

Abstract
Chromosomal instability (CIN) is a hallmark of cancer. While low levels of CIN can be tumor promoting, high levels of CIN cause cell death and tumor suppression. The widely used chemotherapeutic paclitaxel (Taxol™) exerts its anti-cancer effects by increasing CIN above a maximally tolerated threshold. One significant outstanding question is whether the p53 tumor suppressor is required for the cell death and tumor suppression caused by high CIN. Both p53 loss and reduction of the mitotic kinesin CENP-E cause low CIN. Combining both genetic insults in the same cell leads to high CIN. Here we test whether high CIN causes cell death and tumor suppression even in the absence p53. Despite a surprising sex-specific difference in tumor spectrum and latency in p53 heterozygous animals, these studies demonstrate that p53 is not required for high CIN to induce tumor suppression. Pharmacological induction of high CIN results in equivalent levels of cell death due to loss of essential chromosomes in p53+/+ and p53−/− cells, further demonstrating that high CIN elicits cell death independently of p53 function.
IMPLICATIONS:
These results provide support for the efficacy of anti-cancer therapies that induce high CIN, even in tumors that lack functional p53.
Introduction
Defects during mitosis have been observed in cancer cells for over 100 years (1). These defects result in chromosome missegregation and aneuploid daughter cells, which contain an abnormal set of chromosomes that is not a multiple of the haploid karyotype. Continuous chromosome missegregation over multiple divisions results in CIN. Both aneuploidy and CIN are common in diverse cancer types (2–4). The prevalence of aneuploidy and CIN in cancer led to the hypothesis that they are tumor promoting (5,6), which has been tested in numerous animal models that exhibit aneuploidy with or without CIN (2,7–11). These animals have diverse cancer phenotypes, even among those with similar levels of aneuploidy and CIN. Interphase effects of ‘mitotic’ genes whose alteration results in CIN are likely to have substantial impacts on tumor phenotypes (11). Nevertheless, the overarching conclusion from these models is that the effects of aneuploidy on tumors depend on the rate of CIN; while a low level of CIN can aid tumor evolution through gain of oncogenes or loss of tumor suppressors, high rates of CIN cause cell death and tumor suppression. Combining two independent insults that each cause low CIN in the same cell results in high CIN and is sufficient for cell death and tumor suppression (12–15). Thus, high CIN results in increased lethality as compared to low CIN. Cell death could occur due to loss of both copies of an essential chromosome (16) or through activation of the p53 tumor suppressor (17–19).
Our recent studies with the best-selling chemotherapy drug in history, paclitaxel (Taxol™), suggest these findings in cell culture and animal models are clinically relevant (20). At typically used concentrations in cell culture, paclitaxel causes mitotic arrest followed by death in sensitive cells (21,22). However, paclitaxel does not reach sufficient concentrations in primary breast cancers to cause substantial mitotic arrest (20). Instead, clinically relevant concentrations of paclitaxel in cultured breast cancer cells cause chromosome missegregation on multipolar spindles after a brief mitotic delay. Importantly, paclitaxel also causes an increase in multipolar spindles in all patient tumors tested (20). These results support the conclusion that paclitaxel exerts its anti-cancer effects by increasing the rate of CIN above a maximally tolerated threshold. It remains to be determined whether other clinically relevant microtubule poisons, such as vinca alkaloids, eribulin, and ixabepilone, exert their anti-cancer effects by inducing mitotic arrest or via CIN due to multipolar spindles. Other therapies intended to induce chromosome missegregation, such as inhibitors of the mitotic checkpoint kinase Mps1 (23–25), are currently in clinical trials (NCT02366949, NCT03328494). One important outstanding question is whether p53, the protein that is most commonly mutated or lost in human cancer, is required for high rates of CIN to cause cell death and tumor suppression.
p53 is a transcription factor that regulates a complex network of genes whose expression results in cell cycle arrest, senescence, apoptosis and/or ferroptosis (26). For this reason, p53 protein levels are normally kept low due to continuous ubiquitination and degradation (27). p53 is stabilized following a variety of cellular stresses, including DNA damage, oxidative stress, hypoxia, nutrient deprivation and oncogene activation. p53 is mutated or lost in ~50% of human cancers (26,28). Homozygous loss of p53 results in rapid onset of tumors, predominantly lymphomas (29–31). Animals heterozygous for p53 also have a well-characterized increase in tumors, predominantly sarcomas, which occur with substantially later onset than the lymphomas in p53 null animals [~18 months; (17,29–41)]. A subset of tumors that form in p53 heterozygous animals maintain one wild type copy of p53 (28,42–44), as do some human tumors (28,42,43,45). At least in some cases, the retained copy of p53 remains functional (46,47). Reduction of p53 results in tumors in both male and female animals with no sex-specific differences reported.
Both heterozygous and homozygous loss of p53 have been reported to induce aneuploidy and CIN (32,34,36,48–50). p53 limits expression of the mitotic checkpoint gene Mad2 (51,52). The mitotic checkpoint (also known as the spindle assembly checkpoint) is a major cell cycle control mechanism that ensures euploidy by preventing chromosome segregation until all chromosomes are stably attached to spindle microtubules [reviewed in (53,54)]. Overexpression of Mad2, as caused by reduction of p53, results in hyperstabilization of the microtubules that attach chromosomes to the mitotic spindle, resulting in lagging chromosomes, CIN, and aneuploidy (55,56).
To increase levels of CIN in p53 deficient animals, we reduced expression of the mitotic kinesin CENtromere associated Protein-E (CENP-E). CENP-E is one of several proteins that link microtubules to chromosomes through their kinetochores (54). CENP-E congresses chromosomes near spindle poles (polar chromosomes) along detyrosinated microtubules toward the spindle equator (57). CENP-E facilitates full activation of the mitotic checkpoint, at least in part through its role in maximizing recruitment of the mitotic checkpoint genes Mad1, Mad2 and BubR1 to unattached kinetochores (58,59). Loss of CENP-E results in low CIN due to missegregation of one or a few chromosomes every four divisions (59). Though CENP-E is an essential gene, heterozygous loss of CENP-E is sufficient to produce polar chromosomes, aneuploidy and low CIN, which does not increase the rate of cell death (60,61). Aged CENP-E heterozygous mice develop spleen and lung tumors at low frequency (60). However, reducing CENP-E in the presence of another insult that also causes CIN results in a higher rate of CIN, leading to cell death and tumor suppression (12,13,60). Here we test whether high CIN requires p53 to achieve this anti-tumor response.
One limitation of many CIN model systems is that the modified genes have moonlighting roles outside of mitosis, confounding the conclusions. CENP-E is particularly well suited for testing the impact of CIN because its functional roles are almost assured to be limited to mitosis, when it is present. CENP-E protein accumulates in late G2 and is degraded at the end of mitosis (62). It is not detectable in non-dividing tissues, and reduction of CENP-E does not result in DNA damage or structural rearrangements of chromosomes (60). Currently, the only known function of CENP-E is in maintaining proper chromosome segregation.
Here we combined reduction of p53 with heterozygous loss of CENP-E to test the importance of p53 in cell death and tumor suppression caused by high CIN. Heterozygous loss of p53 was not sufficient to induce CIN. However, osteosarcomas in p53+/− female mice lost the remaining copy of p53 with high frequency and exhibited low CIN. Reduction of CENP-E in these tumors led to high CIN, cell death, and increased tumor latency. Moreover, pharmacologically inducing high CIN resulted in equivalent levels of cell death in p53 wild type and p53 null primary Murine Embryonic Fibroblasts (MEFs). Cell death could be rescued by preventing cytokinesis, consistent with high CIN causing cell death due to loss of both copies of one or more essential chromosomes. Surprisingly, we discovered an incidental sex-specific tumor phenotype in which p53 heterozygous female animals exhibit shorter tumor latencies and decreased survival as compared to their male counterparts. These studies demonstrate that p53 is not necessary for the tumor suppressive effects of high CIN.
Materials and Methods
Animals and tissue culture
All animal studies were performed in compliance with all relevant ethical regulations for animal testing and research. This study was approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison. Animals were maintained on a C57Bl/6 background. MEFs were isolated from E14.5 embryos and maintained in DMEM (Life Technologies, Carlsbad, CA) containing 200 mM L-glutamine, 50 μg/mL penicillin-streptomycin, 10 mM nonessential amino acids, 100 mM sodium pyruvate, 1 mM β-mercaptoethanol, and 15% fetal calf serum (FCS) at 10% CO2, 3% O2, and 37°C. Reversine was used at a concentration of 500 nM. Metaphase spreads were prepared from the spleens of 5 month mice. Half of each spleen was saved for histological diagnosis in 10% phosphate buffered formalin for 24 hours, and then rinsed and replaced with 70% EtOH. The other half was homogenized using forceps and vigorous pipetting in PBS. The cells were then pelleted and resuspended in lymphocyte media (RPMI [HyClone], 66 ng/mL gentamicin, 7.4 ng/mL PHA, 50 ng/mL LPS, 10% FBS, 5 ng/mL colchicine) in blood culture tubes. Spleens were cultured 8 hours at 10% CO2 and 37°C. Splenocytes were then pelleted and resuspended in pre-warmed 75 mM KCL and incubated 45 minutes at 10% CO2 and 37°C to swell cells. Cells were fixed with 3:1 ratio of methanol to glacial acetic acid and stored at 4°C. To prepare slides, fixed lymphocytes were washed 2x with 3:1 ratio of methanol to glacial acetic acid and then resuspended in ~400 uL of fixative and dropped onto slides from a distance of 4 ft. Slides were dried on a 75°C heat block for 30s and incubated for 3 min in 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS. Slides were mounted in Vectashield, (Vector Labs, Burlingame, CA) mounting medium.
Immunofluorescence
Primary MEFs were washed with MTSB (100 mM 1,4-piperazinediethanesulfonic acid, pH 6.9, 30% glycerol, 1 mM ethylene glycol tetraacetic acid, and 1 mM MgSO4) and permeabilized in MTSB plus 0.05% Triton X-100 for 30 seconds at room temperature. Fixation was performed in MTSB plus 4% formaldehyde and 0.1% glutaraldehyde for 10 min at room temperature. Coverslips were blocked overnight in Triton Block (0.2 M glycine, 2.5% fetal bovine serum [FBS], and 0.1% Triton X-100 in 1× PBS). Primary antibody incubation (α-tubulin YL1/2, 1:1000, Bio-Rad; pericentrin, 1:500, Abcam) was performed for 1 hour at room temperature in Triton Block. Coverslips were washed 3× in PBS plus 0.1% Triton X-100 and incubated in Alexa Fluor–conjugated secondary antibodies diluted 1:200 in Triton Block for 45 min at room temperature. Coverslips were washed 3× in PBS plus 0.1% Triton X-100, incubated for 3 min in 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS, rinsed 2× in PBS, and mounted in Vectashield (Vector Labs, Burlingame, CA) mounting medium.
Paraffin-embedded sections of 5-μm thickness were first deparaffinized in xylenes 3 × 10 min, rinsed in 100% ethanol, and hydrated in a series of 100, 95, and 70% ethanol for 5 min each, followed by 5 min in double-distilled H2O. Antigen retrieval was performed for 30 minutes in a beaker at 100°C in citrate buffer (10 mM citric acid plus 0.05% Tween-20, pH 6.7). Slides were then washed in H2O and blocked overnight at 4°C in a humidified chamber in TBST plus 10% bovine serum albumin and 1% goat serum. Primary antibody incubation (α-tubulin YL1/2, 1:200, Serotec, Hercules, CA; cleaved caspase-3, 1:200, Cell Signaling) was performed overnight at 4°C in a humidified chamber in blocking buffer. Slides were washed 3 × 5 minutes in TBST and incubated in secondary antibodies (Alexa Fluor, 1:200 in TBST) for 1 hour at room temperature. After three subsequent washes, slides were treated for 5 minutes with 0.05% Sudan Black B in 70% ethanol. Slides were then rinsed with H2O and then incubated in 5 μg/mL DAPI in PBS for 10 min, washed 2× in PBS, and mounted using Vectashield (Vector Labs).
Images were acquired using a Nikon Eclipse Ti-E inverted microscope with a Hamamatsu ORCA Flash 4.0 camera using 40× (0.75 NA), 60x (1.4 NA), or 100× (1.4 NA) objectives. Image acquisition, analysis, and processing were performed using Nikon Elements AR. Autoquant was used for deconvolution.
LOH testing
DNA was extracted from formalin fixed tumors using QIAamp DNA FFPE tissue kit (Qiagen). To prevent confounding effects from preferential amplification of the smaller product, agreement was required from reciprocal primer sets, one of which produced a smaller product for the wild type allele and one of which produced a smaller product for the null allele. To obtain a longer wild type product and shorter null product, PCR and qPCR were performed using the following primer sequences with an annealing temperature of 59.2°C: p53–142bp-Forward: GAAGGAAATTTGTATCCCGAGTATCTG. p53–142bp-Reverse: CATCAGTCTAGGCTGGAGTCAAC. Neo-PGK-108bp-Forward: CTTTACGGTATCGCCGCTCC. Neo-PGK-108bp-Reverse: GATCATCAATTTCTGCAGACTTACAGC. To obtain a shorter wild type product and longer null product, PCR and qPCR were performed using the following primer sequences with an annealing temperature of 62°C: p53–118bp-Forward: GAAGGAAATTTGTATCCCGAGTATCTG. p53–118bp-Reverse: GTCTCTAAGACGCACAAACCAAAAC. Neo-PGK-146bp-Forward: AAGAGCTTGGCGGCGAATGG. Neo-PGK-146bp-Reverse: GATCATCAATTTCTGCAGACTTACAGC. qPCR was carried out in triplicate in a reaction volume of 12.5 μl using 6.25 μl of 2x Power SYBR Green master mix (Bio-Rad), 150 nM of each amplification primer and 10 ng of purified genomic template DNA. An initial denaturation step of 95°C for 3 minutes was followed by 40 cycles of 95°C denaturation for 15 seconds and an annealing/extension step of 30 second at 59.2 or 62°C. Post-amplification melt curve analysis was performed for each reaction product to confirm the absence of non-specific amplification products. Changes in fold amplification were analyzed by the comparative ΔΔCt method. Relative amplification of the null and wild type allele were used to create a fold change of p53 null:p53 wildtype wherein larger values indicate a higher percentage of cells with LOH..Tumors which showed disagreement between PCR and qPCR were excluded. 88% of tumors could be reliably genotyped.
Immunoblotting
Primary MEFs from ~70% confluent 6 wellplates were washed with PBS, harvested in 100 μL of PBS and 100 μL of 2x sample buffer, boiled for 10 minutes and stored at −80°C. Samples were run on 10% acrylamide gels, transferred to nitrocellulose, and blocked in 5% milk in TBST for 1 hour at room temperature before incubation with primary antibodies, which were diluted in 2% BSA + 0.02% sodium azide in PBS. Secondary antibodies were diluted in 5% milk in TBS + 0.1% Tween 20. Primary antibody dilutions: CENP-E 1:500 (63), p53 1:300 (1C12; Cell Signalling Technologies), actin 1:500 (JLA20; Developmental Studies Hybridoma Bank). Secondary antibodies were diluted 1:10,000 (Licor IRDye).
Timelapse microscopy
Cells were incubated under 10% CO2 flow at ~30 mL/minute in a heated, humidified chamber at 37°C. Images were acquired at 5 minute intervals with a 20×/0.75 NA objective for 48 hours. Individual frames were assembled in Nikon Elements and exported as .mov files. Cell death was calculated by dividing the number of cells that died during the course of the movie by the average number of cells in frame for each condition.
Statistical analysis
Significant differences were determined using a two-tailed student’s t test (mitotic defects, cell death, number of daughter cells) log rank test (Kaplan-Meier survival curves), or Fisher’s exact test (tumor spectra). Animals that did not have a histologically confirmed tumor were censored from tumor free survival curves. For osteosarcoma-free survival curve, animals that maintained their wild type copy of p53 were censored.
Results
Homozygous—but not heterozygous—loss of p53 causes lagging chromosomes in vitro and in vivo
To test whether p53 is required for high CIN to cause cell death, we crossed Trp53 (hereafter p53) deficient mice [Jax 002101; (30)] with mice heterozygous for CENP-E (61). Two rounds of mating produced six genotypes that were wild type for p53 (wild type and CENP-E+/−), heterozygous for p53 (CENP-E+/+;p53+/− and CENP-E+/−;p53+/−), or null for p53 (CENP-E+/+;p53−/− and CENP-E+/−;p53−/−). These strains were maintained in a C57Bl/6 background. As expected, p53 null females were born at a reduced frequency compared to their male counterparts (Table S1) due to a Y-chromosome independent failure of neural tube closure (64–66). p53 null animals were used in subsequent crosses with p53 heterozygous animals to generate sufficient CENP-E+/+;p53−/− and CENP-E+/−;p53−/− animals for analysis.
To measure CIN in vitro, the incidence of mitotic cells with polar and lagging chromosomes was scored in primary MEFs prepared from E14.5 mouse embryos since these mitotic phenotypes represent previously validated measures of CIN (59,61,67). As expected, reduced expression of CENP-E did not impact expression of p53, nor did loss of p53 affect CENP-E levels (Fig. S1A). Reduction of CENP-E provides a well characterized model of low CIN due to an increase in polar chromosomes (13,59,61). Consistent with this, a significant increase in polar chromosomes was observed in MEFs heterozygous for CENP-E, irrespective of the p53 status of the MEFs (an increase of 8.1-12.1% over wild type, considered low CIN; Fig. 1A–B). Homozygous loss of p53 produced an increase in lagging chromosomes, which trail behind the segregating masses of chromatin in anaphase and telophase, as compared to wild type MEFs (an increase of 8.6-10.0% over wild type, low CIN; Fig. 1C–D). Despite previous reports that p53 heterozygous animals exhibit CIN, heterozygous loss of p53 was not sufficient to substantially increase the frequency of lagging chromosomes (difference of 1.9-2.3% from wild type; Fig. 1D). Nor did CENP-E heterozygosity substantially impact the incidence of lagging chromosomes (difference of 2.3-2.6% from wild type; Fig. 1D). Thus, an increase in lagging chromosomes requires loss of both copies of p53. Heterozygous loss of CENP-E increased the rate of mitotic defects in the context of p53 heterozygosity, with CENP-E+/−;p53+/− MEFs showing an 8.7% higher rate of mitotic defects than CENP-E+/+;p53+/− MEFs (Fig. 1E) due to an increase in polar chromosomes (Fig. 1B). Overall, CENP-E+/−;p53+/+ MEFs, CENP-E+/−;p53+/− MEFs, and CENP-E+/+;p53−/− MEFs showed very similar rates of low CIN (19.9-21.7%), which were 10.4-12.3% higher than wild type MEFs (Fig. 1E). However, CIN occurred due to different mechanisms. CIN is driven by polar chromosomes in CENP-E+/− MEFs and lagging chromosomes in p53−/− cells. MEFs in which heterozygous loss of p53 was the only genetic alteration showed a similar rate of mitotic errors to that of wild type MEFs (difference of 1.7 +/− 2.3%; Fig. 1E). Only homozygous loss of p53 in combination with heterozygous loss of CENP-E increased the rate of CIN ≥8% above the low rate of CIN conferred by heterozygous loss of CENP-E, which we define here as high CIN (Fig. 1E). Thus, CENP-E+/−;p53−/− MEFs showed high CIN while CENP-E+/−;p53+/+, CENP-E+/−;p53+/−; and CENP-E+/+;p53−/− MEFs showed low CIN.
Figure 1: Homozygous—but not heterozygous—loss of p53 causes CIN via lagging chromosomes in vitro and in vivo.
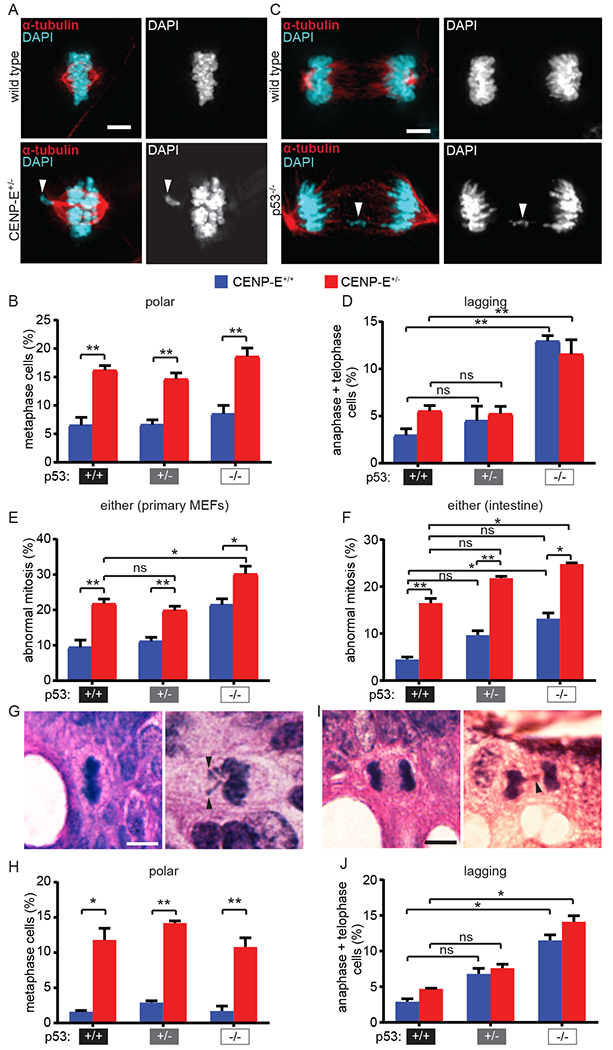
(A-E) CIN measurements in female primary MEFs. (A) Primary MEF images showing normal metaphase (top) and metaphase with a polar chromosome (bottom, arrowhead). Scale bar, 5 μm. (B) Quantification of metaphase cells with polar chromosomes in primary MEFs. n>35 metaphase cells from each of 5 independent experiments. (C) Primary MEF images of normal anaphase (top) and anaphase with a lagging chromosome (bottom, arrowhead). Scale bar, 5 μm. (D) Quantification of anaphase cells with lagging chromosomes in primary MEFs. n>35 anaphase and telophase cells from each of 5 independent experiments. (E) Quantification of total polar and lagging chromosomes observed in female primary MEFs. n=5 independent experiments. (F-J) CIN measurements in intestines from male mice. (F) Quantification of total mitotic defects observed in mouse intestine. n=3 animals per genotype. G) Normal metaphase (left) and metaphase cell with polar chromosomes (right, arrowheads) in H&E stained mouse intestine. Scale bar, 5 μm. (H) Quantification of metaphase cells with polar chromosomes in mouse intestine. n≥55 metaphase cells from each of 3 animals per genotype. (I) Normal anaphase (left) and an anaphase with a lagging chromosome (right, arrowhead) in H&E stained mouse intestine. Scale bar, 5 μm. (J) Quantification of anaphase cells with a lagging chromosome in mouse intestine. n≥30 anaphase and telophase cells from each of 3 male animals per genotype. Error bars indicate SEM. * indicates p<0.05, ** indicates p<0.01.
To measure CIN in vivo, the frequency of abnormal mitotic figures was quantitated in intestine from 5 month old mice, a standard timepoint for CIN assessment (32,60). Intestine was chosen for analysis as one of the few normal tissues in which cells continuously proliferate, allowing assessment of mitosis. As expected, animals heterozygous for CENP-E exhibited an increase in polar chromosomes at similar frequencies regardless of their p53 status (an increase of 9.2-12.6% above wild type, low CIN; Fig. 1F–H). Similar to in vitro data, homozygous loss of p53 led to a substantial increase in lagging chromosomes in intestinal cells (8.6-11.2%, low CIN; Fig. 1I–J), while heterozygous loss of p53 resulted in a more modest increase (3.9-4.7%) that was not statistically significant (Fig. 1I–J). Considering all mitotic defects together, only CENP-E+/−;p53−/− intestine exhibited a ≥8% increase in the rate of mitotic defects as compared to CENP-E+/− intestine, consistent with high CIN (Fig. 1F). Thus, heterozygous loss of CENP-E and homozygous loss of p53 cause similar rates of low CIN in vitro and in vivo, but through distinct mechanisms. Combining both of these insults results in higher CIN than in cells with either p53 loss or CENP-E haploinsufficiency alone.
Reduction of CENP-E does not increase CIN in lymphomas in p53−/− mice
Germline mutations in p53, which cause Li-Fraumeni syndrome, are invariably heterozygous, reducing the clinical relevance of p53−/− animals (68). However, we initially tested p53−/− mice since we anticipated that reduction of CENP-E would increase the rate of CIN in the tumors that form in these animals, thereby providing a test of the impact of high CIN in the absence of p53. p53−/− animals primarily develop early onset lymphomas [at ~ 5 months of age; (29–31)]. We anticipated that lymphomas which formed in CENP-E+/−;p53−/− animals would have higher rates of mitotic defects and CIN than lymphomas in p53−/− animals with a wild type complement of CENP-E. This increase in CIN would allow us to test the requirement for p53 in cell death and tumor suppression. However, lymphomas from CENP-E+/−;p53−/− and CENP-E+/+;p53−/− animals showed very similar levels of mitotic errors (difference of 2.1%-4.4%; Supplementary Fig. S1B–S1C). Since the tumors that formed in CENP-E+/−;p53−/− and CENP-E+/+;p53−/− animals had equivalent levels of CIN, this model system did not permit a test of the impact of different levels of CIN on tumor latency.
CENP-E heterozygosity did not elevate CIN in the context of lymphomas that formed in p53 null animals. One potential reason lymphomas of both CENP-E genotypes had indistinguishable rates of CIN is that the incidence of polar chromosomes is higher in lymphomas (Supplementary Fig. S1B–S1C) than in nontransformed intestine in CENP-E+/+;p53−/− mice (Fig. 1H), potentially due to an increase in multipolar spindles, which can produce polar chromosomes after spindle pole focusing [Supplementary Fig. S1D–S1E; (69)]. Consistent with all tumors showing an equivalent level of CIN, we did not observe a survival difference between CENP-E+/+;p53−/− and CENP-E+/−;p53−/− animals (Supplementary Fig. S1F–S1G). Splenocytes from neoplastic spleens exhibited substantial aneuploidy as compared to non-neoplastic splenocytes, with modal chromosome numbers that deviated from the diploid number of 40 chromosomes, independent of CENP-E status (Supplementary Fig. S1H). Thus, the increase in CIN and aneuploidy from CENP-E reduction was overshadowed by the increase already seen in neoplastic tissue, resulting in equivalent levels of CIN and aneuploidy in tumors in p53−/− mice irrespective of CENP-E genotype. Therefore, this model could not be used to test if p53 is required for the tumor suppressive effects of high CIN, necessitating the use of other models (Supplementary Fig. S1I).
p53 heterozygous mice show sex-specific differences in tumorigenesis
Since p53 null animals proved unsuitable to test whether high CIN requires p53 for cell death and tumor suppression, we turned to p53 heterozygous animals. Though heterozygous loss of p53 is insufficient to induce mitotic errors and CIN, it has previously been shown that a portion of tumors that form in p53+/− animals lose their wild type copy of p53 (hereafter designated p53 loss of heterozygosity, LOH), resulting in tumors that are effectively p53 null. The frequency of p53 LOH varies by tissue type in both mouse and human (43,70). Consistent with these previous results, 68% of the tumors that arose in our cohort of p53+/− animals exhibited p53 LOH, with the highest frequency of LOH (83%) observed in osteosarcomas (Table 1). We predicted that CENP-E+/+; p53−/LOH tumors would show low CIN and CENP-E+/−;p53−/LOH tumors would exhibit high CIN, permitting a test of whether high CIN requires p53 to be tumor suppressive.
Table 1.
Rates of p53 LOH vary by tumor type and sex
| tumor type (# female, male tumors) | CENP-E+/+ tumors with p53 LOH | CENP-E+/− tumors with p53 LOH | ||
|---|---|---|---|---|
| female # (%) | male # (%) | female # (%) | male # (%) | |
| osteosarcoma (29, 6) | 13 (87) | 1 (33) | 13 (93) | 2 (67) |
| histiocytic sarcoma (4, 9) | 1 (100) | 1 (50) | 2 (67) | 4 (57) |
| soft tissue sarcoma (2, 5) | 0 (0) | 0 (0) | 1 (50) | 0 (0) |
| lymphoma (3, 2) | 0 (0) | 1 (100) | 2 (67) | 1 (100) |
| other (4, 7) | 1 (50) | 1 (50) | 2 (100) | 2 (40) |
| total (42, 29) | 15 (83) | 4 (36) | 20 (83) | 9 (50) |
In the course of these studies, we observed unanticipated but strong sex-specific effects in p53 heterozygotes. Both overall and tumor free survival were extended in p53+/− male animals as compared to p53+/− female animals, by a median of ~5 months (Fig. 2A and Supplementary Fig. S2A). Male and female p53+/− mice also exhibited striking differences in tumor spectra (Fig. 2B). In the mice that developed tumors, there were significantly more osteosarcomas in p53 heterozygous females and more soft tissue sarcomas in p53 heterozygous males (Fig. 2B; osteosarcomas: p<0.005, FDR<5%, soft tissue sarcomas: p<0.05, FDR=5% Fisher’s exact test). CENP-E reduction did not alter the predominant tumor type in p53 heterozygous females (osteosarcomas) but altered the ratio of soft tissue sarcomas and histiocytic sarcomas in males (Fig. S2E). Importantly, Li-Fraumeni patients show a similar sex-specific difference in which tumor onset occurs more rapidly in females than in males (71–73).
Figure 2: Sex-specific survival advantage in male p53+/− mice.
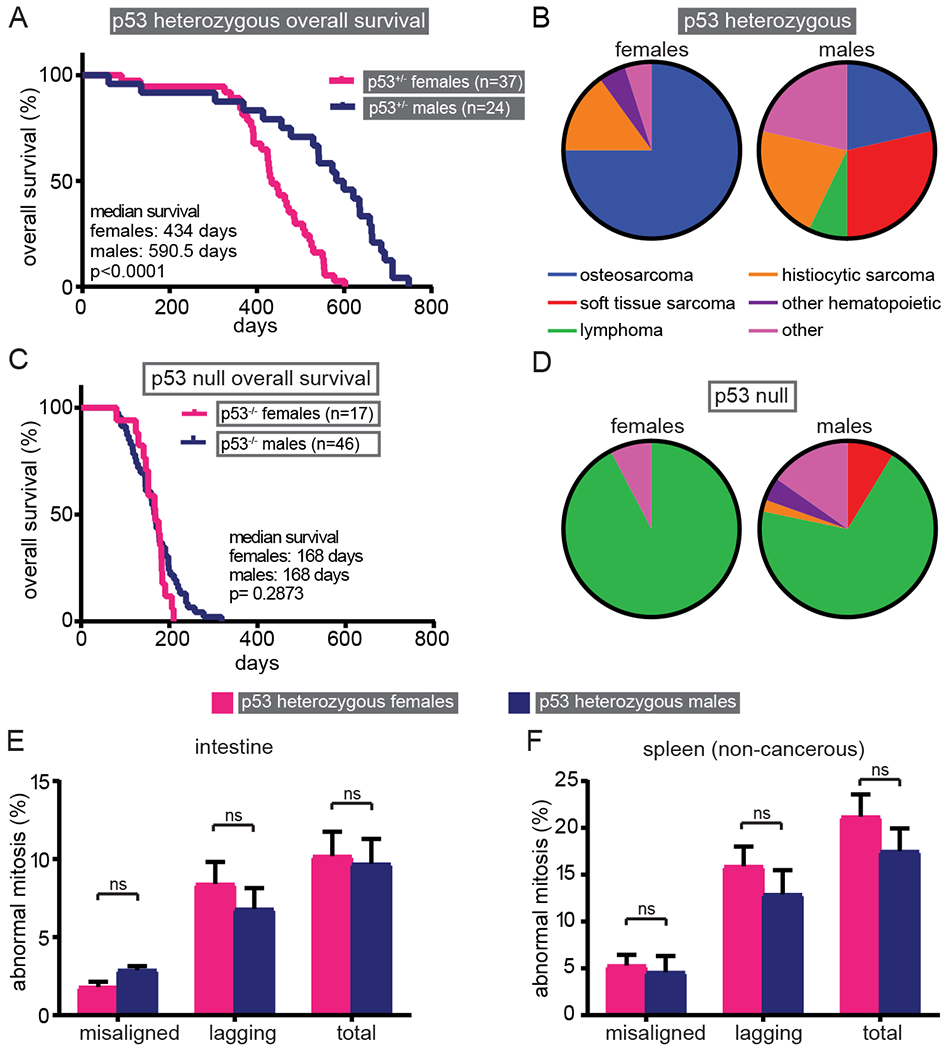
(A) Kaplan-Meier curve showing p53+/− male mice have increased overall survival compared to p53+/− female mice. (B) p53 heterozygous females acquire significantly more osteosarcomas (p<0.005, FDR<5%), while p53 heterozygous males have a diverse tumor spectrum. 20 of 37 females (54%) and 14 of 24 males (58%) developed a single tumor. No p53+/− animal developed more than one tumor. Data are shown only for those mice that developed a tumor. (C) Kaplan-Meier curve showing p53 null animals do not exhibit sex-specific survival differences. (D) p53 null animals primarily develop lymphomas, regardless of sex. 12 of 17 females (71%) developed tumors. 11 females had a single tumor and one female had two tumors. 38 of 46 males (83%) developed tumors. 31 had a single tumor and seven had two tumors. (E-F) Quantification of mitotic defects in p53 heterozygous H&E stained intestine (E) and non-cancerous spleen (F) indicates that the sex-specific survival disparity is not due to differences in CIN. n>35 metaphase and >30 anaphase and telophase cells from each of 3 (E) or 4 (F) animals per sex. Error bars indicate SEM.
The survival difference between male and female p53 heterozygous animals cannot be attributed to differences in tumor spectra, since males had longer survival even when comparing among the same tumor type. For instance, the median tumor free survival for animals developing osteosarcomas was 424 days in females and 621 days in males (Supplementary Fig. S2B). Interestingly, human osteosarcomas also occur earlier in females than in males (74). Histiocytic sarcomas also formed earlier in female mice, with a median latency of 548 days in females and 664 days in males (Supplementary Fig. S2C). Therefore, p53 heterozygous animals exhibit dramatic sex-specific differences in tumor spectra and latency, necessitating that survival comparisons in these animals are made within single sex groups.
In contrast to p53 heterozygous animals, a sex-specific survival disparity was not observed in mice null for p53 (Fig. 2C and Supplementary Fig. S2D). Consistent with this, tumor spectra in p53 null females and males was similar, with lymphomas predominating in both sexes (92.3% and 71.1% respectively, Fig. 2D). Lymphomas also predominated in CENP-E+/−;p53−/− males and females (Fig. S2F). Therefore, conclusions regarding survival in p53 null animals can be made based on the entire cohort, without the need for sex-specific comparison.
Reduction of CENP-E is not protective in contexts where it does not induce high CIN
Since heterozygous loss of p53 did not induce lagging chromosomes, doubly heterozygous CENP-E+/−;p53+/− cells did not have substantially more mitotic defects than CENP-E+/−;p53+/+ cells and did not exhibit high CIN (Fig. 1E–F). Since the tumor phenotype in CENP-E+/− mice (60) is dwarfed by the tumor phenotype caused by p53 heterozygosity (29–31), and we did not increase CIN over the low rate found in CENP-E+/− mice, this cohort did not permit a comparison of low CIN and high CIN and we did not anticipate differences in tumor free survival between p53+/− animals based on CENP-E genotype (Supplementary Fig. S1I). Indeed, reduction of CENP-E had no significant impact on survival of p53 heterozygous male or female animals (Supplementary Fig. S3A–S3B), where it did not induce high CIN. Thus, this model also failed to offer a test of the impact of high CIN on tumors.
Female osteosarcomas offer a test of whether p53 is required for high CIN to mediate tumor suppression
We expected that p53 LOH would confer low CIN in CENP-E wild type tumors and high CIN in CENP-E heterozygous tumors. Therefore, we reasoned that these CENP-E+/+;p53−/LOH and CENP-E+/−;p53−/LOH tumors would permit a test of whether p53 is required for cell death and tumor suppression caused by high CIN. Tumors that had undergone p53 LOH in CENP-E+/−;p53+/− and CENP-E+/+;p53+/− animals were identified by genotyping (Supplementary Fig. S4A–S4B). Rates of p53 LOH were generally higher in female than male p53+/− animals (83% versus 45%; Table 1). Since rates of p53 LOH are higher in tumors that occur earlier (46,47), this offers an explanation for the shorter survival of female animals. However, sex-specific differences in CIN were not observed in intestine or non-cancerous spleen in p53 heterozygous animals (difference 0.5-2.0%; Fig. 2E and F) or in primary MEFs (1.4 +/− 1.4%; Fig. S2G). This suggests that LOH occurs through a mechanism other than whole chromosome loss, such as somatic recombination, which was previously shown to be the primary source of p53 LOH in murine tumors (75–77). This type of recombination event results in p53 LOH without introducing additional mutations.
Distinct tumor types show different times to tumor onset, with lymphomas occurring substantially earlier than sarcomas. Therefore we focused on a single tumor type. The largest cohort of tumors, female osteosarcomas, showed prevalent p53 LOH (90%, Table 1). Interestingly, humans with germline p53 mutations also develop osteosarcomas at increased frequency [12.6% versus <1% sporadic; (74,78)]. Thus, we focused on female osteosarcomas to examine whether p53 LOH was sufficient to induce low CIN via lagging chromosomes. To test this, we quantified mitotic defects in CENP-E+/−;p53−/LOH and CENP-E+/+;p53−/LOH female osteosarcomas (Fig. 3A–C). p53 LOH conferred a substantial increase in lagging chromosomes as compared to p53+/− cells that maintained a wild type copy of p53 (20.1% p53−/LOH vs 6.8% p53+/−), irrespective of CENP-E status. As expected, polar chromosomes were increased in CENP-E+/−;p53−/LOH tumors relative to CENP-E+/+;p53−/LOH tumors (17.5% CENP-E+/−;p53−/LOH vs 8.0% CENP-E+/+;p53−/LOH; Fig. 3A, C). In total, CENP-E+/−;p53−/LOH tumors exhibited substantially (≥8%) higher levels of mitotic defects than p53−/LOH tumors with a full complement of CENP-E, consistent with high CIN (39.1% in CENP-E+/−;p53−/LOH vs 28.1% in CENP-E+/+;p53−/LOH Fig. 3C, right). Since the CENP-E+/−;p53−/LOH osteosarcomas exhibited substantially higher CIN than the CENP-E+/+;p53−/LOH tumors, a comparison of animals bearing these tumors permitted us to examine the effect of high CIN on cell death and tumor-free survival in the absence of p53 (Supplementary Fig. S1I).
Figure 3: High CIN is tumor suppressive after p53 LOH.
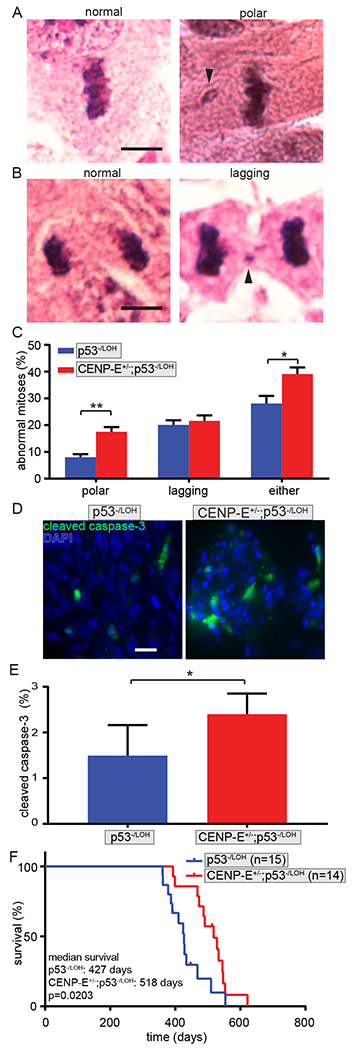
(A) Normal metaphase cell (left) and metaphase cell with a polar chromosome (right, arrowhead) in H&E stained mouse osteosarcoma. Scale bar, 5 μm. (B) Normal anaphase cell (left) and anaphase cell with lagging chromosome (right, arrowhead) in female osteosarcoma. Scale bar, 5 μm. (C) Quantification of polar and lagging chromosomes showing that reduction of CENP-E causes high CIN in p53−/LOH female osteosarcomas. n>25 metaphase and 18 anaphase and telophase cells from each of 3 p53−/LOH and 5 CENP-E+/−; p53−/LOH osteosarcomas. (D) Cleaved caspase-3 staining in p53−/LOH (left) and CENP-E+/−;p53−/LOH (right) osteosarcomas. Scale bar, 20 μm. (E) Quantification of cleaved caspase-3 positive cells in female osteosarcomas with LOH showing that CENP-E+/−; p53−/LOH osteosarcomas with high CIN also have elevated cell death. n>600 cells from each of 5 p53−/LOH and 7 CENP-E+/−; p53−/LOH osteosarcomas. (F) Kaplan-Meier curve showing increased osteosarcoma free survival in female animals with CENP-E+/−;p53−/LOH osteosarcomas as compared to p53−/LOH animals with a wild type complement of CENP-E and a lower rate of CIN. Animals whose osteosarcoma maintained the wild type copy of p53 are censored. Error bars indicate SEM. * indicates p<0.05, ** indicates p<0.01.
High CIN causes cell death and tumor suppression without functional p53
Though increasing the rate of CIN in cells with wild type p53 increases the rate of cell death, it was not known whether the proapoptotic activity of p53 is necessary for the elevated lethality. To test this in tumors with distinct levels of CIN, cleaved caspase-3 activity, which is indicative of apoptotic cell death, was compared in CENP-E+/+;p53−/LOH (low CIN) and CENP-E+/−;p53−/LOH (high CIN) female osteosarcomas (Fig. 3D and E). CENP-E+/−;p53−/LOH osteosarcomas had a 61% increase in the percentage of cleaved caspase-3 positive cells (Fig. 3D and E). This is similar to the increase previously observed when increasing CIN in the presence of two copies of p53 (12,13). Thus, CENP-E+/−;p53−/LOH osteosarcomas with high CIN showed an increase in the rate of apoptotic cell death that was independent of p53.
Since high CIN increased the rate of cell death, even in the absence of p53, and high CIN and an increased rate of cell death have previously been associated with tumor suppression (12–15), we predicted that high CIN due to reduction of CENP-E would extend osteosarcoma-free survival in female animals whose tumors had p53 LOH. Consistent with this, CENP-E reduction was protective against osteosarcoma formation, with CENP-E+/−;p53−/LOH (high CIN) osteosarcomas developing a median of 91 days later than CENP-E+/+;p53−/LOH (low CIN) osteosarcomas (Fig. 3F). The delay in tumor onset due to reduction of CENP-E was a general feature of osteosarcomas, which have a 90% rate of p53 LOH (Table 1, Supplementary Fig. S3C). Importantly, reduction of CENP-E also caused a delay in tumor onset when examining all sarcomas exhibiting p53 LOH (Supplementary Fig. S3D). This survival advantage following p53 LOH demonstrates that p53 function is not required for high CIN to exhibit tumor suppressive effects (Supplementary Fig. S1I).
p53 is not required for pharmacologically induced high CIN to cause cell death
Our finding that cell death and tumor suppression can occur independently of p53 is relevant, since several current and emerging anti-cancer treatments, including Mps1 inhibitors (NCT02366949, NCT03328494), induce high CIN and p53 status is a potential biomarker for response (15,20,79–81). Mps1 is a dual specificity kinase that is required for accurate mitotic checkpoint signaling and chromosome segregation (82). We wanted to determine if the p53−independent cell death we observed was translatable to the mechanisms of action of these drugs. To test this, we treated p53+/+ and p53−/− primary MEFs with the Mps1 inhibitor reversine (24). As reported previously, reversine caused high CIN due to lagging and bridge chromosomes in anaphase and telophase cells in both p53+/+ and p53−/− primary MEFs (increases of 45.5-47.5% in the percentage of cells with mitotic defects; Fig. 4A and B). At the chosen concentration (500 nM), the number of affected chromosomes per cell was similar in p53+/+ and p53−/− cells, consistent with equivalent rates of high CIN being induced in both cell types (Fig. 4C). Inducing uniformly high rates of chromosome missegregation allowed us to test the requirement of p53 in high CIN-mediated cell death. Timelapse microscopy was used to measure rates of cell death with and without reversine (Fig. 4D and E). p53+/+ and p53−/− primary MEFs exhibited similar extents of cell death in response to chromosome missegregation caused by reversine (Fig. 4E), demonstrating that p53 is not required for pharmacologically induced high rates of CIN to cause cell death.
Figure 4: p53 is not required for high rates of CIN to cause cell death or tumor suppression.
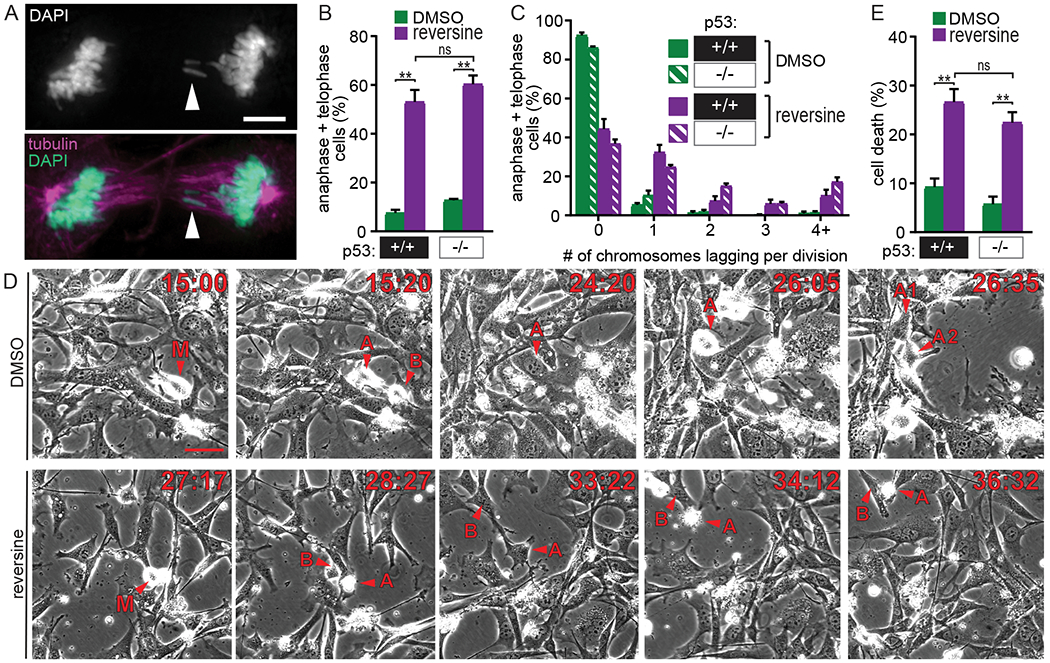
(A-C) The Mps1 inhibitor reversine causes equivalent CIN in p53+/+ and p53−/− primary MEFs by inducing high rates of anaphase defects. (A) p53+/+ primary MEF with two lagging chromosomes (arrowhead) after treatment with 500 nM reversine. Scale bar, 5 μm. (B) Quantification showing the percentage of anaphase and telophase cells with lagging chromosomes and/or chromosome bridges in primary MEFs +/− reversine. n>200 anaphase and telophase cells from ≥3 independent experiments. (C) Quantitation of the number of lagging and/or bridge chromosomes per anaphase or telophase cell. n > 200 anaphase and telophase cells from n≥3 experiments. (D-E) High CIN caused by reversine treatment results in equivalent levels of cell death in p53+/+ and p53−/− primary MEFs. (D) Selected frames from 48 hour timelapse imaging of p53−/− primary MEFs. Scale bar, 50 μm. Top: DMSO-treated mother cell (M) enters mitosis and produces daughter cells A and B. Daughter cell A enters mitosis (t=26:05). Daughter cell B exits the field of view (not shown). Bottom: Reversine-treated mother cell (M) enters mitosis. Daughter cell A dies (t=34:12), while daughter cell B survives but does not undergo a subsequent mitosis. (E) Quantification of cell death observed during 48 hour timelapse. n > 250 cells observed per condition. Error bars indicate SEM. * indicates p<0.05, ** indicates p<0.01.
Aside from activation of p53, the other mechanism proposed to account for cell death due to high CIN is loss of critical genes on essential chromosomes (16). To test if cell death caused by high CIN could be rescued by preventing loss of essential chromosomes, we prevented cytokinesis by including the microtubule poison colcemid. Timelapse microscopy revealed that mitosis in cells treated with reversine alone predominantly resulted in the formation of two daughter cells, one or both of which frequently died, while addition of colcemid with reversine produced a single daughter cell after cytokinesis failure that generally survived for the duration of the experiment (Supplementary Fig. S5A–D). Preventing cytokinesis reduced cell death in both p53+/+ and p53−/− primary MEFs, supporting the conclusion that high rates of CIN cause cell death independent of p53 through loss of essential chromosomes.
Discussion
Abnormal mitotic divisions have divergent consequences depending on the level of chromosome missegregation. While low CIN is tolerable and weakly tumor promoting, when CIN is increased to higher levels either genetically or pharmacologically, cell death and tumor suppression occur (12–16,56,60). There is evidence that commonly used cancer therapies including paclitaxel and emerging therapies such as Mps1 inhibitors cause CIN (20,79,80). It was not clear, however, if increasing CIN would benefit patients with a compromised p53 pathway. Since p53 is mutated or absent in approximately half of human cancers, a requirement for p53 activity would represent a significant limitation of this treatment approach. We therefore tested whether p53 was required for high CIN to cause cell death and subsequent tumor suppression. This was complicated by the lack of CIN in p53 heterozygous animals and the uniformly high CIN in p53 null lymphomas, which made these straightforward models insufficient to experimentally determine whether p53 is necessary for high CIN to cause tumor suppression (Fig. S1I). The sex-specific differences in tumor phenotypes in p53 heterozygous animals presented an additional, unanticipated hurdle. Ultimately, comparison of CENP-E+/+;p53−/LOH and CENP-E+/−;p53−/LOH tumors, which exhibit low CIN and high CIN, respectively, in a sex-matched cohort was necessary to test whether high CIN can suppress tumors in the absence of functional p53. Our results demonstrate that cell death occurs independently of p53 both in primary MEFs and in murine sarcomas, whether CIN is induced genetically or pharmacologically. These data support the conclusion that increasing CIN is a viable cancer treatment strategy, even in patients with compromised p53.
Prior studies have provided evidence both that p53 mutations can cause resistance to paclitaxel in patients (83–85), and that the S47 p53 polymorphism can cause increased sensitivity to paclitaxel (86). p53 status is not currently used clinically as a biomarker to predict sensitivity to paclitaxel. Thus, our data showing p53 is not required for the therapeutic efficacy of high CIN support current clinical practice. These findings also suggest that emerging chemotherapies that increase CIN, such as Mps1 inhibitors, can be clinically effective independent of p53 status.
p53 has been proposed to initiate cellular senescence or death following CIN or aneuploidy. While evidence has been presented that aneuploidy is more tolerated in contexts in which p53 signaling is impaired (17–19), recent work has shown that chromosome missegregation does not necessarily activate p53 or cause cell cycle delay (87). Single cell sequencing data suggests that structural aneuploidy triggers p53-dependent cell cycle arrest, while whole chromosome aneuploidy does not (88). Thus, while p53 recognizes structurally altered chromosomes, presumably because of DNA damage, it does not directly recognize whole chromosome gains and losses. Missegregation of whole chromosomes, particularly lagging chromosomes, can subsequently result in DNA damage and other cellular stresses, such as proteomic imbalance. Because p53 responds to cell stress, certain types of CIN could trigger p53-dependent cell cycle arrest or apoptosis. It is possible that p53-competent cells may have a subtly lower CIN threshold for cell death than p53 null or mutant cells. However, at the levels of CIN in this study, we were not able to observe a subtle threshold difference, and find that p53 function is not necessary for high CIN to cause cell death. Development of additional technical tools will be necessary to further elucidate tissue-specific CIN thresholds as well as the mechanism of cell death in p53 null contexts.
The protection against tumor formation in CENP-E+/−;p53−/LOH osteosarcomas was also a general feature of sarcomas. Reduction of CENP-E caused high CIN in p53−/LOH osteosarcomas and we anticipate this also occurred in the additional sarcomas. However, our cohort of 128 p53+/− animals contained only one CENP-E+/−p53−/LOH and two CENP-E+/−;p53−/LOH female non-ostersarcomas (Table 1), and this small sample size was inadequate to permit a rigorous test.
CENP-E loss is a likely cause of CIN in cancer. Like all mitotic checkpoint genes, CENP-E is infrequently mutated in human cancer. CENP-E is tightly cell cycle regulated, which complicates assessment of CENP-E expression in cancer (62). Compared to normal tissue, CENP-E often appears overexpressed, but this is likely due to the increased proliferative index of tumors relative to normal tissue. The chromosomal region containing CENP-E (4q24) is lost in 7.5-50% of human cancers, including lung, ovarian, testicular, uterine/cervical cancers and lymphomas (89). This is similar to the 4-50% of cancers lacking the chromosomal region containing p53 (17p13) and greater than the 0-15% of cancers lacking an equivalent region on chromosome three with a similar number of genes [3q24, (89)]. Moreover, heterozygous loss of CENP-E causes gain and loss of whole chromosomes without structural rearrangements, a type of aneuploidy which occurs commonly in human cancer (89). Thus, reduction of CENP-E is a frequent occurrence in cancer, underscoring the relevance of this model system.
Our analysis revealed a previously unreported sex disparity in the survival of p53 heterozygous animals. This was quite surprising, since this mouse model (30) was originally created 25 years ago and has been extensively used for survival-based studies (30,32–34,36,90–99). Although sex-specific survival differences were not previously reported in mice, this finding is consistent with human data showing that males with germline p53 mutations have longer overall and tumor free survival than females, even when sex-biased cancers are excluded from analysis (71–73). Additionally, a single nucleotide polymorphism in MDM2 that increases its abundance and decreases p53 levels also accelerates tumors specifically in women, suggesting that the sex-specific difference is dependent on p53 levels rather than a specific mutation (100). In mice, this sex-specific difference is of sufficient magnitude to necessitate sex-specific comparisons be made in p53 heterozygous survival studies, potentially meriting reanalysis of previous data sets.
p53 null animals primarily develop thymic lymphomas. Here we discovered that these lymphomas had uniformly high levels of CIN, regardless of their CENP-E status. The indistinguishable levels of CIN made it impossible to test the question of whether high(er) levels of CIN lead to cell death and tumor suppression in this context, since there are no control thymic lymphomas with lower rates of CIN. Experiments in p53 heterozygous animals generated only a single CENP-E+/+;p53−/LOH lymphoma to use as a control, which was insufficient for rigorous analysis. Nevertheless, this supports previous data suggesting that distinct tissue contexts have different tolerances for CIN (10,101,102). The current data showing that p53 null lymphomas have high CIN suggest that thymus is one of the more tolerant tissue types, consistent with previous results (102). One intruiging possibility is that lowering the rate of CIN would increase the growth rate of p53−/− lymphomas, and that further increasing the rate of CIN would be lethal.
A number of previous studies have tested the impact of CIN-inducing mutations in p53 heterozygous animals [reviewed in (11)]. Most of these reported no effect or decreased survival due to the CIN-causing mutation. One caveat to these studies is that the CIN genes altered have known—and potentially unknown—functions outside of mitosis. Perturbations of their known interphase functions, including in DNA damage responses, cell death pathways and transcriptional regulation, are tumor promoting. The current data indicate that a second caveat is the dramatically decreased tumor latency in female p53 heterozygous animals. This unexpectedly significant variable could have substantially affected tumor development and latency in cohorts consisting of unbalanced groups of males and females.
p53 is an important determinant of cellular response to a variety of cellular stresses. Its role in cell death suggested that p53 function may be necessary for the therapeutic efficacy of treatments that exert their anti-cancer effects by increasing the rate of chromosome missegregation. However, here we show that p53 is not necessary for high CIN to cause cell death or tumor suppression, validating the widespread therapeutic potential of increasing CIN above a maximally tolerated threshold.
Supplementary Material
Acknowledgements
We thank the Experimental Pathology Laboratory and the University of Wisconsin Translational Research Initiatives in Pathology laboratory, in part supported by UWCCC grant P30 CA014520, for use of their facilities and services. We thank Dr. Mark Burkard for critical evaluation of the manuscript and members of the Burkard and Weaver laboratories for useful discussions. This work was supported, in part, by NIH grant R01CA140458 (B.Weaver) and NRSA award T32CA009135 (L. Funk).
Footnotes
The authors declare no conflicts of interest.
References
- 1.von Hansemann D Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Virchow’s Arch Path Anat 1890;119:299–326 [Google Scholar]
- 2.Zasadil LM, Britigan EM, Weaver BA. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol 2013;24:370–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 2012;13:528–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duijf PH, Schultz N, Benezra R. Cancer cells preferentially lose small chromosomes. Int J Cancer 2013;132:2316–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boveri T The Origin of Malignant Tumors by Theodor Boveri; Translated by Boveri Marcella. Williams and Wilkins, Baltimore, 1929 1914 [Google Scholar]
- 6.Boveri T Ueber mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Vehr d phys med Ges zu Wurzburg, NF (available in English translation at: http://8edevbiocom/articlephp?ch=4&id=24) 1902;Bd. 35 [Google Scholar]
- 7.Simon JE, Bakker B, Foijer F. CINcere Modelling: What Have Mouse Models for Chromosome Instability Taught Us? Recent Results Cancer Res 2015;200:39–60 [DOI] [PubMed] [Google Scholar]
- 8.Duijf PH, Benezra R. The cancer biology of whole-chromosome instability. Oncogene 2013;32:4727–36 [DOI] [PubMed] [Google Scholar]
- 9.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009;10:478–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet 2008;24:457–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funk LC, Zasadil LM, Weaver BA. Living in CIN: Mitotic Infidelity and Its Consequences for Tumor Promotion and Suppression. Dev Cell 2016;39:638–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zasadil LM, Britigan EM, Ryan SD, Kaur C, Guckenberger DJ, Beebe DJ, et al. High rates of chromosome missegregation suppress tumor progression, but do not inhibit tumor initiation. Mol Biol Cell 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A 2013;110:E4134–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A 2009;106:19108–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maia AR, de Man J, Boon U, Janssen A, Song JY, Omerzu M, et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann Oncol 2015;26:2180–92 [DOI] [PubMed] [Google Scholar]
- 16.Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A 2004;101:8699–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A 2010;107:14188–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010;188:369–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burds AA, Lutum AS, Sorger PK. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci U S A 2005;102:11296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med 2014;6:229ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008;14:111–22 [DOI] [PubMed] [Google Scholar]
- 22.Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A 1980;77:1561–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt M, Budirahardja Y, Klompmaker R, Medema RH. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep 2005;6:866–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 2010;190:73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wengner AM, Siemeister G, Koppitz M, Schulze V, Kosemund D, Klar U, et al. Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol Cancer Ther 2016;15:583–92 [DOI] [PubMed] [Google Scholar]
- 26.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017;170:1062–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296–9 [DOI] [PubMed] [Google Scholar]
- 28.Mulligan LM, Matlashewski GJ, Scrable HJ, Cavenee WK. Mechanisms of p53 loss in human sarcomas. Proc Natl Acad Sci U S A 1990;87:5863–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, , Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215–21 [DOI] [PubMed] [Google Scholar]
- 30.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994;4:1–7 [DOI] [PubMed] [Google Scholar]
- 31.Purdie CA, Harrison DJ, Peter A, Dobbie L, White S, Howie SE, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 1994;9:603–9 [PubMed] [Google Scholar]
- 32.Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 2009;16:475–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalitsis P, Fowler KJ, Griffiths B, Earle E, Chow CW, Jamsen K, et al. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes Chromosomes Cancer 2005 [DOI] [PubMed] [Google Scholar]
- 34.Chi YH, Ward JM, Cheng LI, Yasunaga J, Jeang KT. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int J Cancer 2009;124:1483–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee M, Ge G, Zhang N, Edwards DG, Sumazin P, Sharan SK, et al. MMTV-Espl1 transgenic mice develop aneuploid, estrogen receptor alpha (ERalpha)-positive mammary adenocarcinomas. Oncogene 2014;33:5511–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukherjee M, Ge G, Zhang N, Huang E, Nakamura LV, Minor M, et al. Separase loss of function cooperates with the loss of p53 in the initiation and progression of T- and B-cell lymphoma, leukemia and aneuploidy in mice. PLoS One 2011;6:e22167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foijer F, Xie SZ, Simon JE, Bakker PL, Conte N, Davis SH, et al. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci U S A 2014;111:13427–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, et al. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol 2016;18:100–10 [DOI] [PubMed] [Google Scholar]
- 39.Coelho PA, Bury L, Shahbazi MN, Liakath-Ali K, Tate PH, Wormald S, et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 2015;5:150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, et al. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci U S A 2015;112:E6321–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cowley DO, Muse GW, Van Dyke T. A dominant interfering Bub1 mutant is insufficient to induce or alter thymic tumorigenesis in vivo, even in a sensitized genetic background. Mol Cell Biol 2005;25:7796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh S, Simon M, Meybohm I, Jantke I, Jonat W, Maass H, et al. Human breast cancer: frequent p53 allele loss and protein overexpression. Hum Genet 1993;90:635–40 [DOI] [PubMed] [Google Scholar]
- 43.Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, et al. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene 1997;14:865–71 [DOI] [PubMed] [Google Scholar]
- 44.Vaslet CA, Messier NJ, Kane AB. Accelerated progression of asbestos-induced mesotheliomas in heterozygous p53+/− mice. Toxicol Sci 2002;68:331–8 [DOI] [PubMed] [Google Scholar]
- 45.Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep 2019;28:1370–84.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venkatachalam S, Tyner SD, Pickering CR, Boley S, Recio L, French JE, et al. Is p53 haploinsufficient for tumor suppression? Implications for the p53+/− mouse model in carcinogenicity testing. Toxicol Pathol 2001;29 Suppl:147–54 [DOI] [PubMed] [Google Scholar]
- 47.Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, et al. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. Embo j 1998;17:4657–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pati D, Haddad BR, Haegele A, Thompson H, Kittrell FS, Shepard A, et al. Hormone-induced chromosomal instability in p53-null mammary epithelium. Cancer Res 2004;64:5608–16 [DOI] [PubMed] [Google Scholar]
- 49.Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, et al. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 1993;8:2457–67 [PubMed] [Google Scholar]
- 50.Fukasawa K, Wiener F, Vande Woude GF, Mai S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene 1997;15:1295–302 [DOI] [PubMed] [Google Scholar]
- 51.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011;19:701–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Britigan EM, Wan J, Zasadil LM, Ryan SD, Weaver BA. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol Biol Cell 2014;25:2761–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lischetti T, Nilsson J. Regulation of mitotic progression by the spindle assembly checkpoint. Mol Cell Oncol 2015;2:e970484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 2003;112:407–21 [DOI] [PubMed] [Google Scholar]
- 55.Kabeche L, Compton DA. Checkpoint-independent stabilization of kinetochore-microtubule attachments by mad2 in human cells. Curr Biol 2012;22:638–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowald K, Mantovan M, Passos J, Buccitelli C, Mardin BR, Korbel JO, et al. Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep 2016;15:2679–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barisic M, Silva E Sousa R, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, et al. Microtubule detyrosination guides chromosomes during mitosis. Science 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abrieu A, Kahana JA, Wood KW, Cleveland DW. CENP-E as an essential component of the mitotic checkpoint in vitro. Cell 2000;102:817–26 [DOI] [PubMed] [Google Scholar]
- 59.Weaver BA, Bonday ZQ, Putkey FR, Kops GJ, Silk AD, Cleveland DW. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol 2003;162:551–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007;11:25–36 [DOI] [PubMed] [Google Scholar]
- 61.Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, et al. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell 2002;3:351–65 [DOI] [PubMed] [Google Scholar]
- 62.Brown KD, Coulson RM, Yen TJ, Cleveland DW. Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling continuous synthesis with specific degradation at the end of mitosis. J Cell Biol 1994;125:1303–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown KD, Wood KW, Cleveland DW. The kinesin-like protein CENP-E is kinetochore-associated throughout poleward chromosome segregation during anaphase-A. J Cell Sci 1996;109 (Pt 5):961–9 [DOI] [PubMed] [Google Scholar]
- 64.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995;10:175–80 [DOI] [PubMed] [Google Scholar]
- 65.Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol 1995;5:931–6 [DOI] [PubMed] [Google Scholar]
- 66.Chen X, Watkins R, Delot E, Reliene R, Schiestl RH, Burgoyne PS, et al. Sex difference in neural tube defects in p53-null mice is caused by differences in the complement of X not Y genes. Dev Neurobiol 2008;68:265–73 [DOI] [PubMed] [Google Scholar]
- 67.Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 2001;153:517–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malkin D Li-fraumeni syndrome. Genes Cancer 2011;2:475–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009;460:278–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.French JE, Lacks GD, Trempus C, Dunnick JK, Foley J, Mahler J, et al. Loss of heterozygosity frequency at the Trp53 locus in p53-deficient (+/−) mouse tumors is carcinogen-and tissue-dependent. Carcinogenesis 2001;22:99–106 [DOI] [PubMed] [Google Scholar]
- 71.Hwang SJ, Lozano G, Amos CI, Strong LC. Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am J Hum Genet 2003;72:975–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82:1932–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu CC, Shete S, Amos CI, Strong LC. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni syndrome. Cancer Res 2006;66:8287–92 [DOI] [PubMed] [Google Scholar]
- 74.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009;115:1531–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boley SE, Anderson EE, French JE, Donehower LA, Walker DB, Recio L. Loss of p53 in benzene-induced thymic lymphomas in p53+/− mice: evidence of chromosomal recombination. Cancer Res 2000;60:2831–5 [PubMed] [Google Scholar]
- 76.Hulla JE. Chromosome 11 allelotypes reflect a mechanism of chemical carcinogenesis in heterozygous p53-deficient mice. Carcinogenesis 2001;22:1891. [DOI] [PubMed] [Google Scholar]
- 77.Blackburn AC, McLary SC, Naeem R, Luszcz J, Stockton DW, Donehower LA, et al. Loss of heterozygosity occurs via mitotic recombination in Trp53+/− mice and associates with mammary tumor susceptibility of the BALB/c strain. Cancer Res 2004;64:5140–7 [DOI] [PubMed] [Google Scholar]
- 78.Correa H Li-Fraumeni Syndrome. J Pediatr Genet 2016;5:84–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bakhoum SF, Kabeche L, Wood MD, Laucius CD, Qu D, Laughney AM, et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun 2015;6:5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov 2014;4:1281–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maia ARR, Linder S, Song JY, Vaarting C, Boon U, Pritchard CEJ, et al. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br J Cancer 2018;118:1586–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tighe A, Staples O, Taylor S. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 2008;181:893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rosell R, González-Larriba JL, Alberola V, Molina F, Monzó M, Benito D, et al. Single-agent paclitaxel by 3-hour infusion in the treatment of non-small cell lung cancer: links between p53 and K-ras gene status and chemosensitivity. Semin Oncol 1995;22:12–8 [PubMed] [Google Scholar]
- 84.Anelli A, Brentani RR, Gadelha AP, Amorim De Albuquerque A, Soares F. Correlation of p53 status with outcome of neoadjuvant chemotherapy using paclitaxel and doxorubicin in stage IIIB breast cancer. Ann Oncol 2003;14:428–32 [DOI] [PubMed] [Google Scholar]
- 85.Zha Y, Gan P, Liu Q, Yao Q. TP53 Codon 72 Polymorphism Predicts Efficacy of Paclitaxel Plus Capecitabine Chemotherapy in Advanced Gastric Cancer Patients. Arch Med Res 2016;47:13–8 [DOI] [PubMed] [Google Scholar]
- 86.Basu S, Barnoud T, Kung CP, Reiss M, Murphy ME. The African-specific S47 polymorphism of p53 alters chemosensitivity. Cell Cycle 2016;15:2557–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, et al. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell 2017;41:638–51.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Soto M, Raaijmakers JA, Bakker B, Spierings DCJ, Lansdorp PM, Foijer F, et al. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep 2017;19:2423–31 [DOI] [PubMed] [Google Scholar]
- 89.Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. http://cgapncinihgov/Chromosomes/Mitelman 2010 [Google Scholar]
- 90.Almeida MQ, Muchow M, Boikos S, Bauer AJ, Griffin KJ, Tsang KM, et al. Mouse Prkar1a haploinsufficiency leads to an increase in tumors in the Trp53+/− or Rb1+/− backgrounds and chemically induced skin papillomas by dysregulation of the cell cycle and Wnt signaling. Hum Mol Genet 2010;19:1387–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DelBove J, Kuwahara Y, Mora-Blanco EL, Godfrey V, Funkhouser WK, Fletcher CD, et al. Inactivation of SNF5 cooperates with p53 loss to accelerate tumor formation in Snf5(+/−);p53(+/−) mice. Mol Carcinog 2009;48:1139–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Delbridge AR, Pang SH, Vandenberg CJ, Grabow S, Aubrey BJ, Tai L, et al. RAG-induced DNA lesions activate proapoptotic BIM to suppress lymphomagenesis in p53-deficient mice. J Exp Med 2016;213:2039–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gendronneau G, Lemieux M, Morneau M, Paradis J, Tetu B, Frenette N, et al. Influence of Hoxa5 on p53 tumorigenic outcome in mice. Am J Pathol 2010;176:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Houde VP, Donzelli S, Sacconi A, Galic S, Hammill JA, Bramson JL, et al. AMPK beta1 reduces tumor progression and improves survival in p53 null mice. Mol Oncol 2017;11:1143–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lacey D, Strasser A, Bouillet P. TNF-induced chronic inflammation does not affect tumorigenesis driven by p53 loss. Cell Death Dis. England 2017. p e2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lawler J, Miao WM, Duquette M, Bouck N, Bronson RT, Hynes RO. Thrombospondin-1 gene expression affects survival and tumor spectrum of p53-deficient mice. Am J Pathol 2001;159:1949–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Loffler KA, Mould AW, Waring PM, Hayward NK, Kay GF. Menin and p53 have non-synergistic effects on tumorigenesis in mice. BMC Cancer 2012;12:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Oers JM, Edwards Y, Chahwan R, Zhang W, Smith C, Pechuan X, et al. The MutSbeta complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene 2014;33:3939–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang Z, He Y, Deng W, Lang L, Yang H, Jin B, et al. Atf3 deficiency promotes genome instability and spontaneous tumorigenesis in mice. Oncogene 2018;37:18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, et al. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res 2006;66:5104–10 [DOI] [PubMed] [Google Scholar]
- 101.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet 2020;21:44–62 [DOI] [PubMed] [Google Scholar]
- 102.Foijer F, Albacker LA, Bakker B, Spierings DC, Yue Y, Xie SZ, et al. Deletion of the. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.