

Abstract
The balanced microbiological system is a significant hallmark of piglet health. One of the crucial factors affecting intestinal microbiota is the host’s genetics. This study explored the difference in the diversity of jejunal microbiota between Saba (SB) and Landrace (LA) piglets. Nine Saba and nine Landrace piglets were fed with sow’s milk until day 35. Jejunal contents were harvested for 16S rRNA sequencing. The birth weight, body weight, and average daily gain of Saba piglets were lower than those of Landrace piglets (p < 0.01). Firmicutes were the main phylum in Saba and Landrace piglets, and the Saba piglets had a higher (p < 0.05) abundance of Bacteroidetes compared with Landrace piglets. The two most abundant genera were Lactobacilli and Clostridium XI in the jejunum of Landrace and Saba piglets. Compared with Landrace piglets, the Saba piglets had significantly lower (p < 0.05) abundance of Veillonella, Streptococcus, and Saccharibacteria genera incertae sedis. The functional prediction showed that “d-glutamine and d-glutamate metabolism” and “one carbon pool by folate” pathways were enriched in Saba piglets, while “limonene and pinene degradation”, “tryptophan metabolism”, and “sulfur relay system” pathways were enriched in Landrace piglets. In summary, the growth performance was higher for Landrace piglets compared with Saba piglets due to their genetic characteristics. The rich diversity and fewer infection-associated taxa were observed in Saba piglets, partially accounting for their higher adaptability to environmental perturbations than Landrace piglets. Furthermore, different pig breeds may regulate their health through different metabolic pathways.
Key words: Saba piglets, Landrace piglets, jejunal content, the 16S rRNA gene, diversity
Introduction
The healthy creature intestine is home to microorganisms (Eckburg et al. 2005; Ley et al. 2006; Lozupone et al. 2012). Although microbiota resides in the intestines, it plays a critical role in the digestion and absorption of nutrients, maturation of immune system, anti-colonization, and stimulation of diverse host functions (Turnbaugh et al. 2006; Levy et al. 2017). The jejunum is a significant site for nutrient absorption (Martinez-Guryn et al. 2019). The jejunal microbiota is closely related to amino acid metabolism (Dai et al. 2010) and lipid deposition (Li et al. 2019).
Previous studies indicated that the host’s genetics shapes the microbial repertoire (Goodrich et al. 2014; Goodrich et al. 2016). It was discovered that the intestinal microbiota in exotic pig breeds varies from Chinese indigenous pig breeds (Yang et al. 2014). To further explore this observation, two pig breeds with different host genetics (Saba and Landrace) were selected as the subjects in this study. The Saba pig is an indigenous breed in Chuxiong of Yunnan Province, China, and it is on the list of National Conservation Program for Chinese Indigenous Livestock Germplasm. Saba pigs grow slow, but this breed is characteristic of a high propensity for meat quality, ability to adapt to the environment, and disease resistance (Jeong et al. 2014; Diao et al. 2019). In contrast, the Landrace breed was commercially selected over generations for rapid growth and enhanced carcass yield (Briggs 1983).
Before birth the intestine of newborns is believed to be free of microbes (Turnbaugh and Turnbaugh 2008). Due to contact with sows and exposure to the surrounding environment, a complex microbial community rapidly colonizes the newborn mammal (Frese et al. 2015). The balanced microbiological system (diverse intestinal microbes) is a significant hallmark of piglet health (Patil et al. 2019). Suckling piglets are an essential stage in the life of pigs, and thus more attention should be paid to the intestinal microbiota of piglets. The 35-day-old piglets easy to cause any diseases or dramatic internal environmental changes, they are about to wean; therefore, we selected 35-day-old Saba and Landrace suckling piglets as the subjects of this study. A comparison of their jejunal microbiota diversity will help comprehend the composition and functionality of gut microbiota in Chinese indigenous pigs.
Experimental
Materials and Methods
Animals and samples collection. All Saba and Landrace pigs were raised on a commercial farm in Chuxiong of Yunnan Province, China. Three Saba and three Landrace sows of third parity were selected for this study. They lived in six enclosures in an environmentally controlled room and were fed with the National Research Council (NRC) diet without antibiotics. After parturition, all piglets from every sow were placed in a single enclosure and fed by sow’s milk until day 35 (35 d). From each sow, three piglets were randomly selected, and their birth weight and 35 d body weight were recorded, and the average daily gain of both groups was calculated. Piglets were sacrificed, and the content from the middle of the jejunum was collected for 16S rRNA sequencing analysis.
DNA extraction and PCR amplification. Based on the manufacturer’s instructions, the QIAamp® Fast DNA Stool Mini Kit (Qiagen, Cat No.: 19593) was used to extracted Genomic DNA from 18 samples. The V3-V4 region of the bacterial 16S ribosomal RNA genes was amplified following the method of Fadrosh and coworkers (Fadrosh et al. 2014).
Illumina MiSeq PE250 sequencing. The Qubit® 2.0 (Invitrogen, USA) was used to quantify DNA in the samples for library preparation. During the amplification, the barcodes were introduced by the ligated primers, which included sequencing adaptor, barcode, and sequence binding to V3-V4 region. The libraries were sequenced on the MiSeq platform (Illumina, Inc., CA, USA). All jejunal content samples from 18 piglets were subjected to 16S rRNA sequencing; however, one sample from Saba piglets and two samples from Landrace piglets failed to build a database.
Processing of sequencing data. The sequencing data analysis referred to the method of Li and coworkers (Li et al. 2019). Trimming of barcodes and primers was performed using Pandaseq (https://github.com/neufeld/pandaseq/releases/tag/v2.8.1), followed by the quality control (e.g., the lengths of reads and an average base quality) using Fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). 16S rDNA tags between 220 bp and 500 bp, with no more than three ambiguous N, were kept, and the average Phred score of bases was no worse than 20 (Q20). The copy number of tags was enumerated, and the redundant tags were removed. Only the tags with a frequency higher than 1, which are more reliable in general, were clustered into Operational Taxonomic Units (OTUs). Each OTU had a representative tag. OTUs were clustered with a criterion of 97% similarity using the Uparse (http://drive5.com/uparse/), with chimeric sequences identified and removed using the Userach (version 7.0). Each representative tag was assigned to taxa by RDP Classifier (http://rdp.cme.msu.edu/) against the RDP database (http://rdp.cme.msu.edu/) using a confidence threshold of 0.8.
The OTU profiling table and alpha/beta diversity were also achieved by Python scripts of QIIME. Alpha diversity was the species diversity in each sample, including community abundance (Chao1 index), the diversity (Shannon and Simpson index), the phylogenetic diversity index (PD whole tree), and coverage (Good’s coverage values). QIIME software was used to calculate the samples’ alpha diversity index based on the OTU results and to generate the corresponding dilution curve. The Bray-Curtis distance was calculated to estimate the dissimilarity in the community structure, which was visualized using principal coordinates analysis (PCoA). Analysis of similarities (ANOSIM) was performed in the Mothur v1.380. We determined the strength of these groups using multiresponse permutation procedures (MRPP). Both analyses were performed in the PC-ORD. In addition to p-values, PC-ORD generated T and A values for all comparisons in the MRPP. T was a measure of separation between groups, with more negative values indicating a stronger separation. Group homogeneity was described by A and was scaled between 0 and 1.
The linear discriminant analysis (LDA) effect size (LEfSe) method (p < 0.05, LDA > 2) was used to identify the most differentially abundant OTUs between groups, with the LDA obtained by a pair-wise computation. The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) based on a closed-reference operational taxonomic unit (OTU) was used to predict the abundances of functional categories in the Kyoto Encyclopedia of Genes and Genomes (KEGG) ortholog (KO). The correlation coefficients between KEGG pathways and bacterial compositions were calculated using Pearson’s correlation test in GraphPad Prism 7.
Statistical analysis. The experimental data, including growth performance and microbiota abundances, were analyzed with the SPSS 22.0 software. Grade and quantitative data were compared with the t-test between the two groups.
We used Spearman’s test to estimate the correlation between KEGG pathway and jejunal microbial composition and host growth performance. P < 0.05 was deemed to statistical significance.
Results
Growth performance of Saba and Landrace piglets. The growth performance of Saba and Landrace piglets is shown in Table I. The birth weight of Landrace piglets was significantly higher than Saba piglets (p < 0.001). On day 35, the body weight and average daily gain (p < 0.001) of Landrace piglets were higher than Saba piglets (p < 0.001).
Table I.
Growth performance of Saba and Landrace piglets.
| SB | LA | |
|---|---|---|
| Birth weight (kg) | 0.76 ± 0.20B | 1.99 ± 0.14A |
| Body weight (kg) | 4.69 ± 1.14B | 10.22 ± 0.57A |
| Average daily gain (kg) | 0.11 ± 0.03B | 0.24 ± 0.02A |
| Stem length (cm) | 37.00 ± 4.84B | 50.00 ± 2.27A |
| Height at withers (cm) | 22.75 ± 1.83B | 29.00 ± 1.6A |
| Chest measurement (cm) | 37.88 ± 3.48B | 48.38 ± 1.41A |
| Chest depth (cm) | 10.75 ± 0.71B | 14.38 ± 2.07A |
| Abdominal girth (cm) | 38.75 ± 3.85B | 49.50 ± 1.60A |
| Cannon circumference (cm) | 8.06 ± 0.56B | 10.13 ± 0.35A |
Different superscripts in the same column indicate significant difference (p < 0.001)
SB – Saba piglets, LA – Landrace piglets
Gut microbiota DNA sequence data and quality control. Sequencing of the amplicons of the 16S rRNA gene at MiSeq generated 884,982 clean reads (mean length of 415 bp) with 368,167,415 base pairs in total, yielding an average of 58,999 clean reads (55,547– 63,409), and 24,544,494 base pairs (22,667,528 bp – 26,990,140 bp) per sample (Table II). Out of the high-quality sequences, about 99.53% were between 420 and 460 bp for these two breeds.
Table II.
Description of the assembly results of jejunum microbiota from piglets.
| Sample name | Clean Reads | Bases (bp) | Q20 (%) | Q30 (%) | GC (%) | Average length (bp) |
|---|---|---|---|---|---|---|
| LA-1 | 58278 | 24288798 | 0.9602 | 0.8873 | 0.5229 | 416 |
| LA-2 | 55975 | 23383577 | 0.9591 | 0.8855 | 0.5188 | 417 |
| LA-3 | 62396 | 25557801 | 0.9696 | 0.9081 | 0.527 | 409 |
| LA-4 | 56565 | 23778906 | 0.9614 | 0.8922 | 0.5365 | 420 |
| LA-5 | 55547 | 22929087 | 0.9671 | 0.9054 | 0.5221 | 412 |
| LA-6 | 56336 | 23469885 | 0.9612 | 0.8928 | 0.5234 | 416 |
| LA-7 | 57139 | 23771848 | 0.96 | 0.8902 | 0.5332 | 416 |
| SB-1 | 63393 | 26990140 | 0.9609 | 0.8917 | 0.5154 | 425 |
| SB-2 | 55959 | 22667528 | 0.9697 | 0.9119 | 0.5271 | 405 |
| SB-3 | 62484 | 25749078 | 0.9661 | 0.9035 | 0.5185 | 412 |
| SB-4 | 61527 | 26100871 | 0.9594 | 0.8867 | 0.5117 | 424 |
| SB-5 | 56907 | 24136231 | 0.9602 | 0.8897 | 0.5123 | 424 |
| SB-6 | 63409 | 26890638 | 0.958 | 0.8848 | 0.5496 | 424 |
| SB-7 | 58920 | 23815087 | 0.9656 | 0.9029 | 0.5306 | 404 |
| SB-8 | 60147 | 24637940 | 0.9626 | 0.8966 | 0.5253 | 409 |
SB – Saba piglets, LA – Landrace piglets
Diversity in jejunal microbiota of Saba and Landrace piglets. We revealed that the jejunal microbiome was different in Saba and Landrace piglets. The USEARCH algorithm was used to cluster at a 0.97 similarity level, and the clustered sequences were filtered by a chimera. We obtained 489 and 365 OTUs (Fig. 1) from Saba and Landrace piglets, respectively. In total, 254 OTUs were shared by Saba and Landrace piglets. The alpha diversity index of the samples is shown in Table III. The PD whole tree in Saba piglets (17.76) was significantly higher (p < 0.05) than in Landrace piglets (13.31). The Chao1 index (242.85 vs. 229.04), the observed species index (139.75 vs. 124.75), and Shannon (3.06 vs. 2.84) and Simpson (0.79 vs. 0.76) indexes for microbiota from Saba piglets are higher than those from Landrace piglets, but the statistical significance (p > 0.05) was not noticed. Besides, we compared the beta diversities between all the samples (Table IV). The test statistic (R) of Multi Response Permutation Procedure (MRPP) was 0.031 (p = 0.102) on the weighted UniFrac, and 0.034 (p = 0.005) on the unweighted Uni-Frac. Also, the test statistic (R) of ANOSIM was 0.131 (p = 0.01; Fig. 2) on the unweighted UniFrac. Using the unweighted UniFrac metric, the Principal Coordinates Analysis (PCoA) showed a clear separation between Saba and Landrace piglet samples (Fig. 2).
Fig. 1.
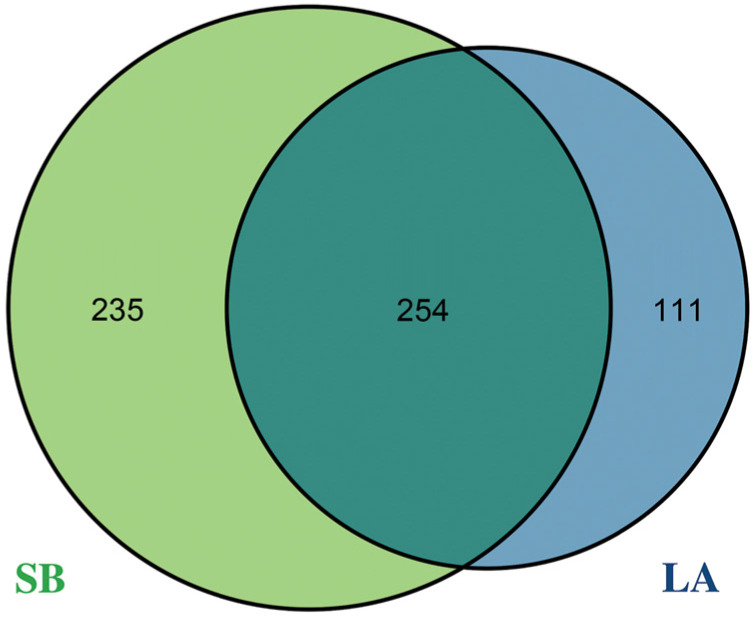
Venn diagram of OTUs clustered at 97% sequence identity of microbiotas from Saba and Landrace piglets. The number of overlapping parts is the total number of OTUs between the groups, while the numbers in non-overlapping parts indicate the number of unique OTUs for each group. SB – Saba piglets, LA – Landrace piglets.
Table III.
Alpha diversity in jejunal microbiota between Saba and Landrace piglets.
| LA | SB | p value | |
|---|---|---|---|
| Chao1 index | 229.04 ± 38.23 | 242.85 ± 10.92 | 0.531 |
| The observed_species index | 124.75 ± 24.27 | 139.75 ± 36.57 | 0.520 |
| PD_whole_tree | 13.31 ± 1.98 | 17.76 ± 2.17 | 0.023 |
| Shannon index | 2.84 ± 0.42 | 3.06 ± 0.89 | 0.671 |
| Simpson index | 0.76 ± 0.077 | 0.79 ± 0.096 | 0.665 |
| Goods_coverage | 0.998 ± 0.00015 | 0.999 ± 0.00024 | 0.149 |
SB – Saba piglets, LA – Landrace piglets
Table IV.
MRPP of the 16S rRNA gene between Saba and Landrace piglets.
| A | Observe Delta | Expect Delta | Significance | |
|---|---|---|---|---|
| The weighted_unifrac | 0.0312699850241734 | 0.276682987004487 | 0.285614136784429 | 0.102 |
| The unweighted_unifrac | 0.0338353054352021 | 0.551581668259676 | 0.570898182641762 | 0.00 |
SB – Saba piglets, LA – Landrace piglets
Fig. 2.

Principal coordinate analysis (PCoA) illustrated bacterial community structures based on Bray-Curtis distances. On the PCoA plot, each color represents one group. Unweighted and weighted PCoA of β-diversity measures of all samples. PCOA1 (19.67%) and PCOA2 (13.63%).
Comparison of jejunal microbiota of Saba and Landrace piglets. The jejunal bacterial taxa were diversified between Saba and Landrace piglets at the phylum level (Fig. 3A). Among these taxa, Firmicutes occurred with the highest abundance within the jejunal microbiota of Landrace piglets (95.82%), followed by Proteobacteria (1.67%), and Bacteroidetes (0.052%). Similarly, the higher relative abundance of Firmicutes (97.6%, p > 0.05) than Bacteroidetes (1.14%, p < 0.05), and a lower proportion of Proteobacteria (0.71%, p > 0.05) were observed in microbiota of Saba piglets.
Fig. 3.

Community composition of the jejunum microbial of Saba and Landrace piglets at the phylum (A) and genus (B) levels, respectively. Data are expressed as means + MSE, *p < 0.05.
The abundance of bacterial species within jejunal taxa is shown in Fig. 3B. The two most abundant genera were Lactobacilli and Clostridium XI, accounting for 28.0% and 42.24% of the jejunal species in Landrace piglets, respectively. Compared with the microbiota of Landrace piglets, the jejunal microbial of Saba piglets had a higher abundance of Lactobacilli (36.81%) and a lower abundance of Clostridium XI (40.02%), but the difference was not statistically significant (p > 0.05). Moreover, Veillonella (0.58% vs. 2.34%), Streptococcus (0.23% vs. 1.32%), and Saccharibacteria genera incertae sedis (0.19% vs. 1.04%) of Saba piglets were remarkably lower than in microbiota of Landrace piglets (p < 0.05).
Differences of bacterial taxa between Saba and Landrace piglets. The different number of OTUs was observed between the jejunal microbiota of Saba and Landrace piglets (Fig. 4). There was one main phylum (Firmicutes) and two genera (Coprococcus and Parabacteroides) significantly enriched in jejunal microbiota of Saba piglets. Also, multiple biomarkers were significantly enriched in jejunal microbiota of Landrace piglets, including two phyla (Candidatus Saccharibacteria and Proteobacteria), two classes (Epsilonproteobacteria and Gammaproteobacteria), two orders (Campylobacterales and Pasteurellales), eight families (Fusobacteriaceae, Leuconostocaceae, Actinomycetaceae, Enterococcaceae, Campylobacteraceae, Dermatophilaceae, Streptococcaceae, and Pasteurellaceae), thirteen genera (Enterococcus, Actinomyces, Fusobacterium, Weissella, Pediococcus, Campylobacter, Oribacterium, Sharpea, Tonsilliphilus, Pasteurella, Saccharibacteria genera incertae sedis, Streptococcus, and Actinobacillus). Furthermore, the increase in the abundance of the phylum Candidatus Saccharibacteria was represented by an increased abundance of the genus Saccharibacteria genera incertae sedis (Fig. 5).
Fig. 4.

Alteration of the relative abundance of bacteria in the Saba and Landrace piglets using linear discriminant analysis effect size (LEfSe). Each bar represents the log 10 effect size (LDA score) for a specific taxon. A longer bar represents a higher LDA score. Only taxa meeting an LDA significant threshold of 2 are shown. These taxa showed a statistically significant difference between the Saba and Landrace piglets (p < 0.05 by the Wilcoxon test); each color represents one group.
p – phylum, c – class, o – order, f – family, and g – genus.
Fig. 5.

A cladogram showed a comparison of the bacterial microbial profiles from Saba and Landrace piglets.
p – phylum, c – class, o – order, f – family, and g – genus.
Correlation between microbiota and growth performance. The correlation between jejunal microbiota and host growth performance was shown in Fig. 6. The Coprococcus was negatively correlated with the body weight (p = 0.046, R = –0.53), average daily gain (p = 0.046, R = –0.53), stem length (p = 0.018, R = –0.61), height at withers (p = 0.0063, R = –0.68), chest measurement (p = 0.046, R = –0.53), chest depth (p = 0.012, R = –0.64), abdominal girth (p = 0.045, R = –0.53), and cannon circumference (p = 0.011, R = –0.65). The Parabacteroides was negatively correlated with the cannon circumference (p = 0.048, R = –0.52). The Tonsilliphilus was positively correlated with the body weight (p = 0.025, R = 0.58), average daily gain (p = 0.022, R = 0.59), stem length (p = 0.026, R = 0.58), height at withers (p = 0.013, R = 0.64), chest measurement (p = 0.0031, R = 0.72), chest depth (p = 0.0015, R = 0.74), abdominal girth (p = 0.0053, R = 0.69), and cannon circumference (p = 0.022, R = 0.72). The Saccharibacteria genera incertae sedis was positively correlated with the stem length (p = 0.036, R = 0.55), chest measurement (p = 0.038, R = 0.54), and abdominal girth (p = 0.031, R = 0.56). The Enterococcus was positively correlated with the body weight (p = 0.0097, R = 0.65), average daily gain (p = 0.015, R = 0.62), stem length (p = 0.049, R = 0.52), height at withers (p = 0.0048, R = 0.70), chest measurement (p = 0.0066, R = 0.68), chest depth (p = 0.0069, R = 0.68), abdominal girth (p = 0.0070, R = 0.67), and cannon circumference (p = 0.0052, R = 0.71). The Pediococcus was positively correlated with the body weight (p = 0.023, R = 0.59), average daily gain (p = 0.021, R = 0.59), stem length (p = 0.026, R = 0.58), height at withers (p = 0.013, R = 0.64), chest measurement (p = 0.0025, R = 0.72), chest depth (p = 0.0021, R = 0.74), abdominal girth (p = 0.0048, R = 0.69), and cannon circumference (p = 0.018, R = 0.63). The Weissella was positively correlated with the body weight (p = 0.033, R = 0.56), average daily gain (p = 0.033, R = 0.56), stem length (p = 0.038, R = 0.54), height at withers (p = 0.011, R = 0.66), chest measurement (p = 0.0039, R = 0.70), chest depth (p = 0.0029, R = 0.73), abdominal girth (p = 0.0078, R = 0.66), and cannon circumference (p = 0.022, R = 0.62). The Streptococcus was positively correlated with the body weight (p = 0.0024, R = 0.74), average daily gain (p = 0.0020, R = 0.75), stem length (p = 0.0037, R = 0.71), height at withers (p = 0.0073, R = 0.67), chest measurement (p = 0.0011, R = 0.77), chest depth (p = 0.0099, R = 0.65), abdominal girth (p = 0.00097, R = 0.78), and cannon circumference (p = 0.0038, R = 0.72). The Sharpea was positively correlated with the body weight (p = 0.023, R = 0.59), average daily gain (p = 0.023, R = 0.59), height at withers (p = 0.0060, R = 0.69), chest measurement (p = 0.0096, R = 0.66), chest depth (p = 0.0079, R = 0.67), abdominal girth (p = 0.010, R = 0.65), and cannon circumference (p = 0.0099, R = 0.66). The Campylobacter was positively correlated with the body weight (p = 0.0034, R = 0.72), average daily gain (p = 0.0031, R = 0.73), stem length (p = 0.0012, R = 0.78), height at withers (p = 0.011, R = 0.65), chest measurement (p = 0.0022, R = 0.75), chest depth (p = 0.0012, R = 0.78), abdominal girth (p = 0.0013, R = 0.77), and cannon circumference (p = 0.0066, R = 0.69).
Fig. 6.

Heatmap analysis of the correlation between microbiota and growth performance.
KEGG pathway and their correlation with microbiota. To assess the jejunal microbiota’s metabolic potential, we performed Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al. 2013; Javurek et al. 2016). KEGG pathway (L3 hierarchy) analysis is shown in Fig. 7. The “d-glutamine and d-glutamate metabolism”, and “one carbon pool by folate” pathway were enriched in Saba piglets. The “limonene and pinene degradation”, “tryptophan metabolism”, and “sulfur relay system” were enriched in Landrace piglets.
Fig. 7.

KEGG enrichment analysis of the difference within groups at the L3 hierarchy.
We used Spearman’s correlation heatmap (Fig. 8) to study the correlation between the jejunal microbiota and the KEGG pathway. The “d-glutamine and d-glutamate metabolism” pathway was positively correlated with the presence of Firmicutes (family, p = 0.0080, R = 0.67), while negatively correlated with Fusobacteriaceae (family, p = 0.020, R = –0.60), and Fusobacterium (genus, p = 0.017, R = –0.62). The pathway of “one carbon pool by folate” was negatively correlated with Fusobacteriaceae (family, p = 0.049, R = –0.52) and Fusobacterium (genus, p = 0.048, R = –0.52). The pathway “tryptophan metabolism” was negatively correlated with Coprococcus (genus, p = 0.023, R = –0.59), but positively correlated with Gammaproteobacteria (class, p = 0.024, R = 0.59), Enterococcaceae (family, p = 0.0089, R = 0.66), Proteobacteria (family, p = 0.022, R = 0.59), and Enterococcus (genus, p = 0.031, R = 0.56).
Fig. 8.

Pearson’s correlation analysis of microorganisms and signal pathways in Saba and Landrace piglets. Heatmap analysis of the correlation between microorganisms and signal pathways. Correlations with p < 0.05 are shown. Blue represents a significant negative correlation (p < 0.05), red represents a significant positive correlation (p < 0.05), and white represents no significant correlation (p > 0.05). The number represents the value of R (p < 0.05).
Discussion
The Landrace pig from Denmark is a typical commercial pig breed of fast growth and high carcass yield (Briggs 1983). The previous research reported that the body weight of Landrace piglets was 1.68 kg and 6.52 kg on day 1 and day 27, respectively (Li et al. 2013). In contrast, the Saba pig is an indigenous breed from China, with a relatively slow growth rate. In our study, the birth weight, body weight (day 35), and average daily gain of Landrace piglets (1.99 kg, 10.22 kg, and 0.24 kg/d, respectively) were higher than those of Saba piglets (0.76 kg, 4.69 kg, and 0.11 kg/d, respectively). The data indicated that the growth performance of Landrace piglets was higher than Saba piglets. Previous studies have shown that growth performance and intestinal microbes were different in Jinhua pigs and Landrace pigs of the same age (Xiao et al. 2018). In addition, our results show that the Coprococcus and Parabacteroides were negatively correlated with the growth performance, while the Tonsilliphilus, Enterococcus, Pediococcus, Weissella, Streptococcus, Campylobacter, Saccharibacteria genera incertae sedis, and Sharpea were positively associated with the growth performance. The above results suggested that the composition of intestinal microbiota was significantly and closely connected with the pig breed.
It is generally believed that intestinal microorganisms have abundant metabolic profiles to maintain their basic life and have a considerable impact on host growth and health (Turnbaugh et al. 2006). The accumulating evidence suggested that diet (Pluske 2013), environment (Thompson et al. 2008), and host’s genetics (Büsing and Zeyner 2015; Hancox et al. 2015) can affect the composition of intestinal microbiota. The previous research (Yang et al. 2014) revealed that the percentages of Firmicutes and Bacteroidetes in the Chinese indigenous pig breeds (Xiaomeishan, Meishan, and Bama sows) were higher than those of exotic breeds (Landrace, Yorkshire, and Duroc sows). It is consistent with our finding that the Firmicutes, Proteobacteria, and Bacteroidetes dominated in the jejunum of both pig breeds. Furthermore, the percentage of Bacteroidetes in Saba piglets was significantly higher than in Landrace piglets. Saba piglets are obese, and Landrace piglets are lean. A previous study has shown that fat deposition is positively correlated with the presence of Bacteroidetes and Firmicutes within the intestinal microbiota (Turnbaugh et al. 2006). Nevertheless, the mechanisms between intestinal microbiota and fat deposition are still unclear, and further study is needed.
At the genus level, the two most numerous genera in the pig’s small intestine were Lactobacillus and Clostridium (Crespo-Piazuelo et al. 2018). Our study demonstrated that Clostridium XI is the most numerous bacteria in jejune of both Saba and Landrace piglets. We speculate that this was principally due to the adaptation of the microbial system for milk nutrition, as sucking lambs have a large proportion of Clostridium XI in the intestinal microbial community (Bi et al. 2019). The Lactobacilli were usually considered beneficial bacteria responsible for more effective anti-inflammation and out-competing microbiota competences (Etzold et al. 2014). Therefore, some Lactobacillus species have been used as substitutes for antibiotics for growth promotion. The average daily gain (ADG) of weaned piglets fed with Lactobacillus plantarum PFM 105 was significantly improved after three weeks (Wang et al. 2019). Again, two Lactobacillus strains (Lactobacillus frumenti and Lactobacillus gasseri LA39), when taken orally, could significantly prevent stress-induced diarrhea caused by early weaning of piglets (Hu et al. 2018). In line with that, in this study, the abundance of Lactobacillus in Saba piglets was higher than in Landrace piglets.
Furthermore, some taxa are recognized as negatively correlated with host health. Veillonella is present in piglets infected with porcine epidemic diarrhea virus (PEDV), caused by the disorder of intestinal amino acid metabolism and energy metabolism (Huang et al. 2018). Also, Veillonella and Streptococcus can increase the secretion of inflammatory cytokine and the production of anti-microbial peptides (AMP), resulting in improved mucosal thickness and epithelial barrier function in 3D-reconstructed human gingiva (Shang et al. 2018). In the present study, a higher proportion of Lactobacilli and a lower proportion of Streptococcus and Veillonella in the microbiome of Saba piglets, suggested that Saba piglets might have stronger disease resistance. Overall, our data indicated that the pig breed could influence the microbiome composition.
The gut microbiome is beneficial for pigs, contributing to improved vitamin K production, cellulose fermentation, and increased resistance to pathogens (Kim and Isaacson 2015; Stokes 2017; Yang et al. 2017). PICRUSt analysis of jejunal microbiotas’ metabolic potential showed that metabolic pathways were significantly different in Saba and Landrace piglets. It is noteworthy that the different amino acid metabolism pathways were enriched in Saba and Landrace piglets. The “d-glutamine and d-glutamate metabolism” pathway was enriched in Saba piglets, while “tryptophan metabolism” pathway was enriched in Landrace piglets. The “d-glutamine and d-glutamate metabolism” pathway participates in the inhibition of lipid peroxidation and quenches free radicals during oxidative stress (Qu et al. 2020). It has been reported that tryptophan is related to the immune response regulation, inflammation, and oxidative stress (Anesi et al. 2019; Liu et al. 2019). Therefore, different pig breeds might have regulated their health through different metabolic pathways.
The central part of amino acids absorption is the small intestine (Wu 1998). The catabolism of arginine and lysine in the jejunum could exceed their transport into intestinal cells (Dai et al. 2010). This phenomenon may be due to the role of intestinal microorganisms. Besides, owing to the deficiency of several key enzymes, the threonine, tryptophan, histidine, lysine, and methionine cannot be metabolized by porcine intestinal cells in the presence of amino acids at physiological concentrations (Chen et al. 2007). However, the histidine, glutamate, threonine, and lysine were utilized by microbiota of the porcine small intestine (Dai et al. 2010). The above results indicated that jejunal microorganisms could participate in amino acid metabolism. In our study, the Firmicutes enriched in the jejunal microbiome of Saba piglets were positively associated with the “d-glutamine and d-glutamate metabolism” pathway. In contrast Fusobacteriaceae and Fusobacterium that enriched the jejunal microbiome of Landrace pigs were negatively correlated with the above pathway. Finally, Coprococcus from the jejunal microbiome of Saba piglets was negatively correlated with the “tryptophan metabolism” pathway, and Enterococcaceae, Enterococcus, Proteobacteria, and Gammaproteobacteria from Landrace piglets were positively correlated with tryptophan metabolism. Therefore, these taxa may be related to the metabolism of amino acids in jejunum.
Conclusions
In summary, the growth performance was higher for Landrace piglets compared to Saba piglets due to their different genetic characteristics. The rich diversity and fewer infection-associated taxa were observed in Saba piglets, partially accounting for their high adaptability to environmental perturbations compared to Landrace piglets. Several taxa in the jejunum of Saba and Landrace piglets were associated with “d-glutamine and d-glutamate metabolism” and “tryptophan metabolism” respectively, suggesting that pig breeds may regulate their health through different metabolic pathways. Although the interaction between pig and microbiota needs further extensive investigations, our study would shed more light on the functional exploration and resource development of local pig intestinal microbiota in China.
Acknowledgments
This research was funded by the National Key Research and Development Program of China (2018YFD0500401), Key Program of Yunnan province Natural Science Foundation of China (2018FA021), Nature Science Foundation of China (U1802234, 31760645).
Footnotes
Conflict of interest
The authors do not report any financial or personal connections with other persons or organizations, which might negatively affect the contents of this publication and/or claim authorship rights to this publication.
ORCID
Hongbin Pan https://orcid.org/0000-0002-9289-2434
Literature
- Anesi A, Rubert J, Oluwagbemigun K, Orozco-Ruiz X, Nöthlings U, Breteler MMB, Mattivi F. Metabolic profiling of human plasma and urine, targeting tryptophan, tyrosine and branched chain amino acid pathways. Metabolites. 2019. November 01;9(11):261 10.3390/metabo9110261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y, Cox MS, Zhang F, Suen G, Zhang N, Tu Y, Diao Q. Feeding modes shape the acquisition and structure of the initial gut microbiota in newborn lambs. Environ Microbiol. 2019. July;21(7): 2333–2346. 10.1111/1462-2920.14614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs HM. International pig breed encyclopedia. Indianapolis (USA): Elanco Products Company; 1983. [Google Scholar]
- Büsing K, Zeyner A. Effects of oral Enterococcus faecium strain DSM 10663 NCIMB 10415 on diarrhoea patterns and performance of sucking piglets. Benef Microbes. 2015. January 01;6(1):41–44. 10.3920/BM2014.0008 [DOI] [PubMed] [Google Scholar]
- Chen L, Yin YL, Jobgen WS, Jobgen SC, Knabe DA, Hu WX, Wu G. In vitro oxidation of essential amino acids by jejunal mucosal cells of growing pigs. Livest Sci. 2007. May;109(1–3):19–23. 10.1016/j.livsci.2007.01.027 [DOI] [Google Scholar]
- Crespo-Piazuelo D, Estellé J, Revilla M, Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, Fernández AI, Ballester M, Folch JM. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep. 2018. December;8(1):12727 10.1038/s41598-018-30932-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai ZL, Zhang J, Wu G, Zhu WY. Utilization of amino acids by bacteria from the pig small intestine. Amino Acids. 2010. November; 39(5):1201–1215. 10.1007/s00726-010-0556-9 [DOI] [PubMed] [Google Scholar]
- Diao S, Huang S, Chen Z, Teng J, Ma Y, Yuan X, Chen Z, Zhang H, Li J, Zhang Z. Genome-wide signatures of selection detection in three south China indigenous pigs. Genes (Basel). 2019. May 07; 10(5):346 10.3390/genes10050346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005. June 10;308(5728):1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzold S, Kober OI, MacKenzie DA, Tailford LE, Gunning AP, Walshaw J, Hemmings AM, Juge N. Structural basis for adaptation of lactobacilli to gastrointestinal mucus. Environ Microbiol. 2014. March;16(3):888–903. 10.1111/1462-2920.12377 [DOI] [PubMed] [Google Scholar]
- Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6 10.1186/2049-2618-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese SA, Parker K, Calvert CC, Mills DA. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome. 2015. December;3(1):28 10.1186/s40168-015-0091-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, Ley RE. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016. May;19(5):731–743. 10.1016/j.chom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014. November; 159(4):789–799. 10.1016/j.cell.2014.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox LR, Le BM, Richards PJ, Guillou D, Dodd CE, Mellits KH. Effect of a single dose of Saccharomyces cerevisiae var. boulardii on the occurrence of porcine neonatal diarrhoea. Animal. 2015;9(11):1756–1759. 10.1017/S1751731114002687 [DOI] [PubMed] [Google Scholar]
- Hu J, Ma L, Nie Y, Chen J, Zheng W, Wang X, Xie C, Zheng Z, Wang Z, Yang T, et al. A microbiota-derived bacteriocin targets the host to confer diarrhea resistance in early-weaned piglets. Cell Host Microbe. 2018. December;24(6):817–832.e8. 10.1016/j.chom.2018.11.006 [DOI] [PubMed] [Google Scholar]
- Huang MZ, Wang SY, Wang H, Cui DA, Yang YJ, Liu XW, Kong XJ, Li JY. Differences in the intestinal microbiota between uninfected piglets and piglets infected with porcine epidemic diarrhea virus. PLoS One. 2018. February 15;13(2):e0192992 10.1371/journal.pone.0192992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javurek AB, Spollen WG, Ali AMM, Johnson SA, Lubahn DB, Bivens NJ, Bromert KH, Ellersieck MR, Givan SA, Rosenfeld CS. Discovery of a novel seminal fluid microbiome and influence of estrogen receptor alpha genetic status. Sci Rep. 2016. March;6(1):23027 10.1038/srep23027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HS, Kim DW, Chun SY, Sung S, Kim HJ, Cho S, Kim H, Oh SJ. Native Pig and Chicken Breed Database: NPCDB. Asian-Australas J Anim Sci. 2014. October;27(10):1394–1398. 10.5713/ajas.2014.14059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Isaacson RE. The pig gut microbial diversity: understanding the pig gut microbial ecology through the next generation high throughput sequencing. Vet Microbiol. 2015. June;177(3–4):242–251. 10.1016/j.vetmic.2015.03.014 [DOI] [PubMed] [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013. September;31(9):814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Blacher E, Elinav E. Microbiome, metabolites and host immunity. Curr Opin Microbiol. 2017. February;35:8–15. 10.1016/j.mib.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006. February;124(4):837–848. 10.1016/j.cell.2006.02.017 [DOI] [PubMed] [Google Scholar]
- Li X, Cao Z, Yang Y, Chen L, Liu J, Lin Q, Qiao Y, Zhao Z, An Q, Zhang C, et al. Correlation between jejunal microbial diversity and muscle fatty acids deposition in broilers reared at different ambient temperatures. Sci Rep. 2019. December;9(1):11022 10.1038/s41598-019-47323-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wu Z, Ren G, Zhao Y, Liu D. Expression patterns of insulin-like growth factor system members and their correlations with growth and carcass traits in Landrace and Lantang pigs during postnatal development. Mol Biol Rep. 2013. May;40(5):3569–3576. 10.1007/s11033-012-2430-1 [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen X, Liu Y, Chen T, Zhang Q, Zhang H, Zhu Z, Chai Y, Zhang J. Metabolomic study of the protective effect of Gandi capsule for diabetic nephropathy. Chem Biol Interact. 2019. December;314:108815 10.1016/j.cbi.2019.108815 [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012. September;489(7415):220–230. 10.1038/nature11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guryn K, Leone V, Chang EB. Regional diversity of the gastrointestinal microbiome. Cell Host Microbe. 2019. September;26(3): 314–324. 10.1016/j.chom.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil Y, Gooneratne R, Ju XH. Interactions between host and gut microbiota in domestic pigs: a review. Gut Microbes. 2020. May 3; 11(3):310–334. 10.1080/19490976.2019.1690363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluske JR. Feed- and feed additives-related aspects of gut health and development in weanling pigs. J Anim Sci Biotechnol. 2013. December; 4(1):1 10.1186/2049-1891-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Gao H, Sun J, Tao L, Zhang Y, Zhai J, Song Y, Hu T, Li Z. Identification of key metabolites during cisplatin-induced acute kidney injury using an HPLC-TOF/MS-based non-targeted urine and kidney metabolomics approach in rats. Toxicology. 2020. February; 431:152366 10.1016/j.tox.2020.152366 [DOI] [PubMed] [Google Scholar]
- Shang L, Deng D, Buskermolen JK, Janus MM, Krom BP, Roffel S, Waaijman T, van Loveren C, Crielaard W, Gibbs S. Multi-species oral biofilm promotes reconstructed human gingiva epithelial barrier function. Sci Rep. 2018. December;8(1):16061 10.1038/s41598-018-34390-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes CR. The development and role of microbial-host interactions in gut mucosal immune development. J Anim Sci Biotechnol. 2017. December;8(1):12 10.1186/s40104-016-0138-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Wang B, Holmes AJ. The immediate environment during postnatal development has long-term impact on gut community structure in pigs. ISME J. 2008. July;2(7):739–748. 10.1038/ismej.2008.29 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI. An invitation to the marriage of metagenomics and metabolomics. Cell. 2008. September;134(5):708–713. 10.1016/j.cell.2008.08.025 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006. December;444(7122):1027–1031. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- Wang T, Teng K, Liu Y, Shi W, Zhang J, Dong E, Zhang X, Tao Y, Zhong J. Lactobacillus plantarum PFM 105 promotes intestinal development through modulation of gut microbiota in weaning piglets. Front Microbiol. 2019. February 5;10:90 10.3389/fmicb.2019.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G. Intestinal mucosal amino acid catabolism. J Nutr. 1998. August 01;128(8):1249–1252. 10.1093/jn/128.8.1249 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Kong F, Xiang Y, Zhou W, Wang J, Yang H, Zhang G, Zhao J. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep. 2018. December;8(1):5985 10.1038/s41598-018-24289-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, Yang M, Zhang Z, Chen C, Huang L. Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front Microbiol. 2017. August 15;8:1555 10.3389/fmicb.2017.01555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Bian G, Su Y, Zhu W. Comparison of faecal microbial community of lantang, bama, erhualian, meishan, xiaomeishan, duroc, landrace, and yorkshire sows. Asian-Australas J Anim Sci. 2014. June 1;27(6):898–906. 10.5713/ajas.2013.13621 [DOI] [PMC free article] [PubMed] [Google Scholar]