

Abstract
Cardiomyocyte apoptosis is a key factor in the deterioration of cardiac function after coronary microembolization (CME). Nicorandil (NIC) affects myocardial injury, which may be related to the inhibition of apoptosis. However, the specific mechanism of cardioprotection has not been elucidated. Therefore, we analyzed the impact of NIC on cardiac function in rats subjected to CME and its effect on the high‐temperature requirement peptidase 2/X‐linked inhibitor of apoptosis protein/poly ADP‐ribose polymerase (HtrA2/XIAP/PARP) pathway. Sprague Dawley rats were divided into four groups: Sham, CME, CME + NIC, and CME + UCF. Echocardiography was performed 9 hours after CME. Myocardial injury markers were evaluated in blood samples, and the heart tissue was collected for hematoxylin‐eosin staining, hematoxylin basic fuchsin picric acid staining staining, TdT‐mediated DUTP nick end labeling (TUNEL) staining, Western blot analysis of the HtrA2/XIAP/PARP pathway, and transmission electron microscopy. NIC ameliorated cardiac dysfunctioncaused by CME and reduced serum levels of CK‐MB and LDH. In addition, NIC decreased myocardial microinfarct size and apoptotic index. NIC reduced the Bax/Bcl‐2 ratio, levels of cleaved caspase 3/9, cytoplasmic HtrA2, and cleaved PARP, and increased the level of XIAP. The effects of NIC were similar to those of the HtrA2 inhibitor, UCF101. This study demonstrated that NIC reduces CME‐induced myocardial injury, reduces mitochondrial damage, and improves myocardial function. The reduction in cardiomyocyte apoptosis by NIC may be mediated by the HtrA2/XIAP/PARP signaling pathway.
Keywords: apoptosis, coronary microembolization, HtrA2, nicorandil, PARP, XIPA
The NIC improves myocardial function in CME rats by reduction cardiomyocyte apoptosis, and the reduction may be mediated by the HtrA2/XIAP/PARP signaling pathway.

Abbreviations
- AI
apoptotic index
- AMI
acute myocardial infarction
- CK‐MB
creatine kinase isoenzyme
- CME
coronary microembolization
- Cyt c
cytochrome c
- DAPI
2‐(4‐Amidinophenyl)‐6‐indolecarbamidine dihydrochloride
- FITC
fluoresceine isothiocyanate
- HBFP
hematoxylin basic fuchsin picric acid staining
- HE
hematoxylin‐eosin
- HtrA2
high‐temperature requirement peptidase 2
- LDH
lactate dehydrogenase
- LVEDd
left ventricular end‐diastolic diameter
- LVEF
left ventricular ejection fraction
- LVFS
left ventricular fractional shortening
- NIC
nicorandil
- PARP
poly ADP‐ribose polymerase
- PCI
percutaneous coronary intervention
- SD
Sprague Dawley
- TUNEL
TdT‐mediated DUTP nick end labeling
- XIAP
X‐linked inhibitor of apoptosis protein
1. INTRODUCTION
Coronary microembolization (CME) is a common complication of atherosclerotic plaque rupture during acute myocardial infarction (AMI) and percutaneous coronary intervention (PCI). CME resulting in microinfarction, myocardial injury, decreased coronary blood flow reserve. Clinical manifestations of CME include decreased myocardial contractility and arrhythmia. 1 , 2 CME affects the prognosis of PCI patients with AMI and reduces the benefit of reperfusion therapy. However, effective measures to prevent and treat CME are not currently available. 3 , 4 , 5 , 6 , 7 Previous studies have demonstrated that apoptosis is an important mechanism of CME‐induced myocardial injury, and inhibiting apoptosis can attenuate the extent of myocardial injury. 8 , 9 High‐temperature requirement peptidase 2 (HtrA2) is a serine protease located in the inner mitochondrial membrane space. HtrA2 is essential for mitochondrial quality control and the maintenance of mitochondrial homeostasis. HtrA2 can combine with the X‐linked inhibitor of apoptosis protein (XIAP), reducing the degradation of caspase 3/7 and promoting DNA fragmentation through poly ADP‐ribose polymerase (PARP), thus activating promoting apoptosis. 10 , 11 , 12 , 13
Nicorandil is a mitochondrial ATP‐sensitive potassium channel activator, which effectively protects cells against ischemia‐reperfusion injury. 14 The protective activity of NIC is most likely related to its anti‐inflammatory and antiapoptotic effects. Clinical studies have shown that NIC reduces the incidence of no‐reflow after primary PCI in AMI patients and preserves cardiac function. 15 However, the molecular mechanism of this effect has not been elucidated. Therefore, the goal of this study was to determine the impact of NIC on cardiomyocyte apoptosis and the role of HtrA2/XIAP/PARP signaling pathway in the mechanism of the NIC‐mediated improvement of cardiac function in rats subjected to CME. The obtained results can provide evidence supporting the application of NIC in the prevention and treatment of CME.
2. MATERIALS AND METHODS
2.1. Animals
Eight‐ to 10‐week‐old healthy Sprague Dawley (SD) rats (250‐300 g) of both sexes were acquired from the Experimental Animal Center of Guangxi Medical University. Experimental protocols involving the use of animals were reviewed and approved by the Animal Experiment Ethics Committee of Guangxi Medical University (No. 201907019).
2.2. Model and grouping
The rats were randomly divided into four groups: saline treatment (Sham), CME, NIC treatment (CME + NIC), HtrA2 inhibition (CME + UCF). The CME model was generated as described previously. 16 , 17 Animals were anesthetized by intraperitoneal injection of sodium pentobarbital (30‐40 mg/kg). After establishing artificial airway and ventilation, the skin and rib structures were incised along the left midclavicular line between the 3‐5 intercostal space, and the fascia, muscle, and soft tissue structure were separated. The ascending aorta was gently clamped with hemostatic forceps, and a 0.1 mL aliquot of normal saline containing about 4000 microspheres (diameter 45 μm, Polysciences, Warrington, PA, USA) was injected from the apical left ventricular area. The aorta was clamped for approximately 10 seconds (approximately 20 cardiac cycles). After the release of the clamp, tissue layers were sutured to close the chest cavity. In the Sham group, a corresponding volume of normal saline was injected into the ventricle, and the rest of the protocol was the same as in the CME group. In the CME + NIC group, NIC (5 mg/kg, Sihuan Kebao Pharmaceutical Co., Ltd., Beijing, China) was injected intraperitoneally (i.p.) 15 minutes before the injection of microspheres, and in the In CME + UCF group, UCF101 (1.5 μmol/kg; Sigma‐Aldrich, St. Louis, MO, USA) was injected i.p. 15 minutes before the injection of microspheres.
2.3. Measurement of cardiac function
Since previous studies documented that cardiac function in rats is significantly reduced 6‐12 hours after CME, 18 echocardiography was performed done 9 hours after sham operation or the induction of CME. The rats were anesthetized by an i.p. injection of 1% pentobarbital (40 mg/kg) to prevent their movement during image acquisition. The animals were fixed in the supine position, and a 10 MHz frequency probe of the MyLabSeven ultrasound system (Esaote, Genoa, Italy) was placed on the left front wall of the rat, facing the heart. Ultrasound imaging included the long axis of the left ventricle and the apical four‐chamber and two‐chamber views of the heart. The values of the left ventricular end‐diastolic diameter (LVEDd), LV fractional shortening (LVFS), LV ejection fraction (LVEF), and cardiac output were measured over three cardiac cycles and averaged. Echocardiography was performed blindly by an echocardiography physician.
2.4. Collection of blood and heart tissue samples
After the echocardiographic examination, a 2 mL blood sample was taken from the abdominal aorta of the rat. The blood was allowed to stand for more than 1 hour and centrifuged at 1200 g for 15 minutes at 4°C. The serum was collected and kept at −80°C until use. After blood collection, rats were sacrificed to collect cardiac tissue. After washing with ice‐cold normal saline, the atria and surrounding tissues were removed, and the ventricle was cut into several parts. One part was fixed in 10% neutral formalin for 24‐48 hours, paraffin‐embedded, sectioned and stained with hematoxylin‐eosin (HE) or hematoxylin basic fuchsin picric acid staining (HBFP). A second part was cut into 1 mm3 pieces and fixed with 2.5% glutaraldehyde for electron microscopy. The remaining portions of the heart were used for Western blotting; they were snap‐frozen in liquid nitrogen and stored at −80°C.
2.5. Detection of serum levels of CK‐MB and LDH
Creatine kinase isoenzyme (CK‐MB) was determined by the DGKC optimized colorimetric method kit, and lactate dehydrogenase (LDH) by the IFCC rate method kit (both kits from Zhicheng Biotechnology, Shanghai, China) using an automatic biochemical analyzer (Hitachi 7600‐020; Hitachi High‐Technologies Corporation, Tokyo, Japan).
2.6. Measurement of myocardial microinfarct size
The HBFP staining detects myocardial ischemia or infarct area at an early stage and is used to assess the area of microinfarction. Red color indicates myocardial ischemia or infarct, whereas yellow indicates normal myocardium. HBFP‐stained sections were analyzed using the DMR‐Q550 imager (Leica Microsystem, Wetzlar, Germany). Leica Qwin analysis software was used to randomly assign 10 fields of view (×100) in each sample and measure the infarct area. The average value of the infarct area divided by the total analyzed area was calculated. 19
2.7. Apoptosis assay
The magnitude of apoptosis was determined using the TUNEL assay kit (Roche Applied Science, Indianapolis, IN, USA). In this assay, all nuclei were stained in blue by 2‐(4‐Amidinophenyl)‐6‐indolecarbamidine dihydrochloride (DAPI), and apoptotic nuclei were stained green by fluorescein. Three sections of each specimen were examined by counting stained nuclei at 200× magnification in 10 randomly selected fields. Cardiomyocyte apoptotic index (AI) was calculated according to the formula: 17
2.8. Western blot analysis
Total protein was extracted from the left ventricular myocardium with RIPA lysis buffer, and protein concentration was determined using the BCA (bicinchoninic acid) method. Mitochondrial protein was extracted and measured as described by Ott. 20 , 21 After SDS‐PAGE gel electrophoresis, the proteins were transferred onto a PVDF membrane (Millipore, Bedford, MA, USA), and the membrane was placed in a blocking solution at room temperature for 1 hour. Subsequently, the membranes were incubated at 4°C with primary antibodies against cleaved caspase 9, cleaved caspase 3, Bax, Bcl‐2, HtrA2, XIPA, cleaved PARP, COX‐IV, and GAPDH (all antibodies from Abcam, Cambridge, MA, USA). The antibodies were diluted 1:1000. The next day, membranes were washed with TBST and incubated with goat anti‐rabbit IgG HL (HRP) (Abcam), diluted 1:10 000, at room temperature for 1 hour. Subsequently, 200‐300 μL of ECL chemiluminescence solution were added, and the membrane was placed in the FluorChemFC3 imaging system (ProteinSimple, Santa Clara, CA, USA). The intensities of bands were converted to gray‐scale values using Image J software. The relative expression of the target protein was calculated by dividing its gray‐scale value by the value of the internal control.
2.9. Statistical analysis
Data were analyzed by the SPSS 23.0 software (IBM Corporation, Armonk, New York, USA). The results are expressed as the mean ± standard deviation. Student's t‐test or ANOVA were used to compare the differences in outcomes between the groups. P < .05 was considered statistically significant.
3. RESULTS
3.1. The CME model
HE staining of myocardial tissue sections in the CME group showed transparent polyethylene microspheres lodged in the capillaries. Around the microspheres, the cardiomyocyte cytoplasm was dark red and condensed, the nucleus was shrunk or broken, and numerous inflammatory cells were present. The HE staining of the CME + NIC and CME + UCF groups showed the same as the CME group. In the Sham group, there was a small amount of inflammatory cell infiltration, and the myocardium did not exhibit the changes observed in the CME group (Figure 1).
FIGURE 1.
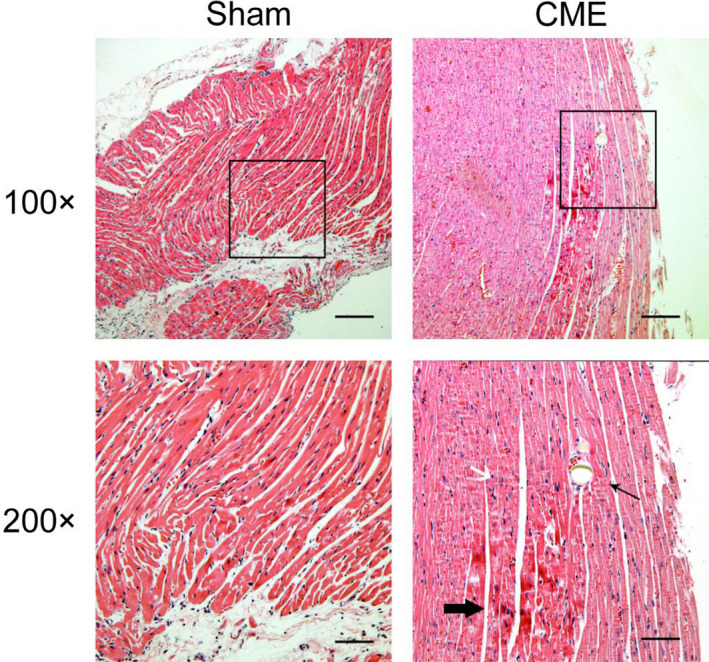
Hematoxylin‐eosin staining of the myocardium (100×, Scale bar = 200 μm; 200×, Scale bar = 100 μm). Microspheres, microinfarctions, and inflammatory cell infiltration are apparent in coronary microembolization (CME), whereas a small amount of inflammatory cell infiltration can be seen in the Sham group. The thin arrow indicates a microsphere in the microcoronary circulation, and the thick arrow indicates a microinfarct below the microsphere
3.2. NIC improved cardiac function after CME
Echocardiogram showed that LVEDd increased, whereas LVEF, LVFS, and cardiac output decreased after CME (P < .05). These changes demonstrate that CME induces cardiac dysfunction. NIC ameliorated cardiac dysfunction caused by CME, and a similar effect was seen in the CME + UCF group (P < .05) (Figure 2).
FIGURE 2.
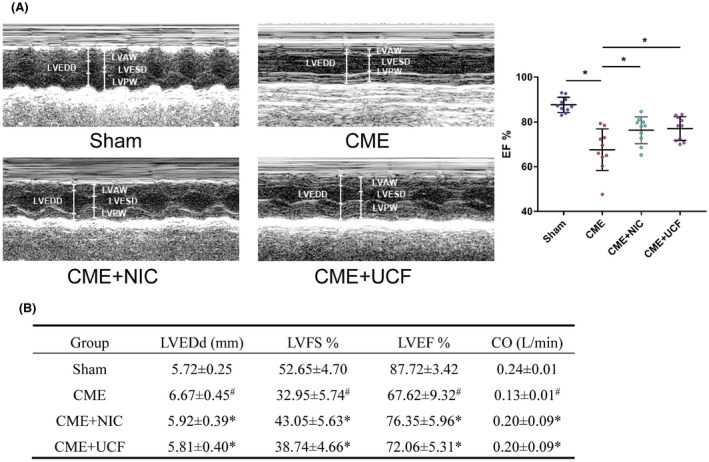
Representative M‐mode echocardiograms (A) and echocardiographic measurement values (B) in the four groups (n = 10 in each group). # P < .05 vs Sham, *P < .05 vs coronary microembolization (CME)
3.3. NIC reduced myocardial injury after CME
The serum concentration of CK‐MB and LDH in the CME group was significantly higher than in the Sham group (P < .05). NIC and UCF suppressed the increase in the level of these markers (P < .05) (Table 1).
TABLE 1.
The serum CK‐MB and LDH concentrations ( ± s)
| Group | n | CK‐MB (U/L) | LDH (U/L) |
|---|---|---|---|
| Sham | 10 | 772.50 ± 101.68 | 614.50 ± 130.10 |
| CME | 10 | 3075.70 ± 562.51* | 2396.90 ± 482.60* |
| CME + NIC | 10 | 1383.30 ± 272.54 # | 1967.30 ± 338.16 # |
| CME + UCF | 10 | 1526.30 ± 737.64 # | 1867.40 ± 327.20 # |
Abbreviations: CK‐MB, creatine kinase isoenzyme; CME, coronary microembolization; LDH, lactate dehydrogenase; NIC, nicorandil.
P < .05 compared with Sham.
P < .05 compared with CME.
3.4. NIC reduced myocardial microinfarct size after CME
To determine the size of microinfarcts, paraffin sections of myocardial tissue were stained with HBFP. The microinfarct area was not observed in the Sham group. In the CME, CME + NIC, and CME + UCF groups, multiple red‐stained infarcts were widely distributed. There was no evident infarcted area in the Sham group (Figure 3). Infarct size in CME, CME + NIC, and CME + UCF groups was 13.24 ± 2.04%, 8.01 ± 1.51%, and 7.76 ± 1.04%, respectively. In comparison with the CME group, the infarct area in rats treated with NIC or UCF was significantly reduced (P < .05) (Figure 3).
FIGURE 3.
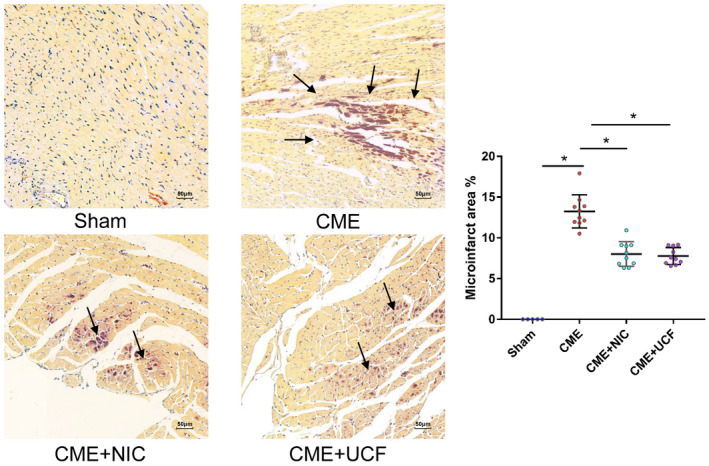
Hematoxylin basic fuchsin picric acid staining of myocardium shows microinfarct after modeling. (magnification 200×; bar = 50 μm) (Sham = 5, CME = 10, CME + NIC = 10, CME + UCF = 10). Normal myocardium is stained yellow, whereas ischemic or infarcted myocardium is stained dark red. Arrow points to the microinfarct area. There was no clear red infarct area in the Sham group. The microinfarct area in the CME + NIC and CME + UCF groups was significantly reduced. *P < .05
3.5. The effect of NIC on cardiomyocyte mitochondrial ultrastructure
The ultrastructure of the myocardium was analyzed by transmission electron microscopy. In the Sham group, the myofibrillar architecture was clear, and the mitochondrial membranes were essentially intact. In contrast, mitochondria in the CME group were significantly swollen, and their cristae were broken. However, the mitochondrial structure in the CME + NIC and CME + UCF groups remained essentially intact. Although they were slightly swollen, there was no evidence of the breakage of the cristae (Figure 4).
FIGURE 4.

Transmission electron micrograph of rat myocardium (magnification: 30 000×, scale bar = 1 μm). The arrows indicate mitochondria. The mitochondrial membrane in the Sham group was essentially intact. The mitochondria in the coronary microembolization (CME) group were significantly swollen, and the ridges were broken. The mitochondria in the CME + NIC group were slightly swollen, and the structure was appeared intact. The changes in CME + NIC were similar to CME + UCF (n = 5 in all groups)
3.6. NIC attenuated myocardial apoptosis after CME
Apoptotic index (AI) measured by the TUNEL assay was 1.56 ± 0.78%, 25.34 ± 1.45%, 17.55 ± 1.96%, 19.32 ± 1.94% in the Sham, CME, CME + NIC, and CME + UCF groups, respectively. In comparison with the Sham group, AI was significantly increased after CME (P < .05), and the AI in the CME + NIC and CME + UCF groups was significantly lower than in the CME group (P < .05). There was no statistical difference in AI between the CME + NIC and CME + UCF groups (Figure 5).
FIGURE 5.
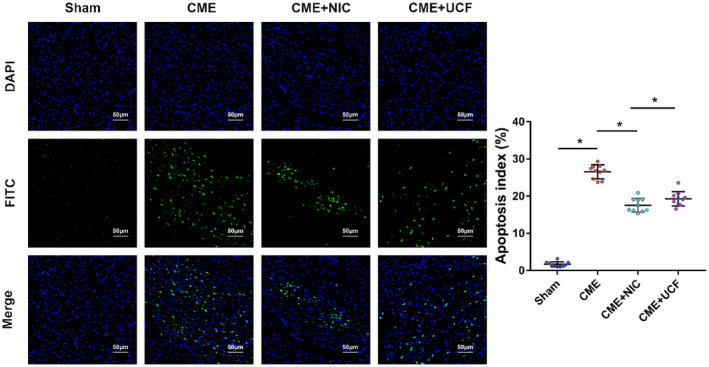
TUNEL staining shows myocardial apoptosis (magnification: 200×, scale bar = 50 μm) Blue fluorescence: nuclei stained by DAPI; green fluorescence: apoptotic nuclei stained by TUNEL (fluoresceine isothiocyanate (FITC). Apoptotic index (AI) in the CME group was significantly higher than in the Sham group, whereas the AI in the CME + NIC and CME + UCF groups was significantly lower than in the CME group. n = 10 in all groups; *P < .05
3.7. Expression of apoptosis‐related proteins
Protein expression levels were measured in all groups by Western blotting. In the CME group, the Bax/Bcl‐2 ratio, the relative level of cleaved caspase 3 and cleaved caspase 9 were elevated (P < .05). In comparison with the CME group, CME + NIC and CME + UCF groups showed decreased levels of cleaved caspase 3, cleaved caspase 9, and Bax, as well as increased expression of Bcl‐2 (P < .05). The difference between the CME + NIC and CME + UCF groups was not statistically significant (P < .05) (Figure 6).
FIGURE 6.
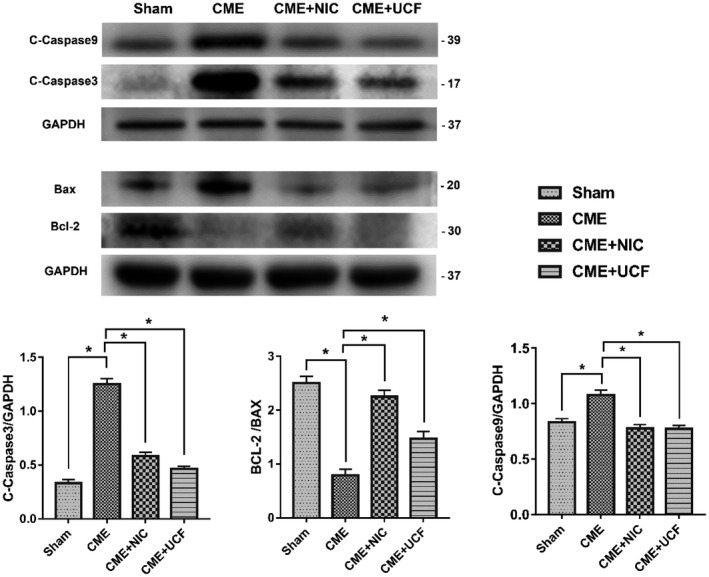
Impact of nicorandil on the expression of apoptosis‐related proteins (n = 6 or 8); *P < .05 vs Sham; # P < .05 vs coronary microembolization (CME)
3.8. The effect of NIC on the HtrA2/XIAP/PARP pathway
The expression of HtrA2 protein was determined in mitochondria and cytoplasm of cardiomyocytes. The level of HtrA2 in the cytoplasm of the CME group was significantly higher than in the Sham group, whereas the level of HtrA2 in the mitochondria was decreased (P < .05) (Figure 7A). Western blotting documented that CME increased the cytoplasmic levels of HtrA2 and cleaved PARP protein but decreased the expression of XIAP (all P < .05). Treatment with NIC suppressed these changes, significantly decreasing cytoplasmic HtrA2 and cleaved PARP and increasing XIAP (all P < .05). A similar effect was achieved with UCF (Figure 7B).
FIGURE 7.
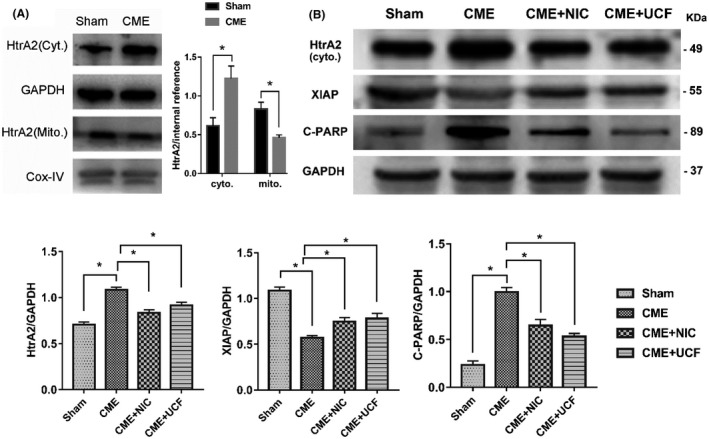
Effect of nicorandil on the HtrA2/XIAP/PARP signaling pathway. A, The effect of CME on HtrA2 by Western blotting. B, The effect of nicorandil on HtrA2/XIAP/PARP by Western blotting (n = 6 or 8 per group). Cyto., cytoplasmic, Mito., mitochondrial. *P < .05
4. DISCUSSION
This obtained results demonstrate CME in rats results in myocardial injury, decreased cardiac function, and cardiomyocyte apoptosis. Administration of NIC prior to CME reduced apoptosis and mitochondrial damage, thereby limiting myocardial injury and improving cardiac function. The mechanism underlying the effects of NIC likely involves. This cardioprotective effect of NIC may be mediated by a reduction in the translocation of HtrA2 into the cytoplasm and the suppression of the HtrA2/XIAP/PARP signaling, resulting in the inhibition of the activation of caspases and apoptosis.
Coronary microembolization is a recurrent consequence of PCI and the burst of unstable plaque in acute coronary syndrome. Unlike proximal epicardial coronary artery occlusion, CME causes myocardial microinfarction, but its effect on the degree of LV systolic dysfunction does not correlate with the magnitude of cardiomyocyte necrosis. 22 , 23 , 24 Cardiac cell apoptosis around microvascular infarction plays a key role in the progressive deterioration of cardiac function caused by myocardial injury after CME, and inhibiting apoptosis can ameliorate CME‐induced myocardial injury. 9 , 25 , 26 In this study, the rats subjected to CME showed elevated levels of myocardial injury markers, cardiac dysfunction, microinfarction, and increased cardiomyocyte apoptosis, consistent with the diverse pathophysiological manifestations of CME.
Mitochondria are the center of cell energy metabolism and form the hub of signaling pathways regulating cell survival and apoptosis. Mitochondrial permeability transition plays an important role in cell apoptosis and necrosis. Physical and chemical factors, hypoxia, and absence of nutrients trigger changes in mitochondrial membrane permeability, resulting in the release of proapoptotic proteins from mitochondria into the cytoplasm. After signal transduction, a series of cascade reactions are activated to promote cell apoptosis, characterized by nuclear pyknosis, cell shrinkage, cell membrane blebbing, and DNA fragmentation. Because the intracellular content is not released, inflammation is not induced. However, if the damage is severe and lasts for a long time, the membrane permeability continues to increase, causing the cells and the organelles to deform and swell. Finally, the cells rupture and the contents are discharged, often causing inflammation. Although the molecular effectors of apoptotic cell death have been extensively investigated over the past 30 years, enhancing the understanding of the importance of this process in cell biology, until recently, necrotic cell death was considered to generate similar effects. The regulation process of the molecule is defined as necroptosis. Mitochondria are important mediators of apoptosis and regulatory necrosis. 27 , 28
The mitochondrial apoptotic pathway is the endogenous signal pathway. The pro‐apoptotic members Bad / Bak of the BCL‐2 protein family form an oligomer complex, which is then inserted into the outer mitochondria membrane, changing the permeability of the mitochondrial membrane in response to damage signals. 29 , 30 Smac (second mitochondria‐derived activator of caspases), apoptosis‐inducing factor, and cytochrome c (Cyt c) are then released, and caspase 9 is recruited to the apoptosome composed of Cyt c, dATP, and Apaf‐1. These events initiate the execution of the mitochondrial pathway‐mediated apoptosis by activating caspase 3. This study documented that the mitochondria in the CME group were significantly swollen and had an abnormal structure. Additionally, the Bax/Bcl‐2 ratio in the CME group was elevated, and the level of cleaved caspases 9 and 3 was increased, indicating that the mitochondrial apoptosis pathway was activated. The administration of NIC before CME reduced mitochondrial damage, lowered the Bax/Bcl‐2 ratio, and decreased the level of cleaved caspases 9 and 3. Together, these results indicate that NIC inhibits the mitochondrial apoptotic pathway.
The HtrA family of proteins is a class of heat shock‐induced serine proteases whose main role is to degrade misfolded proteins in the cytoplasm. The precursor of HtrA2 is located in the mitochondria, but the activated and mature HtrA2 translocates to the cytoplasm where it binds and inactivates IAP, such as XIAP, c‐IAP1, and C‐IAP2, which degrade caspases. 31 , 32 , 33 , 34 , 35 Inactivation of these inhibitors increases caspase activity, potentiating the cleavage of PARP. The limited ability of PARP to repair DNA breaks promotes DNA fragmentation during apoptosis. 36 , 37 , 38 , 39 This study documented that the cytoplasmic HtrA2 was significantly increased after CME, whereas its level in the mitochondria was decreased. This finding implies that HtrA2 migrates from mitochondria to cytoplasm due to the myocardial injury produced by CME. Additionally, the expression of XIAP decreased, and that of cleaved‐PARP increased, suggesting that the HtrA2/XIAP/PARP mitochondrial apoptosis pathway may participate in CME‐induced myocardial injury.
Nicorandil is a selective opener of the mitochondrial ATP‐dependent potassium channel (mitoKATP). Many clinical trials have shown that NIC significantly improves the no‐reflow in PCI patients. 40 , 41 , 42 , 43 In animal experiments, NIC pretreatment decreased infarct size in the ischemia‐reperfusion model, reduced myocardial stunning, improved systolic function, and lowered the incidence of arrhythmia. 44 , 45 , 46 , 47 , 48 , 49 These cardioprotective effects are likely linked to the reduction in cardiomyocyte apoptosis and inflammation. Su et al 50 documented that oral NIC pretreatment ameliorates CME‐induced myocardial injury and improves cardiac function by inhibiting myocardial inflammation. Akao et al 46 demonstrated that NIC inhibits apoptosis induced by oxidative stress in cardiomyocytes in vitro. He et al 17 showed that oral NIC pretreatment effectively inhibits CME‐induced cardiomyocyte apoptosis and improves cardiac function in rats. Numerous studies analyzed the molecular mechanism of action of NIC, 44 , 51 , 52 , 53 , 54 but the specific mechanism by which it affects the HtrA2 pathway involved in mitochondrial apoptosis remains unclear. This work showed that the LVFS, LVEF, and cardiac output in the NIC group were improved in comparison with the CME group. Additionally, CK‐MB and LDH were lower, and the mitochondrial swelling was less pronounced, indicating that NIC protects against myocardial injury generated by CME. These findings are in agreement with previous studies suggesting that the mitochondrial apoptosis pathway participates in CME‐induced myocardial injury and may be inhibited by NIC. 17 The analysis of the key proteins of the HtrA2 signaling pathway showed that NIC decreases the cytoplasmic level of HtrA2 and cleaved‐PARP and upregulates the expression of XIAP in a manner similar to the HtrA2 inhibitor UCF101. Thus, the hypothesis can be advanced that NIC, acting as a mitoKATP activator, may suppress apoptosis by inhibiting the HtrA2/XIAP/PAR pathway, thereby reducing myocardial damage and improving cardiac function.
This study has some limitations. First, the physical properties of the microspheres used to induce CME are not completely consistent with the components causing microembolization clinically, such as fragments of atherosclerotic plaques, neutrophils, and platelet aggregates. Thus, it cannot be assumed that they strictly simulate pathophysiological changes consequent to CME. Second, the small size of rats necessitates the opening of the chest to inject microspheres for CME modeling, and this procedure affects cardiopulmonary function. Although the Sham group was included, the actual damage to heart function by microspheres was more serious than in clinical CME. Third, although the selection of rats was randomized, no special attention was given to gender balance. Therefore, it is impossible to eliminate gender differences in responses to drugs.
5. CONCLUSIONS
In summary, NIC reduces CME‐induced myocardial injury, decreases myocardial mitochondrial damage, and improves cardiac function. The possible mechanism of NIC action involves the reduction in CME‐induced cardiomyocyte apoptosis by affecting the HtrA2/XIAP/PARP signaling pathway. This pathway may become a new target for CME prevention and therapy.
DISCLOSURES
The authors declare no conflict of interest. The funders had no role in the design of the study, the collection, analyses, or interpretation of data, the writing of the manuscript, or the decision to publish the results.
ETHICS APPROVAL AND INFORMED CONSENT
Experimental protocols involving animals have been reviewed and approved by the Animal Experiment Ethics Committee of Guangxi Medical University.
ACKNOWLEDGMENTS
We thank Dr Yuhan Sun and Dr Xiantao Wang for their suggestions for manuscript revision. We are also grateful to Hong Luo, a technician in the Pathology Department, for her assistance with pathologic analyses.
Zheng J, Long M, Qin Z, Wang F, Chen Z, Li L. Nicorandil inhibits cardiomyocyte apoptosis and improves cardiac function by suppressing the HtrA2/XIAP/PARP signaling after coronary microembolization in rats. Pharmacol Res Perspect. 2021;9:e00699 10.1002/prp2.699
Funding information
This research was funded by the Project for innovative Research Team in Guangxi Natural Science foundation (2018GXNSFGA281006), Guangxi Zhuang Autonomous Region Health and Family Planning Commission Self‐Funded Scientific Research Project (Z2016312), and Open Project of Guangxi Key Laboratory of Precision Medicine in Cardio‐Cerebrovascular Diseases Control and Prevention (GXXNXG201902)
DATA AVAILABILITY STATEMENT
The data supporting the results of this research are available on reasonable request from corresponding author.
REFERENCES
- 1. Heusch G, Skyschally A, Kleinbongard P. Coronary microembolization and microvascular dysfunction. Int J Cardiol. 2018;258:17‐23. [DOI] [PubMed] [Google Scholar]
- 2. Sezer M, Umman S. [Role of leucocytes in microvascular malperfusion in reperfused acute myocardial infarction]. Anatol J Cardiol. 2008;8(1):48–50. [PubMed] [Google Scholar]
- 3. Salinas P, Jimenez‐Valero S, Moreno R, et al. Update in pharmacological management of coronary no‐reflow phenomenon. Cardiovasc Hematol Agents Med Chem. 2012;10(3):256‐264. [DOI] [PubMed] [Google Scholar]
- 4. Kloner RA. The importance of no‐reflow/microvascular obstruction in the STEMI patient. Eur Heart J. 2017;38(47):3511‐3513. [DOI] [PubMed] [Google Scholar]
- 5. Ito H. No reflow phenomenon in coronary heart disease. J Cardiol. 2001;37(Suppl 1):39‐42. [PubMed] [Google Scholar]
- 6. Basso C, Rizzo S, Thiene G. The metamorphosis of myocardial infarction following coronary recanalization. Cardiovasc Pathol. 2010;19(1):22‐28. [DOI] [PubMed] [Google Scholar]
- 7. Dols JL, van Zanten AP. Clinical implications of differences between two recommended procedures for determination of aspartate aminotransferase. Clin Chem. 1983;29(3):523‐526. [PubMed] [Google Scholar]
- 8. Wang J, Chen H, Su Q, Zhou Y, Liu T, Li L. The PTEN/Akt signaling pathway mediates myocardial apoptosis in swine after coronary microembolization. J Cardiovasc Pharmacol Ther. 2016;21(5):471‐477. [DOI] [PubMed] [Google Scholar]
- 9. Wang XT, Lu YX, Sun YH, He WK, Liang JB, Li L. TAK‐242 protects against apoptosis in coronary microembolization‐induced myocardial injury in rats by suppressing TLR4/NF‐kappaB signaling pathway. Cell Physiol Biochem. 2017;41(4):1675‐1683. [DOI] [PubMed] [Google Scholar]
- 10. Liu D, Wu L, Wu Y, et al. Heat shock factor 1‐mediated transcription activation of Omi/HtrA2 induces myocardial mitochondrial apoptosis in the aging heart. Aging. 2019;11(20):8982‐8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang K, Yuan Y, Liu X, et al. Cardiac specific overexpression of mitochondrial Omi/HtrA2 induces myocardial apoptosis and cardiac dysfunction. Sci Rep. 2016;6:37927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Briedigkeit L, Müller‐Plathe O, Schlebusch H, Ziems J. Recommendations of the German Working Group on medical laboratory testing (AML) on the introduction and quality assurance of procedures for Point‐of‐Care Testing (POCT) in hospitals. Clin Chem Lab Med. 1999;37(9):919‐925. [DOI] [PubMed] [Google Scholar]
- 13. Hu Y, Bi Y, Yao D, Wang P, Li Y. Omi/HtrA2 protease associated cell apoptosis participates in blood‐brain barrier dysfunction. Front Mol Neurosci. 2019;12:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdel‐Raheem IT, Taye A, Abouzied MM. Cardioprotective effects of nicorandil, a mitochondrial potassium channel opener against doxorubicin‐induced cardiotoxicity in rats. Basic Clin Pharmacol Toxicol. 2013;113(3):158‐166. [DOI] [PubMed] [Google Scholar]
- 15. Ji Z, Zhang R, Lu W, Ma G, Qu Y. The effect of nicorandil in patients with acute myocardial infarction undergoing percutaneous coronary intervention: a systematic review and meta‐analysis. Ir J Med Sci. 2020;189(1):119‐131. [DOI] [PubMed] [Google Scholar]
- 16. Liang J, Li L. The protective effect of activating Nrf2/HO‐1 signaling pathway on cardiomyocyte apoptosis after coronary microembolization in rats. BMC Cardiovasc Disord. 2017;17(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He W, Su Q, Liang J, Sun Y, Wang X, Li L. The protective effect of nicorandil on cardiomyocyte apoptosis after coronary microembolization by activating Nrf2/HO‐1 signaling pathway in rats. Biochem Biophys Res Comm. 2018;496(4):1296‐1301. [DOI] [PubMed] [Google Scholar]
- 18. Wang XT, Wu XD, Lu YX, et al. Potential Involvement of MiR‐30e‐3p in Myocardial Injury Induced by Coronary Microembolization via Autophagy Activation. Cell Physiol Biochem. 2017;44(5):1995‐2004. [DOI] [PubMed] [Google Scholar]
- 19. Thielmann M, Dörge H, Martin C, et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor‐alpha, and sphingosine. Circ Res. 2002;90(7):807‐813. [DOI] [PubMed] [Google Scholar]
- 20. Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two‐step process. Proc Natl Acad Sci U S A. 2002;99(3):1259‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu HR, Gao F, Tao L, et al. Antiapoptotic mechanisms of benidipine in the ischemic/reperfused heart. Br J Pharmacol. 2004;142(4):627‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Appelbaum E, Manning WJ. Science to practice: can the combination of resting first‐pass myocardial perfusion and late gadolinium‐enhanced cardiovascular MR imaging help identify myocardial infarction resulting from coronary microembolization? Radiology. 2009;250(3):609‐611. [DOI] [PubMed] [Google Scholar]
- 23. Carlsson M, Wilson M, Martin AJ, Saeed M. Myocardial microinfarction after coronary microembolization in swine: MR imaging characterization. Radiology. 2009;250(3):703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dörge H, Neumann T, Behrends M, et al. Perfusion‐contraction mismatch with coronary microvascular obstruction: role of inflammation. Am J Physiol Heart Circ Physiol. 2000;279(6):H2587–H2592. [DOI] [PubMed] [Google Scholar]
- 25. Liu T, Zhou Y, Liu YC, et al. Coronary microembolization induces cardiomyocyte apoptosis through the LOX‐1‐dependent endoplasmic reticulum stress pathway involving JNK/P38 MAPK. Can J Cardiol. 2015;31(10):1272‐1281. [DOI] [PubMed] [Google Scholar]
- 26. Zhu HH, Wang XT, Sun YH, et al. MicroRNA‐486‐5p targeting PTEN protects against coronary microembolization‐induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur J Pharmacol. 2019;855:244‐251. [DOI] [PubMed] [Google Scholar]
- 27. Amgalan D, Chen Y, Kitsis RN. Death receptor signaling in the heart: cell survival, apoptosis, and necroptosis. Circulation. 2017;136(8):743‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amgalan D, Garner TP, Pekson R, et al. A small‐molecule allosteric inhibitor of BAX protects against doxorubicin‐induced cardiomyopathy. Nat Cancer. 2020;1(3):315‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157‐183. [DOI] [PubMed] [Google Scholar]
- 30. D'Orsi B, Mateyka J, Prehn JHM. Control of mitochondrial physiology and cell death by the Bcl‐2 family proteins Bax and Bok. Neurochem Int. 2017;109:162‐170. [DOI] [PubMed] [Google Scholar]
- 31. Althaus J, Siegelin MD, Dehghani F, Cilenti L, Zervos AS, Rami A. The serine protease Omi/HtrA2 is involved in XIAP cleavage and in neuronal cell death following focal cerebral ischemia/reperfusion. Neurochem Int. 2007;50(1):172‐180. [DOI] [PubMed] [Google Scholar]
- 32. Bhuiyan MS, Fukunaga K. Activation of HtrA2, a mitochondrial serine protease mediates apoptosis: current knowledge on HtrA2 mediated myocardial ischemia/reperfusion injury. Cardiovasc Ther. 2008;26(3):224‐232. [DOI] [PubMed] [Google Scholar]
- 33. Liu HR, Gao E, Hu A, et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111(1):90‐96. [DOI] [PubMed] [Google Scholar]
- 34. Wang K, Zhang J, Liu J, et al. Variations in the protein level of Omi/HtrA2 in the heart of aged rats may contribute to the increased susceptibility of cardiomyocytes to ischemia/reperfusion injury and cell death: Omi/HtrA2 and aged heart injury. Age. 2013;35(3):733‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bucholz EM, Butala NM, Normand ST, Wang Y, Krumholz HM. Association of guideline‐based admission treatments and life expectancy after myocardial infarction in elderly medicare beneficiaries. J Am Coll Cardiol. 2016;67(20):2378‐2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8(3):613‐621. [DOI] [PubMed] [Google Scholar]
- 37. Virág L, Robaszkiewicz A, Rodriguez‐Vargas JM, Oliver FJ. Poly(ADP‐ribose) signaling in cell death. Mol Aspects Med. 2013;34(6):1153‐1167. [DOI] [PubMed] [Google Scholar]
- 38. Henning RJ, Bourgeois M, Harbison RD. Poly(ADP‐ribose) polymerase (PARP) and PARP inhibitors: mechanisms of action and role in cardiovascular disorders. Cardiovasc Toxicol. 2018;18(6):493‐506. [DOI] [PubMed] [Google Scholar]
- 39. Russell JC, Szuflita N, Khatri R, Laterra J, Hossain MA. Transgenic expression of human FGF‐1 protects against hypoxic‐ischemic injury in perinatal brain by intervening at caspase‐XIAP signaling cascades. Neurobiol Dis. 2006;22(3):677‐690. [DOI] [PubMed] [Google Scholar]
- 40. Xu L, Wang L, Li K, Zhang Z, Sun H, Yang X. Nicorandil prior to primary percutaneous coronary intervention improves clinical outcomes in patients with acute myocardial infarction: a meta‐analysis of randomized controlled trials. Drug Des Devel Ther. 2019;13:1389‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pang Z, Zhao W, Yao Z. Cardioprotective effects of nicorandil on coronary heart disease patients undergoing elective percutaneous coronary intervention. BMC Cardiovasc Disord. 2017;23:2924‐2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qi Q, Niu J, Chen T, Yin H, Wang T, Jiang Z. Intracoronary nicorandil and the prevention of the no‐reflow phenomenon during primary percutaneous coronary intervention in patients with acute ST‐segment elevation myocardial infarction. Med Sci Monit. 2018;24:2767‐2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niu X, Zhang J, Bai M, Peng Y, Sun S, Zhang Z. Effect of intracoronary agents on the no‐reflow phenomenon during primary percutaneous coronary intervention in patients with ST‐elevation myocardial infarction: a network meta‐analysis. BMC Cardiovasc Disord. 2018;18(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang A, Chen F, Xie Y, Guo Z, Yu Y. Protective mechanism of nicorandil on rat myocardial ischemia‐reperfusion. J Cardiovasc Med. 2012;13(8):511–515. [DOI] [PubMed] [Google Scholar]
- 45. Das B, Sarkar C, Karanth KS. Selective mitochondrial K(ATP) channel activation results in antiarrhythmic effect during experimental myocardial ischemia/reperfusion in anesthetized rabbits. Eur J Pharmacol. 2002;437(3):165‐171. [DOI] [PubMed] [Google Scholar]
- 46. Hirose M, Yano S, Nakada T, Horiuchi‐Hirose M, Tsujino N, Yamada M. Nicorandil ameliorates impulse conduction disturbances during ischemia in isolated arterially perfused canine atria. Int J Cardiol. 2011;146(1):37‐43. [DOI] [PubMed] [Google Scholar]
- 47. Ahmed LA, Salem HA, Attia AS, Agha AM. Pharmacological preconditioning with nicorandil and pioglitazone attenuates myocardial ischemia/reperfusion injury in rats. Eur J Pharmacol. 2011;663(1–3):51‐58. [DOI] [PubMed] [Google Scholar]
- 48. Das B, Sarkar C, Karanth KS. Effects of administration of nicorandil or bimakalim prior to and during ischemia or reperfusion on survival rate, ischemia/reperfusion‐induced arrhythmias and infarct size in anesthetized rabbits. Naunyn Schmiedebergs Arch Pharmacol. 2001;364(5):383‐396. [DOI] [PubMed] [Google Scholar]
- 49. Ohno Y, Minatoguchi S, Uno Y, et al. Nicorandil reduces myocardial infarct size by opening the K(ATP) channel in rabbits. Int J Cardiol. 1997;62(3):181‐190. [DOI] [PubMed] [Google Scholar]
- 50. Su Q, Lv X, Sun Y, Ye Z, Kong B, Qin Z. Role of TLR4/MyD88/NF‐κB signaling pathway in coronary microembolization‐induced myocardial injury prevented and treated with nicorandil. Biomed Pharmacother. 2018;106:776–784. [DOI] [PubMed] [Google Scholar]
- 51. Akao M, Teshima Y, Marbán E. Antiapoptotic effect of nicorandil mediated by mitochondrial atp‐sensitive potassium channels in cultured cardiac myocytes. J Am Coll Cardiol. 2002;40(4):803‐810. [DOI] [PubMed] [Google Scholar]
- 52. Li W, Wu N, Shu W, Jia D, Jia P. Pharmacological preconditioning and postconditioning with nicorandil attenuates ischemia/reperfusion‐induced myocardial necrosis and apoptosis in hypercholesterolemic rats. Exp Ther Med. 2015;10(6):2197‐2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu H, Ye M, Yang J, et al. Nicorandil protects the heart from ischemia/reperfusion injury by attenuating endoplasmic reticulum response‐induced apoptosis through PI3K/Akt signaling pathway. Cell Physiol Biochem. 2015;35(6):2320‐2332. [DOI] [PubMed] [Google Scholar]
- 54. Wang X, Pan J. Nicorandil alleviates apoptosis in diabetic cardiomyopathy through PI3K/Akt pathway. J Cell Mol Med. 2019;23(8):5349‐5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the results of this research are available on reasonable request from corresponding author.