

Abstract
Machine learning (ML) can aid in novel discoveries in the field of viral gene therapy. Specifically, big data gathered through next-generation sequencing (NGS) of complex capsid libraries is an especially prominent source of lost potential in data analysis and prediction. Furthermore, adeno-associated virus (AAV)-based capsid libraries are becoming increasingly popular as a tool to select candidates for gene therapy vectors. These higher complexity AAV capsid libraries have previously been created and selected in vivo; however, in silico analysis using ML computer algorithms may augment smarter and more robust libraries for selection. In this study, data of AAV capsid libraries gathered before and after viral assembly are used to train ML algorithms. We found that two ML computer algorithms, artificial neural networks (ANNs), and support vector machines (SVMs), can be trained to predict whether unknown capsid variants may assemble into viable virus-like structures. Using the most accurate models constructed, hypothetical mutation patterns in library construction were simulated to suggest the importance of N495, G546, and I554 in AAV2-derived capsids. Finally, two comparative libraries were generated using ML-derived data to biologically validate these findings and demonstrate the predictive power of ML in vector design.
Keywords: Machine Learning, AAV, Capsid Libraries, Assembly, Packaging, ANN, SVM
Graphical Abstract
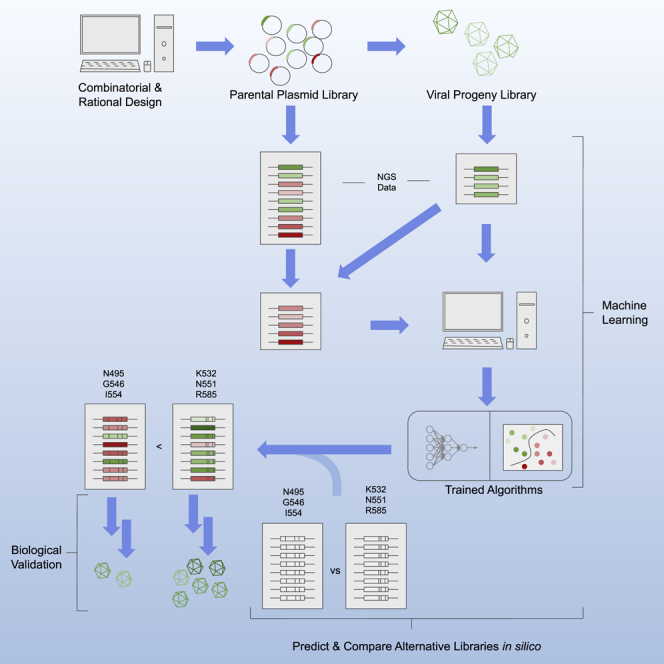
Marques et al. develop a bioinformatics pipeline implementing machine learning to predict whether AAV2-based amino acid sequences will produce viable capsids. The ability to make these predictions may facilitate researchers to optimize AAV gene therapy vector libraries by selecting pools with fewer dead-end variants in silico.
Introduction
Adeno-associated virus (AAV) is a member of the Dependoparvovirus genus in the Parvoviridae family.1 AAV’s non-enveloped capsid consists of 60 viral proteins (VPs).2 These VPs are produced in one of three types (VP1, VP2, and VP3), which assemble in a 1:1:10 stoichiometric ratio, respectively.3 Recently, AAV is proving to be a potent gene therapy vector as demonstrated in several early-stage clinical trials and studies using an animal model of disease.4 A predominant strategy to optimize AAV vectors is to create combinatorial capsid libraries of the cap gene.5
One method to create an AAV capsid library is to use a “virtual family shuffling” protocol. In this approach, multiple variable regions (VRs) within the cap gene are altered based on alignments of numerous (often more than 100) different isolates of AAV.6 These libraries can contain hundreds of millions of unique variants that then may be subjected to selective pressures in a method known as directed evolution, whereupon multiple rounds of selection are used to single out the “fittest” sequence or sequences for the pressures applied.5,7,8 Often this process reduces a highly complex library of millions of sequences to a manageable number of individual sequences that display antibody evasion, tissue tropism, or the capsid assembling properties that were selected.9
During the selection process, the complexity of libraries gets progressively reduced. The original design is dictated by the investigator who determines the number of positions to mutate and the potential residues that each position can be mutated to; this is often more complex than 1 × 1028 unique variants.6 The following plasmid library is an intermediate step that is required to assemble the products into a DNA form that can be transfected into HEK293 cells (a common cell line for AAV production); this is typically restricted to about 1 × 108 unique variants due to the practical limitations of bacterial cells transformation efficiencies.8 Finally, the assembled viral libraries are known to have complexities of approximately 1 × 106 to 1 × 107 variants, which are significantly lower than the parent plasmid libraries.6 It is thought that the drop from plasmid library to viral library is largely due to dead-end variants that do not properly assemble.10 If the investigator can decrease the designed complexity to produce a plasmid library that contains fewer dead-end variants, then it is logical to assume there will be less of a decrease in the viral library complexity, resulting in a more representative viral capsid library that can be more effectively used in directed evolution schemas.
Fortunately, recent advancements in machine learning (ML) computational algorithms may be utilized to interpret the numerous sequences obtained in the pursuit of a more refined capsid library.11 Modern ML techniques are capable of capturing many complex interactions between variables such as thermostability,12,13 solubility,14 three-dimensional structure,15 secondary structure,16 binding-site predictions,17 protein-ligand interaction,18,19 functional class,20,21 subcellular localization,22 and protein-protein interactions.23 ML has been used in tandem with AAV vectors to develop novel approaches to answering previously difficult questions, like ML-guided protein engineering for transcranial optogenetics.24 Moreover, some groups have integrated AAV vector design with computer algorithms using clustering for peptide display25, ML for tropism targeting in Cre-recombinant mice,26 and machine-guided analysis for single amino acid substitutions,27 all to great success using libraries with theoretical complexities of 4 × 106, 3.4 × 1010, and 1.13 × 104 unique variants respectively. These advancements in computational algorithms, met with a widening acceptance in the gene therapy field, facilitate an environment conducive to further expansion of ML applications.
Previously, our group outlined a strategy to integrate the processes of rational capsid mutagenesis based on the biology of AAV with the process of directed evolution to generate highly complex capsid libraries and identify preferential motifs. One such motif in particular, D492G493E494-D499F500 in AAV2 VR-V, exhibited superior characteristics in rounds of in vivo selection.6 In the current project, we now outline a novel strategy to improve vector design, construction, and selection. Specifically, we produce an AAV2-DGE-DF-based combinatorial library and apply dedicated code to demonstrate an application of ML in gene therapy vector design. The backbone of our new library includes characteristics from our previously published library (a DGE-DF motif), monoclonal antibody-evading residues (S384T and Q385N), and proteasome-evading residues (Y444F and S498T).6,28, 29, 30, 31, 32, 33 Overall, this research focuses on producing an AAV2-scaffold combinatorial library based on our previous findings, utilizing advances in next-generation sequencing (NGS), developing code for an ML pipeline to predict viral capsid assembly, and using this trained ML algorithm to suggest improvements for future capsid libraries.
Results
CapLib8 library design
We have used an AAV2-DGE-DF variant as a parent scaffold to derive the CapLib8 library. We sought to identify derivatives of this variant that would maintain high transduction rates while hopefully acquiring targeting specificity. The library produced in this study contains mutations introduced to 33 amino acid residues by using degenerative primers. The design of these particular degenerate positions was driven by the NGS analysis of the original AAV2-based library:6 no amino acid (aa) residues were modified in VR-II, VR-III, or VR-IX as it was deemed detrimental to the overall capsid fitness. The degenerate positions and amino acid residues in the final design are listed in Figures 1A–1F.
Figure 1.

AAV2 capsid library design
(A) AAV2 amino acid residue sequence for VP1 where variable residue mutations (33 residues) and constant residue mutations (9 residues) are highlighted. (B) Three-dimensional model of a VP1 monomeric protein highlighting mutations to the backbone for the DGE-DF motif, as well as the antibody and proteasome evading mutations, S384T, Q385N, Y444F, and S498T. (C) Assembled AAV2 capsid with 60 monomers, highlighting the positions for the DGE-DF motif and antibody evading mutations made to the backbone of the library. (D) VP1 monomeric protein highlighting mutations for the 33 residues within the VRs that will receive variable mutations. (E) Assembled AAV2 capsid with 60 monomers, highlighting the mutated residues within the VRs. (F) Table specifying the 33 codons and potential residue outcomes for each variable residue receiving mutations in this study.
CapLib8 sequence distribution
In order to better understand the properties of the dataset retrieved after NGS, we analyzed the sequences and their distribution to verify their eligibility for ML training. Despite an uneven distribution of reads for the parental plasmid library and the viral progeny library, we found that the high complexity of unique variants was sufficient for training ML algorithms. A calculated 1:1 molar ratio of parental plasmid to viral progeny was anticipated to be sequenced; however, a 1:3 ratio of sequences after NGS and data processing was observed. The distribution of unique variants is visualized in Figure 2A. Specifically, the parental plasmid library resulted in 1.03 × 107 individual reads with 8.27 × 106 unique variants represented. The viral progeny library resulted in 3.28 × 107 individual reads representing 1.47 × 107 unique variants. We concluded that only a small sampling of each group was sequenced because the median coverage per variant was 1. The true complexity of the parental plasmid library is estimated to be ~1.0 × 108 unique variants because 7.44% of the sequenced viral progeny variants can be found in the sequenced parental plasmid library (i.e., 13.43 times deeper sequencing of the parental plasmid library would be needed to have full coverage of the progeny variants in the parental library).
Figure 2.

Library characterization
(A) Series of Venn diagrams depicting the sequence distribution of parental plasmid and viral progeny libraries. Left is a Venn diagram showing the sequencing results after NGS, illustrating how many of the viral progeny variants were not sequenced in the parental plasmid library. Center is a Venn diagram showing the estimated distribution of reads for the parental plasmid and viral progeny libraries, with the assumption that the majority of the viral progeny sequences were sequenced. Right is a Venn diagram representing how a more rational design of libraries would result in an increase in variants expected to make it to the viral progeny library. (B) Sequence logos illustrating the composition of residues of libraries derived from NGS data. Top is the probability distribution for positions from NGS of the parental plasmid library. Bottom is the probability distribution for positions from NGS of the viral progeny library.
A comparison of residue probabilities for the parental plasmid library and viral progeny library is presented in Figure 2B. The differences in residue probabilities between these sequence logos shows the selective process for capsids assembly. When viral progeny sequences are removed from parental plasmid sequences, the resulting pool of variants are thought to be representative of “not assembled” sequences. “Assembled” sequences are those which appear in the viral progeny NGS data. Additionally, we define accuracy in subsequent sections of this paper as the algorithms’ ability to differentiate sequences not used in training that fit into these classes “assembled” and “not assembled.”
Moreover, the gap in theoretical complexity and post-production complexity indicates that designing capsid libraries with a lower theoretical complexity, targeting residues and positions with the intent of increasing the post-production complexity, may result in higher yields of capsid library viral particles, and higher post-production complexity. Specifically, production of capsid libraries should contain as few residues as possible that exhibit high levels of selection in order to decrease the number of dead-end variants, therefore increasing the number of viable capsids and as a result increasing the complexity of the post-production library. ML is one tool that can be utilized to design the next generation of rationally designed AAV capsid libraries that fulfils these criteria.
ML training and tuning
To tune the artificial neural network (ANN), we adjusted for learning rates (LRs) and the number of nodes within a shallow ANN. The script (File S8 in Data S1) is a NumPy-based ANN that written to be used both for deep neural networks and shallow neural networks. A shallow ANN is used in this study to simplify the parameters tested. Figure 3A is a representative graph showing an ANN relative cost function and accuracy curve across 5,000 iterations for one of the training conditions (100 nodes, LR = 0.1). In this project, we define accuracy as the percent of unique variants in the testing/validation set (those sequences not used to train the algorithms) that are correctly classified as “assembled” or “not assembled.” All datasets, training and testing, contain a 1:1 ratio of “assembled” to “not assembled” variants.
Figure 3.

ML training and optimization
(A) Representative graph depicting the decreasing cost and increased accuracy for ANN learning over the course of 5,000 iterations with a LR = 0.01 in a 10-node shallow ANN. (B) Graph of testing accuracies for ANN tested with parameters in a shallow ANN for nodes 1–300 and LR = 0.01–1.0. (C) Table of physical and chemical properties for the 20 common amino acid residues used to represent the training and testing datasets in SVMs. Volume and hydropathy are normalized and represented by continuous values. (D) Bar graph depicting accuracies for 4 SVM kernels (linear, polynomial, RBF, and sigmoidal) and 2 representations (residues and physicochemical). Accuracies are obtained from testing sets distinct from the training sets. Training sets include 50,000 examples of virally isolated DNA and 50,000 examples “not assembled” variants. The testing set includes 100,000 different examples of the viral variants and 100,000 different examples of the “not assembled” variants. (E) Receiver operating characteristics curve for the ANN and SVM optimized models depicting threshold trade-offs for true positive and false positive prediction outcomes where the ANN contains 100 nodes and a LR = 0.1 and the SVM is trained using an RBF kernel and the residues representation. (F) Line graph illustrating the accuracies of ANN and SVM models for different percentiles of copy numbers gathered from NGS of virally isolated DNA. The algorithm hyper-parameters used for training were the optimized ANN (100 nodes, LR = 0.1) and the optimized SVM (RBF kernel with residues representation).
To standardize the ANN training, we performed all tests over 5,000 iterations because this results in a plateau of accuracy improvements while minimizing overfitting of the model to the training set. Training sets consisted of 50,000 unique variants from the “assembled” library and 50,000 unique variants from the “not assembled” variants pool. Testing sets consisted of 100,000 different unique variants from the “assembled” library and 100,000 different unique variants from the “not assembled” variants pool. This follows a 2× validation. Figure 3B shows the distribution of accuracies for LRs and nodes.
Across the conditions that we tested, we found that the highest accuracy occurred with 100 nodes and a LR = 0.1, resulting in a trained ANN with a testing accuracy of 68.18%. In more detail, for sequences that this trained algorithm has not yet seen, the algorithm has a 71.28% accuracy for “assembled” variants and a 65.07% accuracy for “not assembled” variants. A receiver operating characteristics curve visualizes these accuracies in Figure 3E.
Moreover, we found that training the ANN using a stricter dataset by increasing the threshold for the sequencing copy number of each variant to be included in the dataset results in an appreciable increase in accuracy within the training class. We found that predictions for an algorithm trained and testing using the top 1% of copy numbers retrieved (those variants with greater than 21 copies sequenced) yields a model with 76.23% accuracy (Figure 3F). Practically, this trained model can more accurately predict whether capsids variants will be in the top performing class.
To tune the support vector machine (SVM), we examined four different SVM kernels and two types of data representations to determine which kernel and representation captures the trends in our data most accurately. The four SVM kernels we tested include the radial basis function (RBF), linear, polynomial, and sigmoidal kernels. One representation is the binary residue representation, where each position is divided into 20 bits, 1 bit for each of the 20 common residues. Conceptually, this is a form of one-hot representation to represent nominal features.34 The second representation is the physicochemical representation where the physical and chemical properties of each residue are used to train the algorithm. Properties are derived from data gathered by Pommié et al.35 The table in Figure 3C shows the unique profile for the 20 common amino acids broken into 12 bits including volume, hydropathy, polarity, hydrogen donor, hydrogen acceptor, positive charge, negative charge, aliphatic, aromatic, sulfur containing, hydroxyl containing, and amide-containing resides. Volume and hydropathy are normalized.
We used the same randomized distribution of viral progeny library and “not assembled” variants for the training and testing sets for the SVM as we did for the ANN. Figure 3D shows the accuracies for the kernels and representations that were tested.
The residue representation for RBF kernel was found to fit the data most closely without resulting in overfitting. An accuracy of 68.49% using the testing/validation set was found where 72.19% of “assembled” variants and 64.88% of “not assembled” variants were correctly predicted. Figure 3F depicts the accuracies for the optimal hyper-parameter (RBF with residue representation) when used with datasets of different copy numbers. As with the ANN, we found an appreciable increase in accuracy when the threshold of copy numbers was increased. For the most stringent group, the top 1% of high copy number “assembling” variants, the model had an accuracy of 77.39%.
Applying ML algorithms in a single wild-type residue assay
Now that our ML algorithms have been trained, the saved parameters can be used to model the effects of hypothetical library mutation patterns. In this paper, we devise a single wild-type residue assay to determine the relative importance of each mutation position within a combinatorial library. In this assay, computer-simulated libraries are produced where 32/33 mutation positions receive mutations, leaving a single mutation position wild type. This is done for all 33 positions. Additionally, a 34th library containing 33/33 mutations is generated as a reference. The computer-simulated mutations are introduced in accordance with the probabilistic distribution of residues for each position as determined by the degenerative codons used to make the original library (see Figure 1F). Figure 4A depicts the outcome of the single wild-type assay for both the ANN and SVM models.
Figure 4.

ML application and validations
(A) Graph depicting the predicted percent of “assembled” capsids given 34 different hypothetical libraries. The hypothetical libraries are computer-generated libraries consisting of 100,000 sequences using a script that maintains the degenerate codons’ proportional output of residues. One library, “None,” includes no residues restricted to their wild type. The remaining 33 libraries are made such that each of the 33 mutation positions receive a library where it is not mutated. (B) Graph depicting the 2-fold drop in titer for the library containing mutations with the CR residues compared to a library containing mutations to NC residues. Statistical significance was determined using a two-tailed Student’s t test (∗∗∗∗p < 0.00001). (C) Graph showing the difference in the number of “assembled” and “not assembled” variants from the CR and NC mini-library. “Assembled” variants come from the NGS of virally derived DNA. “Not assembled” variants come from the NGS of the plasmid DNA before transfection, where the variants from the virally derived pool are removed. Notably, the theoretical complexity for these libraries is 8,000 unique variants, excluding variants with stop codons; however, both the CR and NC mini libraries did not have all variants sequenced. The CR mini-library had 95.20% of possible variants sequenced and the NC mini-library had 98.40% of possible variants sequenced. Statistical significance was determined using a chi-square test (∗∗∗∗p < 0.00001). (D) Graph depicting the log2 fold change in proportion of residues for the CR mini-library illustrating the selective pressure on these residues. (E) Graph depicting the log2 fold change in proportion of residues for the NC mini-library illustrating the selective pressure on these residues. Values greater than 0 indicate that positive selection occurred, values less than 0 indicate that negative selection occurred, and values equal to 0 indicate that no selection occurred.
We found that computer-simulated libraries with mutation excluding positions 495, 546, and 554 result in a higher percent of predicted “assembled” capsids. These findings suggest that N495, G546, and I554 play a more important role than the other residues in viral assembly and packaging. In other words, these ML-selected residues appear to be more critical than any other positions in our library.
If a partial library were to be synthesized where 30/33 mutation positions received mutations, leaving 3 positions wild type, we would expect a variation in the titer and final complexity. For instance, a partial library where these critical residues (CRs) are left wild type would result in a higher titer and more complex library, compared to a different partial library where these CRs are mutated and any other 3 non-CRs (NCs) are substituted for their wild type.
Validation of ML-predicted residues
To examine the biological validity of the model, we constructed two mini-libraries (3/33 positions receive mutations) based on the residues determined by our ML algorithms. The first mini-library, containing the more CRs (CR mini-library), has mutations to position 495, 546, and 554. The second mini-library, containing examples of NCs (NC mini-library), has mutations to positions 532, 551, and 585. The purpose of this experiment is to illustrate the different characteristics during and after library production.
Figure 4B shows the difference in titers recorded for these libraries when ten 15 cm plates are used for each library using 200 ng of plasmid library DNA per plate. Under identical conditions, this figure illustrates the 2-fold drop in titer for the CRs compared to the NC mini-library.
NGS sequencing of the CR and NC mini libraries was performed before selection (immediately after DNA assembly before transfection) and after selection (amplified viral DNA after viral purification) to determine whether there is a difference in the distribution of assembled variants for the two libraries. Figure 4C illustrates the difference in “assembled” and “not assembled” variants for these libraries. A chi-square test of independence was performed to examine the relation between the CR and NC mini-libraries and the recorded number of unique “assembled” capsids variants. The relation between these variables was significant, X2 (1, n = 15,487) = 954.1545, p < 0.00001. The ML-derived CR mini-library produces significantly fewer “assembled” capsids than the ML-derived NC mini-library.
Moreover, an analysis of selection using the log2 fold change in proportion of sequenced residues is illustrated in Figures 4D and 4E. Log2 fold change values of 0 indicate that no selection occurred since the proportion of that residue would not have changed after selection. A log2 fold change of 1 indicates a positive selective pressure occurred where the proportion of that residue doubled in the process of selection. Finally, a log2 fold change of −1 indicates that a negative selective pressure occurred where the proportion of that residue halved in the process of selection.
We used the magnitude of the log2 fold change in proportion for the following statistical analysis because both negative selection and positive selection are regarded as a selective pressure. Moreover, stop codons are excluded from the statistical analysis because they are expected to be approximately equally selected against for both mini libraries. A two-tailed Student’s t test was performed to determine whether there is a significant difference in the selective pressures for the CR and NC mini-libraries. The magnitude log2 fold change in the 20 residues for each of the 3 positions of the CR mini-library (M = 1.49, SD = 1.1347) compared to the magnitude log2 fold change in the 20 residues for each of the 3 positions of the NC mini-library (M = 0.37, SD = 0.4569) demonstrated a statistically significant increase in selective pressure among the CR mini-library residues, t(59) = 7.02998, p < 0.00001. These findings suggest that the residues targeted in the CR mini-library undergo significantly more selective pressure than the residues in the NC library.
The findings of reduced titer, decreased unique variants assembled, and higher selective pressure for the CR mini-library compared to the NC mini-library confirm the ML algorithms’ abilities to differentiate properties of residues in the selective process of capsid assembly.
Discussion
Directed evolution of AAV combinatorial capsid libraries involves two requisite selection steps: mutant capsid assembly and cell-type-specific targeting. In this study, we developed a pipeline to use NGS data to train ANNs and SVMs geared toward capsid assembly prediction. Additionally, we demonstrated a practical application of a trained ML algorithm in the form of a single wild-type variable residue assay. In this discussion we will review the approaches taken, address some of the concerns with our models, and suggest future steps when using these algorithms for ML in AAV capsid library design.
The “virtual family shuffling” approach to construct AAV combinatorial capsid libraries is based upon simultaneous mutagenesis of multiple capsid surface variable regions (also known as “loops”).6 Since the directed evolution of AAV must navigate rigid constrains imposed by a virus’ biology,10 the majority of the introduced mutations are multidimensional and epistatic (i.e., affecting each other) in nature. Therefore, only a fraction of the available sequence space can be modified without the imposition of the negative selective pressure, and thus only a fraction of the designed and constructed library derives viable capsids. One way to “weed out” dead-end variants is to assemble individual loops as sublibraries, thus introducing an additional selection trait of structural intra-, or interloop three-dimensional compatibility. Although quite effective, this approach introduces additional steps complicating the protocol. In the current project, we set out to design a ML algorithm to identify variable regions’ residues critical for the capsid assembly step, as well as their intra- and interloop dependencies. The D492G493E494-D499F500 motif in AAV2 VR-V was used as a pre-selected molecular parent characterized by high structural and transduction fitness.6
One major potential concern is the grouping of “assembled” and “not assembled” variants. Due to a lack of data from the viral progeny library, it is likely that not all “assembling” variants were sequenced. The implications of this extend to the likely possibility that some sequences used in the “not assembled” pool of variants in fact would be “assembling” variants. Because of the 2× coverage on average of the viral progeny, it is assumed that a majority of assembling sequences were sequenced, and we suspect that this issue results in only a minor proportion of variants in the two pools being miscategorized. Consequently, a deeper sequencing of the viral progeny pool would likely ameliorate this issue. However, even with a deeper understanding of the different classes, it remains possible that some “not assembled” variants may in fact “assemble” given slightly different conditions (e.g., temperature or buffer composition). We concede that our classes are binary and do not reflect the fluidity of classes that represent more closely the truth. Knowing this, our model is still applicable in the conditions tested in this paper and can help in the rationale design of capsid libraries, if not individual sequences.
Another potential problem may stem from frequency divergence or experimental population bottlenecking. While these scenarios to describe the selection of the library may play a role in shifts in population frequency from the parental plasmid library to the viral progeny library, the biological pressures are still thought to be the primary cause of shifts in population distribution at this stage. Data published by Marsic et al.6 support this claim with the finding of numerous dead-end variants when full libraries are attempted to be produced without sublibrary intermediate.10 Moreover, multiple rounds of selection are required to fully capture selective pressures;5,7,8 therefore, even for this dataset where the parental plasmid library has already undergone sublibrary selection, the selection to the viral progeny is effectively the second round of intra-sublibrary interactions and the first round of inter-sublibrary interactions. Overall, we cannot rule out such factors as bottlenecking and frequency divergence as contributing factors, but the biological validation data and these previously published findings support the rationale for this paper’s approach.
To elaborate on current applications of these models, the analysis of this type of selection may be explored through more traditional routes, however, this paper is designed to provide a foundation for a new approach that can be built on to answer novel questions with limited resources. More broadly, the software developed in this study was designed for easy adaption for processing NGS data of binary outcomes after selective pressure in protein libraries. This paper only validates the computer algorithm to predict viral assembly, however, data collected from library selection for antibody evasion, tissue tropism, or other desirable outcomes may be easily fed through our peptide to train the algorithm. Despite the ease of use for the pipeline, biological validation of the algorithm’s predictive capabilities would need to be performed to confirm the model’s use in these applications.
Additionally, our choice to apply the trained ANN and SVM using a single wild-type residue assay allows us to compare drastically different computer algorithms on a comparatively level playing field. This novel approach facilitates the extraction of computationally derived knowledge that might otherwise be locked away in the often-unreadable trained parameters of the algorithms. This is especially true for the ANNs, which are commonly referred to as “black boxes.”36
One aspect to address is the inability to perform a deep enough sequencing to have complete coverage of the parental plasmid library. As stated in the results, a 1:1 ratio of plasmid library to assembled viral progeny library was intended; however, a 1:3 ratio was retrieved after debarcoding and sequence quality control. An average coverage of 10 or more reads per unique sequence would have been sufficient to assume that the entire library had been covered. The 1:1 sequencing ratio was predicated based on our lack of prior empirical evidence about the complexity of the library. In future libraries, we suggest running at least two SP lanes on a NovaSeq 2 × 250 platform to increase the coverage of the plasmid library. An adjusted ratio between 1:1 and 10:1 of parental plasmid to viral progeny DNA would be appropriate. Although we do not know for certain, our 1:3 ratio of reads could have arisen from inaccuracies in the Qubit fluorometric quantification of DNA concentration, incomplete barcoding of sequences due to a more efficient reaction with viral isolated DNA, or differences in the ligation of library adapters to the barcoded DNA. We suggest that an additional step of sequencing using Illumina’s MiniSeq platform could be used to assess the ratio of products and allow the investigator to make the necessary adjustments to ensure the desired ratio on the NovaSeq platform is achieved.
The future direction of our research includes combining a pipeline of ML algorithms to perform in silico selection of libraries using a combined dataset of selective pressures. This may be done by subjecting CapLib8 to individual selective pressures, such as an antibody evading pressure, tropism pressure, and thermostability pressure, each individually. Specifically, the ANN and SVM algorithms designed and validated in this experiment can easily be retooled to predict sequences which might, for example, efficiently package, target hepatocytes, and evade an individual’s specific antibodies to the virus. In all, additional datasets will be useful in expanding the utility of ML in gene therapy vector design.
Finally, our group has designed a website (https://mountainpeak.rc.ufl.edu/) that permits a user to use our trained ML algorithms to predict whether a given capsid sequence will produce a viable virus-like particle. Mountain Peak, also known as the MethOd to UNiTe Artificial INtelligence and Predictive Evaluation of AAV unKnowns, will read in strings of 33 amino acid residues that correlate to the 33 positions that received variable mutations in this project. Because the dataset used to train these algorithms contains the D492G493E494-D499F500 motif, as well as the constant mutations S384T, Q385N, Y444F, and S498T, the predictions output is for a sequence with the variable residues designated by the user and the constant residues that the algorithm trained on, with all other positions remaining wild type. The user may choose between the trained ANN and the trained SVM. Moreover, the user may input either a single sequence, or multiple sequences, so long as each 33-character string is presented on a new line. For large library datasets, we recommend using the command line with the python scripts found in the supplemental files section.
In conclusion, we outline a bioinformatics pipeline to interpret big data collected from highly complex protein libraries and predict classification outcomes. We show practically applicable data that indicates differences in selective pressures for residues previously considered equally variable. Consequently, we suggest an improved rational design for future AAV2-based capsids libraries. In doing so, we illustrate the invaluable role that a marriage between ML and protein libraries can play in helping to solve complex and largely enigmatic biological questions, such as virus-like particle assembly and packaging.
Materials and methods
Library construction
Degenerate PCR
Degenerate PCR primers (see Table S1) were ordered (Eurofins Genomics, Louisville, KY, USA) and PCR was performed using the wild-type AAV2 cap gene from pSub201 as a template and Q5 Hot Start High-Fidelity polymerase (M0494S, New England Biolabs, Ipswich, MA, USA). 25 cycles using the suggested parameters on NEB’s website were followed. PCR products were run on a 1% agarose gel and gel purified using a gel DNA recovery kit (D4007, Zymo Research, Irvine, CA, USA).
Sublibrary plasmid construction
The plasmid pSub201EagApa was used as backbone for the AAV library where the ApaI sites at 3,764 and 4,049 were removed and an EagI site at position 4,373 was added (all silent mutations). The PCR product and pSub201 was digested in CutSmart buffer (B7204S, New England Biolabs, Ipswich, MA, USA) with restriction endonucleases EagI-HF (R3505S, New England Biolabs, Ipswich, MA, USA) and ApaI (R0114S, New England Biolabs, Ipswich, MA, USA) for 25°C overnight and 37°C for 1 h respectively. A DNA isolation kit (D4004, Zymo Research, Irvine, CA, USA) was used to purify the digested products. T4 DNA ligase (M0202S, New England Biolabs, Ipswich, MA, USA) was used to ligate the PCR product to the linearized pSub201EagApa backbone (1 backbone: 3 insert molar ratio) creating a plasmid with the capsid sublibrary inserted. A bacterial transformation using electrocompetent DH10B E. coli from (C640003, Thermo Fisher Scientific, Waltham, MA, USA) where cells were left for 1 h at 37°C in Lysogeny Broth before the antibiotic, carbenicillin, was added. The culture was left to incubate at 37°C overnight. A large-scale plasmid isolation according to Heilig, Elbing, and Brent37 was followed. The procedure described above was completed five times, one for each sublibrary containing one or more VR.
Sublibrary viral preparation
For each of the five libraries, ten 15 cm tissue culture plates were used, each containing 70% confluent HEK293 cells. A PEI Max transfection (NC1038561, Fisher Scientific, Hampton, NH, USA) was performed using 200 ng of library DNA and 29.8 ug of pHelper per 15 cm plate. After 72 h, the HEK293 cells were harvested and AAV was purified using an iodixanol gradient, as described by Zolotukhin et al.38
Parental plasmid library construction
Using non-degenerate primers (Table S1), each sublibrary underwent PCR to generate linear DNA with overlapping regions between the VRs. Q5 Hot Start High-Fidelity polymerase (M0494S, New England Biolabs, Ipswich, MA, USA) was used in an overlap PCR for 18 cycles with an annealing temperature of 60°C to generate combined sublibraries. This was performed two at a time (i.e., A + B = AB; C + D = CD; AB + CD = ABCD; ABCD + E = ABCDE). After every overlap PCR, DNA products were run on a 1% agarose gel and gel purified using a gel DNA recovery kit (D4007, Zymo Research, Irvine, CA, USA). This parental library was digested, purified, ligated, transformed, and subjected to large-scale plasmid isolation identical to the methods described above in the “Sublibrary Plasmid Construction” section. This product was considered the parental plasmid library.
Viral progeny preparation
Steps identical to those described in “Sublibrary Viral Preparation” section were followed using one set of ten 15 cm cell tissues plates and the parental plasmid library as the DNA for transfection. The isolated virus was considered the “assembled” viral library.
Mini-library preparation
Steps identical to those described in “Sublibrary Viral Preparation” section were followed using 15 cm HEK293 cell tissues plates and the mini-library (CR or NC) plasmid DNA as the DNA for transfection.
Library sequencing
Barcoding samples was performed using 10 bp barcoding sequences from Illumina’s adaptor catalog with unique dual indices using i7 bases for forward reads and i5 bases for reverse barcodes. The barcodes were attached to the sequences using the 3′ end of the primers found in Table S1 and added using Q5 Hot Start High-Fidelity polymerase (M0494S, New England Biolabs, Ipswich, MA, USA) over the course of 14 cycles with an annealing temperature of 61°C. After purification using Zymo DNA Clean & Concentrator, samples were sent to the University of Wisconsin-Madison Biotechnology Center and sequenced using paired-end reading (2 × 250) with 1/4 of one SP lane containing the library DNA, loaded on a NovaSeq sequencing platform (Illumina, San Diego, CA, USA). 15% of the loaded DNA was spiked with PhiX DNA to minimize the effects of the largely homologous, non-variable regions. For the mini-library sequencing, the MiSeq v2.0 micro flow cell was used with a 10% spike of PhiX DNA and a calculated 1:1:1:1 of plasmid CR: viral CR: plasmid NC: viral NC samples.
Training ML algorithm
Sequence data preparation
NGS data was debarcoded using the default setting of TagDust2 and subjected to sequence quality controls (threshold Phred assigned Q score of 30 or greater).39 Python 3.7 was used to write several scripts to process the NGS data. AAV-ML-02_MatchPairedEnds_v2.1.py (File S1 in Data S1) was used to ensure that every paired-end sequence had a match. FLASH-1.1.2.11 was used to merge the paired end reads.40 Sequences were then converted from FASTQ to FASTA format, aligned, and VRs were extracted from the reads (discarding the homologous regions) using AAV-ML-04a_CapLib_v2.8.py (File S2 in Data S1). The Unix terminal “comm” command allowed for negative selection of sequences to create a file containing only “not assembled” sequences from the plasmid sequences selected. A final stage of sequence cleaning to use only sequences intended from the NGS primers was performed using AAV-ML-04b_CleanSequences_v2.1.py (File S3 in Data S1). Generating sample sequences with representative of the pre-selection library can be generated with AAV-ML-04c_GeneratePre-Selection-Sequences_v1.1.py (File S4 in Data S1). Training and validation/testing sets were created using the “shuf,” “sed,” and “cat” Unix commands. These datasets were created with a 10:1 ratio of training to testing/validation sequences. Training sets contained 50,000 “assembled” sequences and 50,000 “not assembled” sequences. Testing/validation sets contained 100,000 “assembled” sequences and 100,000 “not assembled” sequences. AAV-ML-05a_MLprep_ANN_v1.5.py and AAV-ML-05b_MLprep_SVMRESIDUES_v2.6.py (Files S5 and S6 in Data S1) were used to convert the 33 variable residues into a binary matrix (values of only 1 or 0) of 660 by 100,000 dimensions for training sets and 660 by 200,000 dimensions for testing/validation sets for the ANN and SVM respectively. AAV-ML-05c_MLprep_SVMPROPERTIES_v1.4.py (File S7 in Data S1) was used to convert the 33 variable residues’ properties representation into a format that can be read by the SVM.
ANN training
The Python 3.7 script AAV-ML-06a_ANN-train_v11.0.py (File S8 in Data S1) was written to train the ANN. Hyperparameters like LR, number of iterations, and number of nodes were tuned to create the neural network. The Python 3.7 script AAV-ML-06b_SVM-train_v1.7.py (File S9 in Data S1) was written to train the SVM. Two different representations (binary residues representation and residues’ physicochemical properties representation) and several kernels (RBF, linear, polynomial, and sigmoidal) were compared to optimize the SVM.
Applying ML algorithms
Using the same degenerate primers to create the plasmid library, an in silico capsid library of amino acid residue sequences were created using script AAV-ML-04d_GenerateHypotheticalCapSeq_v1.1.py (File S10 in Data S1). This script can read in a degenerate primer sequence and randomly create a user-defined number of sequences from the plasmid library, followed by translation into the respective amino acid residue sequence. These residue sequences then followed the schema outlined in “Sequence Data Preparation.” Hypothetical alterations to these primers were performed and processed through the trained ML algorithm to determine which sequences are predicted to form viable capsids. Specifically, 33 sample groups, each with 100,000 sequences, were generated. Each sample was designed to mutate all but one residue position, and this was performed for all 33 positions. Data was gathered using the AAV-ML-07b_ANN_Predict-Type2-Unknown_v4.1.py and AAV-ML-07d_SVM-Predict-Type2-Unknown_v3.0.py (Files S11 and S12 in Data S1) code written to predict what percentage of these hypothetical libraries will form “assembled” capsids for the ANN and SVM trained ML algorithms.
Acknowledgments
This work was funded by NIH/NYI R01 EY024280.
Author contributions
Conceptualization, A.D.M., A.B., M.K., and S.Z.; Methodology, A.D.M., and S.Z.; Software, A.D.M., O.M., and M.K.; Validation, A.D.M., and M.K.; Investigation, A.D.M.; Writing – Original Draft, A.D.M.; Writing – Review & Editing, A.D.M. and S.Z.; Funding Acquisition, S.Z.; Resources, S.Z.; Supervision, A.D.M, O.K., and S.Z.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.11.017.
Supplemental information
References
- 1.Muzyczka, N. and Berns, K. (2001). Parvoviridae: the viruses and their replication. In Fields Virology, In D.M. Knipe, P.M. Howley, D.E. Griffen, R.A. Lamb, M.A. Martin, B. Roizman, and S.E. Straus, ed. (Lippincott Williams and Wilkins), pp. 2327–2359.
- 2.Rose J.A., Maizel J.V., Jr., Inman J.K., Shatkin A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971;8:766–770. doi: 10.1128/jvi.8.5.766-770.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonntag F., Köther K., Schmidt K., Weghofer M., Raupp C., Nieto K., Kuck A., Gerlach B., Böttcher B., Müller O.J. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011;85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinn E., Pacouret S., Khaychuk V., Turunen H.T., Carvalho L.S., Andres-Mateos E., Shah S., Shelke R., Maurer A.C., Plovie E. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015;12:1056–1068. doi: 10.1016/j.celrep.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maheshri N., Koerber J.T., Kaspar B.K., Schaffer D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 6.Marsic D., Govindasamy L., Currlin S., Markusic D.M., Tseng Y.S., Herzog R.W., Agbandje-McKenna M., Zolotukhin S. Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol. Ther. 2014;22:1900–1909. doi: 10.1038/mt.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asuri P., Bartel M.A., Vazin T., Jang J.H., Wong T.B., Schaffer D.V. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol. Ther. 2012;20:329–338. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller O.J., Kaul F., Weitzman M.D., Pasqualini R., Arap W., Kleinschmidt J.A., Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 9.Li W., Asokan A., Wu Z., Van Dyke T., DiPrimio N., Johnson J.S., Govindaswamy L., Agbandje-McKenna M., Leichtle S. Engineering and Selection of Shuffled AAV Genomes: A New Strategy for Producing Targeted Biological Nanoparticles. Mol. Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 10.Grimm D., Zolotukhin S. E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal–Tailored Acceleration of AAV Evolution. Mol. Ther. 2015;23:1819–1831. doi: 10.1038/mt.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang K.K., Wu Z., Arnold F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods. 2019;16:687–694. doi: 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- 12.Giollo M., Martin A.J., Walsh I., Ferrari C., Tosatto S.C. NeEMO: a method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genomics. 2014;15:S7. doi: 10.1186/1471-2164-15-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dehouck Y., Grosfils A., Folch B., Gilis D., Bogaerts P., Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics. 2009;25:2537–2543. doi: 10.1093/bioinformatics/btp445. [DOI] [PubMed] [Google Scholar]
- 14.Khurana S., Rawi R., Kunji K., Chuang G.Y., Bensmail H., Mall R. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics. 2018;34:2605–2613. doi: 10.1093/bioinformatics/bty166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopf T.A., Colwell L.J., Sheridan R., Rost B., Sander C., Marks D.S. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sønderby S.K., Winther O. Protein Secondary Structure Prediction with Long Short Term Memory Networks. arXiv. 2014 arXiv: 1412.7828. [Google Scholar]
- 17.Jiménez J., Doerr S., Martínez-Rosell G., Rose A.S., De Fabritiis G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics. 2017;33:3036–3042. doi: 10.1093/bioinformatics/btx350. [DOI] [PubMed] [Google Scholar]
- 18.Mazzaferro C. 2017. Predicting Protein Binding Affinity With Word Embeddings and Recurrent Neural Networks. bioRxiv. [DOI] [Google Scholar]
- 19.Gomes J., Ramsundar B., Feinberg E.N., Pande V.S. 2017. Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity. arXiv. arXiv: 1703.10603. [Google Scholar]
- 20.Cao R., Freitas C., Chan L., Sun M., Jiang H., Chen Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules. 2017;22:1732. doi: 10.3390/molecules22101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bileschi M.L., Belanger D., Bryant D., Sanderson T., Carter B., Sculley D., DePristo M.A., Colwell L.J. 2019. Using Deep Learning to Annotate the Protein Universe. bioRxiv. [DOI] [PubMed] [Google Scholar]
- 22.Yu C., Chen Y., Lu C., Hwang J. Wiley Online Library; 2006. Prediction of protein subcellular localization Proteins: Structure, Function, and Bioinformatics. [DOI] [PubMed] [Google Scholar]
- 23.Hu J., Liu Z. 2017. DeepMHC: Deep Convolutional Neural Networks for High-performance peptide-MHC Binding Affinity Prediction. bioRxiv. [DOI] [Google Scholar]
- 24.Bedbrook C.N., Yang K.K., Robinson J.E., Mackey E.D., Gradinaru V., Arnold F.H. Machine learning-guided channelrhodopsin engineering enables minimally invasive optogenetics. Nat. Methods. 2019;16:1176–1184. doi: 10.1038/s41592-019-0583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidsson M., Wang G., Aldrin-Kirk P., Cardoso T., Nolbrant S., Hartnor M., Mudannayake J., Parmar M., Björklund T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA. 2019;116:27053–27062. doi: 10.1073/pnas.1910061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravindra Kumar S., Miles T.F., Chen X., Brown D., Dobreva T., Huang Q., Ding X., Luo Y., Einarsson P.H., Greenbaum A. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods. 2020;17:541–550. doi: 10.1038/s41592-020-0799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogden P.J., Kelsic E.D., Sinai S., Church G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366:1139–1143. doi: 10.1126/science.aaw2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochrie M.A., Tatsuno G.P., Christie B., McDonnell J.W., Zhou S., Surosky R., Pierce G.F., Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabinowitz J., Chan Y.K., Samulski R.J. Adeno-associated Virus (AAV) versus Immune Response. Viruses. 2019;11:E102. doi: 10.3390/v11020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong L., Li B., Mah C.S., Govindasamy L., Agbandje-McKenna M., Cooper M., Herzog R.W., Zolotukhin I., Warrington K.H., Jr., Weigel-Van Aken K.A. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markusic D.M., Herzog R.W., Aslanidi G.V., Hoffman B.E., Li B., Li M., Jayandharan G.R., Ling C., Zolotukhin I., Ma W. High-efficiency transduction and correction of murine hemophilia B using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol. Ther. 2010;18:2048–2056. doi: 10.1038/mt.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrs-Silva H., Dinculescu A., Li Q., Deng W.T., Pang J.J., Min S.H., Chiodo V., Neeley A.W., Govindasamy L., Bennett A. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol. Ther. 2011;19:293–301. doi: 10.1038/mt.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabriel N., Hareendran S., Sen D., Gadkari R.A., Sudha G., Selot R., Hussain M., Dhaksnamoorthy R., Samuel R., Srinivasan N. Bioengineering of AAV2 capsid at specific serine, threonine, or lysine residues improves its transduction efficiency in vitro and in vivo. Hum. Gene Ther. Methods. 2013;24:80–93. doi: 10.1089/hgtb.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris D., Harris S. Second Edition. Elsevier Science; 2012. Digital Design and Computer Architecture; p. 712. [Google Scholar]
- 35.Pommié C., Levadoux S., Sabatier R., Lefranc G., Lefranc M. IMGT Standardized Criteria for Statistical Analysis of Immunoglobulin V-REGION Amino Acid Properties. J. Mol. Recognit. 2004;17:17–32. doi: 10.1002/jmr.647. [DOI] [PubMed] [Google Scholar]
- 36.Price W.N. Big data and black-box medical algorithms. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aao5333. eaao5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heilig J.S., Elbing K.L., Brent R. Large-scale Preparation of Plasmid DNA. Curr. Protoc. Nol. Biol. 2001;Chapter 1 doi: 10.1002/0471142727.mb0107s41. Unit 1.7. [DOI] [PubMed] [Google Scholar]
- 38.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 39.Lassmann T. TagDust2: a generic method to extract reads from sequencing data. BMC Bioinformatics. 2015;16:24. doi: 10.1186/s12859-015-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.