

Abstract
In this article we investigate the electrochemical reduction of CO2 at gold electrodes under mildly acidic conditions. Differential electrochemical mass spectroscopy (DEMS) is used to quantify the amounts of formed hydrogen and carbon monoxide as well as the consumed amount of CO2. We investigate how the Faradaic efficiency of CO formation is affected by the CO2 partial pressure (0.1–0.5 bar) and the proton concentration (1–0.25 mM). Increasing the former enhances the rate of CO2 reduction and suppresses hydrogen evolution from proton reduction, leading to Faradaic efficiencies close to 100%. Hydrogen evolution is suppressed by CO2 reduction as all protons at the electrode surfaces are used to support the formation of water (CO2 + 2H+ + 2e– → CO + H2O). Under conditions of slow mass transport, this leaves no protons to support hydrogen evolution. On the basis of our results, we derive a general design principle for acid CO2 electrolyzers to suppress hydrogen evolution from proton reduction: the rate of CO/OH– formation must be high enough to match/compensate the mass transfer of protons to the electrode surface.
Introduction
Carbon monoxide (CO), as a component in syngas, constitutes a C1 building block of high economic importance as it forms the basis for the synthesis of a range of chemicals.1 However, its production from fossil resources is accompanied by the emission of CO2. Electrochemical CO2 reduction to CO has therefore the potential to play an important role in the decarbonization of a significant portion of the chemical industry, provided the used electricity is generated by zero-emission technologies. However, to render electrochemical CO2 reduction economically beneficial, it must proceed both with high Faradaic efficiency and high energy efficiency. That is, although hydrogen is a component of syngas, it is economically more beneficial to produce H2 and CO in two separate processes, which are each optimized for the respective reaction.
Recently, Vennekoetter et al. investigated how both quantities are affected by the electrolyzer design.2 They found that a zero-gap arrangement in which gas diffusion electrodes (GDE) for CO2 reduction and oxygen evolution (OER), respectively, are in direct contact with a Nafion membrane achieves the highest energy efficiency among all tested designs.2 However, the Faradaic efficiency of this electrolyzer was close to zero.2 Since the current is mainly transported by protons through the Nafion membrane, the major reaction occurring at the Ag catalyst is the discharge of protons to hydrogen rather than the reduction of CO2.2 Although CO2 reduction has often been reported to proceed with Faradaic efficiencies close to 100%,3−5 these values are usually achieved at neutral pH, where hydrogen evolution from reduction of water (not of protons) is the competing reaction. As proton reduction has an earlier onset potential than water reduction, CO2 reduction in acidic electrolytes tends to have low Faradaic efficiencies.6,7 Neutral reaction conditions and therefore higher Faradaic efficiencies for CO2 reduction can be achieved when zero-gap electrolyzers are constructed with anion-conducting membranes.8,9 However, the latter also allows the crossover of bicarbonate to the anode, thus leading to the loss of CO2.2,9
Considering the advantages of acidic electrolytes for technical processes (higher conductivity, facile OER kinetics,10 availability of more efficient electrolyzer designs,2,9 and no crossover of HCO3– 2,9), it would be interesting to achieve high Faradaic efficiencies for CO2 reduction in electrolytes of low pH. As there are only a few examples of CO2 reduction under acidic conditions,6,7 we investigate here CO2 reduction at a gold electrode from electrolytes of mild acidity. Using the differential electrochemical mass spectrometry (DEMS) technique,11 in combination with a flow cell,12 we are able to quantify online the amounts of evolved hydrogen and CO formed during or parallel to CO2 reduction. Furthermore, we can monitor the consumption of CO2. Our results indicate that matching the mass transport of protons to the electrode with the CO2 reduction rate can be used to suppress hydrogen evolution in a limited potential range. Our results provide another example of the importance of mass transport to suppress hydrogen evolution and to increase the Faradaic efficiency of CO2 reduction.13
Experimental Section
Chemicals and Instruments
All electrochemical measurements were conducted with an IviumStat (Ivium Technology) with a compliance voltage of 20 V. Potentials were measured against a commercial Ag|AgCl reference electrode (Metrohm). Ar/CO2 gas mixtures featuring a CO2 partial pressure of 0.1, 0.3, and 0.5 bar, respectively, were prepared by adjusting the flow of Ar and CO2 through the electrolyte via a mass flow controller. The electrolyte was prepared from NaClO4 (HPLC grade, Sigma-Aldrich) and HClO4 (suprapur, Merck Millipore).
Roughening of the Gold Electrode
The gold electrode was roughened in an electrolyte of 0.5 M KCl by exposing it to a potential program in which first a value of −0.4 V vs Ag|AgCl is held for 20 s, from which it is then stepped for 5 s to 1.2 V vs Ag|AgCl. After several cycles the electrode turned black and had roughened considerably. The true surface area is determined from the charge passed during gold oxide formation in the potential region between 0.84 and 1.4 V vs Ag|AgCl, assuming a specific capacity of 420 μC/cm2.
DEMS Setup
The DEMS setup used for this study has been described earlier14 and follows the design by Wolter and Heitbaum.11 A schematic drawing of the DEMS setup can be found in a review by Baltruschat.15 The cathode potential of the ion source of the mass spectrometer was set to −27.5 V (vs ground) to avoid fragmentation of CO2 to CO+ during electron impact ionization. As described earlier, this allows us to assign the evolution of a signal in the ionic current for mass 28 to the formation of CO.14
The flow cell used for this study was the dual thin layer cell first introduced by Jusys et al.,12 which allows defined mass transport conditions.12,15 In brief, the electrolyte purged with an Ar/CO2 mixture first flows through the compartment of the working electrode. Electrochemical products formed at the working electrode are swept along with the electrolyte. In the second compartment the electrolyte flows over a porous Teflon membrane. The latter rests on a steel frit for mechanical support and is in direct contact with the vacuum of the mass spectrometer. Because of its surface tension, water cannot penetrate the pores of the Teflon membrane—however, volatile compounds in the electrolyte evaporate and are detected via mass spectroscopy. From there the electrolyte flows out of the cell and is discarded. The flow rate of the electrolyte is controlled by a syringe pump.
Calibration
Calibration of the experimental setup for hydrogen was achieved by evolving H2 from the blank electrolyte of 0.5 M NaClO4 containing 1 mM HClO4 under the same experimental conditions under which the actual experiment was conducted. In the absence of CO2, hydrogen evolution at the polycrystalline gold electrode proceeds with 100% Faradaic efficiency. According to eq 1a, the ratio of the Faradaic current due to hydrogen formation (IF(H2)) divided by the number of transferred electrons (z), Faraday’s constant (F), and the signal in the ionic current for mass 2 (II(2), that is, the response of the mass spectrometer for mass (2) constitutes the calibration constant for hydrogen K*(2).
| 1a |
Once K*(2) is known, the signal in the ionic current for mass 2 measured during the CO2 reduction experiment can be converted into the partial Faradaic current for hydrogen formation.
The calibration for CO is achieved by the electrochemical oxidation of CO. Because of the electrochemical consumption of CO, the signal in the ionic current for mass 28 turns negative after baseline correction. The calibration constant for CO (K*(28)) is obtained according to eq 1b from the ratio of the Faradaic current for CO oxidation IF(COox) and the (negative) ionic current for mass 28, II(28), excluding the potential region where the gold surface is oxidized. As CO oxidation yields CO2, we can determine the calibration constant K*(44) for CO2 from the positive signal in the ionic current for mass 44, II(44) and IF(COox), according to eq 1c.
| 1b |
| 1c |
Results and Discussion
Figure 1 shows DEMS experiments of CO2 reduction on a roughened polycrystalline gold electrode with a roughness factor of 20.3 in an electrolyte of 0.5 M NaClO4 + 1 mM HClO4. The top panels (A–C) show in black the measured CV when the electrolyte is purged with an Ar/CO2 mixture featuring CO2 partial pressures of 0.1 bar (A), 0.3 bar (B), and 0.5 bar (C), respectively. The middle panels below (D–F) show the ionic current for mass 2 as a function of the applied potential. The lower panels (G–I) show the ionic current for mass 28, which is proportional to the electrochemically evolved amount of CO.14 The mass spectroscopic traces were measured in parallel to the electrochemical trace.
Figure 1.

DEMS data for the electrochemical CO2 reduction at a polycrystalline gold electrode with a roughness factor of 20.3 (exposed geometric surface: area 0.283 cm2). (A–C) Measured CV (black) and CV predicted from the amounts of evolved H2 and CO (red); (D–F) ionic current for mass 2 corresponding to the CVs in (A)–(C), respectively; (G–I) ionic current for mass 28 corresponding to the CVs in (A)–(C), respectively. Electrolyte: 0.5 M NaClO4 containing 1 mM HClO4 purged with Ar/CO2 mixtures featuring different CO2 partial pressures: 0.1 bar (A, D, and G), 0.3 bar (B, E, and H), and 0.5 bar (C, F, and I). Sweep rate: 20 mV/s. Flow rate: 5 μL/s.
The red curves in Figure 1A–C is the sum of the partial Faradaic currents of hydrogen and CO formation. Both quantities were determined from the ionic currents for masses 2 (shown in Figure 1D–F) and 28 (shown in Figure 1G–I) via eqs 1a and 1b, respectively. The Faradaic current expected from the sum of the partial Faradaic current of H2 and CO formation shows good overlap with the overall Faradaic current measured by the potentiostat below −0.5 V vs Ag|AgCl. That is, for all Ar/CO2 mixtures, the formed amounts of H2 and CO determined mass spectroscopically account for the charge passed in the potential region of CO2 reduction. This suggests that no other products than H2 and CO are formed, which is also supported by the absence of any signal in the ionic current for masses 16, 27, and 30, indicative of methane, ethylene, and acetaldehyde, respectively. Therefore, our results obtained in mildly acidic electrolytes do not differ from CO2 reduction at gold at higher pH.4,5
As the reaction is conducted under steady flow, a constant current due to mass transport limited proton reduction is expected in Figure 1.6,7,12,15 This behavior is observed both in the Faradaic current (Figure 2A) and the ionic current for mass 2 (Figure 2B) when an argon-purged electrolyte of 0.5 M NaClO4 containing 1 mM HClO4 is used. The reduction process occurring when the potential is scanned below −0.6 V vs Ag|AgCl can be assigned exclusively to the evolution of hydrogen. This is not only indicated by the ionic current for mass 2 that mirrors the behavior of the Faradaic current but also dictated by logic as this is the only possible reduction reaction in the absence of other electroactive species in the electrolyte. Once a potential of −0.95 V vs Ag|AgCl is passed in the negative-going direction, hydrogen evolution enters a steady value, indicative of a mass transport limited current.
Figure 2.

DEMS data of an experiment in which the electrolyte of 0.5 M NaClO4 containing 1 mM HClO4 was purged with Ar. (A) CV; (B) ionic current for mass 2. Sweep rate: 20 mV/s. Flow rate: 5 μL/s. Working electrode: polycrystalline gold.
In Figure 1, a signal evolves in the ionic current for mass 2 as a potential of −0.6 V vs Ag|AgCl is passed. Hence, the presence of CO2 does not affect the onset potential of proton reduction. However, diffusion limitation as in Figure 2 is not achieved. This does not become evident from the CVs shown in Figure 1A–C, but from the ionic current for mass 2, which has a peak at −0.9 V vs Ag|AgCl and goes through a minimum at −1.28 V vs Ag|AgCl, the current of which decreases as the CO2 partial pressure increases. In parallel, the CO formation rate increases (i.e., ionic current for mass 28) with increasing CO2 partial pressure. In addition, the peak in the formation rate of hydrogen at −0.9 V vs Ag|AgCl coincides with the onset of CO formation at −0.85 V vs Ag|AgCl, suggesting that CO2 reduction suppresses hydrogen evolution. The same behavior is observed for electrolytes with lower proton concentration, as shown in Figures S1–S3 of the Supporting Information. Although neither CO nor H2 evolution enter diffusion limitation, the CVs in Figure 1 appear to feature a limiting current. This is due to the increasing partial current as a result of CO formation, which compensates the decrease due to decreasing H2 evolution.
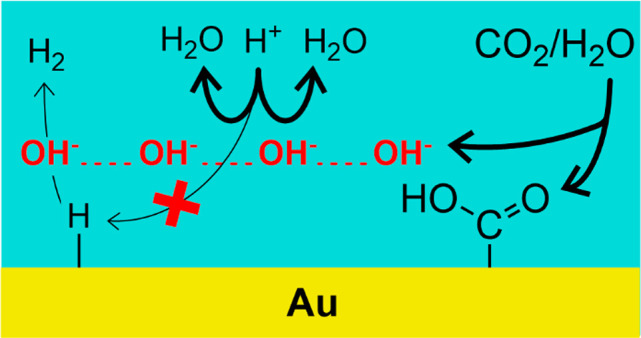
As shown in reactions 2a and 2b, the reduction of CO2 to CO leaves an oxygen atom in oxidation state −II, which will react with protons or water to form water or two OH– ions.
| 2a |
| 2b |
In reaction 2a CO2 reacts with water instead of protons because we expect a local pH of 7 or higher at the electrode surface. That is, at the onset of CO2 reduction, the reduction of protons has nearly reached diffusion limitation, which indicates a local pH of close to 7. Because of the OH– formed in reaction 2a and because of water reduction, the local pH is bound to rise beyond 7 once the capacity of the CO2/HCO3– buffer is exhausted. Only in the very early stages of CO2 reduction, proton discharge is not yet fully diffusion limited, which leaves some protons for reaction 2b to proceed. Since OH– formed via reaction 2a diffuses away from the electrode surfaces, it can intercept protons before they reach the electrode (reaction 3), which are then no longer available to support hydrogen evolution.
| 3 |
A similar mechanism was previously invoked to explain lack of hydrogen evolution in mildly acidic electrolytes parallel to oxygen reduction.16,17 The combination of reactions 2a and 3 is equivalent to reaction 2b, but with the important distinction that protons do not directly react with CO2 but rather with the OH– generated. Such a mechanism is in agreement with the experimental observation that CO2 reduction is pH-independent,18 that is, the relevant hydrogen donor for CO2 is water, not protons.19,20 The mechanistic interpretation for this observation is that CO2 is activated by electron transfer, decoupled from proton transfer, leading to a negatively charged (or polarized) CO2 intermediate, bound to the gold surface.21
In principle, the observations made in Figure 1 could also be interpreted as the competition of reduced CO2 species22 and protons for the same adsorption sites at the gold electrode.23 Following the argumentation of Chaplin and Wragg,23 adsorption of CO2 species, which becomes increasingly favorable as overpotential or CO2 partial pressure, blocks the adsorption of protons and therefore their reduction to H2. However, if proton reduction was a surface-limited process, the rate of proton reduction should decrease with decreasing roughness of the gold electrode. The opposite is observed in Figure S4, where the experiment of Figure 1 is repeated at a smooth gold electrode. With decreasing roughness factor, proton reduction increases, thus ruling out a mechanism based on competitive adsorption. As the CO formation rate is limited by the reaction kinetics, it scales with the true surface area and decreases with the electrode roughness. However, the availability of protons is limited by mass transport, which scales with the geometric surface area of the electrode. Although the flux of protons remains constant, less are used during CO formation, which leaves more protons for hydrogen evolution.
The competition for protons has a positive effect on the Faradaic efficiency for CO formation, which can reach 100% even in mildly acidic electrolytes. This is shown in Figure 3, which compares the Faradaic efficiency for CO formation as a function of potential for CO2 reduction from electrolytes with four different proton concentrations and three different CO2 partial pressures. The data from which the Faradaic efficiency in Figure 3 was calculated are shown in Figure 1 and Figures S1–S3 (Supporting Information). For any given proton concentration, the Faradaic efficiency increases with increasing CO2 partial pressure (panels A–C). This is due to both an increase of the CO formation rate and the increased suppression of hydrogen evolution. Of course, a higher proton concentration increases the rate of hydrogen evolution, and thereby lowers the Faradaic efficiency of CO formation. While the Faradaic efficiency of CO formation at the smooth electrode (c.f. Figure S5) shows the same behavior as that in Figure 3, the absolute values are consistently lower than those at the roughened gold electrode.
Figure 3.

Faradaic efficiency of CO2 reduction at a polycrystalline gold electrode with a roughness factor of 20.3 (negative-going scan only). The electrolyte was an aqueous solution of 0.5 M NaClO4 containing 1 mM (black), 0.63 mM (red), 0.4 mM (blue), and 0.25 mM (magenta) of HClO4, respectively. The electrolyte was purged with Ar/CO2 mixtures featuring different CO2 partial pressures: 0.1 bar (A), 0.3 bar (B), and 0.5 bar (C). Sweep rate: 20 mV/s. Flow rate: 5 μL/s. The corresponding DEMS data from which the Faradaic efficiency was calculated are shown in Figure 1 and Figures S1–S3.
In Figure 3, the potential dependence of the Faradaic efficiency has a bell shape, which reaches its maximum in the potential range between −1.2 and −1.3 V vs Ag|AgCl, from which it decreases as the potential is made more negative.
This behavior can be understood from Figure 1D–F (as well as Figures S1–S3 in the Supporting Information), where the amounts of evolved hydrogen start to increase again as the potential is scanned below −1.28 V vs Ag|AgCl. In this potential region hydrogen evolution due to water reduction begins to take place,6,7 which is the dominant reason for the decreasing Faradaic efficiency. However, it also decreases because the CO formation rate either drops (Figure 1G,H) or its potential-dependent increase begins to flatten out (Figure 1I).
Closer inspection of Figures S1–S3 in the Supporting Information show that the formation rate of CO, that is, the partial Faradaic current due to CO formation, exceeds the decrease in the rate of proton discharge. That is, fewer protons are consumed during CO2 reduction than the CO formation rate suggests. In the potential range prior to water reduction, we can quantify the flux FCO2(H+) of protons that are consumed during CO2 reduction from eq 4
| 4 |
where If(2) is the partial Faradaic current of hydrogen evolution determined from the ionic current for mass 2, If,diff(2) is the diffusion-limited current of proton reduction when no CO2 reduction takes place, F is the Faraday constant, and z is the number of transferred electrons. The difference (If,diff(2) – If(2)) enters eq 4 because the current of protons diffusing to the surface (i.e., If,diff(2)) minus the protons reacting to hydrogen (i.e., If(2)) represent the current of protons that participate in a different reaction (i.e., CO2 reduction). In a similar way we can calculate from eq 5 the flux of CO formed during CO2 reduction (FCO2(CO))
| 5 |
where If(28)represents the partial Faradaic current of CO formation. Furthermore, we can determine from the ionic current for mass 44 (c.f. Figure S6) the flux of CO2, F(CO2), that is consumed parallel to CO formation. Figure 4 shows FCO2(H+) divided by 2, FCO2 (CO), and F(CO2) as a function of the applied potential.
Figure 4.

Top panels: Flux of protons divided by 2 (black), of CO2 (blue), and of CO (red) that are consumed and produced during CO2 reduction, respectively. Bottom panels: Proton deficit (violet) and CO2 surplus (olive). The electrolyte was 0.5 M NaClO4 + 1 mM HClO4 purged with an Ar/CO2 gas mixture featuring a CO2 partial pressure of 0.1 bar (A and D), 0.3 bar (B and E), and 0.5 bar (C and F). Working electrode: Au(pc) (roughness factor, 20.3; exposed geometric surface, area 0.283 cm2). Sweep rate: 20 mV/s. Flow rate: 5 μL/s.
Since the formation of 1 molecule of CO consumes 1 molecule of CO2 and 2 protons, the black, red, and blue curves in Figure 4 should overlap. At low overpotentials this is indeed the case in Figures 4A–C, where because of the low reaction rate, the CO formation rate is low. However, at higher overpotentials in Figure 4B,C, where the formation rate of CO is higher because of the higher CO2 pressure, only a fraction of the consumed CO2 is actually reduced to CO. On the other hand, the number of protons participating in the reduction of CO2 is not large enough to support the observed CO formation rate. We can calculate this proton deficit, DH+, from eq 6:
| 6 |
and the surplus of CO2 consumption SCO2 from eq 7:
| 7 |
With increasing CO formation rate, both DH+ and SCO2 (lower panels of Figure 4) increase and overlap with good agreement. The same behavior is observed in Figures S7–S9 of the Supporting Information, which present the same data as Figure 4 for electrolytes with a proton concentration of 0.63, 0.4, and 0.25 mM, respectively. The fact that DH+ and SCO2 overlap quite well suggests that the proton deficit is compensated by CO2 forming bicarbonate near the electrode surface. That is, OH– formed during CO2 reduction via reaction 2a is not only neutralized by protons as in reaction 3 but also reacted with CO2 to form bicarbonate according to reaction 8.
| 8 |
The good overlap between that DH+ and SCO2 also means that we can completely account for the mass balance of CO2 consumption. That is, CO2 is consumed as a result of CO formation and the reaction with OH– formed during CO2 reduction as well as water reduction.
We show in the Supporting Information that SCO2 equals the rate of reaction 8 (i.e., rate of bicarbonate formation), whereas the consumption of protons during CO formation (i.e., FCO2(H+)) equals twice the rate of CO formation (rate of reaction 2a) minus the rate of bicarbonate formation (rate of reaction 8). This implies that complete suppression of proton reduction requires a CO formation rate that is equal to or exceeds the mass transport rate of protons to the electrode surface. Only then is the formation rate of OH–, which forms according to reaction 2a along with CO, sufficient to intercept all protons diffusing toward the electrode surface. We believe that this is a key guiding principle for designing an efficient electrolyzer for electrochemical CO2 reduction in acid media: it must accommodate such high CO2 reduction rates that the OH– formed as a byproduct can neutralize all protons that would otherwise participate in hydrogen evolution.
With reaction 8 we can also understand why we observe in Figure 1 that the CO formation rate flattens out or decreases as the potential decreases below −1.4 V vs Ag|AgCl. At this potential water reduction leads to the additional formation of OH–, resulting in the consumption of CO2. As shown in Figures S10–S12, this leads even under mass transport control to a significant drop of the CO2 partial pressure at the electrode surface. The potential-dependent increase of the rate constant of CO2 reduction cannot compensate for the effect of the decreasing local CO2 concentration. As a result, the current due to CO2 reduction drops. This is not limited to the potential region of water reduction. Also, in the potential region of proton reduction the CO formation rate increases slightly in Figures S1–S3 with increasing proton concentration. This is remarkable as protons are not involved in the rate-determining step, which means that their concentration should not affect the rate of CO2 reduction.22,18−21 However, a higher proton concentration means that a larger share of OH– formed during CO2 reduction is neutralized via reaction 3. Therefore, a higher proton concentration maintains a higher local CO2 concentration at the electrode surface and supports indirectly a higher CO formation rate.
Conclusion
We have shown in this article that CO2 reduction suppresses hydrogen evolution from proton reduction. Despite the less negative onset potential for proton reduction compared to that for CO2 reduction, a Faradaic efficiency for CO formation close to 100% can be achieved in mildly acidic electrolytes. The effect of the surface roughness of the electrode shows that this phenomenon does not arise because protons and CO2 compete for the same adsorption sites. It is rather due to the consumption of protons in an acid/base reaction with OH– formed during CO2 reduction. When all protons react off with OH– before they can reach the electrode surface, their discharge is suppressed entirely. However, OH– can also react with CO2 to bicarbonate, which manifests itself in our experiments in a higher CO2 consumption rate than expected from the rate of CO formation. Therefore, it is not sufficient to match the mass transport of protons with the formation rate of OH–, which equals the formation rate of CO, to suppress hydrogen evolution entirely. Furthermore, bicarbonate formation reduces the partial pressure of CO2 at the electrode surface. This becomes particularly severe once a potential is reached at which water reduction sets in. The increased consumption of CO2 due to the formation of OH– from water reduction leads eventually to a drop in the CO formation rate. However, also prior to water reduction bicarbonate formation reduces the CO2 partial pressure and therefore the CO formation rate. In electrolytes with low proton concentration the effect is slightly larger since a larger share of OH– reacts to bicarbonate but not to water. Therefore, the proton concentration influences indirectly the rate of CO2 reduction via the local CO2 concentration, although they do not participate in the rate-determining step.
Suppressed proton reduction as described in this article is interesting as it leads to Faradaic efficiencies close to 100% for CO2 reduction in acidic electrolytes. Although this finding is limited in our work to small proton concentrations and to low partial pressures of CO2, our results suggest that proton reduction can be suppressed also in electrolytes with significantly higher proton concentrations if the CO2 reduction rate can be increased accordingly. Under industrial conditions this might be achieved by conducting CO2 reduction under high CO2 pressures and by the use of GDEs featuring high roughness factors. A high surface area (higher roughness factor), enhanced mass transport of CO2 to the electrode surface (GDE), and higher local CO2 concentrations (high CO2 pressure) increase the CO2 reduction current that can be achieved per geometric electrode area. Since the mass transport of protons scales with the geometric surface area of the electrode, the OH– formed as byproduct of CO2 reduction can neutralize all protons that arrive at the catalyst surface and that would otherwise participate in hydrogen evolution. On the other hand, a sufficiently high proton concentration is required so that the formed OH– is neutralized predominantly by protons and not by CO2. Because of higher conductivity, better OER kinetics,10 better electrolyzer design,2,9 and the absence of HCO3– crossover,2,9 acidic electrolytes could be beneficial for technical processes. Our experimental results are of particular relevance for CO2 reduction electrodes that follow the design principle of oxygen depolarized cathodes.24 More research is need to determine whether they are of similar consequence when the catalyst layer of a GDE is in direct contact with a solid polymer electrolyte.25
Acknowledgments
We gratefully acknowledge financial support from The Netherlands Organization for Scientific Research (NOW) and Shell Global Solutions in the framework of the Advanced Research Center Chemical Building Blocks Consortium (ARC-CBBC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10397.
Partial Faradaic currents for CO and H2 formation; fluxes of CO2, H+, and CO for electrolytes with different proton concentrations; DEMS data and Faradaic efficiencies at a smooth gold electrode; local CO2 partial pressure (PDF)
Author Present Address
# Analytische Chemie II, Ruhr-Universität Bochum, Universitätsstr. 150, Bochum 44780, Germany.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Rostrup-Nielsen J. R. Syngas in perspective. Catal. Today 2002, 71 (3–4), 243–247. 10.1016/S0920-5861(01)00454-0. [DOI] [Google Scholar]
- Vennekoetter J.-B.; Sengpiel R.; Wessling M. Beyond the catalyst: How electrode and reactor design determine the product spectrum during electrochemical CO2 reduction. Chem. Eng. J. 2019, 364, 89–101. 10.1016/j.cej.2019.01.045. [DOI] [Google Scholar]
- a Hoshi N.; Kato M.; Hori Y. Electrochemical reduction of CO2 on single crystal electrodes of silver Ag(111), Ag(100) and Ag(110). J. Electroanal. Chem. 1997, 440 (1–2), 283–286. 10.1016/S0022-0728(97)00447-6. [DOI] [Google Scholar]; b Clark E. L.; Ringe S.; Tang M.; Walton A.; Hahn C.; Jaramillo T. F.; Chan K.; Bell A. T. Influence of Atomic Surface Structure on the Activity of Ag for the Electrochemical Reduction of CO 2 to CO. ACS Catal. 2019, 9 (5), 4006–4014. 10.1021/acscatal.9b00260. [DOI] [Google Scholar]
- Cave E. R.; Montoya J. H.; Kuhl K. P.; Abram D. N.; Hatsukade T.; Shi C.; Hahn C.; Nørskov J. K.; Jaramillo T. F. Electrochemical CO2 reduction on Au surfaces: mechanistic aspects regarding the formation of major and minor products. Phys. Chem. Chem. Phys. 2017, 19 (24), 15856–15863. 10.1039/C7CP02855E. [DOI] [PubMed] [Google Scholar]
- Mezzavilla S.; Horch S.; Stephens I. E. L.; Seger B.; Chorkendorff I. Structure Sensitivity in the Electrocatalytic Reduction of CO 2 with Gold Catalysts. Angew. Chem. 2019, 131 (12), 3814–3818. 10.1002/ange.201811422. [DOI] [PubMed] [Google Scholar]
- Ooka H.; Figueiredo M. C.; Koper M. T. M. Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media. Langmuir 2017, 33 (37), 9307–9313. 10.1021/acs.langmuir.7b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-García P.; Kovács N.; Grozovski V.; Gálvez-Vázquez M. d. J.; Vesztergom S.; Broekmann P. Toward CO2 Electroreduction under Controlled Mass Flow Conditions: A Combined Inverted RDE and Gas Chromatography Approach. Anal. Chem. 2020, 92 (6), 4301–4308. 10.1021/acs.analchem.9b04999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Aeshala L. M.; Rahman S. U.; Verma A. Effect of solid polymer electrolyte on electrochemical reduction of CO2. Sep. Purif. Technol. 2012, 94, 131–137. 10.1016/j.seppur.2011.12.030. [DOI] [Google Scholar]; b Hori Y.; Ito H.; Okano K.; Nagasu K.; Sato S. Silver-coated ion exchange membrane electrode applied to electrochemical reduction of carbon dioxide. Electrochim. Acta 2003, 48 (18), 2651–2657. 10.1016/S0013-4686(03)00311-6. [DOI] [Google Scholar]
- Delacourt C.; Ridgway P. L.; Kerr J. B.; Newman J. Design of an Electrochemical Cell Making Syngas (CO+H2) from CO2 and H2O Reduction at Room Temperature. J. Electrochem. Soc. 2008, 155 (1), B42. 10.1149/1.2801871. [DOI] [Google Scholar]
- Giordano L.; Han B.; Risch M.; Hong W. T.; Rao R. R.; Stoerzinger K. A.; Shao-Horn Y. pH dependence of OER activity of oxides: Current and future perspectives. Catal. Today 2016, 262, 2–10. 10.1016/j.cattod.2015.10.006. [DOI] [Google Scholar]
- Wolter O.; Heitbaum J. Differential Electrochemical Mass Spectroscopy (DEMS) - a New Method for the Study of Electrode Processes. Berichte der Bunsengesellschaft für physikalische Chemie 1984, 88 (1), 2–6. 10.1002/bbpc.19840880103. [DOI] [Google Scholar]
- Jusys Z.; Massong H.; Baltruschat H. A New Approach for Simultaneous DEMS and EQCM: Electro-oxidation of Adsorbed CO on Pt and Pt-Ru. J. Electrochem. Soc. 1999, 146 (3), 1093–1098. 10.1149/1.1391726. [DOI] [Google Scholar]
- Goyal A.; Marcandalli G.; Mints V. A.; Koper M. T. M. Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions. J. Am. Chem. Soc. 2020, 142 (9), 4154–4161. 10.1021/jacs.9b10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondue C. J.; Koper M. T.M. A DEMS approach for the direct detection of CO formed during electrochemical CO2 reduction. J. Electroanal. Chem. 2020, 875, 113842. 10.1016/j.jelechem.2020.113842. [DOI] [Google Scholar]
- Baltruschat H. Differential electrochemical mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15 (12), 1693–1706. 10.1016/j.jasms.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Benn E. E.; Gaskey B.; Erlebacher J. D. Suppression of Hydrogen Evolution by Oxygen Reduction in Nanoporous Electrocatalysts. J. Am. Chem. Soc. 2017, 139 (10), 3663–3668. 10.1021/jacs.6b10855. [DOI] [PubMed] [Google Scholar]
- Chen W.; Liao L. W.; Cai J.; Chen Y.-X.; Stimming U. Unraveling Complex Electrode Processes by Differential Electrochemical Mass Spectrometry and the Rotating Ring-Disk Electrode Technique. J. Phys. Chem. C 2019, 123 (49), 29630–29637. 10.1021/acs.jpcc.9b09952. [DOI] [Google Scholar]
- Ringe S.; Morales-Guio C. G.; Chen L. D.; Fields M.; Jaramillo T. F.; Hahn C.; Chan K. Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on Gold. Nat. Commun. 2020, 11 (1), 33. 10.1038/s41467-019-13777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.; Kortlever R.; Kas R.; Birdja Y. Y.; Diaz-Morales O.; Kwon Y.; Ledezma-Yanez I.; Schouten K. J. P.; Mul G.; Koper M. T. M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. 10.1038/ncomms9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (32), E4585–93. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdja Y. Y.; Pérez-Gallent E.; Figueiredo M. C.; Göttle A. J.; Calle-Vallejo F.; Koper M. T. M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4 (9), 732–745. 10.1038/s41560-019-0450-y. [DOI] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6 (20), 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Chaplin R. P. S.; Wragg A. A. Effects of process conditions and electrode material on reaction pathways for carbon dioxide electroreduction with particular reference to formate formation. J. Appl. Electrochem. 2003, 33 (12), 1107–1123. 10.1023/B:JACH.0000004018.57792.b8. [DOI] [Google Scholar]
- Moussallem I.; Jörissen J.; Kunz U.; Pinnow S.; Turek T. Chlor-alkali electrolysis with oxygen depolarized cathodes: history, present status and future prospects. J. Appl. Electrochem. 2008, 38 (9), 1177–1194. 10.1007/s10800-008-9556-9. [DOI] [Google Scholar]
- Higgins D.; Hahn C.; Xiang C.; Jaramillo T. F.; Weber A. Z. Gas-Diffusion Electrode for Carbon Dioxide Reduction: A new Paradigm. ACS Energy Letters 2019, 4 (1), 317–324. 10.1021/acsenergylett.8b02035. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.