify additional potential CCL22 receptors in FLS because it is possible that alternate receptors/pathways are involved in this mechanism (Scanzello, 2017).
To our knowledge a link between CCR3 and S100A12 remains uncharacterized; however, activation of CCR3 is able to drive the expression of a number of pro-inflammatory pathways (Zhu et al., 2018) and therefore it is possible that CCR3 may act directly or indirectly on S100A12, although this would need to be directly examined in future mechanistic studies. It is also important to note that when we knocked down levels of CCR3 in OA FLS derived from patients with high SF CCL22 levels, we did not observe an impact on S100A12 expression. This suggests that once this pathway becomes activated it can potentially operate in a feedforward state in which CCL22-CCR3 interaction is no longer required. Although it is possible that another cell surface receptor is upregulated in this pro-inflammatory state that takes over from CCR3, a simpler scenario is that both these factors (CCL22 and S100A12) are driven at the transcriptional level by a distinct pathway that becomes active (e.g., TNFα) that overrides the CCL22-CCR3 cascade. Although interesting, these hypotheses would require significant experimentation to clarify the underlying mechanisms.
Another interesting observation that should be discussed is the difference in CCL22 production in FLS ex vivo versus in vitro. In normal synovium that was freshly digested, CDH-11-positive FLS cells were negative for CCL22 expression, yet after 1 passage in vitro, they expressed CCL22. This suggests that simply removing the cells from their normal microenvironment may act as a pro-inflammatory stimuli. It also may be possible that there exists an inhibitory molecule to CCL22 in vivo, which is downregulated and/or diluted in the in vitro environment. Although the current study was not designed to test this hypothesis, examining this observation in more detail may be important to understand differences in cell behavior in vivo versus in vitro and may help provide insight into how FLS react to the microenvironment.
Another important finding of the current study was that FLS derived from patients with OA did not all respond similarly to CCL22. We suggest the main reason for this is that patients with OA present with a wide range of CCL22 in their SF. Our results suggest that FLS in vivo respond to SF CCL22 and once CCL22 reaches a threshold level in the FLS microenvironment, a feedforward loop is activated wherein the same FLS upregulate CCL22. This explains why FLS derived from patients with OA with high SF levels of CCL22 were not responsive to additional exogenous CCL22. This also suggests that CCL22 produced by FLS may act in an autocrine fashion, signaling through CCR3 to upregulate S100A12. This type of feedforward mechanism in the synovium is reminiscent of the vicious cycle of inflammation and cartilage degeneration observed in patients with OA. We are therefore curious how generalizable this observation would be to other pro-inflammatory cytokines in OA; it would be interesting to examine FSL sensitivity to different mediators in the context of the concentration of the mediator in the in vivo environment. If patient cells show heterogeneity based on “reprogramming” by the environment they are derived from, this may at least partially explain why there exists such as range of response to stimuli in vitro between published studies.
In conclusion, we have demonstrated that CCL22 can induce a pro-inflammatory response in FLS inhibiting IL-4 and IL-10 while promoting S100A12 expression thought CCR3. The ability of CCL22 to trigger this response is in part dependent on the level of CCL22 in the SF within the joint the FLS were isolated from.
Limitations of the study
Additional mechanistic studies employing in vitro and in vivo techniques would be required to fully elucidate the role of CCL22 in the synovium and validate that signaling through CCR3 and/or additional receptors is involved in synovial inflammation and OA. Although the evidence presented in this study suggests that non-canonical CCL22/CCR3 signaling does take place in the context of FLS cells, it is possible that additional cell types not accounted for in vivo may express CCR4 (e.g., macrophage) that drive the majority of the inflammatory process observed. CCR3, CCR4, and CCL22 transgenic mouse models may help shed light on many of the questions left unanswered in the current study.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roman Krawetz (rkrawetz@ucalgary.ca).
Materials Availability
All data generated/analyzed in this study are included in this published article and its supplemental information.
Data and Code Availability
The published article includes all data generated/analyzed in this study.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
The authors would also like to thank the University of Calgary Flow Cytometry Core Facility (Laurie Kennedy and Yiping Liu) for their assistance. We also thank the Southern Alberta Organ and Tissue Donation Program for collection of normal tissue samples. We would also like to note that primary data and text from the current study were also included in the first author's PhD thesis (G.R.), archived by the University of Calgary in 2018.
Funding: Acknowledgment of funding from Natural Sciences and Engineering Research Council of Canada, Canada Foundation for Innovation (CFI), and Grace Glaum Professorship (R.J.K.). G.R. was supported by an Arthritis Society PhD Studentship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
G.R., N.A., P.R., J.P., R.J.K.: Substantial contributions to the conception or design of the work, or the acquisition, analysis, or interpretation of data for the work. G.R. and R.J.K.: Drafting the work; N.A., P.R., and J.P.: Revising the work critically for important intellectual content. R.J.K.: Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101943.
Supplemental information
References
- Appleton C.T.G., Usmani S.E., Pest M.A., Pitelka V., Mort J.S., Beier F. Reduction in disease progression by inhibition of transforming growth factor α-CCL2 signaling in experimental posttraumatic osteoarthritis. Arthritis Rheumatol. 2015;67:2691–2701. doi: 10.1002/art.39255. [DOI] [PubMed] [Google Scholar]
- Attur M., Samuels J., Krasnokutsky S., Abramson S.B. Targeting the synovial tissue for treating osteoarthritis (OA): where is the evidence? Best Pract. Res. Clin. Rheumatol. 2010;24:71–79. doi: 10.1016/j.berh.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Atukorala I., Kwoh C.K., Guermazi A., Roemer F.W., Boudreau R.M., Hannon M.J., Hunter D.J. Synovitis in knee osteoarthritis: a precursor of disease? Ann. Rheum. Dis. 2016;75:390–395. doi: 10.1136/annrheumdis-2014-205894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekhuizen M., Gierman L.M., van Spil W.E., Van Osch G.J.V.M., Huizinga T.W.J., Saris D.B.F., Creemers L.B., Zuurmond A.M. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 2013;21:918–922. doi: 10.1016/j.joca.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Blanpain C., Migeotte I., Lee B., Vakili J., Doranz B.J., Govaerts C., Vassart G., Doms R.W., Parmentier M. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood. 1999;94:1899–1905. [PubMed] [Google Scholar]
- Bochner B.S., Bickel C.A., Taylor M.L., MacGlashan D.W., Gray P.W., Raport C.J., Godiska R. Macrophage-derived chemokine induces human eosinophil chemotaxis in a CC chemokine receptor 3- and CC chemokine receptor 4-independent manner. J. Allergy Clin. Immunol. 1999;103:527–532. doi: 10.1016/s0091-6749(99)70481-1. [DOI] [PubMed] [Google Scholar]
- Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Das N., Schmidt T.A., Krawetz R.J., Dufour A. Proteoglycan 4: from mere lubricant to regulator of tissue homeostasis and inflammation. Bioessays. 2019;41:1800166. doi: 10.1002/bies.201800166. [DOI] [PubMed] [Google Scholar]
- Dayer J.-M., Williamson S.J., Croft A.P., Buckley C.D., Chizzolini C. Matrix metalloproteinases (MMPs) and cytokines in rheumatology. In: Brinckerhoff C.E., editor. Matrix Metalloproteinases in Health and Disease. World Scientific; 2017. pp. 123–155. [Google Scholar]
- Flytlie H.A., Hvid M., Lindgreen E., Kofod-Olsen E., Petersen E.L., Jørgensen A., Deleuran M., Vestergaard C., Deleuran B. Expression of MDC/CCL22 and its receptor CCR4 in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Cytokine. 2010;49:24–29. doi: 10.1016/j.cyto.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Glyn-Jones S., Palmer A.J.R., Agricola R., Price A.J., Vincent T.L., Weinans H., Carr A.J. Osteoarthritis. Lancet. 2015;386:376–387. doi: 10.1016/S0140-6736(14)60802-3. [DOI] [PubMed] [Google Scholar]
- Goda S., Kato Y., Domae E., Hayashi H., Tani-Ishii N., Iida J., Ikeo T. Effects of JNK1/2 on the inflammation cytokine TNF-α-enhanced production of MMP-3 in human dental pulp fibroblast-like cells. Int. Endod. J. 2015;48:1122–1128. doi: 10.1111/iej.12411. [DOI] [PubMed] [Google Scholar]
- Han M.Y., Dai J.J., Zhang Y., Lin Q., Jiang M., Xu X.Y., Liu Q. Identification of osteoarthritis biomarkers by proteomic analysis of synovial fluid. J. Int. Med. Res. 2012;40:2243–2250. doi: 10.1177/030006051204000622. [DOI] [PubMed] [Google Scholar]
- Harris Q., Seto J., O’Brien K., Lee P.S., Kondo C., Heard B.J., Hart D.A., Krawetz R.J. Monocyte chemotactic protein-1 inhibits chondrogenesis of synovial mesenchymal progenitor cells: an in vitro study. Stem Cells. 2013;31:2253–2265. doi: 10.1002/stem.1477. [DOI] [PubMed] [Google Scholar]
- Heard B.J., Fritzler M.J., Preston Wiley J., McAllister J., Martin L., El-Gabalawy H., Hart D.A., Frank C.B., Krawetz R. Intraarticular and systemic inflammatory profiles may identify patients with osteoarthritis. J. Rheumatol. 2013;40:1379–1387. doi: 10.3899/jrheum.121204. [DOI] [PubMed] [Google Scholar]
- Huh Y.H., Lee G., Song W.H., Koh J.T., Ryu J.H. Crosstalk between FLS and chondrocytes is regulated by HIF-2α-mediated cytokines in arthritis. Exp. Mol. Med. 2015;47:e197. doi: 10.1038/emm.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T., Chantry D., Raport C.J., Wood C.L., Nishimura M., Godiska R., Yoshie O., Gray P.W. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J. Biol. Chem. 1998;273:1764–1768. doi: 10.1074/jbc.273.3.1764. [DOI] [PubMed] [Google Scholar]
- Imai T., Nagira M., Takagi S., Kakizaki M., Nishimura M., Wang J., Gray P.W., Matsushima K., Yoshie O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 1999;11:81–88. doi: 10.1093/intimm/11.1.81. [DOI] [PubMed] [Google Scholar]
- Iyer S.S., Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012;32:23–63. doi: 10.1615/critrevimmunol.v32.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski C.L., Leonard C., Salo P., Krawetz R.J. CCL2 but not CCR2 is required for spontaneous articular cartilage regeneration post-injury. J. Orthop. Res. 2019;37:2561–2574. doi: 10.1002/jor.24444. [DOI] [PubMed] [Google Scholar]
- Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J.-P., Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- Kindon N., Andrews G., Baxter A., Cheshire D., Hemsley P., Johnson T., Liu Y.Z., McGinnity D., McHale M., Mete A. Discovery of AZD-2098 and AZD-1678, two potent and bioavailable CCR4 receptor antagonists. ACS Med. Chem. Lett. 2017;8:981–986. doi: 10.1021/acsmedchemlett.7b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.M., Kiener H.P., Agarwal S.K., Noss E.H., Watts G.F.M., Chisaka O., Takeichi M., Brenner M.B. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr. Rheumatol. Rep. 2013;15:323. doi: 10.1007/s11926-013-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiessen A., Conaghan P.G. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res. Ther. 2017;19:18. doi: 10.1186/s13075-017-1229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R.E., Tran P.B., Das R., Ghoreishi-Haack N., Ren D., Miller R.J., Malfait A.-M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. U S A. 2012;109:20602–20607. doi: 10.1073/pnas.1209294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotla Zarebska J., Chanalaris A., Driscoll C., Burleigh A., Miller R.E., Malfait A.M., Stott B., Vincent T.L. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthr. Cartil. 2017;25:406–412. doi: 10.1016/j.joca.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M., Sakai T., Hiraiwa H., Hamada T., Omachi T., Ono Y., Inukai N., Ishizuka S., Matsukawa T., Oda T. Role of S100A12 in the pathogenesis of osteoarthritis. Biochem. Biophys. Res. Commun. 2012;422:508–514. doi: 10.1016/j.bbrc.2012.05.036. [DOI] [PubMed] [Google Scholar]
- Neogi T. Structural correlates of pain in osteoarthritis. Clin. Exp. Rheumatol. 2017;35:75–78. [PubMed] [Google Scholar]
- Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013;21:1145–1153. doi: 10.1016/j.joca.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu H., Lepus C.M., Wang Q., Wong H.H., Lingampalli N., Oliviero F., Punzi L., Giori N.J., Goodman S.B., Chu C.R. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 2017;76:914–922. doi: 10.1136/annrheumdis-2016-210426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G., Lutz I., Railton P., Wiley J.P., McAllister J., Powell J., Krawetz R.J. Serum and synovial fluid cytokine profiling in hip osteoarthritis: distinct from knee osteoarthritis and correlated with pain. BMC Musculoskelet. Disord. 2018;19:39. doi: 10.1186/s12891-018-1955-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G., Whittaker J.L., Leonard C., De Rantere D., Pang D.S.J., Salo P., Fritzler M., Kapoor M., de Koning A.P.J., Jaremko J.L. CCL22 is a biomarker of cartilage injury and plays a functional role in chondrocyte apoptosis. Cytokine. 2019;115:32–44. doi: 10.1016/j.cyto.2018.11.030. [DOI] [PubMed] [Google Scholar]
- Robinson W.H., Lepus C.M., Wang Q., Raghu H., Mao R., Lindstrom T.M., Sokolove J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016;12:580–592. doi: 10.1038/nrrheum.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanzello C.R. Chemokines and inflammation in osteoarthritis: insights from patients and animal models. J. Orthop. Res. 2017;35:735–739. doi: 10.1002/jor.23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellam J., Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010;6:625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- Sokolove J., Lepus C.M. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013;5:77–94. doi: 10.1177/1759720X12467868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.-Y., Hu K.-Z., Yin Z.-S. Inhibition of the p38-MAPK signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. Cytokine. 2017;90:135–143. doi: 10.1016/j.cyto.2023.156223. [DOI] [PubMed] [Google Scholar]
- Wang L.C., Zhang H.Y., Shao L., Chen L., Liu Z.H., He X., Gong W.X. S100A12 levels in synovial fluid may reflect clinical severity in patients with primary knee osteoarthritis. Biomarkers. 2013;18:216–220. doi: 10.3109/1354750X.2013.766262. [DOI] [PubMed] [Google Scholar]
- White J.R., Lee J.M., Dede K., Imburgia C.S., Jurewicz A.J., Chan G., Fornwald J.A., Dhanak D., Christmann L.T., Darcy M.G. Identification of potent, selective non-peptide CC chemokine receptors-3 antagonist that inhibits eotaxin-, eotaxin-2-, and monocyte chemotactic protein-4-induced eosinophil migration. J. Biol. Chem. 2000;275:36626–36631. doi: 10.1074/jbc.M006613200. [DOI] [PubMed] [Google Scholar]
- Wojdasiewicz P., Poniatowski Ł.A., Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:1–19. doi: 10.1155/2014/561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshie O., Matsushima K. CCR4 and its ligands: from bench to bedside. Int. Immunol. 2015;27:11–20. doi: 10.1093/intimm/dxu079. [DOI] [PubMed] [Google Scholar]
- Zhao X.Y., Yang Z.B., Zhang Z.J., Zhang Z.Q., Kang Y., Huang G.X., Wang S.W., Huang H., Liao W.M. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis) Osteoarthr. Cartil. 2015;23:1405–1411. doi: 10.1016/j.joca.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Zhu X., Liu K., Wang J., Peng H., Pan Q., Wu S., Jiang Y., Liu Y. C-C chemokine receptor type 3 gene knockout alleviates inflammatory responses in allergic rhinitis model mice by regulating the expression of eosinophil granule proteins and immune factors. Mol. Med. Rep. 2018;18:3780–3790. doi: 10.3892/mmr.2018.9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated/analyzed in this study.