

Abstract
Optimal DNA damage response is associated with ADP‐ribosylation of histones. However, the underlying molecular mechanism of DNA damage‐induced histone ADP‐ribosylation remains elusive. Herein, using unbiased mass spectrometry, we identify that glutamate residue 141 (E141) of variant histone H2AX is ADP‐ribosylated following oxidative DNA damage. In‐depth studies performed with wild‐type H2AX and the ADP‐ribosylation‐deficient E141A mutant suggest that H2AX ADP‐ribosylation plays a critical role in base excision repair (BER). Mechanistically, ADP‐ribosylation on E141 mediates the recruitment of Neil3 glycosylase to the sites of DNA damage for BER. Moreover, loss of this ADP‐ribosylation enhances serine‐139 phosphorylation of H2AX (γH2AX) upon oxidative DNA damage and erroneously causes the accumulation of DNA double‐strand break (DSB) response factors. Taken together, these results reveal that H2AX ADP‐ribosylation not only facilitates BER repair, but also suppresses the γH2AX‐mediated DSB response.
Keywords: ADP‐ribosylation, base excision repair, H2AX, PARP1
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
H2AX ADP‐ribosylation upon oxidative DNA damage not only promotes recruitment of BER glycosylase Neil3, but also suppresses γH2AX formation and associated double‐strand break response signaling.
Introduction
The integrity of genomic DNA is constantly challenged by endogenous and exogenous hazards generating numerous DNA lesions in human cells daily. These DNA lesions, if not repaired precisely and timely, can lead to genomic instability (Mills et al, 2003; Motoyama & Naka, 2004; Gorgoulis et al, 2005; Tubbs & Nussenzweig, 2017). To deal with the risks of DNA damage, cells use a complicated molecular network to sense, signal, and repair DNA lesions, aka DNA damage response (DDR) (Zhou & Elledge, 2000; Harper & Elledge, 2007; Jackson & Bartek, 2009; Lord & Ashworth, 2012). One of the major components of DDR involves chromatin remodeling at the DNA lesions (Bao & Shen, 2007; Wang et al, 2007; Lukas et al, 2011; Soria et al, 2012; Price & D'Andrea, 2013), which is primarily achieved with histone modifications (Iizuka & Smith, 2003; van Attikum & Gasser, 2005; Vidanes et al, 2005; van Attikum & Gasser, 2009; Messner & Hottiger, 2011; Xu et al, 2012). The most prominent histone modifications induced by DNA damage occur on H2AX, a variant of canonical histone H2A (Rogakou et al, 1998; Fernandez‐Capetillo et al, 2004; Ikura et al, 2007; Xiao et al, 2009; Jiang et al, 2010; Yuan et al, 2010; Pan et al, 2011; Revet et al, 2011; Mattiroli et al, 2012; Sone et al, 2014; Ikura et al, 2015).
In response to DNA damage, especially DNA double‐strand breaks (DSBs), H2AX is rapidly phosphorylated at serine 139 (S139) by PI3‐like kinases including ATM, ATR, and DNA‐PK (Burma et al, 2001; Ward & Chen, 2001; Celeste et al, 2003; Stiff et al, 2004). The phosphorylated H2AX is also known as γH2AX, which is recognized by the downstream mediators such as MDC1 assists in the recruitment of repair machinery for DSB repair (Paull et al, 2000; Celeste et al, 2003; Stucki et al, 2005; Lou et al, 2006). In addition to phosphorylation, other posttranslational modifications (PTMs) on histones, such as ubiquitination (Pan et al, 2011; Mattiroli et al, 2012), SUMOylation (Corujo & Buschbeck, 2018), methylation (Sone et al, 2014), and acetylation (Jiang et al, 2010), also regulate chromatin status adjacent to DNA lesions and facilitate DNA damage repair.
Among all damage signals, ADP‐ribosylation is one of the earliest signals generated at DNA lesions, and histones due to their proximity with the DNA function as primary substrates of ADP‐ribosylation in response to DNA damage (Ogata et al, 1980; Messner et al, 2010; Messner & Hottiger, 2011; Leidecker et al, 2016). Protein ADP‐ribosylation is catalyzed by a group of poly(ADP‐ribosyl)ation polymerases (aka PARPs) (Hassa et al, 2006). These enzymes utilize NAD+ as the ADP‐ribose donor and transfer ADP‐ribose moiety to protein substrates (Kim et al, 2005). In human cells, 17 PARPs have been identified (Liu & Yu, 2015), and many of them are known to mediate ADP‐ribosylation in response to DNA damage (Kraus, 2015). In particular, PARP1 mediates the majority of ADP‐ribosylation in response to DNA damage (Leung, 2014). Of note, not all the amino acid residues can accept PARP1‐mediated ADP‐ribosylation. Mass spectrometry analyses of the substrate proteins show that glutamate (E), aspartate (D), tyrosine (Y), and serine (S) residues function as primary targets of PARP1‐mediated ADP‐ribosylation (Blanke et al, 1994; Bock et al, 2015; Bonfiglio et al, 2017; Chapman et al, 2013; Kraus, 2015; Matic et al, 2012; Pedrioli et al, 2018; Zhang et al, 2013). Recent studies have shown that other residues including lysine (K) (Altmeyer et al, 2009; Messner et al, 2010), arginine (R) (Laing et al, 2011), and cysteine (C) (West et al, 1985) may also be ADP‐ribosylated through alternate mechanisms. In addition to PARP1, other PARPs, including PARP 2, 3, 7, and 10, also catalyze ADP‐ribosylation during DDR, and thus may have overlapping functions (Vyas et al, 2014; Wei & Yu, 2016; Liu et al, 2017).
The general biological functions of ADP‐ribosylation in DDR have been studied extensively. It has been shown that ADP‐ribosylation brings huge amounts of negative charge to the chromatin adjacent to DNA lesions, which may facilitate chromatin relaxation (Poirier et al, 1982; Tulin & Spradling, 2003). Moreover, several ADP‐ribosylation binding motifs have been identified, suggesting that ADP‐ribosylation, like phosphorylation, acts as a signal to mediate the recruitment of DNA damage repair machinery to DNA lesions for the repair (Liu & Yu, 2015; Wei & Yu, 2016; Liu et al, 2017). However, like protein phosphorylation, different ADP‐ribosylation sites may mediate different steps of DNA damage repair (Liu et al, 2017). Due to lack of analytic approaches, the biological functions of site‐specific ADP‐ribosylation in DNA damage repair are not well studied. Fortunately, over the past few years, advanced mass spectrometry technologies have revealed more and more ADP‐ribosylation sites (Altmeyer et al, 2009; Messner et al, 2010; Laing et al, 2011; Zhang et al, 2013; Bock et al, 2015; Leidecker et al, 2016; Pedrioli et al, 2018). In this study, using unbiased high‐resolution mass spectrometry and other molecular biology approaches, we characterized the ADP‐ribosylation of E141 of H2AX and provided insights into its critical role in base excision repair (BER).
Results
H2AX is ADP‐ribosylated in response to DNA damage
Since H2AX plays a key role in DNA damage response and protein ADP‐ribosylation is one of the earliest signals at DNA lesions, we asked whether H2AX could act as an acceptor of ADP‐ribose in response to DNA damage. We treated 293T cells with various DNA damaging agents, including H2O2, MMS, MMC, cisplatin, and ionizing radiation (IR), and examined the status of H2AX in the chromatin fractions. Using immunoprecipitation (IP) and Western blotting assays, we found that H2AX was clearly ADP‐ribosylated in response to H2O2 and MMS treatment (Fig 1A). However, simulations with MMC, cisplatin, or IR did not induce noticeable H2AX ADP‐ribosylation, indicating ADP‐ribosylation on H2AX is a specific event induced by oxidative DNA damage (Appendix Fig S1A and B). Previous studies have shown that DNA damage‐induced ADP‐ribosylation is associated with the intensity of oxidative damage (Martello et al, 2013; Bilan et al, 2017). Here, we found that the ADP‐ribosylation on H2AX was also remarkably increased along with the increased level of oxidative stress (Appendix Fig S2).
Figure 1. H2AX is ADP‐ribosylated in response to DNA damage.
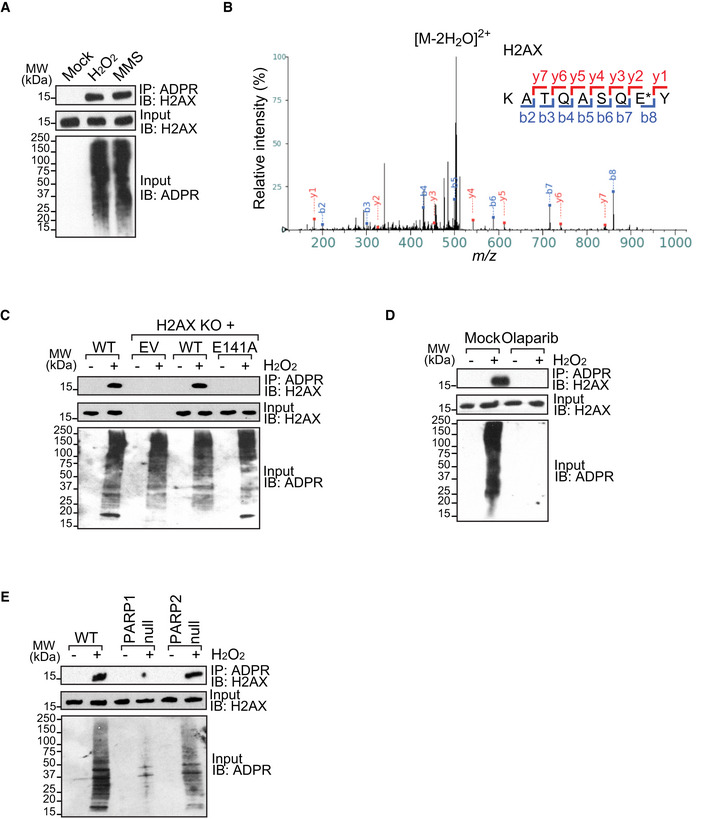
- DNA damage induces H2AX ADP‐ribosylation. 293T cells were treated with H2O2 (2 mM in PBS, 5 min), MMS (1 mM in medium, 30 min), or mock (PBS, 30 min). ADP‐ribosylated proteins were IPed with anti‐ADP‐ribose (anti‐ADPR) antibody. ADP‐ribosylated H2AX was examined by Western blotting using anti‐H2AX antibody.
- H2AX is ADP‐ribosylated at glutamate 141 (E141). shPARG HCT116 cells were treated with H2O2 (2 mM in PBS, 5 min) and ADP‐ribosylated residues were tagged by a hydroxamic acid derivative with an addition of 15.0109 Da, an increment that can be readily distinguished by mass spectrometry. Fragmentation of the NH2OH‐derivatized peptides yielded typical b‐ and y‐ion series, allowing easy localization of ADP‐ribosylation sites. Glutamate 141 (E141) is indicated by an asterisk.
- The E141A mutation abolishes the ADP‐ribosylation of H2AX. Empty vector (EV), wild type (WT), or the E141A mutant (E141A) of H2AX was expressed in U2OS H2AX knockout cells that were treated with H2O2. ADP‐ribosylated proteins were IPed with anti‐ADPR antibody. ADP‐ribosylated H2AX was examined by Western blotting using anti‐H2AX antibody.
- ADP‐ribosylation on H2AX is suppressed by olaparib treatment (1 µM, 1 h). 0.1% DMSO in medium for 1 h as mock. The samples were examined by IP and Western blotting.
- PARP1, but not PARP2, mediates the ADP‐ribosylation on H2AX. Wild‐type (WT) cells, PARP1‐null cells, or PARP2‐null cells were treated with H2O2.
Source data are available online for this figure. Source data are available online for this figure.
Next, to distinguish whether this modification is poly(ADP‐ribosyl)ation (PolyADPR), oligo(ADP‐ribosyl)ation (OligoADPR), or mono(ADP‐ribosyl)ation (MonoADPR), the anti‐ADP‐ribose (anti‐ADPR) antibody used in the tests can recognize pan‐ADPR (Appendix Fig S3A). In addition, we purchased anti‐pan‐ADPR binding reagent from Millipore. Again, we identified the ADP‐ribosylation of H2AX in response to oxidative damage (Appendix Fig S3B). Interestingly, with longer exposure, we observed smeared bands on ADP‐ribosylated H2AX, indicating that the polymer form of ADP‐ribosylation may exist on H2AX (Fig EV1A). Finally, we were able to use anti‐PAR antibody to detect ADP‐ribosylated H2AX, although the signals were relatively weak (Fig EV1B). Collectively, these results suggest that oxidative damage induces ADP‐ribosylation on H2AX might be very short PAR chains or oligo(ADP‐ribosyl)ation.
Figure EV1. Oligo(ADP‐ribose) or short PAR chains are synthesized on H2AX following DNA damage.
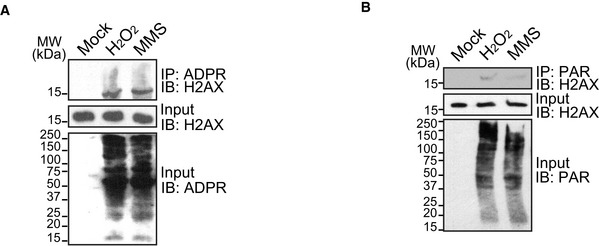
- Smeared band of ADP‐ribosylated H2AX was observed with longer exposure. 293T cells were treated with H2O2 (2 mM in PBS, 5 min), MMS (1 mM in medium, 30 min), or mock (PBS, 30 min). ADP‐ribosylated proteins were immunoprecipitated with anti‐ADPR antibody and examined by Western blotting using anti‐H2AX antibody.
- 293T cells were treated with H2O2 (2 mM in PBS, 5 min), MMS (1 mM in medium, 30 min), or mock (PBS, 30 min). PARylated proteins were immunoprecipitated with anti‐PAR antibody. PARylated‐H2AX was examined by Western blotting using anti‐H2AX antibody.
Source data are available online for this figure.
To exclude the cell line‐specific ADP‐ribosylation, we used HCT116 cells to further validate the ADP‐ribosylation of H2AX. Since ADP‐ribosylation is a reversible posttranslational modification and removal of PARylation is mainly mediated by PARG, knockdown PARG in HCT116 cells suppresses dePARylation (Leung, 2014; Kraus, 2015; Liu & Yu, 2015; Kassab et al, 2020). HCT116 shPARG cells were treated with H2O2 or MMS, and we found that H2AX was ADP‐ribosylated using IP and Western blotting (Appendix Fig S4A), Additionally, to exclude off‐targeted effects of knockdown PARG, we lysed the HCT116 cells with 0.5% SDS buffer which is sufficient to denature the enzymatic activity of PARG in cell lysates. Again, we still observed the ADP‐ribosylation of H2AX (Appendix Fig S4B).
Next, to explore the sites of ADP‐ribosylation on H2AX, an unbiased high‐resolution mass spectrometry was performed using HCT116 shPARG cell lysates treated with H2O2 (Zhang et al, 2013). The ADP‐ribosylated residues were tagged by a hydroxamic acid derivative with an addition of 15.0109 Da, an increment that can be readily distinguished by mass spectrometry. Fragmentation of the NH2OH‐derivatized peptides yielded typical b‐ and y‐ion series, allowing easy localization of ADP‐ribosylation sites. The results identified ADP‐ribosylation of H2AX on glutamate 141 (E141), and these data have been reported by Dr. Yonghao Yu group in Nature Methods in 2013 (Fig 1B).
To further validate the ADP‐ribosylation at E141 in cells, CRISPR/Cas9 was used to delete H2AX in U2OS cells. The deficient cells were reconstituted with either empty vector (EV) or wild‐type H2AX (WT) (Appendix Fig S5A). We also mutated the glutamate 141 into alanine (the E141A mutant) and reconstructed into H2AX knockout U2OS cells (Appendix Fig S5A). The expression levels of reconstituted wild‐type H2AX (H2AX KO + WT) and the E141A mutant (H2AX KO + E141A) were very similar. We also compared and found the level of exogenous H2AX was very similar to that of endogenous H2AX (Appendix Fig S5B). Importantly, the E141A mutant was properly incorporated into the chromatin (Appendix Fig S5C). Unlike the wild‐type H2AX, the E141A mutant abolished DNA damage‐induced ADP‐ribosylation (Fig 1C). Moreover, the DNA damage‐induced ADP‐ribosylation event on H2AX was also suppressed by PARP inhibitor (olaparib) treatment (Fig 1D). Since olaparib suppresses the enzymatic activities of PARP1 and PARP2, we further examined the ADP‐ribosylation of H2AX in PARP1‐ and PARP2‐deficient cells, and found that lack of PARP1 abolished DNA damage‐induced ADP‐ribosylation on H2AX (Fig 1E), suggesting that PARP1 mediates this ADP‐ribosylation event in the cells. Collectively, these results suggest that the E141 of H2AX is ADP‐ribosylated in response to DNA damage.
The ADP‐ribosylation at E141 of H2AX mediates BER
Next, we explored the biological function of this ADP‐ribosylation event. We treated cells with H2O2 or MMS and performed MTT assays to evaluate short‐term cell viability. We found that cells lacking H2AX were hypersensitive to H2O2 or MMS treatment (Fig 2A), which is consistent with earlier studies (Revet et al, 2011). Interestingly, cells only expressing the E141A mutant were also hypersensitive to H2O2 or MMS treatment (Fig 2A). To further validate these results, we performed cell colony formation assays to examine long‐term cell viability under similar genotoxic stress. Again, cells lacking H2AX or only expressing the E141A mutant were hypersensitive to the H2O2 or MMS treatment (Fig 2B).
Figure 2. The ADP‐ribosylation at E141 of H2AX mediates BER.

- U2OS cells (WT) and U2OS H2AX knockout cells reconstituted with empty vector (H2AX KO + EV), wild‐type H2AX (H2AX KO + WT), or the E141A mutant H2AX (E141A) were treated with H2O2 or MMS. Cell viability was measured after 24 h using MTT assay. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01.
- U2OS cells (WT) and U2OS H2AX knockout cells reconstituted with empty vector (H2AX KO + EV), wild‐type H2AX (H2AX KO + WT), or the E141A mutant (E141A) H2AX were treated with H2O2 (2 mM in PBS, 5 min) or MMS (1 mM in medium, 30 min); subsequently, the cells were cultured in fresh DMEM for 2 weeks. The numbers of colony formation were counted. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01.
- Damaged base repair is suppressed in U2OS cells expressing the E141A mutant (E141A). U2OS cells (WT) and U2OS H2AX knockout cells reconstituted with wild‐type H2AX (H2AX KO + WT), or the E141A mutant H2AX (E141A) were treated with H2O2 (2 mM in PBS, 5 min) or MMS (1 mM in medium, 30 min) and harvested at the indicated recovery time points. Then, FPG‐modified comet assay was performed. NT: no treatment. Representative comet tails were shown. The olive tail moments (OTM) were summarized from at least 50 cells in each experiment. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant and **P < 0.01. Scale bar, 10 μm.
- DNA replication in U2OS cells expressing the E141A mutant (E141A) is suppressed. Cells were treated with H2O2 (2 mM in PBS, 5 min), MMS (1 mM in medium, 30 min), or mock (PBS, 30 min), followed by BrdU incorporation (30 μM in medium, 30 min). Representative images show BrdU‐positive cells in cells expressing WT H2AX or E141A following genotoxic stress. Nuclei were stained with DAPI (blue). The bottom panel histogram shows the percentage of BrdU incorporation in each sample. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. ***P < 0.001. Scale bar, 30 μm.
- Generation of AP sites is suppressed in cells expressing E141A. Cells were treated with 2 mM H2O2 for 5 min or 1 mM MMS for 30 min. AP sites were calculated according to the standard curve. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant and **P < 0.01.
Both H2O2 and MMS induce various types of DNA base lesions as well as single‐strand breaks (SSBs), which are mainly repaired by the base excision repair (BER) pathway. To study the role of ADP‐ribosylation of H2AX in BER, we performed FPG‐modified alkaline comet assay (Collins et al, 1993). Unexpectedly, we found accumulated DNA base damage in cells expressing the E141A mutant (Fig 2C), whereas no DSBs was detected following H2O2 treatment using neutral comet assay (Appendix Fig S6). To explore how the ADP‐ribosylation of H2AX results in accumulation of DNA base damage, we examined and found that the replication recovery was remarkably delayed when cells were treated with H2O2 or MMS. These results indicate that a set of base lesions may not be processed effectively under these conditions, which may also block DNA replication (Fig 2D).
To examine the role of H2AX ADP‐ribosylation in BER, we explored the first step of BER, namely removal of damaged DNA base. Only if damaged bases are removed, BER will proceed and replication will resume. Once the damaged bases are removed from DNA lesions, apurinic/apyrimidinic (AP) sites will be generated. Thus, we measured the number of AP sites in genomic DNA (Appendix Fig S7). AP sites were reduced in cells only expressing the E141A mutant (Fig 2E), suggesting that base lesions may accumulate in these cells and ADP‐ribosylation of H2AX may participate in the first step of BER. Taken together, these results suggest that ADP‐ribosylation at E141 plays an important role in BER.
ADP‐ribosylation mediates the recruitment of Neil3 to DNA lesions
Several glycosylases including OGG1 and Nei‐like (Neil) family enzymes are known to recognize and excise damaged bases (Jacobs & Schar, 2012). Thus, we asked which enzyme could mediate H2AX‐dependent ADP‐ribosylation during BER, and examined the recruitment of these enzymes to DNA lesions using laser microirradiation assays. We observed that OGG1 was quickly recruited to DNA lesions. However, the recruitment of OGG1 was not affected by protein ADP‐ribosylation, indicating that OGG1 does not mediate H2AX ADP‐ribosylation‐dependent BER (Appendix Fig S8).
Interestingly, among three Neil family enzymes, Neil3 was quickly recruited to DNA lesions and the recruitment of Neil3 was abolished upon PARP inhibitor olaparib treatment (Fig 3A and Appendix Fig S9A and B), suggesting that DNA damage‐induced ADP‐ribosylation controls the recruitment of Neil3. In addition, PARG inhibitor treatment had no effect on the recruitment of Neil3 (Appendix Fig S10). Because Neil3 is a nuclear polypeptide with multiple domains, we generated a series of truncation mutants to map the domain that mediates the recruitment of Neil3. Notably, only the C‐terminal domain GRF zinc‐finger motifs of Neil3 (GRFs), but not other domains, was recruited to DNA lesions. Moreover, Neil3 lacking the GRFs could not be recruited, suggesting that the GRFs of Neil3 are necessary and sufficient for the relocation of Neil3 to DNA lesions (Fig 3B). In addition, PARP inhibitor treatment or lack of PARP1 suppressed the relocation of the GRFs (Fig 3C), further confirming that the recruitment of Neil3 to DNA lesions is ADP‐ribosylation dependent. Since laser microirradiation generates a mixture of DNA lesions, including both DSBs and SSBs, we further validated the results using KillerRed system, which mainly induces oxidative DNA damage and BER. Consistently, Neil3 was recruited to the oxidative lesions and the recruitment was mediated by ADP‐ribosylation (Fig 3D). To validate the Neil3 recruitment into DNA damage site is PARP1‐dependent, we also examined the Neil3 recruitment in U2OS TRE PARP1 knockout cells. Again, our results showed that the relocation of Neil3 to the sites of DNA damage was impaired in the PARP1‐deficient cells, demonstrating that Neil3’s recruitment is dependent on the PARP1‐mediated ADP‐ribosylation (Appendix Fig S11).
Figure 3. ADP‐ribosylation mediates the recruitment of Neil3 to DNA lesions.
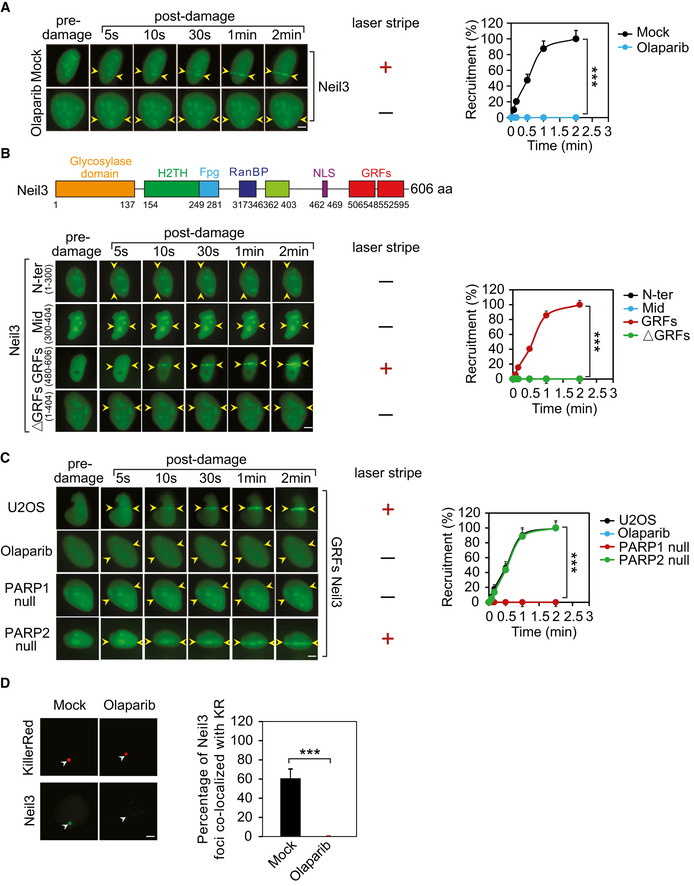
- ADP‐ribosylation mediates the recruitment of Neil3. GFP‐Neil3 was expressed in U2OS cells, and cells were treated with olaparib (1 µM, 1 h) or mock (0.1% DMSO in medium, 1 h). Following laser microirradiation treatment, the recruitment of Neil3 was examined with live‐cell imaging at the indicated time points (left panel). The laser stripe is indicated with yellow arrowheads. The relocation kinetics is shown in the right panel. Data represent mean ± SD from three biologically independent experiments (right panel). At least 20 cells were included in each experiment. P‐values were calculated using Student’s t‐test. ***P < 0.001. Scale bar, 2 μm.
- The GRFs of Neil3 alone are sufficient to be recruited to the sites of DNA damage. Truncated mutants of Neil3 were generated and fused with a GFP tag. Following laser microirradiation treatment, the recruitment of truncated mutants Neil3 was examined with live‐cell imaging at the indicated time points (left panel). The laser stripe is indicated with yellow arrowheads. The relocation kinetics is shown in the right panel. Data represent mean ± SD from three biologically independent experiments (right panel). At least 20 cells were included in each experiment. P‐values were calculated using Student’s t‐test. ***P < 0.001. Scale bar, 2 μm.
- PARP inhibitor treatment (olaparib: 1 µM, 1 h) or lack of PARP1 suppresses the relocation of the GRFs of Neil3 to the DNA lesions. The laser stripe is indicated with yellow arrowheads. The relocation kinetics is shown in the right panel. Data represent mean ± SD from three biologically independent experiments (right panel). At least 20 cells were included in each experiment. P‐values were calculated using Student’s t‐test. ***P < 0.001. Scale bar, 2 μm.
- The recruitment of Neil3 to the oxidative lesions is inhibited by olaparib treatment (1 µM, 1 h). 0.1% DMSO in medium for 1 h as mock. Oxidative damage was induced by KillerRed (KR) system with the exposure to a 15‐W SYLVANIA cool white fluorescent bulb for 10 min. Scale bar, 2 μm. The foci were indicated with white arrowheads. Right panel: the percentage of Neil3 foci co‐localized with KR was quantified. Mean value with SD is from 20 cells. P‐values were calculated using Student’s t‐test. ***P < 0.001.
Neil3 recognizes ADP‐ribosylation of H2AX
Next, we investigated whether Neil3 directly recognizes ADP‐ribosylation. We generated recombinant full‐length Neil3 and GRFs Neil3, and found that both the full‐length Neil3 and GRFs Neil3 bind to PAR in vitro (Fig 4A). To determine whether Neil3 recognizes H2AX ADP‐ribosylation in cells, we performed tandem co‐immunoprecipitation (IP) and Western blotting assays, and found that Neil3 interacted with ADP‐ribosylated H2AX but not with the E141A mutant (Fig 4B). Moreover, the GRFs of Neil3 are required for the interaction (Fig 4C and D).
Figure 4. Neil3 recognizes ADP‐ribosylation of H2AX.
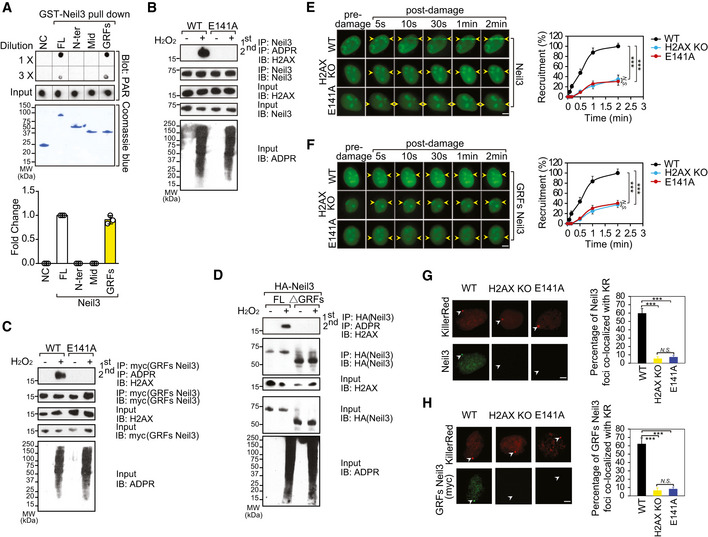
-
AThe GRFs of Neil3 recognize PAR. The recombinant GST fusion proteins were incubated with PAR, and the protein–PAR complex was pulled down by glutathione agarose beads. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐PAR antibody (top panel). FL represents full‐length Neil3, N‐ter represents N‐terminal Neil3 (1–300 aa), Mid represents middle Neil3 (300–404 aa), and GRFs represent C‐terminal Neil3 GRFs domain (480–606 aa). Recombinant GST protein was used as the negative control (NC). The GST fusion proteins were also examined by the SDS–PAGE followed with Coomassie blue staining (middle panel). The relative PAR binding in the pull‐down assay was summarized in the graph (bottom panel). Values are mean ± SD of three assays.
-
BTandem co‐immunoprecipitation and Western blotting assays demonstrate that Neil3 interacts with ADP‐ribosylated H2AX but not the E141A mutant H2AX.
-
C, DThe GRFs of Neil3 are sufficient and required for the interaction with ADP‐ribosylated H2AX. Tandem co‐immunoprecipitation and Western blotting assays were used as in Fig 4B.
-
E, FThe relocation kinetic of full‐length Neil3 or the GRFs of Neil3 to DNA lesions was analyzed. GFP‐tagged full‐length Neil3 or the GRFs of Neil3 was expressed in the indicated cells, and relocation kinetics was monitored in a time course following laser microirradiation (left panel). The laser stripe is indicated with yellow arrowheads. The relocation kinetics is shown in the right panel. Data represent mean ± SD from three biologically independent experiments (right panel). At least 20 cells were included in each experiment. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant and ***P < 0.001. Scale bars, 2 μm.
-
G, HThe recruitment of full‐length Neil3 or the GRFs of Neil3 to the KR‐induced oxidative lesions is mediated by H2AX ADP‐ribosylation (left panel). Scale bars, 2 μm. The foci were indicated with white arrowheads. Scale bars, 0.5 μm. Right panel: the percentage of Neil3 or GRFs Neil3 foci co‐localized with KR was quantified. Mean value with SD is from 20 cells. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant and ***P < 0.001.
Source data are available online for this figure. Source data are available online for this figure.
Our results indicated that the modification on H2AX was likely oligo(ADP‐ribosyl)ation. To further determine whether PAR chains are recognized by Neil3 or GRFs Neil3, we examined the binding between Neil3 or GRFs Neil3 and different length of PAR chains, and found that neither Neil3 nor GRFs Neil3 bounds to MonoADPR in vitro. Furthermore, six units of ADPR were sufficient to be recognized by Neil3 or GRFs Neil3, suggesting Neil3 or GRFs Neil3 could bind to OligoADPR. Collectively, it is likely that H2AX is oligo(ADP‐ribosyl)ated in response to oxidative damage, which mediates the recruitment of Neil3 (Fig EV2A–D).
Figure EV2. Neil3 or GRFs Neil3 recognizes OligoADPR.
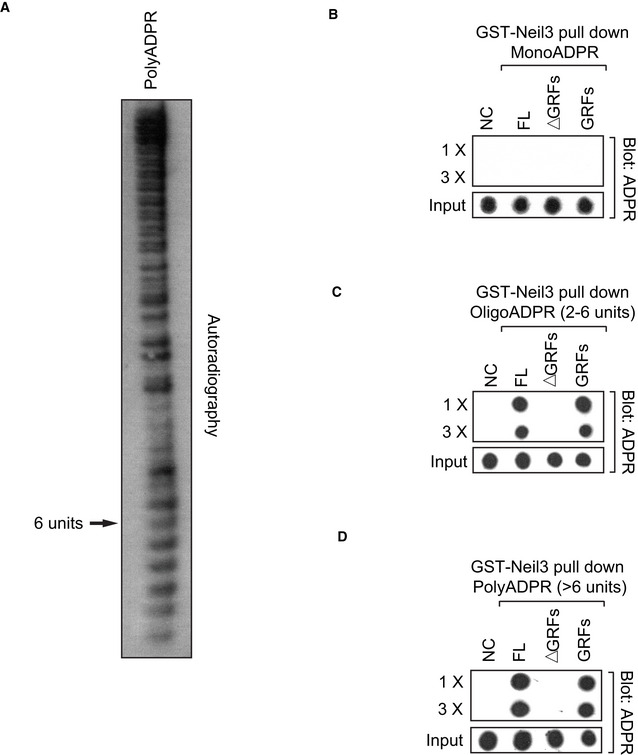
- PAR fractionations were characterized and visualized on 15% native polyacrylamide gel using [32P]‐NAD+ as donor in PAR synthesis.
- Neil3 or GRFs Neil3 does not bind to MonoADPR. The recombinant GST fusion proteins were incubated with MonoADPR, and the protein–MonoADPR complex was pulled down by glutathione agarose beads. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐ADPR antibody. Recombinant GST protein was used as the negative control (NC). FL represents full‐length Neil3, ∆GRFs represent full‐length Neil3 without GRFs domain (1–480 aa), and GRFs represent C‐terminal Neil3 GRFs domain (480–606 aa).
- OligoADPR (less than 6 ADPR units) is sufficient to be recognized by Neil3 or GRFs Neil3. The recombinant GST fusion proteins were incubated with OligoADPR (2–6 units), and the protein–OligoADPR complex was pulled down by glutathione agarose beads. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐ADPR antibody. Recombinant GST protein was used as the negative control (NC). FL represents full‐length Neil3, ∆GRFs represent full‐length Neil3 without GRFs domain (1–480 aa), and GRFs represent C‐terminal Neil3 GRFs domain (480–606 aa).
- PolyADPR (>6 units) can bind with Neil3 or GRFs Neil3. The recombinant GST fusion proteins were incubated with PolyADPR (>6 units), and the protein–PolyADPR complex was pulled down by glutathione agarose beads. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐ADPR antibody. Recombinant GST protein was used as the negative control (NC). FL represents full‐length Neil3, ∆GRFs represent full‐length Neil3 without GRFs domain (1–480 aa), and GRFs represent C‐terminal Neil3 GRFs domain (480–606 aa).
Neil3 differs from other Neil family enzymes in harboring a unique C‐terminal domain which contains two GRF zinc‐finger motifs. GRF is named after three highly conserved residues “GRXF”. Zf‐GRF domain has about 50 residues and is widely distributed in eukaryote. Moreover, zf‐GRFs are found in a number of DNA‐binding proteins involved in DNA/RNA metabolism including APE2, TOP3A, and Neil3. The alignments of zf‐GRF family show that they share high degree of homology (Appendix Fig S12A and B). Two GRFs in Neil3 have been known to recognize various nucleic acids, such as DNA or RNA, and ssDNA has been identified as the most predominant substrate of Neil3. Since Neil3 can bind to ssDNA and PAR in vitro, to further examine the interaction between Neil3 and PAR, we performed a competition binding assays. In these assays, 30‐mer ssDNA was used to compete with 30‐mer PAR in an increased molar ratio up to 50:1 (Appendix Fig S13A). The ssDNA was unable to compete out the interaction between PAR and Neil3. However, when using an increased amount of 30‐mer PAR to compete with 30‐mer ssDNA, we found that PAR easily disrupted the interaction between Neil3 and ssDNA (Appendix Fig S13B). These results suggest that the binding affinity between Neil3 and PAR is much stronger than that between Neil3 and ssDNA. To further understand this molecular event, we modeled the structure of human Neil3‐OligoADPR‐H2AX C‐terminus (the last ten amino acids of H2AX: KKATQASQEY) complex based on the known structures of APE2 and Neil3 (Liu et al, 2013; Wallace et al, 2017) (Appendix Fig S13C). Our model suggests the second GRF of Neil3 might function as a PAR‐binding module to recognize H2AX E141 ADP‐ribosylation.
To examine whether the ADP‐ribosylated H2AX mediated the recruitment of Neil3 to DNA lesions, we examined the relocation of Neil3 in cells lacking H2AX and in cells only expressing the E141A mutant. Compared to wild‐type H2AX cells, the recruitment of Neil3 was impaired in cells lacking H2AX or only expressing E141A mutant (Fig 4E). Similar recruitment kinetics was observed with the GRFs of Neil3 (Fig 4F). Of note, PARylation was not affected in H2AX‐deficient cells (Appendix Fig S14). Moreover, the recruitment results were further validated in the KillerRed system (Fig 4G and H). Collectively, these results demonstrate that Neil3 recognizes ADP‐ribosylated H2AX for its relocation to DNA lesions.
ADP‐ribosylation of H2AX suppresses γH2AX
The E141 of H2AX is an evolutionarily conserved residue in the C‐terminal S‐Q‐E‐Y motif of H2AX. In this motif, Ser139 (S139) is also phosphorylated by PI3‐like kinases in response to DNA damage. Although the ADP‐ribosylation at E141 is close to the S139, we did not observe the phosphorylation of S139 when E141 was ADP‐ribosylated, nor was ADP‐ribosylation found in the S139 phosphorylated H2AX (aka γH2AX) (Fig EV3A–C). Consistently, we also observed phosphorylated S139 in the absence of E141 ADP‐ribosylation in our mass spectrometry analysis. Thus, phosphorylation of S139 and ADP‐ribosylation of E141 may be mutually exclusive events during DNA damage response.
Figure EV3. E141 ADP‐ribosylation and S139 phosphorylation do not co‐exist in H2AX following DNA damage.
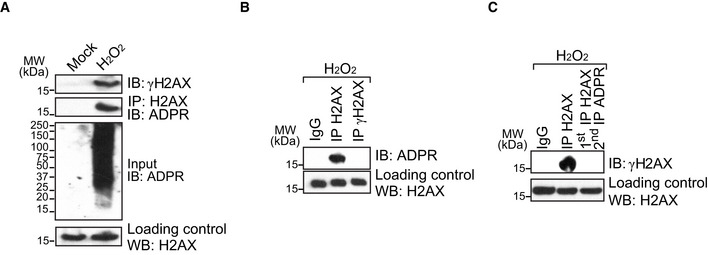
-
AH2O2 treatment induces both phosphorylation and ADP‐ribosylation of H2AX (H2O2: 2 mM in PBS, 5 min; mock: PBS, 30 min). For detection of H2AX ADP‐ribosylation, cell extracts were IPed with anti‐H2AX and Western blotting with anti‐ADPR. For the detection of γH2AX, the cell lysates were examined by Western blotting with anti‐γH2AX antibody.
-
B, CH2O2‐induced ADP‐ribosylation and phosphorylation do not co‐exist in H2AX. Cells were treated with H2O2 (2 mM in PBS, 5 min). The chromatin fraction was extracted and examined by IP and Western blotting with the indicated antibodies.
To explore whether the ADP‐ribosylation regulates γH2AX formation, we treated cells with H2O2 and suppressed PARP1 using PARP inhibitor. Interestingly, the global levels of γH2AX were increased as observed by Western blotting and immunostaining assays (Fig 5A). The similar result was also observed in the H2O2‐treated PARP1 knockout cells (Appendix Fig S15A and B). To exclude any indirect effect of PARP inhibitor treatment on γH2AX levels, we examined the level of γH2AX in cells expressing the E141A mutant. Again, loss of ADP‐ribosylation on H2AX promoted the phosphorylation of S139 (Fig 5B). Using the KillerRed system, we found that local γH2AX was enriched at DNA lesions when cells only expressed the E141A mutant, suggesting that the ADP‐ribosylation at E141 likely blocks the phosphorylation of S139. However, loss of ADP‐ribosylation in the E141A mutant abolishes the suppression thereby facilitating phosphorylation of H2AX by PI3‐like kinases (Fig 5C). In contrast, when cells were pre‐treated with PI3‐like kinase inhibitor to suppress γH2AX, the ADP‐ribosylation of H2AX was not affected (Fig EV4A and B). Moreover, in cells only expressing the S139A mutant, ADP‐ribosylation of H2AX was not affected either (Fig EV4C). Collectively, these results suggest that ADP‐ribosylation of H2AX suppresses phosphorylation of H2AX following oxidative damage. However, phosphorylation of H2AX does not affect ADP‐ribosylation of H2AX.
Figure 5. ADP‐ribosylation of H2AX suppresses γH2AX.
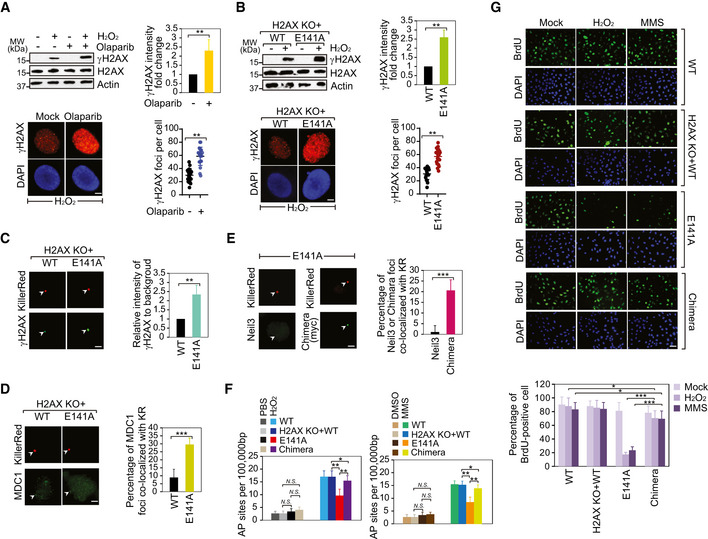
- PARP inhibition facilitates γH2AX following oxidative stress. The cells were pre‐treated with olaparib (1 µM, 1 h) prior to H2O2 stimulation. 0.1% DMSO in medium for 1 h as mock. γH2AX or H2AX was detected using Western blotting and immunostaining assays. The intensity of γH2AX levels was examined by Western blotting normalized by β‐actin and averaged. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01 (left panel). The number of γH2AX foci examined in immunostaining assay was counted by ImageJ software and graphed by GraphPad Prism 8. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01 (right panel). Scale bar, 2 μm.
- The effect of H2AX E141 ADP‐ribosylation on γH2AX following oxidative stress. γH2AX or H2AX was detected using Western blotting and immunostaining assays. The intensity of γH2AX levels was examined by Western blotting normalized by β‐actin and averaged. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01 (left panel). The number of γH2AX foci examined in immunostaining assay was counted by ImageJ software and graphed by GraphPad Prism 8. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. **P < 0.01 (right panel).
- The intensity of γH2AX is increased in cell expressing the E141A mutant following KillerRed‐induced oxidative DNA lesions (left panel). Scale bar, 2 μm. The foci were indicated with white arrowheads. Scale bar, 0.5 μm. Right panel: the relative intensity of γH2AX at the site of tetR (intensity of γH2AX at the site of tetR/intensity of γH2AX area away from tetR) was quantified. Mean value with SD is the relative intensity of γH2AX in 20 cells. P‐values were calculated using Student’s t‐test. **P < 0.01.
- The level of MDC1 at KillerRed‐induced oxidative DNA lesions is increased in the cells expressing the E141A mutant (left panel). Scale bar, 2 μm. The foci were indicated with white arrowheads. Right panel: the percentage of MDC1 foci co‐localized with KR was quantified. Mean value with SD is from 20 cells. P‐values were calculated using Student’s t‐test. ***P < 0.001.
- The recruitment of Neil3/MDC1 chimera but not wild‐type Neil3 to the KR‐induced oxidative lesions in cells expressing the E141A mutant (left panel). Neil3/MDC1 chimera: replaced the GRFs of Neil3 with the tandem BRCT domains of MDC1. Scale bar, 2 μm. The foci were indicated with white arrowheads. Right panel: The percentage of detected protein foci co‐localized with KR was quantified. Mean value with SD is from 20 cells. P‐values were calculated using Student’s t‐test. ***P < 0.001.
- AP sites were measured in cells expressing the E141A mutant or the Neil3/MDC1 chimera, according to the standard curve. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant, *P < 0.05, and **P < 0.01.
- DNA replication of U2OS cell expression with individual wild type (WT), the H2AX KO + WT, the E141A mutant (E141A), or the Neil3/MDC1 Chimera (Chimera) was analyzed by BrdU incorporation following treatment with H2O2, MMS, or mock (PBS). Represented images are shown in the left panel. The graph shows percentages of BrdU‐positive cells expressing WT, E141A, or Chimera (right panel). Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. *P < 0.05 and ***P < 0.001. Scale bar, 30 μm.
Source data are available online for this figure. Source data are available online for this figure.
Figure EV4. DNA damage‐induced Ser139 phosphorylation does not affect ADP‐ribosylation on H2AX.
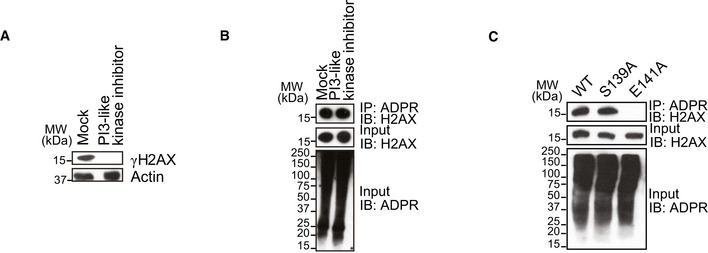
- 293T cells were pre‐treated with pan PI3‐like kinase inhibitor (Torin2: 0.5 µM for 1 h) followed by H2O2 stimulation (2 mM in PBS, 5 min). 0.1% DMSO in medium for 1 h as mock. γH2AX was examined by Western blotting using anti‐γH2AX antibody. Actin was blotted as a control.
- 293T cells were pre‐treated with pan PI3‐like kinase inhibitor (Torin2: 0.5 µM for 1 h) followed by H2O2 stimulation (2 mM in PBS, 5 min). 0.1% DMSO in medium for 1 h as mock. ADP‐ribosylated proteins were immunoprecipitated with anti‐ADPR antibody; ADP‐ribosylated H2AX was examined by Western blotting using anti‐H2AX antibody.
- The Ser139 phosphorylation does not affect the H2AX ADP‐ribosylation. The cells expressing the wild‐type H2AX, the S139A mutant, and the E141A mutant were treated by H2O2 stimulation (2 mM in PBS, 5 min) and then examined by IP and Western blotting with indicated antibodies.
To further dissect the possible crosstalk between phosphorylation of S139 and ADP‐ribosylation of E141, we performed in vitro biochemistry analysis. We examined and found that recombinant PARP1 could directly transfer ADP‐ribose to H2AX peptide, but not the peptide with the E141A mutation (Appendix Fig S16A). Although the S139 phosphorylated H2AX peptide can be ADP‐ribosylated by PARP1, the ATM kinase, one of the key PI3‐like kinases mediated signal transduction in response to DNA damage, cannot further phosphorylate S139 when H2AX peptide is ADP‐ribosylated (Appendix Fig S16B and C). These results suggest that phosphorylation of H2AX may not be able to suppress ADP‐ribosylation; however, ADP‐ribosylation at E141 may block the epitope of S139 motif for its phosphorylation.
Moreover, we also examined the recruitment of MDC1 under oxidative lesion stress in cells lacking ADP‐ribosylation. MDC1 functions as a key downstream partner of γH2AX. Thus, although MDC1 is known to play a role in DSB repair, due to unrestrained γH2AX, MDC1 is mis‐located to oxidative lesions (Fig 5D). We also explored the role of MDC1 in BER. Following H2O2 treatment, we examined the level of 8‐oxo‐dG in the MDC1 deficient cells. However, compared to the MDC1 proficient cells, the level of 8‐oxo‐dG was slightly changed in the MDC1‐deficient cells. Moreover, the level of AP sites in the MDC1‐deficient cells was similar to that of MDC1‐proficient cells. These results suggest that MDC1 may not directly participate in BER. In addition, when we knocked down MDC1 in the cells expressing E141A mutant, we did not find additive defects on BER repair (Fig EV5A–D). Moreover, since γH2AX and MDC1 participates in DSB repair, we examined the role of ADP‐ribosylation of H2AX in IR‐induced DSB repair. However, cells expressing the E141A mutant were insensitive to IR treatment (Appendix Fig S17), indicating that ADP‐ribosylation of H2AX may not have a direct impact on DSB repair.
Figure EV5. Loss of MDC1 has no effect on 8‐oxo‐dG and AP sites.
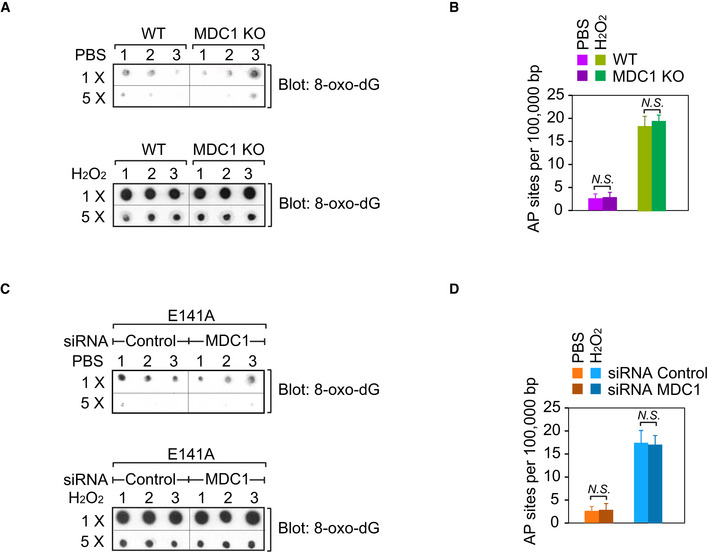
- Wild‐type or MDC1 knockout cells were treated with 2 mM H2O2 for 5 min in PBS (PBS as control), and then the genomic DNA was extracted and equal amount of aliquot was spotted onto a nitrocellulose membrane and then subjected to dot blotting using anti‐8‐oxo‐dG antibody. Triplicate samples were tested.
- AP sites were determined in the wild‐type or MDC1 knockout cells following H2O2 treatment (2 mM H2O2 for 5 min in PBS, PBS as control).
- U2OS cells expressing the E141A mutant were transfected with control siRNA or MDC1 siRNA for 48 h prior to H2O2 treatment (2 mM H2O2 for 5 min in PBS, PBS as control), and then the genomic DNA was extracted and equal amount of aliquot was spotted onto a nitrocellulose membrane and then subjected to dot blotting using anti‐8‐oxo‐dG antibody. Triplicate samples were tested.
- U2OS cells expressing the E141A mutant were transfected with control siRNA or MDC1 siRNA for 48 h prior to H2O2 treatment (2 mM H2O2 for 5 min in PBS, PBS as control), and then AP sites were determined. Values are mean ± SD of three assays. P‐values were calculated using Student’s t‐test. N.S.: nonsignificant.
It has been shown that MDC1 recognizes γH2AX by its BRCT domain. We replaced the GRFs of Neil3 with the tandem BRCT domains of MDC1 (Appendix Fig S18). Interestingly, the chimera was relocated to the oxidative lesions in cells expressing the E141A mutant (Fig 5E). The recruitment was suppressed by PI3‐like kinase inhibitor treatment as well as in cells lacking H2AX, suggesting that the recruitment of chimera is mediated by γH2AX (Appendix Fig S19). Moreover, we expressed the chimera in cells treated with H2O2 or MMS, and found that the expression of chimera rescued the formation of AP sites in cells expressing the E141A mutant, suggesting that the chimera was recruited to DNA lesions by γH2AX and removed damaged base in the absence of H2AX ADP‐ribosylation (Fig 5F). In addition, the expression of chimera also rescued replication in cells lacking H2AX ADP‐ribosylation when the cells were under oxidative stress (Fig 5G). We also examined the role of Neil3 in replication recovery during oxidative damage. Neil3 knockout cells and Neil3 knockout cells reconstituted with the full‐length Neil3 or the ΔGRFs mutant were labeled with BrdU. Following the H2O2 treatment, we examined and found that the DNA replication recovery was blocked in the cells lacking Neil3 or only expressing the ΔGRFs mutant, suggesting that the H2AX ADP‐ribosylation recognized by Neil3 GRFs domain might be essential for replication resuming (Appendix Fig S20).
Taken together, these results demonstrate that H2AX ADP‐ribosylation mediates the recruitment of Neil3 for removal of damaged base during BER (Fig 6).
Figure 6. A model of H2AX ADP‐ribosylation facilitates BER/SSB repair.
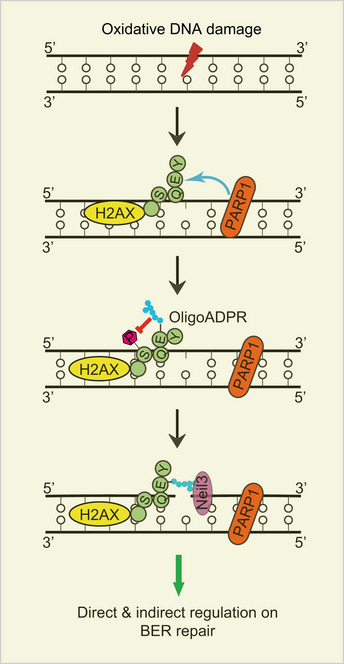
ADP‐ribosylation at E141 mediates the recruitment of Neil3 for removal of damaged base and suppresses phosphorylation at S139 during BER.
Discussion
Oxidative stress is recognized as a potent inducer of protein ADP‐ribosylation; notably, ADP‐ribosylation would be strongly induced following higher oxidative stress (Bilan et al, 2017). In this study, we demonstrate that the E141 of H2AX is ADP‐ribosylated in response to oxidative damage. Moreover, this ADP‐ribosylation site is recognized by the GRFs of Neil3, which is important for BER/SSB repair.
A unique feature of this ADP‐ribosylation site is its close to the S139 phosphorylation site on H2AX. S139 is phosphorylated by PI3‐like kinases during DNA damage response. Generally, γH2AX is also known as the surrogate marker of DSBs (Banath & Olive, 2003). However, S139 of H2AX is also phosphorylated when SSBs are generated (Katsube et al, 2014). In contrast, ADP‐ribosylation of E141 is only occurred in response to oxidative damage. Moreover, ADP‐ribosylation of E141 and phosphorylation of S139 are mutually exclusive events. And suppression of ADP‐ribosylation of E141 promotes the ATM kinase‐mediated phosphorylation of S139, suggesting that ADP‐ribosylation of E141 may negatively regulate the phosphorylation of S139. Since ADP‐ribosylation brings negatively charged phosphate moieties, it is possible that the negative charge changes the topology of the S139 motif and thereby preventing phosphorylation. Indeed, this phenomenon may facilitate SSB repair because S139 phosphorylation is directly recognized by DSB repair machinery such as MDC1. In the event of loss in γH2AX suppression, MDC1 will accumulate at the damage sites. Although γH2AX is known to be induced by oxidative stress, the biological function of γH2AX‐dependent pathway remains elusive. It is possible that MDC1 might be indirectly involved in BER. Thus, ADP‐ribosylation of E141 may indirectly facilitate SSB repair by masking the epitope of the S139 motif of H2AX and suppress any possible misloading of DSB repair factors.
In addition to its indirect role in regulating γH2AX during SSB response, E141 ADP‐ribosylation also directly participates in BER by mediating the recruitment of Neil3. Neil3 is a unique DNA glycosylase that can excise both oxidized purines and pyrimidines, including spiroiminodihydantoin (Sp), guanidinohydantoin (Gh), 2,6‐diamino‐4‐hydroxy‐5‐formamidopyrimidine (FapyG), and 4,6‐diamino‐ 5‐formamidopyrimidine (FapyA) DNA lesions (Liu et al, 2010; Liu et al, 2013; Zhou et al, 2015). In addition to its role in BER, Neil3 is also known to participate in DNA interstrand crosslinks (ICLs) repair (Liu et al, 2013; Zhou et al, 2017; Wu et al, 2019). However, we found that H2AX ADP‐ribosylation is not induced in by ICLs, indicating that H2AX ADP‐ribosylation‐mediated recruitment of Neil3 may not be involved in ICLs repair. Also our results show that H2AX is oligo(ADP‐ribosyl)ated in response to oxidative damage. In addition to binding to the ADP‐ribose units, Neil3 is likely to recognize other motifs on H2AX, which determines the specificity of this particular ADP‐ribosylation site on chromatin and exclusive function of this ADP‐ribosylation event in BER. Future structural analysis will reveal the detailed binding between Neil3 and ADP‐ribosylated H2AX.
Although γH2AX formation happens during SSB repair, it might be a slightly late stage event (Driessens et al, 2009; Katsube et al, 2014). It has been shown that γH2AX may participate in oxidative stress‐induced SSB repair at stalled replication forks (Katsube et al, 2014). However, ADP‐ribosylation of H2AX may be a much earlier event. Consistently, we have shown that phosphorylation and ADP‐ribosylation are mutually exclusive, although these two modifications locate close to each other. Similar to the S‐Q‐E motif in H2AX, there are many other SQE/SQD motifs that can be phosphorylated by PI3‐like kinases. Interestingly, the +2 position E or D may also the acceptors for ADP‐ribosylation. Thus, the functional interaction between phosphorylation and ADP‐ribosylation may occur in other DNA repair mediators/effectors. Here, our results provide the first evidence of such functional interactions between these two important posttranslational modifications during SSB repair process. In addition, the crosstalk between ADP‐ribosylation and phosphorylation can be different in other types of damage response, such as DSBs response. In response to DSBs, phosphorylation of H2AX is clearly observed but not the ADP‐ribosylation. And others have noticed that the E141A mutation may slightly change the epitope of S139 motif and thus affect the robust phosphorylation of H2AX by the PI3‐like kinases during DSB repair (Rogakou et al, 1998; Burma et al, 2001; Celeste et al, 2003). Because of the different damage response, it would be difficult to directly compare the crosstalk between ADP‐ribosylation and phosphorylation of H2AX.
Collectively, our study reveals that E141 of H2AX is ADP‐ribosylated in response to DNA base damage. Moreover, we demonstrate molecular mechanism by which this posttranslational modification event regulates BER/SSB.
Materials and Methods
Cell lines and reagents
U2OS and 293T cells were purchased from American Type Culture Collection (ATCC). U2OS H2AX knockout cells were kindly provided by Dr. Justin W Leung (University of Arkansas for Medical Sciences). U2OS TRE cell line was a kind gift from Dr. Lan Li (Harvard Medical School). Cells were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin–streptomycin (Invitrogen) at 37°C with 5% CO2. HCT116 shGFP and shPARG cells were maintained as previously described (Zhang et al, 2013). Cells were confirmed negative for mycobacteria contamination using the MycoAlert Mycoplasma Detection Kit (Lonza).
To generate the U2OS Neil3 knockout cells or U2OS TRE Neil3 knockout cells, CRISPER/Cas9‐mediated gene editing was performed. Cells were transfected with specific guide RNA (sgRNA) against Neil3, constructed in pSpCas9(BB)‐2A‐Puro (PX459 V2.0) vector (Addgene plasmid ID: 62988). The sgRNA sequence targeted Neil3 was 5'‐TTGACTCATCAGTAGAACTC‐3'. To generate the U2OS TRE PARP1 knockout cells, the sgRNA sequence for PARP1 was 5'‐CCACCTCAACGTCAGGGTGC‐3'. H2AX deficient in U2OS TRE cells was set up through targeted sgRNA against H2AX, and the sequence was 5'‐GGTGGCCTTCTTGCCGCCCG‐3'. Transfection of the cells was carried out using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. The transfected cells were selected in medium containing 1.25 µg/ml puromycin and then sub‐cloned into 96‐well plates 12 h posttransfection. The monoclonal cell lines were identified by Western blotting and Sanger sequencing. The sequence of MDC1 siRNA oligonucleotides was 5'‐ACAGUUGUCCCCACAGCCCdTdT‐3'. The control siRNA used was 5'‐CGUACGCGGAAUACUUCGAdTdT‐3'. Cells were transfected with siRNA duplexes by using Oligofectamine (Invitrogen) as described in the previous report (Stewart et al, 2003).
To reconstitute the empty vector (EV), wild‐type H2AX (WT), or H2AX E141A mutant (E141A, lacking ADP‐ribosylation site on H2AX) into the cells, H2AX‐deficient U2OS cells or H2AX‐deficient U2OS TRE cells were individually transfected with HA‐H2AX or HA‐H2AX E141A, followed by 300 µg/ml hygromycin B selection for 2 weeks. U2OS Neil3 knockout cells stably expressing HA‐full‐length Neil3 or HA‐∆GRFs Neil3 were selected with 300 µg/ml of hygromycin B, respectively. For KillerRed system assays, the Neil3/MDC1 chimera or GRFs Neil3 with myc‐tag was transfected into H2AX‐deficient U2OS TRE cells or H2AX E141A U2OS TRE cells, respectively. The effect of the reconstitution of indicated protein was confirmed by Western blotting.
Anti‐poly(ADP‐ribose) monoclonal antibody (Cat #4335‐MC‐100) was purchased from Trevigen. Anti‐mono‐ADPR binding reagent (Cat #MABE1076) and anti‐pan‐ADPR binding reagent (Cat #MABE1016) were purchased from Millipore. Anti‐β‐actin antibody (Cat #A2228) was purchased from Sigma. Anti‐HA antibody (Cat #MMS‐101R) was from Covance. Anti‐myc antibody (9E10) (Cat #13‐2500) was from Thermo Fisher Scientific. Mouse primary anti‐BrdU antibody (Cat #347580) was purchased from BD biosciences. Anti‐KillerRed antibody (Cat #Ab961) was obtained from Evrogen. Polyclonal anti‐H2AX antibody, monoclonal anti‐γH2AX antibody, anti‐MDC1 antibody, and anti‐ADP‐ribose antibody were generated in‐house. Ni‐SepharoseTM 6 Fast Flow (Cat #17‐5318‐06) and Glutathione SepharoseTM 4B (Cat #17‐0756‐05) were purchased from GE Healthcare. M‐Aminophenyl‐boronic acid‐Agarose (Cat #A8312) was purchased from Sigma. Torin2 used as pan‐PI3‐like kinase inhibitor was purchased from Selleck Chemicals LLC.
Vector constructs
To generate HA‐tagged protein, DNA fragment containing full‐length Neil3, truncated Neil3, wild‐type H2AX, or H2AX E141A was cloned in‐frame with HA tag into pcDNA3.1/Hygro(+) vector. Neil3/MDC1 chimera or GRFs Neil3 with myc‐tag was inserted into pcDNA3.1‐myc vector. Full‐length Neil3 coding sequence was cloned into pFastBac vector with GST tag to generate recombinant protein. Truncated Neil3 with GST tag was cloned into pGEX‐4T‐1 vector. Deletion mutant or point mutation was introduced by Quick change site‐directed mutagenesis and confirmed by Sanger sequencing.
Sample preparation, mass spectrometry analysis, and data analysis
HCT116 shPARG cells were treated with 2 mM H2O2 for 5 min. Samples were prepared and analyzed according to the previous report (Zhang et al, 2013). Samples were analyzed by LC‐MS/MS on a LTQ‐Velos Pro Orbitrap mass spectrometer (Thermo Fischer Scientific) using a top20 method. The isolation window and the minimal signal threshold for MS/MS experiments were set to be 2 Th and 500 counts, respectively. The ReAdW.exe program was used to convert the raw files into the mzXML format (http://sashimi.svn.sourceforge.net/viewvc/sashimi/). MS/MS spectra were searched against a composite database of the human IPI protein database and its reversed complement using the Sequest algorithm. Search parameters allowed for a static modification of 57.02146 Da on cysteine and a dynamic modification of addition of 15.0109 Da to aspartic acid and glutamic acid. Search results were filtered to include < 1% matches to the reverse database by the linear discriminator function using parameters including Xcorr, dCN, missed cleavage, charge state (exclude 1 + peptides), mass accuracy, peptide length, and fraction of ions matched to MS/MS spectra.
Cell proliferation assay
Sensitivity to various DNA damage reagents was determined using MTT assay. Briefly, cells were seeded on 96‐well plates and were treated with the indicated doses of H2O2 for 15 min, MMS for 30 min or IR treatment, either PBS (for H2O2‐treated group) or DMSO (for MMS‐treated group) as mock‐treated control. Then, the cells were given fresh medium and allowed to grow for 24 h. The number of viable cells was determined by adding 20 μl of MTT (thiazolyl blue tetrazolium bromide, 5 mg/ml) (Sigma) into each well, followed by incubation at 37°C for 3 h. The medium was removed and changed into 100 μl DMSO. After incubation at 37°C for 30 min, absorbance was read at 490 nm on a plate reader (BioTek Synergy HT). Each experiment was performed at least in four replications. Data were interpreted as a survival fraction of the untreated control.
Colony formation assay
The cells were plated at about 1,500 cells per well in 6‐well plates and cultured to adhere overnight. Then, cells were treated with 2 mM H2O2 in PBS for 5 min or 1 mM MMS in medium for 30 min. Following treatment, cells were washed with PBS and supplemented with fresh growth media. The cells were cultured for 2 weeks to allow colony formation. The colonies were washed with PBS and fixed with 70% methanol for 10 min, followed by staining with 0.1% crystal violet dissolved in 25% methanol and 75% water. The number of colonies was counted by ImageJ software, and survival graphs were generated from three replicate wells with colony numbers normalized to untreated controls.
DNA replication analysis
Bromodeoxyuridine (BrdU) incorporation assay was performed to analyze the DNA replication. Briefly, exponentially growing cells were plated on coverslips overnight to reach about 50% confluence. H2O2 (2 mM in PBS, 5 min) or MMS (1 mM in medium, 30 min) was added during the course of the experiments. Sequentially, cells were labeled with 30 μM BrdU (Sigma) in fresh DMEM for 30 min. The slides were rinsed with PBS three times and fixed in cold 4% PFA for 10 min, followed by treatment with 0.5% Triton X‐100 for another 10 min. Before blocking, the slides were denatured with 2.5 N HCl for 30 min, washed out by PBS, and then blocked with 8% goat serum/PBS for 30 min at room temperature. Next, the incorporated BrdU was tested with anti‐BrdU antibody for 1 h at room temperature, followed by washing three times with PBS and stained with goat anti‐mouse FITC secondary antibody for 30 min at room temperature. The nuclei were visualized by DAPI staining. Images were captured with an Olympus fluorescence microscope at × 20. ImageJ software was used for processing and quantifying nuclear BrdU‐positive cells in images.
FPG‐modified comet assay
The cells were treated with H2O2 (2 mM in PBS, 5 min) or MMS (1 mM in medium, 30 min) and recovered for indicated time points. Then, cells were harvested, combined with 0.8% low melting agarose and spread onto agarose‐coated slides. After gelling at 4 °C, the slides were then immersed in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% N‐lauroylsarcosine, and 1% Triton X‐100, pH 10.0) for 1 h. Following lysis, the slides were washed three times with enzyme buffer (40 mM HEPES, 0.1 M KCl, 0.5 mM EDTA, and 0.2 mg/ml BSA pH 8.0). After buffer change, the slides were covered with FPG in the enzyme buffer (1 unit per gel), sealed with coverslip, and incubated in a humidified chamber for 45 min at 37°C. The slides were immersed in cold alkaline electrophoresis buffer (300 mM NaOH and 1 mM EDTA, pH > 13) for 30 min to unwind the DNA. Electrophoresis was then performed at 25 V, 300 mA for 20 min. Slides were washed in neutralization buffer (0.4 M Tris–HCl, pH 7.5) for 10 min followed by 100% ethanol for 5 min at room temperature and air‐dried. Finally, slides were stained with 5 μg/ml propidium iodide (PI) and imaged on a fluorescent microscope (Olympus). Average Olive Tail Moment (OTM) was analyzed (50 cells/slide) by using Comet Assay Software Project Casp‐1.2.2 (University of Wroclaw, Poland). All experiments were repeated three times.
Neutral comet assay
For evaluating DNA double‐strand breaks, the neutral version of comet assay was performed. Briefly, the cells were harvested and mixed with 0.8% low melting agarose and layered onto agarose‐coated slides. Slides were then submerged into cold lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris–HCl, 1% N‐lauroylsarcosine and 0.5% Triton X‐100, pH 9.5) for 2 h. After lysis, slides were incubated in electrophoresis buffer (300 mM sodium acetate and 100 mM Tris–HCl, pH 8.3) for 1 h, and then placed in a horizontal gel electrophoresis unit filled with a fresh cold electrophoretic buffer. The slides were electrophoresed at 14 V (0.5 V/cm, 11‐12 mA) for 1 h. Following electrophoresis, slides were neutralized, placed into 100% ethanol, and then air‐dried. Slides were subsequently stained with 5 μg/ml of PI, and images were taken using a fluorescent microscope (Olympus). Average Olive Tail Moment (OTM) was analyzed (50 cells/slide) by using Comet Assay Software Project Casp‐1.2.2 (University of Wroclaw, Poland). All experiments were repeated three times.
AP sites analysis
Indicated cells were cultured in 6‐well plate, and the cells were exposed to 2 mM H2O2 for 5 min or 1 mM MMS for 30 min. Next, the cells were washed three times with PBS, and then genomic DNA was isolated using DNAzol genomic DNA isolation kit (Invitrogen). The concentration and purity of the purified genomic DNA was determined. The AP sites assay was conducted using DNA Damage AP sites assay kit (Colorimetric, Abcam). The number of AP sites was measured and calculated based upon a standard curve generated using ARP standard DNA solutions, according to the instruction of manufacturer. Each group has at least triplicates.
8‐oxo‐dG assay
Indicated cells were cultured in 6‐well plate, and the cells were exposed to 2 mM H2O2 for 5 min. Next, the cells were washed three times with PBS, and then genomic DNA was isolated using DNAzol genomic DNA isolation kit (Invitrogen). The concentration and purity of the purified genomic DNA was determined. Samples were serially diluted fivefold and spotted onto a nitrocellulose membrane, and anti‐8‐oxo‐dG monoclonal antibody was used for dot blotting.
Recombinant protein production
All recombinant truncated Neil3 proteins were expressed in BL21 cells, except full‐length Neil3. Baculoviruses expressing GST‐tagged full‐length Neil3 were prepared using Bac‐to‐Bac system according to the manufacture’s protocols. Proteins were expressed in SF9 insect cells by infection with the Baculoviruses for 2 days. GST fusion proteins were purified using Glutathione Sepharose 4B. His‐tagged proteins were purified using Ni2 +‐NTA chromatography.
Synthesis, purification, and fractionation of PAR
PAR synthesis reaction was carried out in a 2 ml mixture containing 100 mM Tris–HCl pH 8.0, 150 mM NaCl, 10 mM MgCl2, 2 mM NAD+, 5 µM DTT, 50 μg octameric activator oligonucleotide GGAATTCC, and 1 mg PARP1. The reaction was incubated at 37°C for 30 min and stopped by addition of 2 ml ice‐cold 20% TCA. OligoDNA was cleaved by DNase I, and proteins were digested by proteinase K. Following precipitation, the pellet was washed with ice‐cold pure ethanol. PAR was detached from PARP1 protein using 0.5 M KOH/50 mM EDTA and extracted with phenol–chloroform–isoamyl alcohol. The final PAR product was collected in pure water. Then, the concentration was measured and stored at −80°C. PAR fractionation was performed as described in the previous study (Fahrer et al, 2007). Fractions were collected manually according to UV absorbance at 258 nm, followed by precipitated with ethanol, dissolved in water, pooled, and dialyzed. PAR fractionations were characterized and visualized on 15% native polyacrylamide gel using [32P]‐NAD+ as donor in PAR synthesis.
Binding assay of proteins to ADP‐ribose chains
Briefly, 500 nM recombinant proteins were incubated with 500 nM ADP‐ribose chains (MonoADPR, OligoADPR, and PolyADPR) and 25 μl Glutathione Sepharose 4B agarose in the PBS buffer. The reaction mixture was incubated for 1 h followed by extensive wash of beads with PBS. The protein–PAR complex was released by heating at 95°C for 5 min in the SDS‐loading buffer without bromophenol blue. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐ADPR antibody.
Mono‐, Oligo‐, and Poly(ADP‐ribose) generation
To validate the anti‐ADPR antibody, mono‐, oligo‐, and poly(ADP‐ribose) were produced with PARP1 and PARP3 using modified method from the previous reports (Gibson et al, 2017; Lin et al, 2018). One milligram of PARP1 or PARP3 was incubated at 30°C in a 50 μl reaction mixture containing 20 mM Tris pH 8.0, 5 mM MgCl2, 1 mM DTT, and 1 μg octameric activator oligonucleotide GGAATTCC under the following conditions: (i) PARP1 with 250 μM NAD+ for 15 min for poly(ADP‐ribose), (ii) PARP1 with 3 μM NAD+ for 5 min for oligo(ADP‐ribose), and (iii) PARP3 with 250 μM NAD+ for 30 min for mono(ADP‐ribose). All reactions were stopped by heating at 95°C for 5 min in the SDS‐loading buffer without bromophenol blue.
In vitro ATM kinase assay
ATM protein immunoprecipitations were carried out as described (Burma et al, 2001). 293T cells were harvested and lysed in fresh cold NETN100 buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP‐40) containing protease and phosphatase inhibitors (Thermo Fisher scientific). The lysate was cleared by centrifugation, and the supernatant was incubated with 10 μg of anti‐ATM monoclonal antibody MAT3–4G10/8 (Sigma) and protein A/G‐Sepharose beads for 2 h at 4 °C. The beads were intensively washed with high salt buffer and then with kinase buffer (25 mM Tris–HCl pH 7.5, 5 mM beta‐glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, and 10 mM MgCl2). The beads were then incubated in a kinase mixture (20 μl of kinase buffer, 2 μg H2AX peptides, and 2 μl of 200 μM ATP) at 30°C for 20 min. Samples were serially diluted threefold, spotted onto the nitrocellulose membrane, and subjected to dot blotting using anti‐γH2AX antibody.
Competition binding assay
For the DNA/PAR competition assay, 30‐mer ssDNA, the most predominant DNA substrate of Neil3, was used (Liu et al, 2010). Accordingly, the same length PAR (30‐mer) was prepared as described previously (Popp et al, 2013). To examine the preferred binding between PAR and Neil3, approximately 500 nM GST‐Neil3 was incubated with 500 nM biotin‐PAR and 25 μl Glutathione Sepharose 4B agarose in the PBS buffer for 1 h at 4°C, followed by intensive washes with PBS. A competitor unlabeled ssDNA was used to compete with biotin‐PAR with an increased molar ratio up to 50:1. In the interaction between ssDNA and Neil3 test, biotin‐PAR was replaced by 500 nM biotin‐ssDNA in the same experimental condition. A competitor unlabeled PAR was used to compete out biotin‐ssDNA in the molar ratio low to 0.2:1. The dot blotting was performed using streptavidin‐HRP. To confirm the finding, the reciprocal pull‐down assays were also carried out using streptavidin beads to pull down the complex and the products were detected by Western blotting using anti‐GST antibody.
Immunofluorescence staining
U2OS cells or U2OS H2AX E141A cells were cultured on cover slides, to visualize the H2AX foci formation after H2O2 treatment (2 mM, 5 min, then recovery for 30 min) or olaparib treatment (1 μM, 1 h), and cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min, followed by permeabilization in PBS buffer containing 0.5% Triton X‐100 for 10 min. Cells were blocked in 8% goat serum/PBS at room temperature for 1 h, followed by incubation with anti‐γH2AX antibody overnight at 4°C. Next, slides were washed three times with PBS and incubated with rhodamine red‐conjugated anti‐mouse secondary antibody for another 1 h at room temperature. Nuclei were stained with DAPI dye. Images were captured with a fluorescence microscope. The captured images were analyzed for the number of foci per cell using ImageJ software. At least 20 cells were randomly collected from each group.
To examine PARylation on the DNA damage sites, U2OS cells or U2OS H2AX knockout cells were subjected to laser microirradiation, recovered for the indicated time, then the cells were immunostained with anti‐PAR antibody. The fluorescence images were captured using microscope.
KillerRed activation
KillerRed (KR) activation was carried out as described previously. KillerRed, a light‐stimulated ROS‐inducer, was fused to a tet‐repressor (tetR‐KR). TetR‐KR, bound to a TRE cassette (∼90 kb) integrated at a defined genomic locus in U2OS cells, was used to induce ROS damage in heterochromatin (Lan et al, 2014). In brief, activation of KR in bulk cells was done by exposing cells to a 15‐W SYLVANIA cool white fluorescent bulb for 10 min in a stage UVP (Upland). In the case of fluorescent light activation, the rate of light was 15 J/m2/s. Cells were placed under a water bottle (height to light is 15 cm) to prevent an increase of temperature. For staining of KR foci, anti‐KR antibody was used in the assay. Anti‐myc antibody, anti‐Neil3 antibody or anti‐MDC1 antibody were also used to visualize the indicated tested protein, and then followed with the routine immunostaining protocol. Nuclei were stained with DAPI dye. Images were captured with an Olympus fluorescence microscope. The captured images were analyzed for the intensity of foci per cell using ImageJ software. At least 20 cells were randomly collected from each group.
Immunoprecipitation and Western blotting
For ADP‐ribosylated protein pull‐down assay, cells were lysed immediately following DNA damage treatments with SDS lysis buffer (0.5% SDS, 10 mM HEPES pH 8.5, 500 U Benzonase, and 2 mM MgCl2) for 5 min. Supernatants were collected by centrifugation for 5 min at 10,000 g, and the lysates were diluted to 0.1% SDS for immunoprecipitation with anti‐PAR or anti‐ADP‐ribose (anti‐ADPR) antibodies. The immune complex was washed with PBS and resolved in the SDS–PAGE for Western blotting assay using anti‐H2AX antibody. For chromatin fraction preparation, cells were lysed with the NETN100 buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP‐40) with 50 µM GLTN, protease inhibitors, and phosphatase inhibitors (Thermo Fisher Scientific) on ice for 10 min. After high speed centrifugation, insoluble fraction was recovered and resuspended in 0.2 N HCl for 10 min at room temperature. Following another round of high speed centrifugation, the soluble chromatin fraction was neutralized with 1 M Tris–HCl (pH 8.0) for further analysis.
Live‐cell imaging by laser microirradiation
GFP‐tagged constructs were transfected into the indicated cells which were plated on 35‐mm glass bottom dishes. Cellular DNA damages were generated in the nuclei of cultured cells by microirradiation with a pulsed nitrogen laser (Spectra‐Physics; 365 nm, 10 Hz pulse). The laser system was directly coupled to the epifluorescence path of the microscope for time‐lapse imaging and focused through a Plan‐Apochromat 63×/NA 1.40 oil immersion objective. The output of the laser power was set at 50–70% of the maximum as indicated. The green fluorescence strips were recorded at indicated time points and then analyzed with ImageJ software. All results represented images of 20 cells from three independent experiments.
Statistical analysis
Data are represented as mean ± SD as indicated from three independent experiments. Significance of differences was evaluated by Student’s t‐test. N.S.: nonsignificant; *: statistically significant (P < 0.05); **: statistically significant (P < 0.01); and ***: statistically significant (P < 0.001).
Author contributions
XY conceived the project and designed the approach. QC, CB, XW, and YY performed the experiments and analyzed the data. XL conducted the structural analysis. QC, MAK, and XY wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Dr. Justin W Leung at the University of Arkansas for Medical Sciences for sharing the U2OS H2AX knockout cells. We are thankful to Dr. Lan Li in the University of Harvard Medical School for the KillerRed plasmid and U2OS TRE cell line. This work was supported in part by grants from National Institutes of Health (CA132755 and CA130899 to X.Y., R01GM122932 and R35GM134883 to Y.Y.) and Welch Foundation (I‐1800 to Y.Y.). X.Y. is a recipient of Research Scholar Award from Pancreatic Cancer Action Network, The Henry and Marilyn Taub Foundation, and Tower Cancer Research Foundation. Research reported in this publication included work performed in the Integrative Genomics and Bioinformatics Core supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572.
The EMBO Journal (2021) 40: e104542.
Contributor Information
Qian Chen, Email: qchen@coh.org.
Xiaochun Yu, Email: xiaochun_yu@hotmail.com.
Data availability
This study includes no data deposited in external repositories.
References
- Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO (2009) Molecular mechanism of poly(ADP‐ribosyl)ation by PARP1 and identification of lysine residues as ADP‐ribose acceptor sites. Nucleic Acids Res 37: 3723–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banath JP, Olive PL (2003) Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double‐strand breaks. Cancer Res 63: 4347–4350 [PubMed] [Google Scholar]
- Bao YH, Shen XT (2007) Chromatin remodeling in DNA double‐strand break repair. Curr Opin Genet Dev 17: 126–131 [DOI] [PubMed] [Google Scholar]
- Bilan V, Selevsek N, Kistemaker HAV, Abplanalp J, Feurer R, Filippov DV, Hottiger MO (2017) New quantitative mass spectrometry approaches reveal different ADP‐ribosylation phases dependent on the levels of oxidative stress. Mol Cell Proteomics 16: 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanke SR, Huang K, Collier RJ (1994) Active‐site mutations of diphtheria‐toxin ‐ role of tyrosine‐65 in nad binding and Adp‐ribosylation. Biochemistry 33: 15494–15500 [DOI] [PubMed] [Google Scholar]
- Bock FJ, Todorova TT, Chang P (2015) The promise of proteomics for the study of ADP‐ribosylation. Mol Cell 58: 911–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs‐Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, Matic I (2017) Serine ADP‐ribosylation depends on HPF1. Mol Cell 65: 932–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double‐strand breaks. J Biol Chem 276: 42462–42467 [DOI] [PubMed] [Google Scholar]
- Celeste A, Fernandez‐Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675–U651 [DOI] [PubMed] [Google Scholar]
- Chapman JD, Gagne JP, Poirier GG, Goodett DR (2013) Mapping PARP‐1 Auto‐ADP‐ribosylation sites by liquid chromatography‐tandem mass spectrometry. J Proteome Res 12: 1868–1880 [DOI] [PubMed] [Google Scholar]
- Collins AR, Duthie SJ, Dobson VL (1993) Direct enzymatic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis 14: 1733–1735 [DOI] [PubMed] [Google Scholar]
- Corujo D, Buschbeck M (2018) Post‐translational modifications of H2A histone variants and their role in cancer. Cancers 10: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De Deken X, Van Sande J, Dumont JE, Miot F, Corvilain B (2009) Hydrogen peroxide induces DNA single‐ and double‐strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr‐Relat Cancer 16: 845–856 [DOI] [PubMed] [Google Scholar]
- Fahrer J, Kranaster R, Altmeyer M, Marx A, Burkle A (2007) Quantitative analysis of the binding affinity of poly(ADP‐ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res 35: e143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of the genome. DNA Repair 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Gibson BA, Conrad LB, Huang D, Kraus WL (2017) Generation and characterization of recombinant antibody‐like ADP‐ribose binding proteins. Biochemistry 56: 6305–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B et al (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907–913 [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28: 739–745 [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO (2006) Nuclear ADP‐ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70: 789–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Smith MM (2003) Functional consequences of histone modifications. Curr Opin Genet Dev 13: 154–160 [DOI] [PubMed] [Google Scholar]
- Ikura M, Furuya K, Matsuda S, Matsuda R, Shima H, Adachi J, Matsuda T, Shiraki T, Ikura T (2015) Acetylation of histone H2AX at Lys 5 by the TIP60 histone acetyltransferase complex is essential for the dynamic binding of NBS1 to damaged chromatin. Mol Cell Biol 35: 4147–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K et al (2007) DNA damage‐dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 27: 7028–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AL, Schar P (2012) DNA glycosylases: in DNA repair and beyond. Chromosoma 121: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Xu Y, Price BD (2010) Acetylation of H2AX on lysine 36 plays a key role in the DNA double‐strand break repair pathway. FEBS Lett 584: 2926–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassab MA, Yu LL, Yu X (2020) Targeting dePARylation for cancer therapy. Cell Biosci 10: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsube T, Mori M, Tsuji H, Shiomi T, Wang B, Liu Q, Nenoi M, Onoda M (2014) Most hydrogen peroxide‐induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double‐strand breaks. J Biochem 156: 85–95 [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL (2005) Poly(ADP‐ribosyl)ation by PARP‐1: 'PAR‐laying' NAD(+) into a nuclear signal. Gene Dev 19: 1951–1967 [DOI] [PubMed] [Google Scholar]
- Kraus WL (2015) PARPs and ADP‐ribosylation: 50 years … and counting. Mol Cell 58: 902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing S, Koch‐Nolte F, Haag F, Buck F (2011) Strategies for the identification of arginine ADP‐ribosylation sites. J Proteomics 75: 169–176 [DOI] [PubMed] [Google Scholar]
- Lan L, Nakajima S, Wei L, Sun L, Hsieh CL, Sobol RW, Bruchez M, Van Houten B, Yasui A, Levine AS (2014) Novel method for site‐specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res 42: 2330–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I (2016) Serine is a new target residue for endogenous ADP‐ribosylation on histones. Nat Chem Biol 12: 998–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK (2014) Poly(ADP‐ribose): an organizer of cellular architecture. J Cell Biol 205: 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, Huang D, Kraus WL (2018) Generating protein‐linked and protein‐free mono‐, oligo‐, and poly(ADP‐ribose) in vitro. Methods Mol Biol 1813: 91–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Vyas A, Kassab MA, Singh AK, Yu X (2017) The role of poly ADP‐ribosylation in the first wave of DNA damage response. Nucleic Acids Res 45: 8129–8141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Yu X (2015) ADP‐ribosyltransferases and poly ADP‐ribosylation. Curr Protein Pept Sci 16: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MM, Bandaru V, Bond JP, Jaruga P, Zhao XB, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci USA 107: 4925–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MM, Doublie S, Wallace SS (2013) Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat Res‐Fund Mol Mutagen 743: 4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2012) The DNA damage response and cancer therapy. Nature 481: 287–294 [DOI] [PubMed] [Google Scholar]
- Lou ZK, Minter‐Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT et al (2006) MDC1 maintains genomic stability by participating in the amplification of ATM‐dependent DNA damage signals. Mol Cell 21: 187–200 [DOI] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J (2011) More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13: 1161–1169 [DOI] [PubMed] [Google Scholar]
- Martello R, Mangerich A, Sass S, Dedon PC, Burkle A (2013) Quantification of cellular poly(ADP‐ribosyl)ation by stable isotope dilution mass spectrometry reveals tissue‐ and drug‐dependent stress response dynamics. Acs Chem Biol 8: 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I, Ahel I, Hay RT (2012) Reanalysis of phosphoproteomics data uncovers ADP‐ribosylation sites. Nat Methods 9: 771–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK (2012) RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150: 1182–1195 [DOI] [PubMed] [Google Scholar]
- Messner S, Altmeyer M, Zhao HT, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang DZ, Caflisch A, Hottiger MO (2010) PARP1 ADP‐ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38: 6350–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner S, Hottiger MO (2011) Histone ADP‐ribosylation in DNA repair, replication and transcription. Trends Cell Biol 21: 534–542 [DOI] [PubMed] [Google Scholar]
- Mills KD, Ferguson DO, Alt FW (2003) The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 194: 77–95 [DOI] [PubMed] [Google Scholar]
- Motoyama N, Naka K (2004) DNA damage tumor suppressor genes and genomic instability. Curr Opin Genet Dev 14: 11–16 [DOI] [PubMed] [Google Scholar]
- Ogata N, Ueda K, Hayaishi O (1980) Adp‐ribosylation of histone H2b ‐ identification of glutamic‐acid residue 2 as the modification site. J Biol Chem 255: 7610–7615 [PubMed] [Google Scholar]
- Pan MR, Peng G, Hung WC, Lin SY (2011) Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem 286: 28599–28607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Pedrioli DML, Leutert M, Bilan V, Nowak K, Gunasekera K, Ferrari E, Imhof R, Malmstrom L, Hottiger MO (2018) Comprehensive ADP‐ribosylome analysis identifies tyrosine as an ADP‐ribose acceptor site. EMBO Rep 19: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier GG, Demurcia G, Jongstrabilen J, Niedergang C, Mandel P (1982) Poly(Adp‐Ribosyl)Ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci‐Biol 79: 3423–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp O, Veith S, Fahrer J, Bohr VA, Burkle A, Mangerich A (2013) Site‐specific noncovalent interaction of the biopolymer poly(ADP‐ribose) with the werner syndrome protein regulates protein functions. ACS Chem Biol 8: 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, D'Andrea AD (2013) Chromatin remodeling at DNA double‐strand breaks. Cell 152: 1344–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH, Dankort D, Cleaver JE (2011) Functional relevance of the histone gamma H2Ax in the response to DNA damaging agents. Proc Natl Acad Sci USA 108: 8663–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Sone K, Piao L, Nakakido M, Ueda K, Jenuwein T, Nakamura Y, Hamamoto R (2014) Critical role of lysine 134 methylation on histone H2AX for gamma‐H2AX production and DNA repair. Nat Commun 5: 5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966 [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA‐PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64: 2390–2396 [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double‐strand breaks. Cell 123: 1213–1226 [DOI] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin A, Spradling A (2003) Chromatin loosening by poly(ADP)‐ribose polymerase (PARP) at Drosophila puff loci. Science 299: 560–562 [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM (2005) The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 6: 757–765 [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM (2009) Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19: 207–217 [DOI] [PubMed] [Google Scholar]
- Vidanes GM, Bonilla CY, Toczyski DP (2005) Complicated tails: histone modifications and the DNA damage response. Cell 121: 973–976 [DOI] [PubMed] [Google Scholar]
- Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P (2014) Family‐wide analysis of poly(ADP‐ribose) polymerase activity. Nat Commun 5: 4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Berman Z, Mueller GA, Lin YF, Chang T, Andres SN, Wojtaszek JL, DeRose EF, Appel CD, London RE et al (2017) APE2 Zf‐GRF facilitates 3 '‐5 ' resection of DNA damage following oxidative stress. Proc Natl Acad Sci USA 114: 304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Allis CD, Chi P (2007) Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol Med 13: 363–372 [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen JJ (2001) Histone H2AX is phosphorylated in an ATR‐dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Wei H, Yu X (2016) Functions of PARylation in DNA damage repair pathways. Genom Proteom Bioinform 14: 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RE, Moss J, Vaughan M, Liu T, Liu TY (1985) Pertussis toxin‐catalyzed Adp‐ribosylation of transducin ‐ cysteine 347 is the Adp‐ribose acceptor site. J Biol Chem 260: 4428–4430 [PubMed] [Google Scholar]
- Wu RA, Semlow DR, Kamimae‐Lanning AN, Kochenova OV, Chistol G, Hodskinson MR, Amunugama R, Sparks JL, Wang M, Deng L et al (2019) TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567: 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao A, Li HT, Shechter D, Ahn SH, Fabrizio LA, Erdjument‐Bromage H, Ishibe‐Murakami S, Wang B, Tempst P, Hofmann K et al (2009) WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457: 57–U57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ayrapetov MK, Xu C, Gursoy‐Yuzugullu O, Hu YD, Price BD (2012) Histone H2A.Z controls a critical chromatin remodeling step required for DNA double‐strand break repair. Mol Cell 48: 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Adamski R, Chen J (2010) Focus on histone variant H2AX: to be or not to be. FEBS Lett 584: 3717–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J, Ding M, Yu Y (2013) Site‐specific characterization of the Asp‐ and Glu‐ADP‐ribosylated proteome. Nat Methods 10: 981–984 [DOI] [PubMed] [Google Scholar]
- Zhou BBS, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
- Zhou J, Chan J, Lambele M, Yusufzai T, Stumpff J, Opresko PL, Thali M, Wallace SS (2017) NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. Cell Rep 20: 2044–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS (2015) The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res 43: 4039–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
This study includes no data deposited in external repositories.