

Abstract
Degradation and collapse of stalled replication forks are main sources of genomic instability, yet the molecular mechanisms for protecting forks from degradation/collapse are not well understood. Here, we report that human CST (CTC1‐STN1‐TEN1) proteins, which form a single‐stranded DNA‐binding complex, localize at stalled forks and protect stalled forks from degradation by the MRE11 nuclease. CST deficiency increases MRE11 binding to stalled forks, leading to nascent‐strand degradation at reversed forks and ssDNA accumulation. In addition, purified CST complex binds to 5’ DNA overhangs and directly blocks MRE11 degradation in vitro, and the DNA‐binding ability of CST is required for blocking MRE11‐mediated nascent‐strand degradation. Our results suggest that CST inhibits MRE11 binding to reversed forks, thus antagonizing excessive nascent‐strand degradation. Finally, we uncover that CST complex inactivation exacerbates genome instability in BRCA2 deficient cells. Collectively, our findings identify the CST complex as an important fork protector that preserves genome integrity under replication perturbation.
Keywords: CST complex, DNA degradation, genome stability, nascent strand, replication stress
Subject Categories: DNA Replication, Repair & Recombination
CTC1‐STN1‐TEN1, originally identified as telomeric single‐strand binding proteins, join the ranks of fork protecting factors, by virtue of ss/dsDNA junction binding in response to replication stalling.
Introduction
Faithful DNA replication is fundamental to genome integrity maintenance. During the process of DNA replication, replication machinery may encounter obstacles that cause replication forks slowing or stalling, leading to replication stress. Defects in stalled fork restart or stabilization induce genome instability, which is the main source of the pathology of human genetic diseases including cancer, aging, neurological diseases, and developmental defects (Zeman & Cimprich, 2013; Mazouzi et al, 2014).
A panoply of mechanisms protects genome stability in response to fork stalling to guard normal cell proliferation. Slowing or stalled replication generates extensive ssDNA stretches at forks, which are bound by the replication protein A (RPA) complex. RPA binding to ssDNA activates the ataxia telangiectasia and Rad3‐related (ATR) kinase and recruits numerous proteins to facilitate the protection and restart of stalled forks (Flynn & Zou, 2011; Marechal & Zou, 2015). Multiple pathways, including dormant origin firing, replication repriming, translesion synthesis, template switching, and fork reversal, act to facilitate the protection and restart of the stalled forks (Zeman & Cimprich, 2013; Bhat & Cortez, 2018).
Electron microscopy analysis has revealed that various replication stressors can trigger fork remodeling to form a chicken foot‐like physical structure through fork regression (Zellweger et al., 2015; Kolinjivadi et al., 2017). This fork reversal mechanism has emerged as an important means for stabilizing stalled forks and resuming replication following replication perturbation. For this reason, the dynamics of reversed forks has been extensively studied (Neelsen & Lopes, 2015; Quinet et al., 2017b; Rickman & Smogorzewska, 2019). The formation of reversed forks is catalyzed by SMARCAL1, ZRANB3, FANCM, HLTF, FBH1 (Gari et al., 2008; Betous et al., 2012; Kile et al., 2015; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017) and also involves RAD51 (Zellweger et al., 2015). While fork reversal is a protective mechanism to preserve fork stability under replication stress, the regressed arm also provides a target for nuclease‐dependent end resection. Without fork protectors such as BRCA1/2, RAD51, BARD1, and Abro1, reversed forks become vulnerable to nucleases MRE11, EXO1, DNA2, CtIP (Ying et al., 2012; Thangavel et al., 2015; Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Xu et al., 2017; Billing et al., 2018). Uncontrolled fork degradation by these nucleases is detrimental to genome stability and has been linked to both the lethality of BRCA2‐defective embryonic stem cells and the sensitivity of BRCA‐defective cells to certain chemotherapeutic treatments (Schlacher et al., 2011; Ying et al., 2012; Ray Chaudhuri et al., 2016). To date, the molecular mechanism underlying the balance between the protection and resection of regressed arms remains unclear.
The human CST complex, comprising of CTC1, STN1, and TEN1, contains multiple OB‐ fold domains and resembles the RPA70‐RPA32‐RPA14 complex structure (Miyake et al., 2009). CST prefers binding to G‐rich sequences in vitro in a manner dependent on the sequence length (Miyake et al., 2009; Chen et al., 2012). It is capable of binding to an 18‐nt G‐rich ssDNA with high affinity, but the sequence specificity is lost if the oligonucleotide becomes longer (Miyake et al., 2009; Hom & Wuttke, 2017). Interestingly, although CST is unable to bind to dsDNA, ss/dsDNA junctions stabilize CST binding and decrease minimal ssDNA length requirement for CST binding to 10 nt (Bhattacharjee et al., 2017), indicating that CST may bind to special DNA structures with ss/dsDNA junctions in vivo. A set of missense mutations of CTC1 and STN1 genes have been identified in patients with the Coats plus disease, a rare autosomal recessive disorder characterized by bilateral exudative retinopathy, retinal telangiectasias, growth retardation, intracranial calcifications, bone abnormalities, gastrointestinal vascular ectasias, accompanied by common early‐aging pathological features (Anderson et al., 2012; Keller et al., 2012; Simon et al., 2016).
Like its homolog in budding yeast (Cdc13‐Stn1‐Ten1), the well‐characterized and conserved roles of human/mammalian CST are in preserving telomere integrity. CST promotes efficient and complete replication of telomeric DNA to prevent sudden loss of telomeres (Huang et al., 2012; Gu & Chang, 2013). In addition, CST stimulates the priming activity of DNA polymerase α (POLα)‐primase (Lue et al., 2014; Ganduri & Lue, 2017) and mediates C‐strand fill‐in at telomere ends to replenish resected C‐strands (Huang et al., 2012). CST also inhibits telomerase access to telomeres and coordinates G‐ and C‐strand synthesis (Chen et al., 2012).
Recently, a number of telomere‐independent roles of CST have been uncovered. As a downstream effector in the 53BP1 pathway, CST participates in double‐strand break (DSB) repair in a Shieldin‐dependent manner (Barazas et al., 2018; Mirman et al., 2018). CST also plays a role in genome‐wide replication. During normal replication, CST directly associates with the MCM complex and blocks MCM binding to CDT1 to decrease origin licensing in G1‐phase. In addition, CST facilitates origin firing by promoting AND‐1 and POLα chromatin association in S‐phase (Wang et al., 2019). Previously, we have shown that CST associates with G‐ or C‐rich repetitive non‐telomeric sequences in response to fork stalling, probably by binding to ssDNA accumulated at stalled forks (Chastain et al., 2016). CST facilitates fork recovery and its deficiency induces instabilities of these sequences and chromosome fragmentation, indicating that CST is an important player for protecting genome stability under replication stress (Stewart et al., 2012; Chastain et al., 2016). However, the precise molecular mechanism underlying how CST participates in rescuing stalled replication remains unclear.
In this study, we report that CST is located at stalled forks upon hydroxyurea (HU) treatment, and CST deficiency leads to ssDNA accumulation under replication stress. DNA fiber analysis demonstrates that CST depletion induces MRE11 degradation of nascent‐strand DNA that is dependent on fork reversal, suggesting that CST antagonizes nascent DNA degradation at reversed forks. Using purified human CST and human MRE11 proteins, we find that CST binds to the ss/dsDNA structure that mimics the reversed fork, directly blocking MRE11 degradation in vitro. Consistent with our in vitro data, we observe that MRE11 binding to stalled forks is increased upon CST depletion. The fork protection function of CST requires its binding to DNA, and loss of CST binding to DNA leads to nascent‐strand degradation in cells and in vitro. In addition, we uncover that CST inactivation exacerbates genome instability in BRCA2 deficient cells. Collectively, our results suggest that CST binds to reversed forks to inhibit MRE11 access to nascent‐strand DNA, thus acting as a direct fork protector to maintain genome stability in response to replication stress.
Results
CST localizes at stalled forks
We have previously shown that in addition to promoting efficient replication of telomeric DNA, CST is also enriched at G‐ or C‐rich repetitive sequences genome‐wide after replication fork stalling to protect the stability of these sequences (Chastain et al., 2016). However, it is unknown whether such protective function is direct or indirect due to the lack of explicit evidence supporting CST localization at stalled forks. To gain insight into the function of CST in protecting genome stability, we examined the localization of endogenous CST proteins at stalled forks in response to replication stress using the in situ protein interactions at nascent and stalled replication forks (SIRF) assay, which offers sensitive visualization of protein localization at forks at a single‐cell level if the protein‐of‐interest is in close proximity to EdU‐labeled nascent strands (Roy et al., 2018). Briefly, nascent DNA in proliferating cells is labeled with EdU, and the incorporated EdU is then biotinylated by the click reaction. Subsequently, the proximity ligation assay (PLA) is used to visualize the colocalization of the target protein with biotinylated EdU (Fig 1A). To ensure the results were not cell line specific, we performed SIRF in two cell lines U2OS and HCT116. We pulse‐labeled cells with EdU for 8 min, treated them with HU for 3 h and then carried out SIRF analysis using anti‐CTC1 or anti‐STN1 antibody (Fig 1B and C). Since EdU incorporation efficiency might be affected by HU treatment (Roy et al., 2018), we performed the EdU SIRF as previously described (Roy et al., 2018). In agreement with previous report (Roy et al., 2018), we observed slight increase of EdU‐SIRF signal after HU treatment (Appendix Fig S1A). Thus, we normalized CTC1/STN1‐SIRF signals to mean EdU‐SIRF signals before statistical analysis (Fig 1B and C, and Appendix Fig S1A). Very few SIRF positive cells were observed without of EdU, suggesting that SIRF assay was specific for determining protein localization at forks (Fig 1B and C and Appendix Fig S1B). To validate that the antibody specifically recognized the endogenous CTC1 protein, we performed CTC1 SIRF in knockdown cells. Depletion of CTC1 resulted in the dramatic reduction of CTC1 SIRF foci (Appendix Fig S1F), suggesting the observed SIRF signal primarily resulted from antibody specific recognition of the CTC1 protein. As control, we performed SIRF against the DNA polymerase processivity factor proliferating cell nuclear antigen (PCNA). As expected, PCNA localized at active forks and formed distinct SIRF signals in untreated replicating cells (Fig 1B and C, Appendix Fig S1B). We also observed decreased PCNA SIRF foci after HU‐induced fork stalling (Fig 1B and C), consistent with previous findings from iPOND and SIRF assays that the amount of PCNA at stalled forks reduces when compared to that at elongating forks (Sirbu et al., 2011; Roy et al., 2018).
Figure 1.

-
AScheme of SIRF assay. Nascent DNA was pulse‐labeled with EdU. Upon fork stalling, proteins located at stalled forks are in close proximity to EdU. EdU was then biotinylated using click reaction, followed by incubating with biotin and target protein antibodies. PLA amplification was used to visualize the localization of target proteins at EdU‐labeled stalled forks.
-
B, CDetection of CST at stalled and active forks with SIRF assay in U2OS (B) and HCT116 (C) cells. To detect CST at stalled forks, cells were pulse‐labeled with EdU for 8 min, followed by 4 mM HU treatment for 3 h. To detect CST at active forks, SIRF was performed without HU treatment. Representative SIRF images of CTC1, STN1, and PCNA on normal or stalled forks are shown. Scale bars: 10 µm. Three independent experiments were performed and scatter plots from one experiment are shown in the main figure. Scatter plots of two additional biological replicates are included in Appendix Fig S1C (U2OS cells) and Appendix Fig S1D and E (HCT116 cells). Relative SIRF fluorescence intensity was calculated by normalizing to untreated samples and then to their respective EdU‐SIRF signals. Mean value in each sample is labeled in the graph. N: the number of cells analyzed in each sample. P values were calculated by Mann–Whitney test. ***P < 0.001. Red lines indicate the mean values. Error bars: SEM.
In both cell lines, we detected SIRF signals of CTC1 and STN1 in untreated replicating cells, indicating that CST also localizes at unperturbed forks (Fig 1B and C). This is in agreement with previous reports that CST interacts with POLα and facilitates DNA replication (Casteel et al., 2009; Chen et al., 2013; Ganduri & Lue, 2017). SIRF amplification signals from CTC1 and STN1 were increased by HU treatment (Fig 1B and C), suggesting an increased CTC1 and STN1 localization at stalled forks in response to HU treatment.
CST deficiency leads to the accumulation of ssDNA and increases MRE11 degradation of nascent‐strand DNA at stalled forks
The initial outcome of replication stalling is the aberrant ssDNA production at stalled forks, likely caused by the uncoupling of DNA helicase from DNA polymerase machinery. CST is a ssDNA‐binding protein complex and previously has been reported to suppress the formation of aberrant ssDNA at telomeres (Huang et al., 2012; Chen et al., 2013). We therefore examined the effect of CST depletion on genomic ssDNA production after fork stalling. HeLa cells depleted of STN1 (Fig 2A) were treated with HU for 3 h, a condition that induced fork stalling and produced ssDNA (Ercilla et al., 2019). Subsequently, BrdU staining was performed under the non‐denaturing condition to measure ssDNA amount. As shown in Fig 2B, STN1 depletion increased the amount of ssDNA after HU treatment, suggesting that CST loss leads to aberrant ssDNA accumulation (Fig 2B and Appendix Fig S2A).
Figure 2.

- Western blot showing STN1 knock‐down. shLUC (siRNA targeting luciferase) was used as the control.
- CST deficiency causes ssDNA accumulation upon fork stalling. HeLa cells with STN1 knock‐down were incubated with 10 µM BrdU for 48 h, followed by 2 mM HU treatment for 3 h, and subsequently stained with BrdU antibody under a non‐denaturing condition to detect ssDNA. N, the number of cells analyzed in each condition. Scale bars: 20 µm. Relative BrdU fluorescence intensity was quantitated by Image J. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. Error bars, SEM, ***P < 0.001, **P < 0.01. Three independent experiments were performed. Results from two additional biological replicates are included in Appendix Fig S2A.
- Scheme of DNA fiber analysis and representative fiber images.
- DNA fiber analysis of U2OS cells with STN1 knock‐down and its RNAi‐resistant Flag‐STN1 WT re‐expression with and without mirin treatment. Results from biological replicates and corresponding representative fiber images are included in Appendix Fig S2B.
- DNA fiber analysis of BJ cells with STN1 knock‐down by two siRNA sequence with and without mirin treatment. Results from biological replicates are included in Appendix Fig S2C.
- DNA fiber analysis of HCT116 cells with STN1 knock‐down with and without mirin treatment. Results from biological replicates are included in Appendix Fig S2D.
- DNA fiber analysis of U2OS cells showing that SMARCAL1‐mediated fork reversal is needed for nascent‐stand degradation observed in CST‐deficient cells. Results from biological replicates are included in Appendix Fig S2E.
- DNA fiber analysis of U2OS cells showing that ZRANB3‐mediated fork reversal is needed for nascent‐strand degradation observed in CST‐deficient cells. Results from biological replicates are included in Appendix Fig S2F.
Data information: For all DNA fiber experiments in this study, at least two independent treatments and fiber experiments were performed to ensure reproducibility. Results from one experiment are shown in the main figure. Scatter plots indicate IdU/CIdU tract length ratios for individual replication forks. N, the number of DNA fibers analyzed in each condition. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. ***P < 0.001, **P < 0.01, *P < 0.05. Red lines indicate mean values. Error bars: SEM. Western blots showing protein knock‐down in DNA fiber assays are included.
Source data are available online for this figure.
In response to replication stalling, forks may reverse to stabilize stalled forks and promote restart (Quinet et al., 2017b). Reversed forks, if not protected properly, are attacked by nucleases such as MRE11, DNA2 and EXOI, causing excessive degradation of nascent‐strand DNA (Thangavel et al., 2015; Lemacon et al., 2017). The accumulation of ssDNA in CST deficiency cells led us to hypothesize that CST might play a role in protecting nascent‐strand DNA from nucleolytic degradation. To test this hypothesis, we analyzed fork stability using DNA fiber assays (Nieminuszczy et al., 2016; Quinet et al., 2017a). Replication tracks were sequentially labeled with chlorodeoxyuridine (CIdU) and iododeoxyuridine (IdU) for 30 min, followed by 4 mM HU treatment for 5 h to induce fork stalling (Fig 2C). Only bi‐colored tracks were included in analysis. We found that STN1 depletion in U2OS cells caused increased fork degradation, and RNAi‐resistant wild‐type STN1 rescued such degradation in knockdown cells (Fig 2D and Appendix Fig S2B). The presence of the MRE11 inhibitor mirin suppressed such shortening, indicating that STN1 depletion leads to unscheduled MRE11‐mediated degradation of nascent‐strand DNA at stalled forks (Fig 2D). The same phenotype was recapitulated in two other cell lines normal foreskin fibroblast cell line BJ and colon cancer cell line HCT116 (Figs 2E and F, and Appendix Figs S2C and D). Likewise, CTC1 depletion with shRNA caused similar fork degradation (see Fig 5D below). Expression of the RNAi‐resistant CTC1 (res‐CTC1) cDNA fully rescued nascent‐strand shortening in knock‐down cells, confirming that the phenotype was caused specifically by knock‐down (see Fig 5D below).
Figure 5.
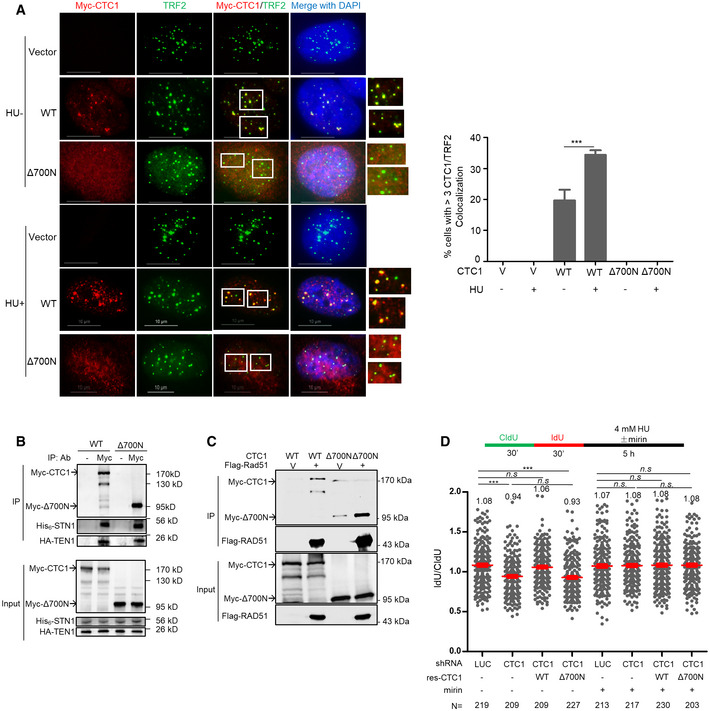
- Deletion of the N‐terminal 700 aa of CTC1 abolishes CTC1 localization at telomeres. U2OS stably expressing vector (V), WT Myc‐CTC1 and Myc‐Δ700N were treated with or without HU and co‐stained with Myc (red) and TRF2 (green) antibodies. Boxed areas are amplified in inserts to indicate CTC1/TRF2 colocalization (yellow).Scale bars: 10 μm. Quantification of percent of cells with > 3 CTC1/TRF2 colocalization foci was from three independent experiments. In each experiment, > 150 cells from each sample were analyzed. P values were calculated by one‐way ANOVA analysis with post hoc Tukey from three independent experiments, ***P < 0.001. Error bars: SEM.
- Δ700N does not affect CST complex formation. HEK293T cells were co‐transfected with Myc‐ CTC1 or Myc‐Δ700N, His6‐STN1, and HA‐TEN1. Co‐IP was performed with Myc antibody to pulldown His6‐ STN1 and HA‐TEN1. Full‐length Myc‐CTC1 was prone to degradation during immunoprecipitation.
- Δ700N retains RAD51 interaction. HEK293T cells were co‐transfected with Myc‐CTC1 or Myc‐Δ700N, His6‐ STN1, HA‐TEN1, and Flag‐RAD51 or vector control (V) and treated with HU (2 mM, 16 h). Co‐IP was performed with Flag antibody to pulldown Myc‐CTC1.
- DNA fiber analysis of CTC1 depleted U2OS cells with RNAi‐resistant WT or Δ700N re‐expression. Two independent experiments were performed. Results from the biological replicate are included in Appendix Fig S4B. Mean values in each sample are listed at the top of the graph. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. N, the number of cells analyzed in each condition, ***P < 0.001. Red lines indicate the mean values. Error bars: SEM.
Source data are available online for this figure.
Nascent‐strand degradation caused by CST deficiency requires fork reversal
Fork reversal through remodeling of stalled forks into four‐way junction structures is catalyzed by SMARCAL1, ZRANB3, and others (Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017). To determine whether the fork protection function of CST relied on fork reversal, we knocked down SMARCAL1 or ZRANB3 in STN1‐depleted cells and measured nascent‐strand degradation. Our results revealed that depletion of SMARCAL1 or ZRANB3 rescued nascent‐strand degradation in STN1‐deficient cells (Figs 2G and H, and Appendix Figs 2E and F). Consistent with previous reports (Coquel et al., 2018; Tonzi & Yin, 2018), depletion of SMARCAL1 slightly increased IdU/CldU ratio in control knock‐down cells (Fig 2G), though the underlying reason is unclear. Regardless, our results suggest that fork reversal is a prerequisite for fork degradation in CST‐deficient cells.
CST deficiency increases MRE11 association to stalled forks, leading to ssDNA accumulation
To determine whether nascent‐strand DNA degradation in CST‐deficient cells was caused by the accumulation of MRE11 at stalled forks, we then performed MRE11 SIRF. SIRF results revealed that STN1 depletion increased the accumulation of MRE11 at stalled forks (Fig 3A and Appendix Fig S3A). Treating STN1 knock‐down cells with mirin rescued ssDNA accumulation (Fig 3B and Appendix Fig S3B), suggesting that the elevated genome instability in CST‐deficient cells was likely caused by the unscheduled MRE11 nucleolytic resection of the nascent‐strand DNA.
Figure 3.
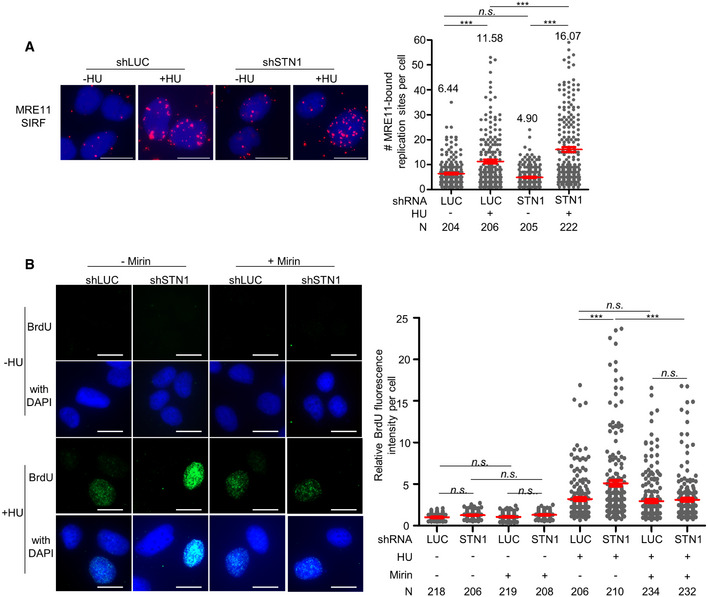
- SIRF detection of MRE11 at stalled forks in STN1‐deficient U2OS cells. Representative SIRF images of MRE11 at normal or stalled replication forks are shown. Scale bars: 10 µm. Two independent experiments were performed. Results from the biological replicate are shown in Appendix Fig S3A. P values were calculated by one‐way ANOVA analysis with post hoc Tukey, ***P < 0.001. Red lines indicate mean values. Error bars: SEM.
- ssDNA accumulation analysis in STN1‐deficient U2OS cells with and without mirin treatment. Representative images of native BrdU staining are shown. Scale bars: 20 µm. Three independent experiments were performed. Results from two additional biological replicates are shown in Appendix Fig S3C. P values were calculated by one‐way ANOVA analysis with post hoc Tukey, ***P < 0.001. Red lines indicate mean values. Error bars, SEM.
Purified CST binds to 5’ overhang and directly blocks MRE11 degradation of DNA
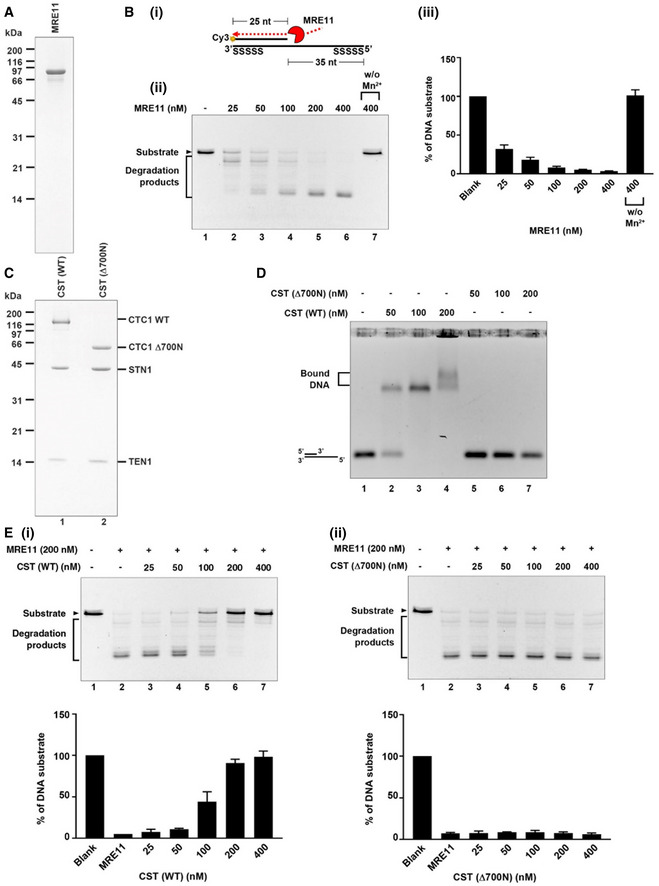
The above observations indicated that CST could protect reversal forks from MRE11 degradation to maintain genome stability. MRE11 possesses the 3’–5’ exonuclease activity and can extensively degrade ss/dsDNA junction structures with 5’ overhangs formed by regressed forks (Kolinjivadi et al., 2017). It has been reported that CST prefers binding to ss/dsDNA junctions in vitro (Bhattacharjee et al., 2017). Hence, we hypothesized that CST might bind to 5’ overhangs of regressed arms to protect nascent‐strand DNA from MRE11 degradation. To test this, we purified the human MRE11 protein (Fig 4A), generated a ss/dsDNA junction structure with a 5’ overhang that mimicked the regressed arm (Fig 4B i), and used the purified human heterotrimeric CST complex and MRE11 proteins to reconstitute nascent‐strand DNA protection in vitro. Recombinant human MRE11 was purified from Expi293F cells as described in Methods (Fig 4A) and then incubated with the ss/dsDNA substrate. In agreement with the previous report, MRE11‐dependent degradation in the 3’ to 5’ direction was observed (Fig 4B ii), and the MRE11 nuclease activity required cofactor Mn2+ (Fig 4B iii, and Appendix Fig S4F) (Paull & Gellert, 1998). Next, we purified the human CST complex from Expi293F cells to near homogeneity (Fig 4C, left lane). To ensure that only the complete protein complexes containing all CST subunits were used in the biochemical assays, affinity‐purified CST proteins were subject to gel filtration chromatography at the final step to remove non‐complexed individual proteins. As shown in Fig 4D, purified wild‐type CST (WT‐CST) complex bound to the 5’ overhang structure (Fig 4D). Intriguingly, when incubating WT‐CST pre‐assembled ss/dsDNA junctions with MRE11, WT‐CST protected DNA from MRE11 degradation in a dosage‐dependent manner (Fig 4E i).
Figure 4.
-
ACoomassie blue stained SDS–PAGE gel (15%) of purified human MRE11.
-
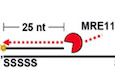
B(i) Scheme shows the nuclease activity of MRE11 in degrading 5’ Cy3‐labeled substrates (25 nt + 60 nt with phosphorothioate bonds on both ends). (ii) The 5’ Cy3‐labeled substrates (80 nM) were incubated with the indicated concentration of MRE11 with or without the Mn2+ cofactor at 37°C for 20 min. Reactions were stopped by SDS and proteinase K and resolved on 27% denatured polyacrylamide gel. (iii) The results are graphed, and error bars represent the standard deviation (± SD) calculated from at least three independent experiments.
-
CCoomassie blue stained SDS–PAGE gel (15%) of purified human CST complex (CTC1 wild‐ type and Δ700N).
-
DDNA‐binding activity of wild‐type CST complex and Δ700N mutant complex. The 5’ Cy3‐ labeled substrates (80 nM) were incubated with the indicated concentrations of CST. Samples were analyzed by 0.8 % agarose gel.
-
EMRE11 degradation analysis. 5’ Cy3‐labeled substrates (80 nM) were pre‐incubated with indicated concentrations of CST wild‐type (i) or Δ700N mutant variant (ii) at 37°C for 5 min. Reactions were completed by adding MRE11 (200 nM) for an additional 20 min incubation and then stopped by SDS and proteinase K. Samples were resolved in 27% denatured polyacrylamide gel. The results are graphed, and error bars represent the standard deviation (± SD) calculated from at least three independent experiments.
Source data are available online for this figure.
To validate the specificity of the ds/ssDNA junction protection by CST from MRE11 degradation, we tested the peculiarities of different components in this protecting system. We first purified human heterotrimeric RPA complex and reconstituted the DNA protection in vitro (Appendix Fig S4A). The result revealed that both of RPA and CST bound to ss/dsDNA substrates vividly with a comparable DNA‐binding affinity (Appendix Fig S4B). As shown above, CST (200 nM) protected DNA from MRE11 degradation with high efficiency (~90%). In contrast, side‐by‐side comparison showed that the same concentration of RPA significantly lacked such ability (only ~15%) (Appendix Fig S4C).
Next, to examine whether the CST inhibitory effect was specific for MRE11, we tested another 3’‐5’ exonuclease, bacterial ExoIII to replace MRE11 in the ds/ssDNA protection assay. Results showed that CST (200 nM) only slightly protected the ds/ssDNA substrate from ExoIII degradation (~37%) while provided efficient protection from MRE11 degradation (~90%) (Fig 4E i and Appendix Fig S4D).
CTC1‐STN1‐TEN1 prefers binding to G‐rich ssDNA in vitro (Chen et al., 2012; Hom & Wuttke, 2017), and our previous studies also showed that CST is significantly enriched at GC‐rich repetitive sequences genome‐wide after fork stalling in cells (Chastain et al., 2016). However, we found that both the GC‐rich and the random DNA sequence at 5’ overhang of the ss/dsDNA junction structure were protected from MRE11 degradation by CST at a similar level (Fig 4Ei and Appendix Fig S4E). Together, these results reveal the specific protection of CST to the 5’‐overhang DNA from MRE11 degradation.
The DNA‐binding ability of CST is required for protecting nascent‐strand degradation
CST is a ssDNA‐binding protein complex containing multiple OB folds (Bryan et al., 2013; Lim et al., 2020), and the previous report reveals that its DNA‐binding ability primarily relies on the OB‐fold domains at the N‐terminus of CTC1 (Miyake et al., 2009). To test the role of CST binding to ssDNA in antagonizing MRE11 degradation of nascent‐strand DNA, we constructed a truncated CTC1 mutant that removed the 700 residues in the N‐terminus harboring the DNA‐binding domain and then purified Δ700N in complex with STN1 and TEN1 (Fig 4C, right lane). In contrast to purified WT‐CST complex, Δ700N‐ST failed to bind to the 5’ overhang structure (Fig 4D) and completely abolished the ability to protect against MRE11 degradation (Fig 4E ii). These results support that CST binding to 5’ overhangs directly may protect ssDNA from MRE11 degradation.
We then tested the ability of Δ700N‐ST in nascent‐strand protection using cell‐based methods. Since CST binds to telomeric DNA, we first tested whether Δ700N localized at telomeres following the expression of Myc‐Δ700N in U2OS cells using retroviral transduction. While the full‐length WT‐CTC1 displayed telomere localization in both untreated and HU‐treated cells, Myc‐Δ700N exhibited diffused staining and failed to colocalize with TRF2, indicating that Δ700N lost telomere binding (Fig 5A), validating our in vitro observation that this mutant does not bind to DNA (Fig 4D). Next, we co‐transfected Myc‐Δ700N with His6‐STN1 and HA‐TEN1 into HEK293T cells and used co‐IP to confirm that Δ700N was still able to form a complex with STN1 and TEN1 in cells (Fig 5B). Since CST interacts with RAD51 in response to HU treatment and this interaction is important for recruiting RAD51 to fragile sites (Chastain et al., 2016), we also tested the effect of Δ700N on RAD51 binding. Previously, we have shown that deleting the N‐terminal 600 amino acids of CTC1 has no effect on CST‐RAD51 interaction while removing the N‐terminal 840 amino acids partially loses RAD51 interaction (Wang & Chai, 2018). When Δ700N was tested, we found that Δ700N largely retained the RAD51 interaction (Fig 5C), consistent with our previous finding that the RAD51‐interacting domain primarily resides within the C‐terminal half of CTC1 (Wang & Chai, 2018). This result also confirms our previous observation that CST interacts with RAD51 in a DNA‐independent manner (Chastain et al., 2016).
We then stably expressed RNAi‐resistant WT‐CTC1 or Δ700N in U2OS cells via retroviral transduction, depleted endogenous CTC1 with shRNA (Appendix Fig S5A) and carried out the DNA fiber analysis to measure fork degradation. While WT‐CTC1 fully rescued nascent‐strand degradation caused by CTC1 knockdown, expressing Δ700N failed to rescue (Fig 5D and Appendix Fig S5B). Thus, our results suggest that CST binding to ssDNA is required for protecting nascent strands from MRE11 attack.
CST depletion elevates genome instability in BRCA2‐deficient cells
BRCA2 is another ssDNA‐binding protein which antagonizes nascent‐strand degradation (Schlacher et al., 2011). Interestingly, CST shares a number of molecular features with BRCA2. They both interact with RAD51, bind to ssDNA, assist RAD51 recruitment to stalled forks (Schlacher et al., 2011; Taglialatela et al., 2017). Additionally, the phenotypes found after CST knock‐down at stalled forks are reminiscent to BRCA2‐deficient cells. These observations prompted us to explore their genetic relationship in genome maintenance. We first analyzed genome instability in cells co‐depleted of STN1 and BRCA2 (Appendix Fig S6A). As expected, depletion of STN1 alone or BRCA2 alone increased genome instability markers including micronuclei and abnormal anaphase bridges (Fig 6A and B). Single depletion of either STN1 or BRCA2 also induced the accumulation of γH2AX‐positive cells (Fig 6C), indicative of increased spontaneous DNA damage that might have arisen from fork instability. When BRCA2 and STN1 were co‐depleted, we observed elevated levels of micronuclei, anaphase bridges, and γH2AX (Fig 6A–C). In addition, co‐depletion significantly reduced BrdU incorporation (Fig 6D), suggesting that CST and BRCA2 double‐deficiency severely impairs DNA synthesis and diminishes the population of replicating cells. Upon HU treatment, either CST or BRCA2 single depletion displayed elevated chromosome aberrations and co‐depletion significantly increased chromosome aberrations (Fig 6E and Appendix Fig S6B). Collectively, these results suggest an additive effect of CST inactivation and BRCA2 deficiency in inducing genome instability. We then made repeated attempts to determine nascent‐strand degradation in double knock‐down cells using the DNA fiber assay. However, efficient co‐depletion of STN1 and BRCA2 resulted in massive detachment of cells from dish, and cells remaining on the dish had abolished STN1 knockdown (Appendix Fig S6C). Since detached cells were removed by washing during the DNA fiber assay, we were unable to obtain data that truly represented fork dynamics in double knock‐down cells.
Figure 6.
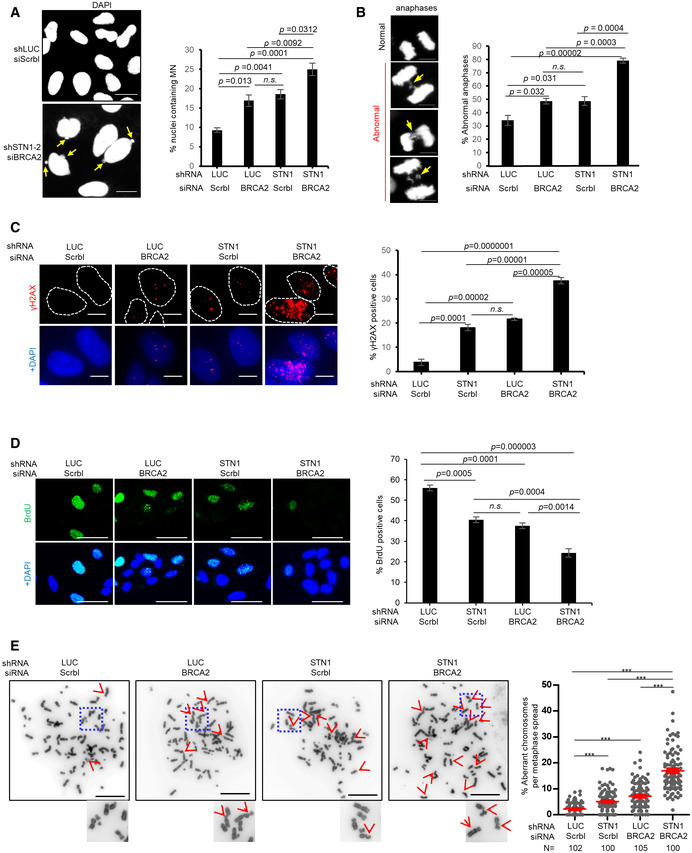
- Micronuclei (MN) formation in BRCA2‐ and STN1‐deficient cells. Co‐depletion was achieved by transfecting siBRCA2 in U2OS cells expressing shSTN1. Representative DAPI staining images are shown. Arrows point to MN. Scale bars: 20 µm. Average percentages of nuclei containing MN from three independent experiments are presented. In each experiment, >300 nuclei were analyzed per sample. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. Error bars: SEM.
- Anaphase bridges (arrows) in BRCA2‐ and STN1‐deficient U2OS cells. Scale bar: 10 µm. Average percentages of anaphase bridges from three independent experiments are presented. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. Error bars: SEM.
- γH2AX induced by BRCA2 knock‐down and STN1 knock‐down in U2OS cells. Nuclei containing ≥ 5 foci were considered as positive γ‐H2AX staining. Results were from three independent knock‐down experiments. In each experiment, > 80 cells were analyzed per sample. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. Error bars: SEM.
- Co‐depletion of STN1 and BRCA2 significantly impairs DNA replication. Scale bar: 50 µm. Results were from three independent knock‐down experiments. In each experiment, > 180 nuclei were analyzed per sample. P values were calculated by one‐way ANOVA analysis with post hoc Tukey from three independent experiments. Error bars: SEM.
- Co‐depletion of STN1 and BRCA2 increases chromosome instabilities. U2OS cells with siBRCA2 and/or shSTN1 knock‐down were treated with HU (2 mM, 3 h). Representative metaphase images show aberrant chromosomes (red arrows). Boxed areas are amplified and shown at the bottom of images. Scale bars: 20 µm. Two independent knock‐down and chromosome spread experiments were performed. Results of the biological replicate are included in Appendix Fig S6B. N, the number of metaphase spreads analyzed in each sample. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. ***P < 0.001. Error bars: SEM.
RAD51 recruitment by CST may provide an alternative means of stalled fork protection
RAD51 is essential for both mediating fork reversal and for protecting reversal fork from degradation (Zellweger et al., 2015; Kolinjivadi et al., 2017; Bhat & Cortez, 2018). Previously, we have observed that CST depletion attenuates RAD51 foci formation and its recruitment to fragile sites (Chastain et al., 2016), indicating that CST may facilitate RAD51 recruitment to stalled forks. To firmly test this, we performed RAD51 SIRF analysis in STN1 knockdown cells and observed that STN1 deficiency partially reduced RAD51 localization at stalled forks (Fig 7A and Appendix Fig S7A). In addition, loss of CST DNA‐binding ability decreased HU‐induced RAD51 foci formation (Fig 7B). Thus, CST binding to DNA is needed for efficient RAD51 recruitment to stalled forks. These results suggest that in addition to directly binding to ds/ssDNA at the regressed arms to block MRE11 degradation, CST recruiting RAD51 to stalled forks may provide an additional protection. Next, we co‐depleted STN1 and RAD51 and found that RAD51 depletion fully restored nascent‐strand stability in CST‐deficient cells (Fig 7C and Appendix Fig S7B). Therefore, RAD51‐mediated fork reversal is a prerequisite for CST protection of nascent‐strand DNA. This observation is consistent with the restoration of nascent‐strand stability after SMARCAL1 or ZRANB3 knock‐down in CST‐deficient cells (see above, Fig 2G and H).
Figure 7.
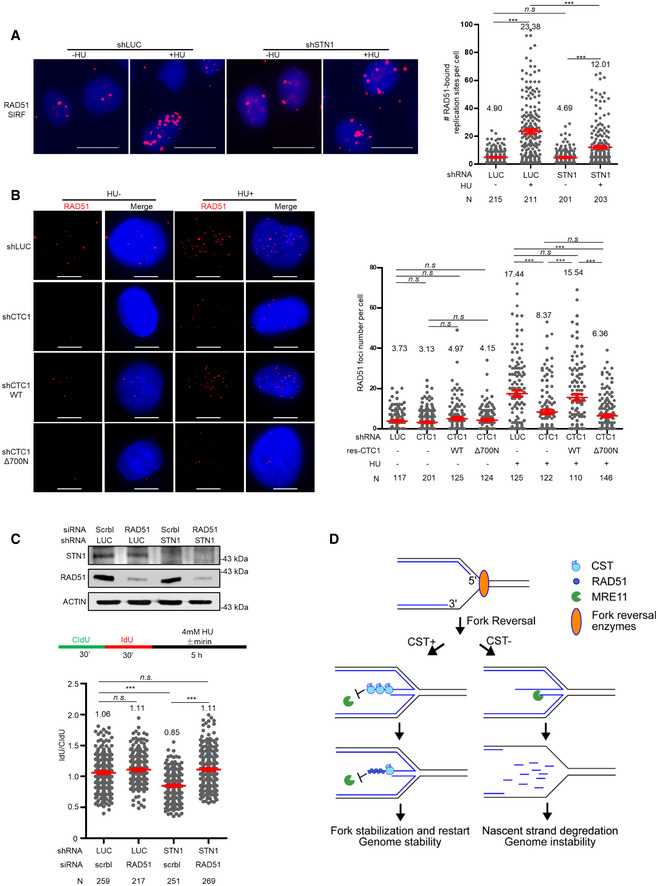
- SIRF analysis of RAD51 binding to forks. Representative RAD51 SIRF images at normal or stalled forks are shown. Scale bars: 10 µm. Two independent treatments were performed and scatter plot of RAD51‐bound replication sites from one experiment is shown. Results from the biological replicate are included in Appendix Fig S7A. Mean values in each sample are listed at the top of the graph. N, number of cells analyzed in each sample in one experiment. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. ***P < 0.001. Red lines indicate the mean values. Error bars: SEM.
- RAD51 foci formation in U2OS CTC1 knock‐down cells was rescued by RNAi‐resistant WT but not by Δ700N. Cells were treated with HU (2 mM, 16 h) and stained with anti‐RAD51. Representative images are shown. Scale bars: 10 µm. Scatter plot of RAD51 foci number with and without HU treatment is shown. Mean values in each condition are listed at the top of the graph. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. ***P < 0.001. N, the number of cells analyzed in each sample in one experiment. Red lines indicate the mean values. Error bars: SEM. Results from biological replicates are included in Appendix Fig S7B.
- DNA fiber analysis of nascent‐strand DNA degradation in U2OS cells with STN1 and RAD51 co‐depletion. Three independent treatments were performed and results from one biological replicate are shown. Results from two additional replicates are shown in Appendix Fig S7C. N, the number of DNA fibers analyzed in each sample in one experiment. P values were calculated by one‐way ANOVA analysis with post hoc Tukey. ***P < 0.001. Red lines indicate the mean values. Error bars: SEM. Western blots showing protein knock‐down in DNA fiber assays are included.
- Model for CST in protecting reversal forks stability. Upon fork stalling, the stalled fork is reversed by SMARCAL1, ZRANB3, or others. CST binds to ssDNA (5’ overhang) at reversed forks to directly block MRE11 access and may also recruit RAD51. Without CST, nascent‐strand DNA is degraded by MRE11, leading to the generation of excessive ssDNA and genome instability.
Source data are available online for this figure.
CST expression level positively correlates with cancer patient survival
Since genome instability gives rise to mutations that drive tumor initiation and development, we then assessed whether increased genome instabilities caused by CST deficiency would impact the outcome of cancer patient survival. Using Kaplan–Meier analysis, we found that breast cancer patients with CTC1 and STN1 expression at the upper tertile showed significant better disease‐free survival, while a worse survival outcome was observed in patients with CTC1 and STN1 expression at the lower tertile (Appendix Fig S8A; Gyorffy et al., 2010; Nagy et al., 2018). Future studies to determine whether CST can be used as a predictive biomarker in these subgroups may be clinically valuable.
Discussion
Protecting stalled forks from excessive nucleolytic resection is critical in resolving replication stress and maintaining genome stability. A number of studies have shown that many homology‐directed repair (HDR) factors, including BRCA1/2, RAD51, and CtIP, have HDR‐ independent roles in protecting fork stability (Hashimoto et al., 2010; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Przetocka et al., 2018). In this study, we provide direct evidence that the ssDNA‐binding complex CST is a new component at stalled forks and functions in limiting excessive nascent‐strand degradation. Using SIRF analysis, we observe CST localization at stalled replication forks. CST deficiency increases MRE11 binding to stalled forks and results in nascent‐strand degradation, leading to ssDNA accumulation and increased genome instability. Using purified CST proteins and a reconstituted biochemical system, we also show that CST binds to 5’ overhangs and directly blocks MRE11 nucleolytic degradation of DNA in vitro, supporting our DNA fiber results. Collectively, results in this study suggest that CST binds to 5’ overhangs formed at reversed forks, and such binding blocks MRE11 access to reversed forks, thus directly restricting MRE11 and perhaps other nucleases from excessively digesting nascent‐strand DNA (Fig 7D). In addition, we uncover the synthetic lethal relationship of CST with BRCA2.
Previously, we have observed that CST depletion attenuates RAD51 recruitment to GC‐rich sites (Chastain et al., 2016). In this study, we provide direct evidence demonstrating that CST deficiency partially reduces RAD51 localization to stalled forks (Fig 7A). Since RAD51 is essential for protecting fork stability (Kolinjivadi et al., 2017), reduced RAD51 binding to stalled forks may also contribute to nascent‐strand degradation in CST‐deficient cells. It is likely that CST may provide two layers of fork protection against unscheduled nuclease attack—it binds to reversed arms and directly blocks MRE11 activity in a RAD51‐independent manner, and meanwhile, it also interacts with RAD51 and recruits RAD51 to stalled forks (Fig 7D). Given the partial colocalization of CST and RAD51 (Chastain et al., 2016), we speculate that CST and RAD51 may protect stalled fork stability both collaboratively and independently.
CST was identified as an RPA‐like protein complex due to its ssDNA‐binding ability and structural similarity to RPA (Miyake et al., 2009; Lim et al., 2020). Despite this similarity, recent discoveries on CST at stalled forks and DSBs suggest that CST is functionally distinct from RPA. Although both CST and RPA can bind to ss/ds DNA junctions, RPA displays weak ability in antagonizing strand degradation (Appendix Fig S4C; Kolinjivadi et al., 2017). Unlike CST, RPA has no role in inhibiting DSB end resection. In fact, RPA has been found to promote end resection (Yan et al., 2011; Cannavo et al., 2013). Although it has been well established that the primary function of RPA binding to DNA is for ATR activation (Marechal & Zou, 2015), there has been no report describing the participation of CST in ATR signaling. While the exact relationship of RPA and CST remains to be determined, we notice that CST shares a number of functional and structural similarities with another genome stability protector, BRCA2. Structurally, both BRCA2 and CST contain multiple OB‐fold domains. Biochemically, they both bind to ssDNA and also interact with RAD51 (Schlacher et al., 2011; Chastain et al., 2016). Functionally, they both assist RAD51 loading to stalled forks and protect nascent‐strand DNA from MRE11 nucleolytic degradation, and CST depletion induces similar genome instabilities like BRCA2 depletion ((Schlacher et al., 2011) and this study). Our findings that CST inactivation further elevates genome instability and severely impairs DNA replication in BRCA2‐deficient cells (Fig 6) suggest an additive effect of CST dysfunction and BRCA2 deficiency. Further molecular investigation is needed to pinpoint the genetic relationship between these two important genome maintenance players.
CTC1‐STN1‐TEN1 interacts with POLα and stimulates POLα priming activity and primase‐to‐polymerase switching (Casteel et al., 2009; Nakaoka et al., 2011; Lue et al., 2014; Ganduri & Lue, 2017). At telomere ends, both CST and POLα participate in filling in C‐strands at resected telomere ends to shorten telomeric G‐overhang lengths (Gu et al., 2012; Huang et al., 2012). At DSBs in BRCA1‐null cells, CST deficiency results in an increase of phosphorylation of RPA S4/S8, indicative of extensive end resection (Mirman et al., 2018). The resemblance of lengthened G‐overhangs to increased DSB end resection after CST loss has led to the proposal that CST may also promote the fill‐in synthesis at resected DSB ends and that the defective fill‐in caused by CST deficiency may give rise to long 3’ overhangs that are suitable for initiating strand invasion and HDR (Barazas et al., 2018; Mirman et al., 2018). Since the same group of nucleases involved in DSB end processing, namely MRE11, DNA2, and EXO1, also resect reversed forks, it is tempting to speculate that CST may stimulate POLα fill‐in activity at reversed forks to antagonize end resection. Interestingly, our results show that CST binding to reversed forks can directly block MRE11 degradation of DNA (Fig 5). While our present data do not exclude the possible involvement of fill‐in synthesis in countering fork degradation, our results uncover a new function of CST in protecting fork stability and offer an alternative model that CST binds to ssDNA at reversed forks and directly inhibits MRE11 nuclease degradation, thereby protecting fork stability in a POLα‐independent manner (Fig 7D). Interestingly, while this manuscript was under review, the cryo‐EM structure of CST was reported, revealing that CST proteins are able to assemble into a decameric super complex when binding a 18‐nt ss telomeric oligo (Lim et al., 2020). We speculate that if such or a similar structure can be formed at reversed forks, CST occupying 5’ overhangs may establish a physical barrier for MRE11 access to the 3’ end. Future structural studies on the CST‐replication fork complex will be helpful to determine the exact nature of such a barrier.
Mutations of CST have been identified in the rare genetic disease known as Coats plus and thus CST has been mostly studied in the context of Coats plus pathogenesis. Our findings identifying CST’s role in protecting the fork stability thus reaffirm CST as an important genome maintenance player. Since the instability of stalled forks is a major source of genome instability in the early stages of tumorigenesis, it is likely that CST dysfunction may contribute to carcinogenesis. Indeed, the Cancer Genomics Atlas database shows that the CST genes are frequently altered in many types of cancers with mutations, deletions, and amplifications (Appendix Fig S8B) (Cerami et al., 2012; Gao et al., 2013). The positive correlation between CST gene expression level and the survival outcome in breast cancer patients (Appendix Fig S8A) suggests that CST may be worth clinical investigation as a potential prognosis biomarker. Investigating whether CST deficiency may promote tumorigenesis in vivo will also be compelling and crucial.
Materials and Methods
Cell culture
HeLa, U2OS, HCT116, BJ, and HEK293T cells were obtained from American Type Culture Collection. Cells were cultured in DMEM media supplemented with 10% cosmic calf serum (Thermo Fisher) at 37 °C containing 5% CO2.
| Cells | Source | Identifier |
|---|---|---|
| U2OS | ATCC | HTB‐96; RRID: CVCL_0042 |
| HeLa | ATCC | CCL‐2; RRID: CVCL_0030 |
| HEK293T | ATCC | CRL‐3216; RRID: CVCL_0063 |
| BJ | ATCC | CRL‐2522; RRID:CVCL_3653 |
| HCT116 | ATCC | CCL‐247; RRID: CVCL_0291 |
Plasmids
RNAi‐resistant pCL‐CTC1‐13xMyc, pcDNA‐HA‐TEN1, and pBabe‐Flag‐STN1 were described previously (Miyake et al., 2009; Huang et al., 2012; Chastain et al., 2016; Wang & Chai, 2018). The pCL‐CTC1Δ700N‐13xMyc plasmid was constructed by PCR amplifying CTC1Δ700 cDNA from pCL‐CTC1‐13xMyc and then cloned into pCL‐puro‐ FLAG‐MYC plasmid using the In‐Fusion® HD cloning kit (Takara). pCI‐neo‐His6‐STN1‐His6 plasmid was constructed by PCR amplifying STN1 cDNA using primers containing His6 sequences, and then cloning into the Xhol and Not1 sites of the pCI‐neo vector (Promega). The pEAK8 Flag‐CTC1, pEAK8 STN1, and pEAK8 TEN1‐(His)6 expression plasmids were gifts from Dr. Liuh‐Yow Chen (Institute of Molecular Biology, Academia Sinica (Chen et al., 2012)). For the co‐expression of STN1 and TEN1, the cDNAs of STN1 and carboxyl‐terminal (His)6‐tagged TEN1 were cloned into pcDNA3.4 vector with a dual CMV promoter. The amino‐terminal Flag‐tagged CTC1Δ700N mutant was generated with deletion PCR mutagenesis by using pEAK8 Flag‐CTC1 as the template. For the expression of human MRE11, the cDNA was amplified by PCR and then cloned into pcDNA3.4 vector to harbor amino‐terminal Flag‐tagged and carboxyl‐terminal (His)6‐tagged MRE11. All constructs were sequenced to ensure sequence accuracy.
Antibodies
The following primary antibodies are used. Primary antibodies, anti‐STN1 (WB 1:1,000, SIRF 1:100, Abcam, ab89250); anti‐CTC1 (WB 1:500, Abcam, ab175074 (discontinued by manufacturer)); anti‐CTC1 (SIRF 1:100, WB 1:500, Abcam, ab230538); anti‐Actin (WB 1:60,000, sigma, A5441); anti‐BrdU reacting with CldU (IF 1:1,000, DNA fiber 1:500, Abcam, ab6326); anti‐BrdU reacting with IdU (DNA fiber 1:50, BD Biosciences, 347580); anti‐Flag (WB 1:2,000, Sigma, F1804); anti‐SMARCAL1 (WB 1:1,000, Bethyl, A301‐616A); anti‐ZRANB3 (WB 1:1,000, Bethyl, A301‐033); anti‐Myc (WB 1:500, Santa cruz, sc‐40); anti‐Myc (WB 1:20,000, IF 1:20,000, Bethyl, A190‐105), anti‐HA (WB 1:10,000, Bethyl, A190‐108); anti‐Flag (WB 1:4,000, Millipore‐Sigma, F7245), anti‐MRE11 (WB 1:2,000, GeneTex, GTX70213); anti‐MRE11 (SIRF 1:200, Abcam, ab214); anti‐TRF2 (IF 1:200, IMGENEX, IMG‐124A); anti‐γH2AX (IF 1:2,000, Active Motif, 39117); anti‐BRCA2 (WB 1:1,000, IF 1:500, Millipore‐Sigma, OP95); anti‐RAD51 (IF 1:10,000. SIRF1:200, Abcam, ab63801); anti‐biotin (SIRF 1:100 Millipore‐Sigma, SAB4200680); anti‐biotin (SRIF 1: 200, Cell Signaling Technology, 5597).
Secondary antibodies: HRP goat anti‐rabbit (WB 1:10,0000, Vector Laboratory, PI‐1000); HRP Goat anti‐mouse (WB 1:5,000, BD Pharmingen, 554002);Goat anti‐Mouse Alexa Fluor 488 (IF 1:1,000, Thermo Fisher Scientific, A11029); Goat anti‐rabbit Alexa Fluor 550 (IF 1:1,000, Thermo Fisher Scientific, 84541); Goat anti‐Rat Alexa Fluor 488 (IF 1:1,000, Thermo Fisher Scientific, AB_2534074);
Chemicals
| Chemicals | Source | Identifier |
|---|---|---|
| X‐tremeGENE™ HP DNA Transfection Reagent | Millipore Sigma | 6366244001 |
| Puromycin | VWR | AAJ61278 |
| Hygromycin B | Millipore Sigma | 400053 |
| Hydroxyurea | Thermo Fisher Scientific | A10831 |
| Camptothecin | Millipore Sigma | 208925 |
| Methyl methanesulfonate | Thermo Fisher Scientific | AC156890050 |
| Mirin | Millipore Sigma | M9948 |
| BrdU | MP Biomedicals | SKU 02100166‐CF |
| IdU | Millipore Sigma | 54‐42‐2 |
| CIdU | MP Biomedicals | 105478 |
| EdU | Lumiprobe | 10540 |
| Biotin‐azide | Jena Bioscience | CLK‐1265‐5 |
| Duolink in situ PLA probe anti‐rabbit plus | Millipore Sigma | DUO92002 |
| Duolink in situ PLA probe anti‐mouse minus | Millipore Sigma | DUO92002 |
| Duolink in situ detection reagents red | Millipore Sigma | DUO92008 |
| Mounting medium with DAPI | Vector Labs | H‐1200 |
| Lipofectamine RNAiMax | Thermo Fisher Scientific | Cat#13778030 |
| Supersignal West Pico | Thermo Fisher Scientific | Cat#34078 |
| Supersignal West Femto | Thermo Fisher Scientific | Cat#34095 |
Software and algorithms
| Software | Source | Website |
|---|---|---|
| GraphPad Prism5 | GraphPad Software | https://www.graphpad.com/scientific‐software/prism;/ RR‐D:SCR_002798 |
| ImageJ | Department of Health and Human Services, NIDA | https://fiji.sc/ RRID: SCR_002285 |
siRNA
| siRNA | Source | Identifier |
|---|---|---|
| Negative Control siRNA | Qiagen | Catalog No. 1022076 |
| siBRCA2 | Qiagen | Catalog No. SI02653434 |
| siSMARCAL1 | Dharmacon | Catalog No. M‐013058‐01‐0005 |
| siZRANB3 | Dharmacon | Catalog No. D‐010025‐03‐005 |
| siRAD51 | Dharmacon | Catalog No. J‐003530‐11 |
shRNA
The above sequences were cloned into pSIREN‐retro as previously described (Chastain et al., 2016).
siRNA Transfections
All the transient gene depletion with siRNAs oligos were carried out using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. Cells transfected with siSMARCAL1 (40 nM), siZRANB3 (40 nM) or siRAD51 (20 nM) in experiments were performed 60 h post transfection. For siBRCA2 (40 nM), cells were transfected twice.
Immunofluorescence (IF) staining
IF was carried out as described previously (Chastain et al., 2016), Briefly, cells were grown on cover slips or chamber slides, then either fixed directly with 4% paraformaldehyde (PFA) in PBS for 15 min, or permeabilized in 0.15% Triton X‐100 in PBS for 2 min at room temperature prior to PFA fixation. Following fixation, cells were permeabilized with 0.15% Triton X‐100 in PBS for 15 min, washed 3 times for 5 min with PBS, blocked with 10% BSA at 37 °C for 1 h in humidified chamber, and then incubated with appropriate primary antibodies for overnight at 4 °C. Samples were then washed with PBS three times, incubated with secondary antibodies at room temperature for 1 h, and finally washed 3 times in PBS. Slides were then treated with cold ethanol series and dried in dark. Nuclei were visualized by counter staining with DAPI mounting medium (Vector Laboratories). Z‐stack images were obtained at a 0.3 µm thickness per slice under Zeiss AxioImager M2 epifluorescence microscope with a 40× or 100× objective. Single Z‐slice or max projection images were selected as representative images.
ssDNA detection (BrdU staining under the non‐denaturing condition)
To detect ssDNA under the non‐denaturing condition, cells were cultured in media supplemented with 20 µM BrdU for 48 h, followed by 2 mM HU treatment for 3 h. After fixation with PFA, cells were permeabilized with 0.15% Triton X‐100 for 15 min, washed 3 times for 5 min with PBS, blocked with 10% BSA at 37°C for 1 h in humidified chamber, and then incubated with anti‐BrdU (Abcam) overnight at 4°C. Regular IF procedures were then followed (see above).
Detection of replicating cells (BrdU staining under the denaturing condition)
To detect replicating cells, cells were cultured in media supplemented with 10 µM BrdU for 1 h, and then rinsed with PBS two times. After fixation with 4% PFA for 15 min, cells were rinsed with PBS three times and then incubated with 1 M HCl at 37°C for 20 min to denature DNA. Subsequently, denatured samples were washed with Na3PO4 (pH7.4) 3 times and then with PBS 5 times. Cells were then permeabilized with 0.15% Triton X‐100 for 15 min. After washing with PBS three times, samples were blocked with 10% BSA at 37°C for 1 h in humidified chamber and then incubated with anti‐BrdU at 4°C for overnight. Regular IF procedures were then followed (see above).
Co‐immunoprecipitation (Co‐IP)
Cells were lysed in lysis buffer (0.1% NP‐40, 50 mM Tris–HCl, pH 7.4, 50 mM NaCl, 2 mM DTT) supplemented with protease inhibitor cocktail (1 mM AEBSF, 0.3 µM aprotinin, 50 µM bestatin, 10 µM E‐64, 10 µM leupeptin, 5 µM pepstain and 1 mM PMSF), sonicated on ice, and centrifuged (13,000 rpm, 15 min, 4°C). The supernatants were precleared and immunoprecipitated with anti‐Myc or anti‐Flag antibody overnight at 4°C with constant rotation. For Myc IP, protein G‐conjugated agarose beads (Roche) were used to do pulldown. For Flag IP, protein G‐conjugated magnetic beads (Thermo Fisher) were used to pulldown according to the manufacturer’s protocol. Precipitates were washed with cold lysis buffer 4 times, then resuspended in lysis buffer with SDS sample loading buffer, boiled for 10 min, and used immediately on SDS–PAGE for immunoblotting.
SIRF assay
SIRF assay was performed as described previously (Roy et al., 2018). Briefly, cells were exponentially grown on chamber slides and labeled with EdU for 8 min. Cells were washed with PBS once and fixed with 2% PFA for 15 min at room temperature. For the condition to induce replication fork stalling, EdU was removed by washing with PBS twice and then treated with 4 mM HU for 3 h before fixation. Slides were washed in Coplin jars 3 times for 5 min each with PBS. After permeabilization with 0.25% Triton X‐100 in PBS for 15 min at room temperature, slides were washed 3 times for 5 min each with PBS, followed by incubation with the click reaction cocktail (2 mM copper sulfate, 10 µM biotin‐ azide, and 100 mM sodium ascorbate) in humid chamber at 37°C for 1 h. Slides were then washed 3 times for 5 min each with PBS and blocked with blocking buffer (10% BSA and 0.1% Triton X‐100 in PBS) at 37°C for 1 h. Primary antibodies were diluted in blocking buffer, dispensed onto slides, and incubated at 4°C in humidified chamber. Slides were then washed with wash buffer A (0.01 M Tris, 0.15 M NaCl and 0.05% Tween‐20, pH7.4) three times for 5 min each, and incubated with Duolink in situ PLA probe anti‐mouse plus and anti‐rabbit minus for 1 h at 37°C. Following wash with wash buffer A (0.01 M Tris, 0.15 M NaCl, and 0.05% Tween 20, pH 7.4) three times for 5 min each, slides were incubated with Duolink ligation mix at 37°C for 30 min, washed again with wash buffer A two times for 2 min each, incubated with Duolink amplification mix at 37°C for 100 min, washed with wash buffer B (0.2 M Tris and 0.1 M NaCl) three times for 10 min each and one time in 0.01x diluted wash Buffer B for 1 min. Lastly, slides were dried in dark and nuclei were visualized by counter staining with DAPI mounting medium (Vector Laboratories). Images were obtained under Zeiss AxioImager M2 epifluorescence microscope with a 100× objective.
DNA fiber analysis
DNA fiber assays were carried out as described previously (Nieminuszczy et al., 2016). Briefly, cells were first labeled with 25 µM CIdU for 30 min, followed by 250 µM IdU for 30 min. CldU/IdU was then removed by washing, and cells were immediately treated with 4 mM HU for 5 h. Mirin (50 µM) was concomitantly added with HU as indicated. Subsequently, cells were harvested, resuspended in PBS at 1,000 cells per µl, lysed in lysis buffer (200 mM Tris–HCl pH7.4, 50 mM EDTA, 0.5% SDS), and then DNA fibers were stretched on glass slides. Following fixation in methanol: acetic acid (3:1), slides were denatured with 2.5 M HCl for 80 min, washed with PBS, and then blocked with 5% BSA in PBS (w/v) for 30 min. Nascent DNA labeled by CIdU and IdU were immunostained with anti‐CldU and anti‐IdU antibodies, then incubated with anti‐rat Alexa Fluor 488 and anti‐mouse Alexa Fluor 568 secondary antibodies (Thermo Fisher Scientific). Coverslips were mounted using mounting medium (Vector Lab). Images were acquired with a Zeiss AxioImager M2 epifluorescence microscope at 40X magnification and analyzed using the Image J software. In all DNA fiber experiments in this study, two or three independent treatments and fiber experiments were performed to ensure reproducibility. Results from one set of experiment are shown in the main figure, and results from biological replicates are provided in Appendix figures. Scatter plots indicate IdU/CIdU tract length ratios for individual replication forks.
Expression and purification of the human CST complex, human MRE11, and RPA
Expression
pEAK8‐CTC1 WT or pEAK8‐CTC1Δ700N and pcDNA3.4‐STN‐TEN1‐(His)6 plasmids were co‐transfected into Expi293F cells according to instruction manual from ExpiFectamine 293 kit (Thermos Fisher Scientific). Cells were harvested by centrifugation after 72 h transfection.
Purification
All protein purification steps were carried out at 4°C. Cells were suspended in buffer A (25 mM Tris–HCl, pH 7.5, 10% glycerol, 0.01% Igepal, 150 mM KCl and 1 mM 2‐ mercaptoethanol) supplemented with protease inhibitors (1 mM PMSF and Benzamidine, 3 μg/ml each of aprotinin, chymostatin, leupeptin, and pepstatin A). Cells were lysed by sonication, followed by centrifugation at 40,000 g for 1 h. Lysates were supplemented with 10 mM imidazole and incubated with Ni2+ NTA‐agarose resin (QIAGEN) for 3 h. The resin was then poured into an open column and washed with buffer A containing 10 mM imidazole twice. The CST complex was eluted with buffer A (without 2‐mercaptoethanol) containing 200 mM imidazole. The eluate was then loaded into the anti‐Flag M2 affinity gel packaged column (Sigma) and washed with buffer A (without 2‐mercaptoethanol) twice. The CST complex was eluted with buffer A (without 2‐mercaptoethanol) containing 3x Flag peptide (100 µg/ml). The affinity‐purified CST complex was then loaded on Superdex 200 increase 10/300 GL column (GE Healthcare) equilibrated with butter A. The peak fractions were pooled and concentrated, then divided into small aliquots and stored at −80°C. The CTC1 Δ700N complex was purified using the same protocol.
For the human MRE11 purification, transient transfection of Flag‐MRE11‐(His)6 plasmid was also performed in Expi293F cells for 72 h. Cells were harvested and suspended with buffer A containing 500 mM KCl and the protease inhibitors as described above. After sonication and centrifugation, lysates were supplemented with 10 mM imidazole and incubated with Ni2+ NTA‐ agarose resin for 3 h. The resin was then poured into an open column and washed with buffer A containing 500 mM KCl and 10 mM imidazole twice. MRE11 was eluted with buffer A (without 2‐ mercaptoethanol) containing 200 mM imidazole. The eluate was then loaded into the anti‐Flag M2 affinity gel packaged column (Sigma) and washed with buffer A (without 2‐mercaptoethanol) twice. The MRE11 protein was eluted with buffer A (without 2‐mercaptoethanol) containing 3x Flag peptide (100 µg/ml), and then the eluate was fractionated in a 1 ml MonoQ column (GE Healthcare), using a 30 ml gradient of 150–1,000 mM KCl in buffer A. The peak fractions containing pure MRE11 were pooled and concentrated, then divided into small aliquots and stored at −80°C.
Other proteins
Human RPA was expressed in E. coli and purified as previously described (Sebesta et al., 2013). Bacterial ExoIII was purchased from New England Biolabs.
DNA substrates
Fluorescence‐labeled 5’ overhang DNA substrate was prepared by annealing the synthetic oligonucleotides described below. 60 nt ssDNA with the modified phosphorothioate bond (labeled with an asterisk) at both ends to prevent the non‐specific nucleases digestion was purchased from IDT:
Oligo 1:
5’‐A*C*G*C*T*GCCGAATTCTACCAGTGCCTTGCTAGGACATCTTTGCCCACCTGCAGGTTC*A*C*C*C*‐3’
Oligo 2 (for G‐rich 5’ overhang only):
5’‐G*A*A*C*G*CAGGGACATTGTGCTGTGCGGTGAGGTTTGTCTTTGCCCACCTGCAGGTTC*A*C*C*C*‐3’
Oligo 3 (25 nt) with modified Cy3 fluorescence dye at the 5’ end was purchase from Genomics: 5’‐Cy3‐GGGTGAACCTGCAGGTGGGCAAAGA‐3’.
Briefly, equal amounts of oligonucleotides (oligo 1 + 3, oligo 2 + 3) were mixed in the annealing buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 100 mM NaCl, and 1 mM DTT) and heated at 80 °C for 3 min. The mixed reaction was subsequently transferred to 65°C for 30 min and cooled down slowly to room temperature. The annealed substrate was purified from a 10% native polyacrylamide gel by electro‐elution and filter‐dialyzed in an Amicon ultra‐4 concentrator (Millipore, NMWL 10 kDa) at 4°C into TE buffer (10 mM Tris–HCl, pH 8.0, and 0.5 mM EDTA). The substrate concentration was quantified by using absorbance at 260 nm and the molar extinction coefficients of the substrate with Cy3 calculated by Molbiotools online software.
Electrophoretic mobility shift assay
The fluorescence‐labeled 5’ overhang DNA substrate (80 nM) was incubated with indicated amounts of the CST complex (CTC1 wild‐type or CTC1 Δ700N) or the RPA complex in 10 µl buffer B (35 mM Tris–HCl, pH 7.5, 1 mM DTT, 100 ng/µl BSA, and 50 mM KCl) at 37°C for 5 min. The reaction mixtures were then electrophoresed on a 0.8 % agarose gel with 1 × TBE buffer (89 mM Tris, 89 mM borate, and 2 mM EDTA, pH 8) at 100 V for 30 min at 4°C. Gels were analyzed by BioSpectrum 810 imaging system with a 533‐587 nm filter (UVP).
MRE11 degradation assay
Fluorescence‐labeled 5' overhang DNA substrate (80 nM) was incubated with indicated amounts of the CST complex (CTC1 wild‐type or CTC1 Δ700N) or the RPA complex in 10 μl buffer B containing 2.5 mM MgCl2, 1 mM ATP, and 1 mM MnCl2 at 37°C for 5 min, followed by incubation of purified human MRE11 (200 nM) at 37°C for 20 min or 40 min. Reactions were stopped by incubation with 2.5 µl stop buffer (50 mM EDTA, 0.4% SDS and 3.2 mg/ml proteinase K) at 37°C for 15 min. Samples were then mixed with the equal volume 2× denature dye (95% formamide, 0.1% Orange G, 10 mM Tris–HCl, pH 7.5, 1 mM EDTA, and 12% Ficoll PM400), heat denatured at 95°C for 10 min, and analyzed on 27% denature TBE‐Urea‐PAGE (7M Urea) with 1× TBE buffer at 300 V for 40 min at 55°C. Gels were analyzed by BioSpectrum 810 imaging system with a 533–587 nm filter. The intensity of DNA was quantified by the Image J software. Images shown represent results of independent experiments repeated at least three times. Data were analyzed and presented with the PRISM 7 software and shown as mean ± SD.
ExoIII degradation assay
Fluorescence‐labeled 5' overhang DNA substrate (80 nM) was incubated with indicated amounts of the CST complex in 10 μl buffer B containing 10 mM MgCl2 at 37°C for 5 min, followed by incubation of bacterial ExoIII (0.01U) at 37°C for 20 min. Reactions were stopped and samples were analyzed on 27% denature TBE‐Urea‐PAGE as described above.
Chromosome breakage assay
Chromosome fragmentation assay was performed as described (Chastain et al., 2016). To quantify the chromosome breakages, metaphase Images were obtained under Zeiss AxioImager M2 epifluorescence microscope with a 100× objective.
Prognostic value analysis
Kaplan–Meier analysis was performed using KM‐plotter online software (Gyorffy et al., 2010) (http://kmplot.com/analysis/). The relationship of gene expression and relapse‐free survival (RFS) (n = 3.955) was evaluated in an integrated multi‐study breast cancer microarray data set containing 13 breast cancer expression profiling data sets from GEO. Kaplan–Meier estimates of RFS (relapse‐free survival) were calculated by setting the software to look for the optimal cut‐off for separation of patients into the higher‐ and lower‐tertile expressing groups. The hazard ratio, log‐rank P value, and number of patients in each group are shown on the KM plot for each gene.
Author contributions
XL performed the majority of experiments except for the in vitro studies. K‐HL purified CST and MRE11 and carried out the in vitro experiments. PBS generated data in Appendix Fig S1F. OS participated in various experiments and collected data. MC performed IF experiments, generated K‐M plots, and collected data. WC and PC conceived and directed the study. XL, K‐HL, PC, and WC wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank Liuh‐Yow Chen (Academia Sinica, Taiwan) for constructs. The work is supported by NIH R01GM112864 and R01CA234266 to W.C., and Academia Sinica, National Taiwan University, and Taiwan Ministry of Science Technology (MOST 108‐2321‐B‐002‐054) to P.C.
The EMBO Journal (2021) 40: e103654.
Contributor Information
Peter Chi, Email: peterhchi@ntu.edu.tw.
Weihang Chai, Email: vchai@luc.edu.
Data availability
This study includes no data deposited in external repositories.
References
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J et al (2012) Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet 44: 338–342 [DOI] [PubMed] [Google Scholar]
- Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R et al (2018) The CST complex mediates end protection at double‐strand breaks and promotes PARP inhibitor sensitivity in BRCA1‐deficient cells. Cell Rep 23: 2107–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu‐Nkansah A, Sirbu BM, Eichman BF, Cortez D (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Cortez D (2018) RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 25: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Wang Y, Diao J, Price CM (2017) Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome‐wide roles of human CST. Nucleic Acids Res 45: 12311–12324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billing D, Horiguchi M, Wu‐Baer F, Taglialatela A, Leuzzi G, Nanez SA, Jiang W, Zha S, Szabolcs M, Lin CS et al (2018) The BRCT Domains of the BRCA1 and BARD1 Tumor Suppressors Differentially Regulate Homology‐Directed Repair and Stalled Fork Protection. Mol Cell 72(1): 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan C, Rice C, Harkisheimer M, Schultz DC, Skordalakes E (2013) Structure of the human telomeric Stn1‐Ten1 capping complex. PLoS One 8: e66756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P, Kowalczykowski SC (2013) Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11‐Rad50‐Xrs2 to DNA end resection. Proc Natl Acad Sci USA 110: E1661–E1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteel DE, Zhuang S, Zeng Y, Perrino FW, Boss GR, Goulian M, Pilz RB (2009) A DNA polymerase‐α·primase cofactor with homology to replication protein A‐32 regulates DNA replication in mammalian cells. J Biol Chem 284: 5807–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastain M, Zhou Q, Shiva O, Whitmore L, Jia P, Dai X, Huang C, Fadri‐Moskwik M, Ye P, Chai W (2016) Human CST facilitates genome‐wide RAD51 recruitment to GC‐rich repetitive sequences in response to replication stress. Cell Rep 16: 1300–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Majerska J, Lingner J (2013) Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev 27: 2099–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Redon S, Lingner J (2012) The human CST complex is a terminator of telomerase activity. Nature 488: 540–544 [DOI] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL et al (2018) SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61 [DOI] [PubMed] [Google Scholar]
- Ercilla A, Feu S, Aranda S, Llopis A, Brynjólfsdóttir SH, Sørensen CS, Toledo LI, Agell N (2019) Acute hydroxyurea‐induced replication blockade results in replisome components disengagement from nascent DNA without causing fork collapse. Cell Mol Life Sci 77: 735–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Zou L (2011) ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 36: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganduri S, Lue NF (2017) STN1‐POLA2 interaction provides a basis for primase‐pol alpha stimulation by human STN1. Nucleic Acids Res 45: 9455–9466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A (2008) Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA 105: 16107–16112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu P, Chang S (2013) Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell 12: 1100–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu P, Min JN, Wang Y, Huang C, Peng T, Chai W, Chang S (2012) CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 31: 2309–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123: 725–731 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom RA, Wuttke DS (2017) Human CST prefers G‐Rich but not necessarily telomeric sequences. Biochemistry 56: 4210–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Dai X, Chai W (2012) Human Stn1 protects telomere integrity by promoting efficient lagging‐strand synthesis at telomeres and mediating C‐strand fill‐in. Cell Res 22: 1681–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller RB, Gagne KE, Usmani GN, Asdourian GK, Williams DA, Hofmann I, Agarwal S (2012) CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer 59: 311–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA (2015) HLTF's ancient HIRAN domain binds 3' DNA ends to drive replication fork reversal. Mol Cell 58: 1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L et al (2017) Smarcal1‐Mediated fork reversal triggers Mre11‐dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell 67(5): 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N et al (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81‐dependent fork rescue in BRCA2‐deficient cells. Nat Commun 8: 860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CJ, Barbour AT, Zaug AJ, Goodrich KJ, McKay AE, Wuttke DS, Cech TR (2020) The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 368: 1081–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue NF, Chan J, Wright WE, Hurwitz J (2014) The CDC13‐STN1‐TEN1 complex stimulates Pol alpha activity by promoting RNA priming and primase‐to‐polymerase switch. Nat Commun 5: 5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Zou L (2015) RPA‐coated single‐stranded DNA as a platform for post‐translational modifications in the DNA damage response. Cell Res 25: 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazouzi A, Velimezi G, Loizou JI (2014) DNA replication stress: causes, resolution and disease. Exp Cell Res 329: 85–93 [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P et al (2017) Replication fork reversal triggers fork degradation in BRCA2‐defective cells. Nat Commun 8: 859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T (2018) 53BP1‐RIF1‐shieldin counteracts DSB resection through CST‐ and Polalpha‐dependent fill‐in. Nature 560: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F (2009) RPA‐like mammalian Ctc1‐Stn1‐Ten1 complex binds to single‐stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell 36: 193–206 [DOI] [PubMed] [Google Scholar]
- Nagy Á, Lánczky A, Menyhárt O, Győrffy B (2018) Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 8: 9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaoka H, Nishiyama A, Saito M, Ishikawa F (2011) Xenopus laevis Ctc1‐Stn1‐Ten1 (xCST) complex is involved in priming DNA synthesis on single‐stranded DNA template in Xenopus egg extract. J Biol Chem 287: 619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220 [DOI] [PubMed] [Google Scholar]
- Nieminuszczy J, Schwab RA, Niedzwiedz W (2016) The DNA fibre technique ‐ tracking helicases at work. Methods 108: 92–98 [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1998) The 3' to 5' exonuclease activity of Mre 11 facilitates repair of DNA double‐strand breaks. Mol Cell 1: 969–979 [DOI] [PubMed] [Google Scholar]
- Przetocka S, Porro A, Bolck HA, Walker C, Lezaja A, Trenner A, von Aesch C, Himmels SF, D'Andrea AD, Ceccaldi R et al (2018) CtIP‐mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol Cell 72: 568–582 [DOI] [PubMed] [Google Scholar]
- Quinet A, Carvajal‐Maldonado D, Lemacon D, Vindigni A (2017a) DNA fiber analysis: mind the gap!. Methods Enzymol 591: 55–82 [DOI] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, Vindigni A (2017b) Replication fork reversal: players and guardians. Mol Cell 68: 830–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, Smogorzewska A (2019) Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol 218: 1096–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Luzwick JW, Schlacher K (2018) SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J Cell Biol 217: 1521–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebesta M, Burkovics P, Juhasz S, Zhang S, Szabo JE, Lee MYWT, Haracska L, Krejci L (2013) Role of PCNA and TLS polymerases in D‐loop extension during homologous recombination in humans. DNA Repair 12: 691–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, Kol N, Barel O, Cesarkas K, Soudack M et al (2016) Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med 213: 1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD 2nd, Wright WE, Price CM (2012) Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J 31: 3537–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R et al (2017) Restoration of replication fork stability in BRCA1‐ and BRCA2‐deficient cells by inactivation of SNF2‐family fork remodelers. Mol Cell 68(2): 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA et al (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonzi P, Yin Y (2018) Translesion polymerase kappa‐dependent DNA synthesis underlies replication fork recovery. eLife 7:e41426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K et al (2017) Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol Cell 67(5): 882–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Brady KS, Caiello BP, Ackerson SM, Stewart JA (2019) Human CST suppresses origin licensing and promotes AND‐1/Ctf4 chromatin association. Life Sci Alliance 2:e201800270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chai W (2018) Pathogenic CTC1 mutations cause global genome instabilities under replication stress. Nucleic Acids Res 46: 3981–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Wu X, Wu L, Castillo A, Liu J, Atkinson E, Paul A, Su D, Schlacher K, Komatsu Y et al (2017) Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev 31: 1469–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Toczylowski T, McCane J, Chen C, Liao S (2011) Replication protein A promotes 5'–>3' end processing during homology‐dependent DNA double‐strand break repair. J Cell Biol 192: 251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T (2012) Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821 [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA (2013) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.