

Abstract
Sepsis accounts for nearly 700 000 deaths in Europe annually and is caused by an overwhelming host response to infection resulting in organ failure. The endothelium is an active contributor to sepsis and as such represents a major target for therapy. During sepsis, endothelial cells amplify the immune response and activate the coagulation system. They are both a target and source of inflammation and serve as a link between local and systemic immune responses. In response to cytokines produced by immune cells, the endothelium expresses adhesion molecules and produces vasoactive compounds, inflammatory cytokines, and chemoattractants, thus switching from an anticoagulant to procoagulant state. These responses contribute to local control of infection, but systemic activation can lead to microvascular thrombosis, capillary permeability, hypotension, tissue hypoxia, and ultimately tissue damage. This review focuses on the role of the endothelium in leucocyte adhesion and transmigration as well as production of reactive oxygen and nitrogen species, microRNAs and cytokines, formation of signalling microparticles, and disseminated intravascular coagulation. We also discuss alterations in endothelial permeability and apoptosis. Finally, we review the diagnostic potential of endothelial markers and endothelial pathways as therapeutic targets for this devastating disease.
Keywords: Sepsis, Endothelium, Mechanism, Diagnosis, Treatment
Graphical Abstract
1. Introduction
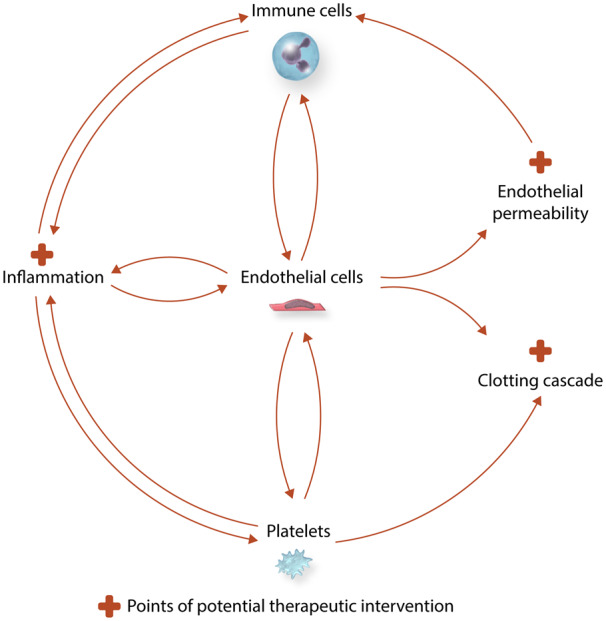
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection1 associated with significant morbidity and mortality. According to the latest reports, each year at least 1.7 million adults in the USA develop sepsis and nearly 265 000 of them die. In Europe, the incidence of sepsis is estimated to be more than 3.4 million cases per year; 700 000 of these patients do not survive the hospitalization and one-third of survivors die during the first year after hospitalization.2 Due to the lack of a quick and specific diagnostic tool and fairly non-specific clinical presentation, the definition of sepsis has undergone few modifications in recent years.1 Clinical features of sepsis reflect the body’s response to infection and include fever, tachycardia, hypotension, and leucocytosis as well as end organ dysfunction, such as acute lung injury, acute kidney injury, encephalopathy, and cardiomyopathy.3 As sepsis is a systemic condition, it affects virtually all organs and tissues, with endothelium being one of the first cell types to encounter and respond to the insult. During sepsis, the two most pronounced roles of endothelial cells (ECs) are to amplify the immune response and to activate the coagulation system. Endothelial activation and/or dysfunction ultimately contribute to end organ damage during sepsis. Furthermore, the endothelium provides a link between local and systemic immune responses, as it is simultaneously a target and a source of inflammation.4 Upon stimulation, ECs express adhesion molecules and produce vasoactive compounds, inflammatory cytokines, and chemoattractants, thus switching from an anticoagulant to procoagulant state.5 While endothelial activation locally aids in fighting the source of infection, systemic activation may result in microvascular thrombosis, capillary permeability, hypotension, tissue hypoxia, and ultimately tissue damage.
2. Endothelium and the pathogen—initial encounter
When there is a breach allowing a pathogen to enter the blood stream, generalized inflammation from exposure to bacterial components and tissue breakdown products occurs. While immune cells ensure an adequate response to the insult, endothelium is also activated and is thought to direct and modulate the inflammatory response.4 During severe inflammation, such as seen in sepsis, the activation of an inflammatory cascade by the pathogen (Figure 1) can lead to auto-amplifying cytokine production, the cytokine storm. Cytokines are a broad category of relatively small proteins (<40 kDa) (interleukins, chemokines, interferons, tumour necrosis factor, and growth factors6) produced and released predominantly by immune cells.7 Infection leads to the activation of the cytokine network, which is comprised of pro-inflammatory cytokines and anti-inflammatory cytokines. The balance between these counter-regulatory pathways eventually determines the net inflammatory activity of the cytokine network.
Figure 1.

Inflammatory cascade in sepsis. Bacterial components activate both immune cells and endothelium inducing cytokine production, which is self-perpetuating. Endothelial cells become activated and express adhesion molecules, to which immune cells bind. This initiates the process of transmigration of immune cells to the site of injury. ROS secreted by immune cells and endothelium further augment the inflammatory response. The combination of these insults leads to shedding of glycocalyx, induction of adhesion molecules, increased endothelial permeability, and endothelial apoptosis. Chemokines secreted by immune cells and endothelium recruit immune cells from the bone marrow. The shift in the eNOS/iNOS balance results in excess NO synthesis and vasodilation.
NF-κB plays a crucial role in the cell (both inflammatory cells and ECs) response to cytokines or bacterial cell wall components [i.e. lipopolysaccharide (LPS)] (Figure 2).8 LPS forms a complex with LPS-binding protein, MD-2, toll-like receptor-4 (TLR-4), and CD14, further initiating intracellular signalling.9 The downstream pathways can be crudely divided in two competitive pathways: TLR4/TRIF/IRF3 and TLR4/MyD88/NF-κB. The TLR4/TRIF/IRF3 pathway involves activation of TRIF, internalization of the TLR4/TRIF complex within endosomes with subsequent activation of interferon regulatory transcription factor-3 (IRF3) and interferon production. At the same time, activation of the TLR4/MyD88/NF-κB pathway leads to phosphorylation of MyD88 and interleukin-1 receptor-associated kinases 1 and 4 (IRAK1 and IRAK4). IRAKs in turn phosphorylate TNF receptor-associated factor 6 (TRAF6), which promotes degradation of IκB and nuclear translocation of NF-κB. TRAF6 is also thought to activate mitogen-activated protein kinases (MAPKs), ultimately resulting in activation of activator protein-1 (AP-1).9 Inflammatory cytokines, such as TNF-α, can activate similar pathways resulting in nuclear translocation of NF-κB, further increasing cytokine production.10 Although immune cells are responsible for most cytokine production during sepsis, ECs are not only the target of cytokines, but are also, via similar pathways,11 able to secrete such cytokines as IL-1β, IL-6 and interferon. While the exact role of endothelial-derived cytokines is unclear, current thought is that ECs contribute to ramping up and modulating the inflammatory cascade and play an important role in activation and fine tuning of the local immune response.12
Figure 2.

Examples of inflammatory pathways within endothelial cells during sepsis (TLR4 and TNF-α). LPS and cytokines trigger intracellular pathways that ultimately lead to the activation of number of transcription factors (NF-κB, AP-1, and IRF3), resulting in increased cytokine production and adhesion molecule expression. These pathways also likely contribute to mitochondrial dysfunction and ROS production (see text for details).
With growing evidence on the important role of the endothelium in sepsis development, much attention has been devoted to its potential as a target for therapeutic intervention. Initially, multiple studies focused on quenching the bacterial components, thus preventing their deleterious effect on the endothelium. Due to its well-described toxic effects, quenching LPS from the membrane of the Gram-negative bacteria with various lipophilic and amphiphilic cations as well as zwitterions has been successfully explored in animal models of LPS-induced endotoxaemia and Escherichia coli sepsis.13,14 However, due to the fact that most of these compounds are cationic amphipaths and potentially can exhibit membrane-perturbing activity, the implementation of most of these quenchers in clinical practice is limited.13 Among drugs with known safety profile that are already being used in clinical practice, colistin showed promising anti-endotoxin effects in vitro15 as well as in a randomized clinical trial investigating its anti-inflammatory effect in LPS-induced endotoxaemia in healthy volunteers.16
As noted above, LPS exerts its effect on endothelium via binding to TLR4; thus, intervening in LPS binding to TLR4 as well as targeting the downstream signalling pathways is another potential treatment for Gram-negative sepsis.17 While designed to be protective by its nature, in cases when the stimulus becomes overwhelming, the TLR4 pathway can contribute to the pathologic downward spiral of sepsis. Therefore, several approaches were suggested for intervening in this pathway. First, a number of TLR4-antagonists were tested. For instance, Eritoran, a structural analogue of lipid A that does not exhibit TLR4-agonistic effects, competitively binds to TLR4-MD2 and prevents LPS from initiating an inflammatory response (i.e. it was shown to block NF-κB activation and TNF-α and IL-6 production in animal and human models of endotoxaemia).18 Another direct TLR4 antagonist, TAK-242, binds directly to Cys747 in the intracellular TLR4 domain and disrupts TLR4 interactions with TIRAP and TRAM.19 While showing promising results in animal models of sepsis,20 it failed, however, to be effective in clinical trials.21 A number of existing agents were shown to alter the levels of TLR4 expression (i.e. chloroquine, ketamine, nicotine, opioids, statins, vitamin D3, lidocaine, glycine, and proton pump inhibitors), with some of them showing auspicious results in animal studies. Randomized controlled trials are needed to evaluate the efficacy of these compounds in clinical practice.22 Finally, an immune cell-derived protein that is highly elevated in sepsis, resistin, was found also to prevent LPS binding to the TLR4 receptor and was suggested as a potential therapeutic modality, but further studies are needed to support this possibility.23
Apart from blocking the TLR4 receptor itself, attempts were made to intervene with various downstream targets. For example, ARF6, a small ATPase activated by the MyD88–ARNO interaction, was shown to induce vascular leak in septicaemia24 and blocking ARF6 with a peptide reversed this effect, improving survival in endotoxic shock.25 Another study attempted to assess vascular permeability in response to LPS after inhibition of either MyD88 or TRIF and demonstrated that blocking TRIF, but not MyD88, had therapeutic potential.26 A number of compounds used in traditional Chinese medicine (such as Houttuynia, tanshinones, Emodin, Ugonin M, LianQinJieDu, Astragalus membranaceus, and Salvia miltiorrhiza) were shown to intervene at various stages of TLR4 pathway and therefore were suggested as potential adjunct therapies for the treatment of Gram-negative sepsis; however, randomized clinical trials are needed to assess their efficacy in clinical setting.17,27,28
3. Endothelium and leucocyte adhesion and transmigration
In response to inflammatory cytokines (TNF-α and IL-1β in particular), the expression of adhesion molecules [selectins, integrins, and members of an immunoglobulin superfamily known as intercellular adhesion molecules (ICAMs) and vascular cell adhesion molecule-1 (VCAM-1)] on the surface of ECs increases dramatically (Figure 1).29 In the initial phase, the Sialyl-Lewis carbohydrate ligands found on leucocytes loosely attach to E-selectin and P-selectin, which allows them to roll on the endothelium. In the second phase, the rolling leucocytes are activated by chemokines locally released by macrophages and ECs to express integrins on their surface, which permits firmer adhesion to ICAM-1 and VCAM-1 and initiates their transendothelial migration into the injured tissues.30 Chemokines that are bound to the endothelium via heparan sulphates form a chemotactic gradient directing further migration of the leucocytes.31 Moreover, the gradient of chemokines (such as CXCL8, CXCL1, CXCL2, and CXCL5) produced by the immune cells and ECs promotes recruitment of the neutrophils from bone marrow reserves and enhances neutrophil adhesion.31 P-selectin on ECs not only captures leucocytes and promotes their rolling but also activates integrins through P-selectin glycoprotein ligand-1 (PSGL-1) and induces further leucocyte activation.32 Other molecules that have been shown to be necessary for transmigration of leucocytes across the endothelial lining are platelet EC adhesion molecule-1 (PECAM-1) and CD99,33,34 with blockade of PECAM-1 function severely impairing transendothelial migration.33
Meanwhile, as inflammatory responses progress, soluble isoforms of the leucocyte recruitment adhesion molecules are shed from cell surfaces and accumulate within the circulating blood plasma.35 Although still a matter of controversy, increasing evidence suggests that shedding serves regulatory roles to dampen inflammation (and specifically to reduce leucocyte–endothelial interactions) and protect the host from excessive collateral damage.35
Measurement of these soluble adhesion molecules has also been proposed as a diagnostic tool for early detection of sepsis as well as severity and outcome prediction. Serum concentrations of adhesion molecules such as E-selectin, P-selectin, ICAM-1, and VCAM-1 increase during sepsis36 and correlate with sepsis severity (as assessed by the Simplified Acute Physiology Score and the number of organ failures) and mortality.37 E-selectin levels rise significantly with the onset of sepsis-related organ dysfunction and decrease after organ dysfunction has resolved.38 In particular, E-selectin was elevated in patients with positive blood cultures indicating that bacteraemia resulted in pronounced endothelial damage and greater shedding of E-selectin.39 When compared to such classical markers of bacterial infection as procalcitonin, measurement of either circulating E- or P-selectin levels had higher sensitivity and specificity for predicting future sepsis development in ICU patients.40 In contrast, while soluble ICAM-1 levels were significantly elevated in patients with sepsis, they also rose (although to a lesser degree) in patients with systemic inflammation without sepsis.41 Nevertheless, soluble ICAM-1 levels directly correlated with severity of sepsis and organ dysfunction.41 Moreover, in patients with multiple organ dysfunction who did not have sepsis, elevation of soluble ICAM-1 levels was much less pronounced.42 While quite sensitive to organ dysfunction and severity of sepsis, a moderate rise in levels of circulating adhesion molecules is also seen in other conditions that are associated with vascular damage (such as trauma or cardiopulmonary bypass), so their use as a biomarker must be interpreted with care.43,44
While adhesion molecule expression is crucial for recruiting immune cells to battle the infection, targeting adhesion molecules in treatment of sepsis remains controversial. Interestingly, one study demonstrated that in a mouse model of polymicrobial sepsis, the use of antibodies against ICAM resulted in an increased recruitment of neutrophils to the site of infection with a concurrent decrease of infiltration in other organs (such as lung, thymus, and spleen).45 Other studies explored the idea of fusing compounds with antibodies against adhesion molecules for endothelium-specific delivery of certain drugs.46,47 Finally, one of the proposed mechanisms of the beneficial effect of inotropes in sepsis is their ability to decrease adhesion molecule expression on the endothelium,48 resulting in reduced transendothelial neutrophil migration.49 Overall, despite the fact that the role of endothelial adhesion molecules in sepsis is undeniable, surprisingly few studies have focused on targeting them in treatment of sepsis.
4. Endothelial permeability in sepsis
A highly selective endothelial barrier is essential to maintain tissue fluid homeostasis and to support normal organ function.50 A main feature of the endothelium in sepsis is an increased permeability or loss of barrier function, resulting in a shift of circulating elements and tissue oedema (Figure 3).51
Figure 3.

Endothelial permeability in sepsis. ROS and bacterial components (i.e. LPS) damage the glycocalyx. LPS and inflammatory cytokines result in disruption of tight junctions (TJ), adherence junctions (AJ), and gap junctions (GJ) via activation of TNF-α and Ang2 pathways. The above effects increase endothelial permeability (see text for details).
Vascular endothelial (VE)-cadherin is the major component of endothelial adherens junctions—tightly regulated protein complexes that join adjacent ECs and prevent leucocyte migration and vascular leak. Inflammatory mediators suppress cAMP/Rac1 signalling, promote Rho activity and activate kinases such as Src and Pyk2,52 which results in VE-cadherin phosphorylation, dissociation from p120 catenin and induces its endocytosis.53 Endocytosis of VE-cadherin by itself is sufficient to induce gaps between ECs, resulting in increased permeability.54 Similarly to adherens junctions, several studies reported disruption of endothelial tight junctions in sepsis as well with reduction of protein levels of occludin and zonula occludens-1.55,56 TNF-α, one of the main cytokines in sepsis as noted above, was shown to cause disruption of claudin-5 at cell–cell junctions of ECs through activation of the NF-κB pathway.57 Another important player in maintenance of the endothelial barrier is gap junctions, formed by connexins, which allow cell–cell flux of small molecules and solutes.58 Inhibition of connexins prevents the increase in endothelial permeability observed in an experimental model of lung injury.58
Several other mechanisms involved in maintenance of the endothelial barrier have been suggested. For example, we showed that decreased expression of DNA polymerase-δ interacting protein 2 (Poldip2) maintains the endothelial barrier in sepsis,59,60 although more work needs to be done to fully define the mechanism. One of the most commonly implicated pathways in vascular permeability is the Ang/Tie2 pathway. Angiopoetin2 (Ang2) is highly up-regulated in the serum of patients with sepsis, correlating with disease severity.61 Current evidence suggests a crucial role of Ang2 in vascular permeability in sepsis. Ang2 expression by ECs is increased upon stimulation by inflammatory cytokines, resulting in disruption of Ang1–Ang2 equilibrium and increasing vascular permeability.62 Endothelial-specific Ang2 overexpression in mice leads to the development of haemodynamic changes resembling sepsis.63 These effects can be abrogated by shifting the Ang1/Ang2 balance towards a normal ratio by injecting mice with Angpt1‐ or Pdgfb‐encoding adeno‐associated virus. Moreover, Ang-2 neutralizing antibody decreases LPS-induced mortality,63 and mice with heterozygous deletion of Ang2 have reduced VCAM-1 expression, inflammatory cell infiltration, and improved survival in sepsis.61
Both Ang1 and Ang2 are known to bind Tie2, a transmembrane endothelial tyrosine kinase receptor.64 Ang1 acts as an agonist, phosphorylating Tie2, and, through Akt activation, inhibiting Foxo1,65 ultimately promoting EC survival and migration and inhibiting vascular leakage.66 Ang2, on the other hand, acts as a Tie2 antagonist by blocking the stabilizing action of Ang1.64 Ang2 exposure results in a formation of a complex between Tie2, αvβ3 integrin, and focal adhesion kinase, leading to focal adhesion kinase phosphorylation, αvβ3 integrin internalization67 and ultimately EC barrier destabilization. Ang2 was also shown to induce VE-cadherin phosphorylation, causing disruption of adherence junctions, although the exact molecular pathway remains unclear.68 Activation of Tie2 with a specific ligand or introducing a stable Ang1 variant improves survival in experimental models of sepsis.69,70 To date most work focused on the antagonistic effects of Ang2 on the Tie2 receptor; recent studies, however, have shown that Ang2 also binds β1 integrin thus destabilizing endothelium even further.71 Moreover, targeting β1 integrin either by heterozygous deletion or inhibitory antibodies exhibited a protective effect on endotoxaemia-induced VE-cadherin redistribution and endothelial permeability.72 These studies, taken together, indicate an important role of the Ang1/Ang2 balance in vascular permeability in sepsis and point towards potential therapeutics to modulate this equilibrium, preventing the downward spiral that is characteristic of sepsis.
Evaluating Ang1/Ang2 equilibrium by measuring levels of soluble Ang2 in the serum of patients with sepsis was investigated as a predictor of outcomes. A satellite study of the ProCESS trial demonstrated significantly higher levels of serum Ang-2 in patients who did not survive sepsis.73 Moreover, another study investigated the predictive value of circulating levels of Ang-2 together with a few other factors (VEGF, TM, vWF) for acute respiratory distress syndrome (ARDS) development in sepsis, which further supported an important role of vascular permeability in general, and Ang2 in particular, in sepsis-induced lung injury.74 Finally, higher levels of soluble Ang-2 were associated with higher likelihood of disseminated intravascular coagulation (DIC), one of the most morbid complications of sepsis.75
Despite the undeniable importance of Ang1/Ang2 equilibrium, only a handful of studies is available focusing on altering this balance in treatment of sepsis. Various compounds stimulating Tie2 (thus mimicking the effect of Ang1) were reported to improve mortality in murine models of sepsis.69,76 However, the short half-life and high rate of non-specific binding of these compounds limits their use in systemic disease like sepsis in larger organisms.77 On the other hand, antibodies and small-interfering RNAs against Ang2 were also shown to delay septic progression in experimental sepsis models; their efficacy, however, was variable.78,79 Finally, a special Tie2-agonistic antibody that also oligomerized Ang2 was able to enhance the effects of Tie2 stimulation by concomitant Ang2 inhibition in vitro and in sepsis mouse model.80 While promising, these results also point towards the need for further studies focusing on possible therapeutics altering Ang1/Ang2 equilibrium in sepsis.
5. Glycocalyx in sepsis
The endothelial glycocalyx has been recognized as a critical regulator of barrier integrity in the endothelium. In a unique position between the blood and vessel wall, the endothelial glycocalyx consists of a membrane-bound negatively charged network of proteoglycans, glycoproteins, glycolipids, glycosaminoglycans (i.e. heparan sulphate), and adherent plasma proteins.81 It actively regulates barrier function via mechanotransduction, and its alteration may lead to augmentation in endothelial hydraulic conductivity and subsequent formation of pulmonary oedema.82
Inflammation in general and sepsis in particular result in changes in the glycocalyx, leading to endothelial damage and microvascular dysfunction.83 Thus, the glycocalyx is an intriguing candidate for assessment of the degree of endothelial damage in sepsis. However, studies focusing on the components of glycocalyx in sepsis remain limited. Syndecan-1, a transmembrane (type I) heparan sulphate proteoglycan, for example was shown to increase in conditions resulting in endothelial damage, including sepsis.84 Plasma levels of heparan sulphate were significantly higher in patients with septic shock85 with markedly higher levels in non-survivors.86 However, heparan sulphate levels were also significantly elevated in patients who underwent neurosurgery and did not have sepsis.86 One of the most promising glycocalyx markers in sepsis is endocan (a soluble dermatan sulphate proteoglycan), which was shown to increase after LPS administration in healthy volunteers,87 and in patients with sepsis was predictive of later development of sepsis-induced lung injury and mortality.88
Shedding of the glycocalyx exposes ECs for leucocyte and platelet adhesion, augmenting inflammation and activating the clotting cascade.89 Several studies focused on maintaining the integrity of glycocalyx during sepsis. The effect of fluid resuscitation, a mainstay therapy in septic shock, on glycocalyx has been studied quite extensively. It was suggested that hypervolemia can be associated with increased glycocalyx degradation in sepsis;90 however, other groups observed no difference in circulating glycocalyx components between patients receiving low or large volumes of fluid resuscitation.91 Based on lower circulating components of glycocalyx in animal studies and a few clinical studies, albumin and fresh frozen plasma were offered as glycocalyx-protective options for intravenous solutions.92,93 Whether or not the observed effect can translate into improved mortality in sepsis is yet to be determined,94 with crystalloids (such as normal saline or Ringer’s lactate solution) currently remaining a preferred means for the resuscitation in septic shock. Such widely used drugs as dexamethasone and doxycycline can exhibit their beneficial effect through suppression of metalloproteinases and decreased shedding of components of the glycocalyx.95 Heparin is also postulated to protect the glycocalyx via inhibition of heparanase, thus decreasing activity of metalloproteinases.96 Finally, inhibition of Ang2 has also been suggested to maintain glycocalyx integrity.80 While it is accepted that disruption of the glycocalyx adds to the organism-wide insult in sepsis, it remains unclear if maintaining or restoring the glycocalyx by itself would have mortality benefit. The issue is complicated by the interconnection of the cascades involved, and the challenge of designing a glycocalyx-specific intervention.
6. Endothelium and the clotting cascade in sepsis
Disseminated intravascular coagulation is one of the gravest complications of sepsis and is associated with extremely high mortality.97 While the exact mechanism and the order of triggering events remains unknown, the endothelium is thought to play a major role in this process (Figure 4). One of the suggested mechanisms is disruption of the EC membrane by membrane attack complex,98 which further augments inflammation and activates the microthrombotic pathway. Activation of the microthrombotic pathway mediates platelet activation and exocytosis of unusually large von Willebrand factor multimers from ECs and initiates microthrombogenesis.98 Under normal conditions, these von Willebrand factor multimers are rapidly cleaved to less active forms by a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13). During overwhelming inflammation in sepsis, however, inflammatory mediators such as IL-6, plasma-free haemoglobin, VWF proteolytic fragments, Shiga toxin, and neutralizing autoantibodies can inactivate ADAMTS-13.99,100 ADAMTS-13 can also be inhibited by plasmin, thrombin, products of activated coagulation, granulocyte elastase released by activated neutrophils as well as neutrophil-derived reactive oxygen species (ROS). Together, these events lead to an acquired ADAMTS-13 deficiency and increased risk of DIC. Finally, in sepsis the endothelium releases increased amounts of plasminogen activator inhibitor-1 (PAI-1), thus suppressing the fibrinolytic pathway. All of this creates an imbalance between prothrombotic and antifibrinolytic pathways, leading to the dissemination of fibrin-rich microvascular thrombi as observed in DIC.101 This results in organism-wide microvascular thrombus formation, depletion of the clotting factors, and bleeding as the most classical presentation of DIC.
Figure 4.

Endothelium and coagulation cascade in sepsis. When endothelial integrity is compromised by membrane attack complexes and endothelial apoptosis, a prothrombotic state ensues: endothelial cells express adhesion molecules and platelets become activated. Activated platelets release a number of active compounds contributing to the inflammatory cascade. Bacterial components, ROS, and cytokines lead to acquired ADAMTS13 deficiency and accumulation of vWF multimers that are secreted from the activated endothelium. Activated endothelium also secretes PAI-1, further contributing to a prothrombotic state (see text for details).
While ECs undoubtedly orchestrate the clotting cascade and platelet activation and aggregation, platelets themselves can secrete a number of cytokines upon activation.102 Most of these cytokines stimulate immune cells and promote immune-cell adhesion to the endothelium.102 A few of them, however, have very distinct effects on endothelium itself. CD40L, for example interacts with CD40 on ECs and promotes chemokine secretion and expression of adhesion molecules.103 Activated platelets also release IL-1(α and/or β) and induce CCL2 (a chemokine) secretion and ICAM-1 expression in ECs.104
Apart from classic tests, such as D-dimer and fibrinogen for establishing a diagnosis of DIC, other players of the clotting cascade have been also suggested for prediction of sepsis severity, DIC development, and mortality. For example, in the aforementioned satellite study of the ProCESS trial, the levels of haemostasis factors, such as VEGF, thrombomodulin (TM), and tissue plasminogen activator, in sepsis were evaluated and circulating levels of all three factors were significantly higher in non-survivors.73 Higher PAI-1 levels were also associated with increased mortality and higher rates of DIC and end organ failure.105
Due to the enormous mortality associated with DIC,97 attempts were made to modulate coagulation in order to treat sepsis and prevent sepsis-induced DIC. The possibility of heparin inducing further bleeding in severe sepsis as well as negative results from a few animal studies106 limited enthusiasm for its use in sepsis. Low-dose heparin, however, showed promising results in a few randomized clinical trials as well as after meta-analysis, with reduced 28-day mortality in severe sepsis and no increase in bleeding rates.107 Inhibition of thrombin (by thrombin inhibitors, decrease in thrombin generation, binding thrombin with AT III, or thrombin degradation by protein C or TM) has shown encouraging results in animal studies, indicating a possible effect on both coagulation and inflammatory pathways within the endothelium.108,109 Follow-up clinical studies, however, showed variable results. Further studies are therefore needed to fully evaluate their role in the treatment of sepsis. Tissue factor pathway inhibitor (TFPI) suppresses the primary steps of thrombin generation and exhibits its anti-inflammatory effect through suppression of thrombin binding to protease-activated receptor-1.110 Use of recombinant TFPI was suggested for treatment of sepsis; however, a few large randomized clinical trials failed to show any improvement in mortality.111,112 Another thrombin antagonist with anti-inflammatory properties is ATIII, which apart from directly inhibiting a number of serine proteases and binding thrombin, can also induce release of prostacyclin, suppress P-selectin, and decrease the expression of inflammatory cytokines.109,113,114 The results of clinical trials with ATIII were overall positive, with most reporting a decrease in 28-day mortality. The effect, however, varied with different doses, concomitant heparin administration, and age of the patient.115,116
Activated protein C inactivates factors Va and VIIIa, thus limiting thrombin generation, and is involved in lysis of the thrombin–TM complex. It also has been shown to have anti-inflammatory qualities by suppressing pro-inflammatory cytokines in activated leucocytes, exhibiting antioxidant properties, displaying antiapoptotic activity, and stabilizing the endothelial barrier.117 The results of the randomized clinical trials with recombinant activated protein C, however, were inconsistent, with some studies showing no improvement in mortality.118,119 While the consensus regarding its efficacy has not been reached, the production of recombinant activated protein C was nevertheless stopped.
Thrombomodulin binds to thrombin and activated protein C. It also has some anti-inflammatory effect by interfering with complement activation and inhibition of leucocyte–endothelial interaction.120 Initial clinical studies of recombinant TM showed improved outcomes in patients with DIC and larger studies are currently underway.121,122 Thus, intervening at various stages of thrombin synthesis and degradation harbours promising potential in treatment of sepsis and sepsis-induced DIC. One has to realize, however, that while the results of many of the clinical trials investigating thrombin inhibitors in sepsis are optimistic, differences in inclusion criteria and reported bleeding events should be noted, which might account for some of the inconsistencies in results.
7. Endothelium and ROS in sepsis
During sepsis, activated immune cells release reactive molecules that are designed to fight pathogens, but on the other hand can cause tissue damage.123 ROS can directly attack ECs, resulting in increased vascular permeability, worsening hypotension, and decreased colloid osmotic pressure of the plasma. Furthermore, they alter oxygen consumption by the tissues, accelerating organ failure.124 Published studies provide substantial evidence that ROS can damage the EC glycocalyx125 and have profound negative effects on endothelial barrier function,126 thereby promoting neutrophil recruitment and trafficking. Both in vitro and in vivo exposure of ECs to ROS results in EC cytoskeletal remodelling and an up-regulated expression of ICAM-1, PECAM-1, VCAM-1, and P-selectin and a subsequent increase of neutrophil adhesion to ECs.127 Alterations in calcium homeostasis and activation of protein kinase C, p38MAPK, and phosphodiesterases have been proposed as mediators of the ROS-induced increase in endothelial permeability.126,128,129 ROS have also been implicated in disruption of tight junction proteins: H2O2 caused redistribution of occludin on the cell surface, limiting its association with zonula occludens-1 and leading to an increase in endothelial permeability.130
ECs themselves also can produce ROS mediated by the mitochondrial electron transport chain, nicotinamide adenine dinucleotide phosphate hydrogen oxidases (NOXs), especially NOX2 and NOX4, uncoupled endothelial nitric oxide synthase (eNOS), and xanthine oxidase.131 At physiological levels, ROS-induced signalling is necessary for maintaining vascular tone by the endothelium and also facilitates angiogenesis and acute inflammatory responses to fight the invading pathogens.132 In pathological settings like sepsis, however, when the ROS response is overwhelming, reduction of ROS production by inhibition of NOX2, for example was shown to be protective against sepsis-induced organ damage.133–135
A main endogenous vasodilator and antiproliferative agent is nitric oxide (NO), which is synthesized in ECs by eNOS. NO stimulates soluble guanylate cyclase to increase the level of cyclic GMP in smooth muscle cells.136 During sepsis, some of the essential cofactors necessary for eNOS activity, such as tetrahydrobiopterin, are depleted via oxidation, resulting in uncoupling of the enzyme, superoxide anion generation, and reduced NO production by eNOS.125 At the same time, iNOS in immune cells and endothelium becomes activated, providing a large amount of NO (Figure 1). The reaction between NO and superoxide anion results in formation of reactive nitrogen species (RNS), such as peroxynitrite (ONOO−), nitrogen dioxide (•NO2), and dinitrogen trioxide (N2O3), which also contribute to endothelial dysfunction.137 Deleterious cellular effects of both ROS and RNS have been attributed to their role in the oxidation of proteins, lipids, and oxidant-induced DNA damage.
Despite the overwhelming ROS production during sepsis, the utility of ROS measurement in diagnosis of sepsis and mortality prediction remains limited. This may be partly due to the instability of ROS, which reduces our ability to accurately measure them in a clinical setting, and the relative non-specificity of the ROS response. Despite those limitations, one study identified a ROS-specific gene expression signature that was able to predict mortality in patients with sepsis.138 Another study investigated ROS-activatable nanoprobes for early diagnosis and sepsis severity prediction in mouse model of sepsis.139
Multiple approaches, however, have been suggested to halt the chain reaction of ROS production. Indeed, both catalase and superoxide dismutase prevented shedding of the glycocalyx and preserved the endothelial barrier in response to hydrogen peroxide in vitro.140 Interestingly, to date there are no trials investigating the therapeutic potential of these molecules. This might be partly due to the challenges with cell-specific delivery of these relatively large proteins. Those challenges are being addressed in animal studies with the use of SOD conjugated with antibodies targeted to endothelial endosomes141 and the mitochondria-targeted antioxidant, MitoTEMPO.142 Positive results of these studies in animal models of endotoxaemia would need, however, further evaluation before their potential use in clinical practice due to the universal role of ROS in normal physiology.
NO, particularly due to dysregulated iNOS activity, has been implicated in organism-wide vasodilatation and increased vascular permeability in sepsis. Pharmacological blockade of NO production mitigates sepsis-induced hypotension in animal models.143,144 Unfortunately, these studies failed to translate into clinical practice after a large-scale multi-centre randomized clinical trial demonstrated that treatment with NG-monomethyl-l-arginine (L-NMMA) (a non-specific NOS inhibitor) increased mortality in sepsis while raising mean blood pressure and decreasing the vasopressor requirement.145 More targeted approaches by attempting to act more specifically on the eNOS/iNOS balance were suggested. One of the most recent suggestions is BH4, which is a crucial co-factor of NOS. The data on exact function of BH4 in sepsis, however, are conflicting, with some studies showing improved mortality with inhibition of BH4 production146 while others show that addition of BH4 improves microcirculation in sepsis.147 Moreover, tetrahydrobiopterin analogues such as sepiapterin, which reduce superoxide generation via uncoupling of eNOS, were shown to protect the endothelium and organ function.148 Therefore, more detailed studies, focusing particularly on the eNOS/iNOS balance in sepsis, are needed.
Finally, vitamin C, due to its recognized antioxidant effect, has been suggested as an adjunctive therapy in sepsis. Vitamin C scavenges ROS and RNS, and as a result becomes oxidized to ascorbate-free radical, which then dismutates to form dehydroascorbic acid.149 Besides the direct effect on ROS and RNS, ascorbate reduces production of ROS and RNS by preventing NOX activation, decreasing expression of iNOS and increasing NO bioavailability.150,151 By decreasing ROS, vitamin C decreases endothelial permeability.149,152 There are also reports from in vitro studies that vitamin C can bind to alpha-adrenergic receptors and augment the effect of vasopressors.153 In animal models of vascular injury due to endotoxaemia, reperfusion injury, ARDS, and burns, vitamin C administration preserved lung barrier function, prevented oedema formation in burns and decreased ROS production.154,155 These promising experimental data combined with the fact that the levels of vitamin C in critically ill patients are markedly decreased,156 made it a compelling candidate for adjunctive therapy in sepsis to supplement the nutrient that has been lost due to increased demand by tissues suffering from oxidative stress. A small phase I trial including 24 ICU patients with severe sepsis showed that administration of vitamin C decreased the extent of the organ failure.157 Other studies used vitamin C in conjunction with either vitamin E158 or thiamine and hydrocortisone159 and demonstrated decreased vasopressor requirement and mortality. A recent large randomized trial focusing on the effects of vitamin C in septic patients with ARDS showed no improvement in organ dysfunction or inflammatory marker levels. While in that study, the 28-day mortality was significantly lower in vitamin C group compared to the placebo group, the authors point out that per study design, mortality was a secondary outcome, and some internal biases could not be excluded.160 Thus, large randomized multi-centre studies are still needed to establish the utility of vitamin C in treatment of sepsis. The relative safety (apart from possible worsening of kidney failure159) and low cost of this drug make it a very attractive therapy.
8. Endothelial cell apoptosis and regeneration in sepsis
Endothelial cell apoptosis is a highly regulated process.161 LPS from Gram-negative bacteria was shown to induce apoptosis in ECs in vitro; however, the effect appeared to depend on experimental conditions.162 Interestingly, no apoptotic effect on human umbilical vein ECs was observed in vitro at concentrations of LPS matching those in patients’ serum during sepsis.162 Besides the direct effect of pathogen-derived substances and particles, ROS and RNS accumulating during sepsis are also toxic and proapoptotic for the endothelium.123 Endothelial apoptosis results in a loss of normal anticoagulant properties of endothelium. The surface of the apoptotic cell exhibits increased tissue factor procoagulant activity, as well as reduced surface TM, heparan sulphate, and TFPI expression.163 This leads to increased thrombin formation by both adherent and detached apoptotic ECs164 and further augments the inflammatory and clotting cascade. Regeneration of ECs in sepsis is an important component of recovery from sepsis and is regulated via HIF-1α and its downstream target Sox17.165 Thus, both EC apoptosis and regeneration play important roles in pathophysiology of sepsis and targeting them might provide future insights into the development of novel therapeutics. For example, a few small studies have shown that both circulating ECs and circulating endothelial progenitor cells were increased during sepsis, with higher numbers of progenitor cells and lower number of ECs associated with improved survival.166 The relatively low number of both circulating ECs and progenitor cells, however, limits their utility as a diagnostic test.
9. Novel directions
9.1 Endothelium, sepsis, and microRNAs
MicroRNAs (miRs) are non-coding endogenous RNAs that target messenger RNA (mRNA) resulting in translational repression.167 miRs have been implicated in various stages of sepsis development (Table 1). For example, miR-146 has been shown to prevent endothelial activation by inhibiting several inflammatory pathways, such as those regulated by NF-κB, MAPK, and AP-1. miR-146 also targets the RNA-binding protein, human antigen R, thus dampening the pro-inflammatory response.169 In addition to miR-146, the NF-κB pathway in the endothelium is regulated by miR-181b via inhibition of importin-α3, which is essential for NF-κB nuclear translocation. miR-181b also reduces the expression of VCAM-1 and E-selectin in ECs.170 Similar to miR-146 and -181 b, miR-155 was shown to suppress endothelial inflammation by down-regulating NF-κB p65 and adhesion molecule expression in TNFα-stimulated ECs.171 In addition, miR-130b appears to inhibit inflammatory gene expression by down-regulating LPS-induced aberrant activation of ERK signalling.172 A possible role of miRs in maintenance of endothelial barrier via regulation of tight junctions has also recently been suggested.179 For instance, ECs from miR-150 deficient mice demonstrated a persistent increase in Ang2 levels, thus resulting in an irreversible increase in vascular permeability. Restoring miR-150 expression in miR-150 knockout mice prevented the production of Ang2 and preserved vascular barrier function, ultimately reducing mortality from sepsis.173 miR-147b was also associated with maintenance of the endothelial barrier after treatment with LPS via targeting the 3′-UTR of the metalloproteinase ADAM15, which favours the increased endothelial permeability during inflammation and sepsis.174
Table 1.
MicroRNAs with potential roles in the endothelium in sepsis
| Potential therapeutic implication | MicroRNA | Pathway affected | Observed effect in sepsis models |
|---|---|---|---|
| Anti-inflammatory effect | miR-146 | NF-κB, MAPK, and AP-1168 human antigen R169 | Decreases inflammatory response |
| miR-181b | Importin-α3170 | Inhibits nuclear translocation of NF-κB; reduces the expression of VCAM-1 and E-selectin | |
| miR-155 | NF-κB p65171 | Decreased expression of ICAM and VCAM | |
| miR-130b | Tpl2172 | Decreased IL-6 and TNF-α expression | |
| Inhibition of endothelial permeability | miR-150 | Angiopoetin2173 | Decreased vascular permeability |
| miR-147b | ADAM15174 | Preservation of endothelial barrier | |
| DIC | miR-30b | PAI-1175 | Unknown |
| miR-181b | PAI-1176 | Unknown | |
| miR-143/145 | PAI-1177 | Unknown | |
| miR-96 | Aquaporin-5178 | Unknown | |
| miR-330 | Aquaporin-5178 | Unknown |
PAI-1 inhibits fibrinolysis and sustains DIC in septic patients, leading to multiple organ failure.180 PAI-1 levels are elevated in the endothelium during severe sepsis and their levels correlate with disease severity.181 Several miRs, such as miR-30b, miR-181b, and miR-143/145 clusters, were shown to regulate PAI-1 levels.175–177 Moreover, miR-96 and miR-330 have been found to contribute to lung injury in LPS-induced DIC in rat models.178 This growing body of evidence pointing towards the importance of miRs in sepsis can provide insights for the development of novel therapeutic targets for maintenance of endothelial barrier and prevention of inflammatory response dysregulation.
9.2 Microparticles and endothelium in sepsis
The endothelium is not only a victim in sepsis but also an active participant that, via its interaction with other cells and tissues, is capable of modulating the inflammation. Well-orchestrated interactions between immune cells and endothelium ensure appropriate responses at times of infection and inflammation. One mechanism of cell–cell communication that has received a significant amount of attention in recent years is microparticles (MPs).182 MPs are 0.2–2 μm cell membrane-derived particles harbouring phosphatidylserine and tissue factor on the outer leaflet and proteins, mRNAs, and miRNAs on the inside that are capable of promoting coagulation and inflammation. Sepsis-induced microvascular injury during sepsis leads to the release of MPs from ECs, red blood cells, monocytes, and platelets into the systemic circulation.183 Specific receptors (such as Annexin I or CD36) are required for the interaction between MPs and target cells;184,185 however, there may be other receptors that have not yet been described. Apart from their role in promoting coagulation and inflammation, MPs may also have some anti-inflammatory effect, with several studies showing decreased immune-cell activation after incubation with platelet-derived MPs.186
Other investigators have reported an increased number of MPs in patients with septic shock187 as well as in animal models of sepsis.188 Endothelium and leucocyte-derived MPs were one of the first markers to be elevated in DIC, with endothelium-derived CD105-MPs and CD31-MPs strongly associated with early DIC in multivariate analysis.189 Overall, the relative stability of the MPs in the blood stream as well as their signalling potential make them a compelling target for developing novel diagnostic tools and therapeutic agents.
10. Conclusions
The body of evidence supporting a crucial role of the endothelium in sepsis is continuously growing. It is clear now that endothelium is involved in both physiologic and pathologic responses in sepsis and targeting those responses has great potential for developing therapeutics for management of this condition. Significant efforts have been already made to investigate the utility of targeting various endothelial pathways activated in sepsis for diagnosis and treatment of sepsis; more detailed studies are needed, however, to unravel the exact mechanisms as well as to further explore potential clinical applications. While some aspects of endothelial activation in sepsis have received much attention and have even been tested in large randomized clinical trials, the knowledge of other pathways lags behind. Translating experimental data from animal models into clinical practice has been particularly challenging, which might be a reflection of the limitations of animal models or the high variability of clinical scenarios underlying sepsis that are difficult to account for during the design of a randomized clinical trial. Overall, the endothelium undergoes dramatic changes during sepsis and remains one of the most compelling targets for therapeutic development. The modalities currently recommended for management of sepsis, however, do little to protect the endothelium or restore endothelial function, leaving much room for future drug development.
Conflict of interest: None of the authors have any financial interests or connections, direct or indirect, or other situations that might raise the question of bias in the work reported or the conclusions, implications or opinions stated.
Funding
This work was supported by National Institutes of Health grants T32HL007745 to support E.V.D. and HL095070 to K.K.G.
References
- 1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC.. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016;315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The Brussels Sepsis Resolution Global Sepsis Alliance, Brussels, March 20th, 2018. (Meeting).
- 3. Nunnally ME, Patel A.. Sepsis—what’s new in 2019? Curr Opin Anaesthesiol 2019;32:163–168. [DOI] [PubMed] [Google Scholar]
- 4. Hack CE, Zeerleder S.. The endothelium in sepsis: source of and a target for inflammation. Crit Care Med 2001;29:S21–S27. [DOI] [PubMed] [Google Scholar]
- 5. Reinhart K, Bayer O, Brunkhorst F, Meisner M.. Markers of endothelial damage in organ dysfunction and sepsis. Crit Care Med 2002;30:S302–S312. [DOI] [PubMed] [Google Scholar]
- 6. Stober VP, Lim YP, Opal S, Zhuo L, Kimata K, Garantziotis S.. Inter-alpha-inhibitor ameliorates endothelial inflammation in sepsis. Lung 2019;197:361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dinarello CA. Historical insights into cytokines. Eur J Immunol 2007;37:S34–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li P, Allen H, Banerjee S, Seshadri T.. Characterization of mice deficient in interleukin-1 beta converting enzyme. J Cell Biochem 1997;64:27–32. [DOI] [PubMed] [Google Scholar]
- 9. Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F.. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines 2017;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu H, Liu J, Li W, Liu G, Li Z.. LncRNA-HOTAIR promotes TNF-alpha production in cardiomyocytes of LPS-induced sepsis mice by activating NF-kappaB pathway. Biochem Biophys Res Commun 2016;471:240–246. [DOI] [PubMed] [Google Scholar]
- 11. Leng B, Tang F, Lu M, Zhang Z, Wang H, Zhang Y.. Astragaloside IV improves vascular endothelial dysfunction by inhibiting the TLR4/NF-kappaB signaling pathway. Life Sci 2018;209:111–121. [DOI] [PubMed] [Google Scholar]
- 12. Chousterman BG, Swirski FK, Weber GF.. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 2017;39:517–528. [DOI] [PubMed] [Google Scholar]
- 13. Miller KA, Suresh Kumar EV, Wood SJ, Cromer JR, Datta A, David SA.. Lipopolysaccharide sequestrants: structural correlates of activity and toxicity in novel acylhomospermines. J Med Chem 2005;48:2589–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho EJ, Doh KO, Park J, Hyun H, Wilson EM, Snyder PW, Tsifansky MD, Yeo Y.. Zwitterionic chitosan for the systemic treatment of sepsis. Sci Rep 2016;6:29739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soon RL, Velkov T, Chiu F, Thompson PE, Kancharla R, Roberts K, Larson I, Nation RL, Li J.. Design, synthesis, and evaluation of a new fluorescent probe for measuring polymyxin-lipopolysaccharide binding interactions. Anal Biochem 2011;409:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matzneller P, Strommer S, Drucker C, Petroczi K, Schorgenhofer C, Lackner E, Jilma B, Zeitlinger M.. Colistin reduces LPS-triggered inflammation in a human sepsis model in vivo: a randomized controlled trial. Clin Pharmacol Ther 2017;101:773–781. [DOI] [PubMed] [Google Scholar]
- 17. Ding J, Liu Q.. Toll-like receptor 4: a promising therapeutic target for pneumonia caused by Gram-negative bacteria. J Cell Mol Med 2019;23:5868–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, Przetak M, Chow J, Gusovsky F, Christ WJ, Rossignol DP.. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther 2003;304:1093–1102. [DOI] [PubMed] [Google Scholar]
- 19. Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M.. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 2009;157:1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sha T, Iizawa Y, Ii M.. Combination of imipenem and TAK-242, a toll-like receptor 4 signal transduction inhibitor, improves survival in a murine model of polymicrobial sepsis. Shock 2011;35:205–209. [DOI] [PubMed] [Google Scholar]
- 21. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J.. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med 2010;38:1685–1694. [DOI] [PubMed] [Google Scholar]
- 22. Wittebole X, Castanares-Zapatero D, Laterre PF.. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflamm 2010;2010:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jang JC, Li J, Gambini L, Batugedara HM, Sati S, Lazar MA, Fan L, Pellecchia M, Nair MG.. Human resistin protects against endotoxic shock by blocking LPS-TLR4 interaction. Proc Natl Acad Sci USA 2017;114:E10399–E10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, Shi DS, Guo J, Smith MC, Grossmann AH, Thomas KR, Li DY.. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature 2012;492:252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis CT, Zhu W, Gibson CC, Bowman-Kirigin JA, Sorensen L, Ling J, Sun H, Navankasattusas S, Li DY.. ARF6 inhibition stabilizes the vasculature and enhances survival during endotoxic shock. J Immunol 2014;192:6045–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dubbert J, Bowers A, Su Y, McClenahan D.. Effect of TRIF on permeability and apoptosis in bovine microvascular endothelial cells exposed to lipopolysaccharide. Vet J 2013;198:419–423. [DOI] [PubMed] [Google Scholar]
- 27. Gao H, Liu X, Sun W, Kang N, Liu Y, Yang S, Xu QM, Wang C, Chen X.. Total tanshinones exhibits anti-inflammatory effects through blocking TLR4 dimerization via the MyD88 pathway. Cell Death Dis 2017;8:e3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yuan R, Huang L, Du LJ, Feng JF, Li J, Luo YY, Xu QM, Yang SL, Gao H, Feng YL.. Dihydrotanshinone exhibits an anti-inflammatory effect in vitro and in vivo through blocking TLR4 dimerization. Pharmacol Res 2019;142:102–114. [DOI] [PubMed] [Google Scholar]
- 29. Martinez-Mier G, Toledo-Pereyra LH, Ward PA.. Adhesion molecules in liver ischemia and reperfusion. J Surg Res 2000;94:185–194. [DOI] [PubMed] [Google Scholar]
- 30. Zhong L, Simard MJ, Huot J.. Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation. FASEB J 2018;32:4070–4084. [DOI] [PubMed] [Google Scholar]
- 31. Kolaczkowska E, Kubes P.. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 32. Mestas J, Ley K.. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med 2008;18:228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muller WA, Randolph GJ.. Migration of leukocytes across endothelium and beyond: molecules involved in the transmigration and fate of monocytes. J Leukoc Biol 1999;66:698–704. [DOI] [PubMed] [Google Scholar]
- 34. Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA.. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 2002;3:143–150. [DOI] [PubMed] [Google Scholar]
- 35. Garton KJ, Gough PJ, Raines EW.. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol 2006;79:1105–1116. [DOI] [PubMed] [Google Scholar]
- 36. Kjaergaard AG, Dige A, Nielsen JS, Tonnesen E, Krog J.. The use of the soluble adhesion molecules sE-selectin, sICAM-1, sVCAM-1, sPECAM-1 and their ligands CD11a and CD49d as diagnostic and prognostic biomarkers in septic and critically ill non-septic ICU patients. APMIS 2016;124:846–855. [DOI] [PubMed] [Google Scholar]
- 37. Duman A, Turkdogan KA, Avcil M, Yenisey C, Ture M, Akoz A, Dagli B, Kapci M, Orun S.. The predictive value of the inflammatory markers P-selectin and MCP1 in determining the length of stay and 30-day survival in the differentiation of sepsis patients. J Pak Med Assoc 2018;68:1321–1326. [PubMed] [Google Scholar]
- 38. Cowley HC, Heney D, Gearing AJ, Hemingway I, Webster NR.. Increased circulating adhesion molecule concentrations in patients with the systemic inflammatory response syndrome: a prospective cohort study. Crit Care Med 1994;22:651–657. [DOI] [PubMed] [Google Scholar]
- 39. Cummings CJ, Sessler CN, Beall LD, Fisher BJ, Best AM, Fowler AA III. Soluble E-selectin levels in sepsis and critical illness. Correlation with infection and hemodynamic dysfunction. Am J Respir Crit Care Med 1997;156:431–437. [DOI] [PubMed] [Google Scholar]
- 40. Vassiliou AG, Mastora Z, Orfanos SE, Jahaj E, Maniatis NA, Koutsoukou A, Armaganidis A, Kotanidou A.. Elevated biomarkers of endothelial dysfunction/activation at ICU admission are associated with sepsis development. Cytokine 2014;69:240–247. [DOI] [PubMed] [Google Scholar]
- 41. Sessler CN, Windsor AC, Schwartz M, Watson L, Fisher BJ, Sugerman HJ, Fowler AA III.. Circulating ICAM-1 is increased in septic shock. Am J Respir Crit Care Med 1995;151:1420–1427. [DOI] [PubMed] [Google Scholar]
- 42. Endo S, Inada K, Kasai T, Takakuwa T, Yamada Y, Koike S, Wakabayashi G, Niimi M, Taniguchi S, Yoshida M.. Levels of soluble adhesion molecules and cytokines in patients with septic multiple organ failure. J Inflamm 1995;46:212–219. [PubMed] [Google Scholar]
- 43. Seekamp A, Jochum M, Ziegler M, van Griensven M, Martin M, Regel G.. Cytokines and adhesion molecules in elective and accidental trauma-related ischemia/reperfusion. J Trauma 1998;44:874–882. [DOI] [PubMed] [Google Scholar]
- 44. Boldt J, Kumle B, Papsdorf M, Hempelmann G.. Are circulating adhesion molecules specifically changed in cardiac surgical patients? Ann Thorac Surg 1998;65:608–614. [DOI] [PubMed] [Google Scholar]
- 45. Zhao YJ, Yi WJ, Wan XJ, Wang J, Tao TZ, Li JB, Wang JF, Deng XM.. Blockade of ICAM-1 improves the outcome of polymicrobial sepsis via modulating neutrophil migration and reversing immunosuppression. Mediators Inflamm 2014;2014:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li R, Kowalski PS, Morselt HWM, Schepel I, Jongman RM, Aslan A, Ruiters MHJ, Zijlstra JG, Molema G, van Meurs M, Kamps J.. Endothelium-targeted delivery of dexamethasone by anti-VCAM-1 SAINT-O-Somes in mouse endotoxemia. PLoS One 2018;13:e0196976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferrer MC, Shuvaev VV, Zern BJ, Composto RJ, Muzykantov VR, Eckmann DM.. ICAM-1 targeted nanogels loaded with dexamethasone alleviate pulmonary inflammation. PLoS One 2014;9:e102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fortenberry JD, Huber AR, Owens ML.. Inotropes inhibit endothelial cell surface adhesion molecules induced by interleukin-1beta. Crit Care Med 1997;25:303–308. [DOI] [PubMed] [Google Scholar]
- 49. Sookhai S, Wang JH, Winter D, Power C, Kirwan W, Redmond HP.. Dopamine attenuates the chemoattractant effect of interleukin-8: a novel role in the systemic inflammatory response syndrome. Shock 2000;14:295–299. [DOI] [PubMed] [Google Scholar]
- 50. Kasa A, Csortos C, Verin AD.. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015;3:e974448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ferro T, Neumann P, Gertzberg N, Clements R, Johnson A.. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol 2000;278:L1107–L1117. [DOI] [PubMed] [Google Scholar]
- 52. Allingham MJ, van Buul JD, Burridge K.. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 2007;179:4053–4064. [DOI] [PubMed] [Google Scholar]
- 53. Radeva MY, Waschke J.. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol 2018;222:e12860. [DOI] [PubMed] [Google Scholar]
- 54. Lee WL, Slutsky AS.. Sepsis and endothelial permeability. N Engl J Med 2010;363:689–691. [DOI] [PubMed] [Google Scholar]
- 55. He Y, Yuan X, Zuo H, Sun Y, Feng A.. Berberine exerts a protective effect on gut-vascular barrier via the modulation of the Wnt/beta-catenin signaling pathway during sepsis. Cell Physiol Biochem 2018;49:1342–1351. [DOI] [PubMed] [Google Scholar]
- 56. Ni J, Lin M, Li JY, Guo J, Zhou Y, Hong J, Zhao G, Lu Z.. Gas6 attenuates sepsis-induced tight junction injury and vascular endothelial hyperpermeability via the Axl/NF-kappaB signaling pathway. Front Pharmacol 2019;10:662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Clark PR, Kim RK, Pober JS, Kluger MS.. Tumor necrosis factor disrupts claudin-5 endothelial tight junction barriers in two distinct NF-kappaB-dependent phases. PLoS One 2015;10:e0120075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parthasarathi K. Endothelial connexin43 mediates acid-induced increases in pulmonary microvascular permeability. Am J Physiol Lung Cell Mol Physiol 2012;303:L33–L42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Forrester SJ, Xu Q, Kikuchi DS, Okwan-Duodu D, Campos AC, Faidley EA, Zhang G, Lassegue B, Sadikot RT, Griendling KK, Hernandes MS.. Poldip2 deficiency protects against lung edema and vascular inflammation in a model of acute respiratory distress syndrome. Clin Sci 2019;133:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hernandes MS, Lassegue B, Hilenski LL, Adams J, Gao N, Kuan CY, Sun YY, Cheng L, Kikuchi DS, Yepes M, Griendling KK.. Polymerase delta-interacting protein 2 deficiency protects against blood-brain barrier permeability in the ischemic brain. J Neuroinflammation 2018;15:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, Karumanchi SA, Shapiro NI, Parikh SM.. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med 2012;40:3034–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leligdowicz A, Richard-Greenblatt M, Wright J, Crowley VM, Kain KC.. Endothelial activation: the Ang/Tie axis in sepsis. Front Immunol 2018;9:838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, Rohwedder I, Hinkel R, Gross L, Lee S, Hu J, Soehnlein O, Franz WM, Sperandio M, Pohl U, Thomas M, Weber C, Augustin HG, Fassler R, Deutsch U, Kupatt C.. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest 2013;123:3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van der Heijden M, van Nieuw Amerongen GP, Chedamni S, van Hinsbergh VW, Johan Groeneveld AB.. The angiopoietin-Tie2 system as a therapeutic target in sepsis and acute lung injury. Expert Opin Ther Targets 2009;13:39–53. [DOI] [PubMed] [Google Scholar]
- 65. Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, Rudge JS.. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev 2004;18:1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fukuhara S, Sako K, Noda K, Zhang J, Minami M, Mochizuki N.. Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol Histopathol 2010;25:387–396. [DOI] [PubMed] [Google Scholar]
- 67. Thomas M, Felcht M, Kruse K, Kretschmer S, Deppermann C, Biesdorf A, Rohr K, Benest AV, Fiedler U, Augustin HG.. Angiopoietin-2 stimulation of endothelial cells induces alphavbeta3 integrin internalization and degradation. J Biol Chem 2010;285:23842–23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zheng W, Nurmi H, Appak S, Sabine A, Bovay E, Korhonen EA, Orsenigo F, Lohela M, D’Amico G, Holopainen T, Leow CC, Dejana E, Petrova TV, Augustin HG, Alitalo K.. Angiopoietin 2 regulates the transformation and integrity of lymphatic endothelial cell junctions. Genes Dev 2014;28:1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, Bockmeyer CL, Parikh SM, Pavenstadt H, Haller H, Shushakova N.. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care 2011;15:R261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alfieri A, Watson JJ, Kammerer RA, Tasab M, Progias P, Reeves K, Brown NJ, Brookes ZL.. Angiopoietin-1 variant reduces LPS-induced microvascular dysfunction in a murine model of sepsis. Crit Care 2012;16:R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, Eklund L, Ivaska J, Alitalo K, Saharinen P.. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun 2015;6:5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hakanpaa L, Kiss EA, Jacquemet G, Miinalainen I, Lerche M, Guzman C, Mervaala E, Eklund L, Ivaska J, Saharinen P.. Targeting beta1-integrin inhibits vascular leakage in endotoxemia. Proc Natl Acad Sci USA 2018;115:E6467–E6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hou PC, Filbin MR, Wang H, Ngo L, Huang DT, Aird WC, Yealy DM, Angus DC, Kellum JA, Shapiro NI, Pro CI.. Endothelial permeability and hemostasis in septic shock: results from the ProCESS Trial. Chest 2017;152:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hendrickson CM, Matthay MA.. Endothelial biomarkers in human sepsis: pathogenesis and prognosis for ARDS. Pulm Circ 2018;8:2045894018769876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Statz S, Sabal G, Walborn A, Williams M, Hoppensteadt D, Mosier M, Rondina M, Fareed J.. Angiopoietin 2 levels in the risk stratification and mortality outcome prediction of sepsis-associated coagulopathy. Clin Appl Thromb Hemost 2018;24:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. David S, Park JK, Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, Shushakova N, Gueler F, Haller H, Kumpers P.. Acute administration of recombinant angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine 2011;55:251–259. [DOI] [PubMed] [Google Scholar]
- 77. Cho CH, Kim KE, Byun J, Jang HS, Kim DK, Baluk P, Baffert F, Lee GM, Mochizuki N, Kim J, Jeon BH, McDonald DM, Koh GY.. Long-term and sustained COMP-Ang1 induces long-lasting vascular enlargement and enhanced blood flow. Circ Res 2005;97:86–94. [DOI] [PubMed] [Google Scholar]
- 78. Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, Santel A, Ghosh CC, Parikh SM, David S.. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med 2014;42:e654–e662. [DOI] [PubMed] [Google Scholar]
- 79. Lomas-Neira J, Venet F, Chung CS, Thakkar R, Heffernan D, Ayala A.. Neutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. Am J Respir Cell Mol Biol 2013;50:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Han S, Lee SJ, Kim KE, Lee HS, Oh N, Park I, Ko E, Oh SJ, Lee YS, Kim D, Lee S, Lee DH, Lee KH, Chae SY, Lee JH, Kim SJ, Kim HC, Kim S, Kim SH, Kim C, Nakaoka Y, He Y, Augustin HG, Hu J, Song PH, Kim YI, Kim P, Kim I, Koh GY.. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med 2016;8:335–355. [DOI] [PubMed] [Google Scholar]
- 81. Weinbaum S, Tarbell JM, Damiano ER.. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 2007;9:121–167. [DOI] [PubMed] [Google Scholar]
- 82. Maniatis NA, Orfanos SE.. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 2008;14:22–30. [DOI] [PubMed] [Google Scholar]
- 83. Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C.. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care 2015;19:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sallisalmi M, Tenhunen J, Yang R, Oksala N, Pettila V.. Vascular adhesion protein-1 and Syndecan-1 in septic shock. Acta Anaesthesiol Scand 2012;56:316–322. [DOI] [PubMed] [Google Scholar]
- 85. Martin L, De Santis R, Koczera P, Simons N, Haase H, Heinbockel L, Brandenburg K, Marx G, Schuerholz T.. The synthetic antimicrobial peptide 19-2.5 interacts with heparanase and heparan sulfate in murine and human sepsis. PLoS One 2015;10:e0143583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Steppan J, Hofer S, Funke B, Brenner T, Henrich M, Martin E, Weitz J, Hofmann U, Weigand MA.. Sepsis and major abdominal surgery lead to flaking of the endothelial glycocalix. J Surg Res 2011;165:136–141. [DOI] [PubMed] [Google Scholar]
- 87. Cox LA, van Eijk LT, Ramakers BP, Dorresteijn MJ, Gerretsen J, Kox M, Pickkers P.. Inflammation-induced increases in plasma endocan levels are associated with endothelial dysfunction in humans in vivo. Shock 2015;43:322–326. [DOI] [PubMed] [Google Scholar]
- 88. Scherpereel A, Depontieu F, Grigoriu B, Cavestri B, Tsicopoulos A, Gentina T, Jourdain M, Pugin J, Tonnel AB, Lassalle P.. Endocan, a new endothelial marker in human sepsis. Crit Care Med 2006;34:532–537. [DOI] [PubMed] [Google Scholar]
- 89. Chappell D, Westphal M, Jacob M.. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr Opin Anaesthesiol 2009;22:155–162. [DOI] [PubMed] [Google Scholar]
- 90. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, Conzen P, Becker BF, Rehm M.. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care 2014;18:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Puskarich MA, Cornelius DC, Tharp J, Nandi U, Jones AE.. Plasma syndecan-1 levels identify a cohort of patients with severe sepsis at high risk for intubation after large-volume intravenous fluid resuscitation. J Crit Care 2016;36:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jacob M, Paul O, Mehringer L, Chappell D, Rehm M, Welsch U, Kaczmarek I, Conzen P, Becker BF.. Albumin augmentation improves condition of guinea pig hearts after 4 hr of cold ischemia. Transplantation 2009;87:956–965. [DOI] [PubMed] [Google Scholar]
- 93. Straat M, Muller MC, Meijers JC, Arbous MS, Spoelstra-de Man AM, Beurskens CJ, Vroom MB, Juffermans NP.. Effect of transfusion of fresh frozen plasma on parameters of endothelial condition and inflammatory status in non-bleeding critically ill patients: a prospective substudy of a randomized trial. Crit Care 2015;19:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Winters ME, Sherwin R, Vilke GM, Wardi G.. What is the preferred resuscitation fluid for patients with severe sepsis and septic shock? J Emerg Med 2017;53:928–939. [DOI] [PubMed] [Google Scholar]
- 95. Cui N, Wang H, Long Y, Su L, Liu D.. Dexamethasone suppressed LPS-induced matrix metalloproteinase and its effect on endothelial glycocalyx shedding. Mediators Inflamm 2015;2015:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM.. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 2012;18:1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Iba T, Levy JH, Raj A, Warkentin TE.. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Clin Med 2019;8: 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kerr H, Richards A.. Complement-mediated injury and protection of endothelium: lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012;217:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Studt J-D, Hovinga JAK, Antoine G, Hermann M, Rieger M, Scheiflinger F, LäMmle B.. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood 2005;105:542–544. [DOI] [PubMed] [Google Scholar]
- 100. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, Moake JL.. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood 2005;106:4199–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Levi M, van der Poll T.. Disseminated intravascular coagulation: a review for the internist. Intern Emerg Med 2013;8:23–32. [DOI] [PubMed] [Google Scholar]
- 102. Thomas MR, Storey RF.. The role of platelets in inflammation. Thromb Haemost 2015;114:449–458. [DOI] [PubMed] [Google Scholar]
- 103. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA.. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998;391:591–594. [DOI] [PubMed] [Google Scholar]
- 104. Gawaz M, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C, Koch W, Schömig A, Neumann F-J.. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis 2000;148:75–85. [DOI] [PubMed] [Google Scholar]
- 105. Hoshino K, Kitamura T, Nakamura Y, Irie Y, Matsumoto N, Kawano Y, Ishikura H.. Usefulness of plasminogen activator inhibitor-1 as a predictive marker of mortality in sepsis. J Intensive Care 2017;5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Taylor FB Jr, Chang AC, Peer GT, Mather T, Blick K, Catlett R, Lockhart MS, Esmon CT.. DEGR-factor Xa blocks disseminated intravascular coagulation initiated by Escherichia coli without preventing shock or organ damage. Blood 1991;78:364–368. [PubMed] [Google Scholar]
- 107. Wang C, Chi C, Guo L, Wang X, Guo L, Sun J, Sun B, Liu S, Chang X, Li E.. Heparin therapy reduces 28-day mortality in adult severe sepsis patients: a systematic review and meta-analysis. Crit Care 2014;18:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Emerson TE Jr, Fournel MA, Redens TB, Taylor FB. Jr. Efficacy of antithrombin III supplementation in animal models of fulminant Escherichia coli endotoxemia or bacteremia. Am J Med 1989;87:27S–33S. [DOI] [PubMed] [Google Scholar]
- 109. OelschläGer C, RöMisch J, Staubitz A, Stauss H, LeithäUser B, Tillmanns H, HöLschermann H.. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood 2002;99:4015–4020. [DOI] [PubMed] [Google Scholar]
- 110. Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflam 2011;2011:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, Light B, Spapen H, Stone J, Seibert A, Peckelsen C, De Deyne C, Postier R, Pettila V, Sprung CL, Artigas A, Percell SR, Shu V, Zwingelstein C, Tobias J, Poole L, Stolzenbach JC, Creasey AA; OPTIMIS Trial Study Group. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA 2003;290:238–247. [DOI] [PubMed] [Google Scholar]
- 112. Wunderink RG, Laterre PF, Francois B, Perrotin D, Artigas A, Vidal LO, Lobo SM, Juan JS, Hwang SC, Dugernier T, LaRosa S, Wittebole X, Dhainaut JF, Doig C, Mendelson MH, Zwingelstein C, Su G, Opal S; CAPTIVATE Trial Group. Recombinant tissue factor pathway inhibitor in severe community-acquired pneumonia: a randomized trial. Am J Respir Crit Care Med 2011;183:1561–1568. [DOI] [PubMed] [Google Scholar]
- 113. Neviere R, Tournoys A, Mordon S, Marechal X, Song FL, Jourdain M, Fourrier F.. Antithrombin reduces mesenteric venular leukocyte interactions and small intestine injury in endotoxemic rats. Shock 2001;15:220–225. [DOI] [PubMed] [Google Scholar]
- 114. Yamashiro K, Kiryu J, Tsujikawa A, Honjo M, Nonaka A, Miyamoto K, Honda Y, Tanihara H, Ogura Y.. Inhibitory effects of antithrombin III against leukocyte rolling and infiltration during endotoxin-induced uveitis in rats. Invest Ophthalmol Vis Sci 2001;42:1553–1560. [PubMed] [Google Scholar]
- 115. Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H.. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: an observational nationwide study. J Thromb Haemost 2014;12:1470–1479. [DOI] [PubMed] [Google Scholar]
- 116. Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, Keinecke HO, Warren BL, Opal SM; the KyberSept Investigators. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost 2006;4:90–97. [DOI] [PubMed] [Google Scholar]
- 117. Mosnier LO, Zlokovic BV, Griffin JH.. The cytoprotective protein C pathway. Blood 2007;109:3161–3172. [DOI] [PubMed] [Google Scholar]
- 118. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD; PROWESS-SHOCK Study Group. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012;366:2055–2064. [DOI] [PubMed] [Google Scholar]
- 119. Wiedermann CJ. When a single pivotal trial should not be enough-the case of drotrecogin-alfa (activated). Intensive Care Med 2006;32:604–604; Author reply 610–602. [DOI] [PubMed] [Google Scholar]
- 120. Iba T, Aihara K, Watanabe S, Yanagawa Y, Takemoto M, Yamada A, Yang D.. Recombinant thrombomodulin improves the visceral microcirculation by attenuating the leukocyte-endothelial interaction in a rat LPS model. Thromb Res 2013;131:295–299. [DOI] [PubMed] [Google Scholar]
- 121. Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, Hirayama A, Matsuda T, Asakura H, Nakashima M, Aoki N.. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost 2007;5:31–41. [DOI] [PubMed] [Google Scholar]
- 122. Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, Hoste E, Levy H, Hirman J, Levi M, Daga M, Kutsogiannis DJ, Crowther M, Bernard GR, Devriendt J, Puigserver JV, Blanzaco DU, Esmon CT, Parrillo JE, Guzzi L, Henderson SJ, Pothirat C, Mehta P, Fareed J, Talwar D, Tsuruta K, Gorelick KJ, Osawa Y, Kaul I.. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med 2013;41:2069–2079. [DOI] [PubMed] [Google Scholar]
- 123. Prauchner CA. Oxidative stress in sepsis: pathophysiological implications justifying antioxidant co-therapy. Burns 2017;43:471–485. [DOI] [PubMed] [Google Scholar]
- 124. Parihar A, Parihar MS, Milner S, Bhat S.. Oxidative stress and anti-oxidative mobilization in burn injury. Burns 2008;34:6–17. [DOI] [PubMed] [Google Scholar]
- 125. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, Hernandez G, Murray P, De Backer D; ADQI XIV Workgroup The endothelium in sepsis. Shock 2016;45:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Usatyuk PV, Vepa S, Watkins T, He D, Parinandi NL, Natarajan V.. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid Redox Signal 2003;5:723–730. [DOI] [PubMed] [Google Scholar]
- 127. Gaboury JP, Anderson DC, Kubes P.. Molecular mechanisms involved in superoxide-induced leukocyte-endothelial cell interactions in vivo. Am J Physiol 1994;266:H637–H642. [DOI] [PubMed] [Google Scholar]
- 128. Shasby DM, Lind SE, Shasby SS, Goldsmith JC, Hunninghake GW.. Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood 1985;65:605–614. [PubMed] [Google Scholar]
- 129. Suttorp N, Weber U, Welsch T, Schudt C.. Role of phosphodiesterases in the regulation of endothelial permeability in vitro. J Clin Invest 1993;91:1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS.. H(2)O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol 2000;279:C21–C30. [DOI] [PubMed] [Google Scholar]
- 131. Cai H, Harrison DG.. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 132. Craige SM, Kant S, Keaney JF Jr.. Reactive oxygen species in endothelial function—from disease to adaptation. Circ J 2015;79:1145–1155. [DOI] [PubMed] [Google Scholar]
- 133. Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K, Morrow JP.. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2017;2:e94248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Fisher AB, Dodia C, Chatterjee S, Feinstein SI.. A peptide inhibitor of NADPH oxidase (NOX2) activation markedly decreases mouse lung injury and mortality following administration of lipopolysaccharide (LPS). Int J Mol Sci 2019;20:2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hernandes MS, D’Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, Castro-Faria-Neto HC, Cunha FQ, Britto LR, Bozza FA.. The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation 2014;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Vanhoutte PM. Endothelial dysfunction and vascular disease. Verh K Acad Geneeskd Belg 1998;60:251–266. [PubMed] [Google Scholar]
- 137. McQuaid KE, Keenan AK.. Endothelial barrier dysfunction and oxidative stress: roles for nitric oxide? Exp Physiol 1997;82:369–376. [DOI] [PubMed] [Google Scholar]
- 138. Bime C, Zhou T, Wang T, Slepian MJ, Garcia JG, Hecker L.. Reactive oxygen species-associated molecular signature predicts survival in patients with sepsis. Pulm Circ 2016;6:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wang H, Yu D, Li B, Liu Z, Ren J, Qu X.. Ultrasensitive magnetic resonance imaging of systemic reactive oxygen species in vivo for early diagnosis of sepsis using activatable nanoprobes. Chem Sci 2019;10:3770–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Singh A, Ramnath RD, Foster RR, Wylie EC, Friden V, Dasgupta I, Haraldsson B, Welsh GI, Mathieson PW, Satchell SC.. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One 2013;8:e55852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Shuvaev VV, Han J, Tliba S, Arguiri E, Christofidou-Solomidou M, Ramirez SH, Dykstra H, Persidsky Y, Atochin DN, Huang PL, Muzykantov VR.. Anti-inflammatory effect of targeted delivery of SOD to endothelium: mechanism, synergism with NO donors and protective effects in vitro and in vivo. PLoS One 2013;8:e77002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR.. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol 2014;306:F734–F743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nava E, Palmer RM, Moncada S.. The role of nitric oxide in endotoxic shock: effects of NG-monomethyl-L-arginine. J Cardiovasc Pharmacol 1992;20(Suppl. 12):S132–S134. [DOI] [PubMed] [Google Scholar]
- 144. Ullrich R, Scherrer-Crosbie M, Bloch KD, Ichinose F, Nakajima H, Picard MH, Zapol WM, Quezado ZM.. Congenital deficiency of nitric oxide synthase 2 protects against endotoxin-induced myocardial dysfunction in mice. Circulation 2000;102:1440–1446. [DOI] [PubMed] [Google Scholar]
- 145. Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R.. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 2004;32:21–30. [DOI] [PubMed] [Google Scholar]
- 146. Chuaiphichai S, Starr A, Nandi M, Channon KM, McNeill E.. Endothelial cell tetrahydrobiopterin deficiency attenuates LPS-induced vascular dysfunction and hypotension. Vascul Pharmacol 2016;77:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Dumbarton TC, Maxan A, Farah N, Sharawy N, Zhou J, Nantais J, Lehmann C.. Tetrahydrobiopterin improves microcirculation in experimental sepsis. Clin Hemorheol Microcirc 2017;67:15–24. [DOI] [PubMed] [Google Scholar]
- 148. Legrand M, Kandil A, Payen D, Ince C.. Effects of sepiapterin infusion on renal oxygenation and early acute renal injury after suprarenal aortic clamping in rats. J Cardiovasc Pharmacol 2011;58:192–198. [DOI] [PubMed] [Google Scholar]
- 149. Wilson JX. Mechanism of action of vitamin C in sepsis: ascorbate modulates redox signaling in endothelium. Biofactors 2009;35:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wu F, Tyml K, Wilson JX.. Ascorbate inhibits iNOS expression in endotoxin- and IFN gamma-stimulated rat skeletal muscle endothelial cells. FEBS Lett 2002;520:122–126. [DOI] [PubMed] [Google Scholar]
- 151. Mortensen A, Lykkesfeldt J.. Does vitamin C enhance nitric oxide bioavailability in a tetrahydrobiopterin-dependent manner? In vitro, in vivo and clinical studies. Nitric Oxide 2014;36:51–57. [DOI] [PubMed] [Google Scholar]
- 152. May JM, Harrison FE.. Role of vitamin C in the function of the vascular endothelium. Antioxid Redox Signal 2013;19:2068–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Dillon PF, Root-Bernstein RS, Lieder CM.. Antioxidant-independent ascorbate enhancement of catecholamine-induced contractions of vascular smooth muscle. Am J Physiol Heart Circ Physiol 2004;286:H2353–H2360. [DOI] [PubMed] [Google Scholar]
- 154. Fisher BJ, Seropian IM, Kraskauskas D, Thakkar JN, Voelkel NF, Fowler AA III, Natarajan R.. Ascorbic acid attenuates lipopolysaccharide-induced acute lung injury. Crit Care Med 2011;39:1454–1460. [DOI] [PubMed] [Google Scholar]
- 155. Tanaka H, Lund T, Wiig H, Reed RK, Yukioka T, Matsuda H, Shimazaki S.. High dose vitamin C counteracts the negative interstitial fluid hydrostatic pressure and early edema generation in thermally injured rats. Burns 1999;25:569–574. [DOI] [PubMed] [Google Scholar]
- 156. Borrelli E, Roux-Lombard P, Grau GE, Girardin E, Ricou B, Dayer J, Suter PM.. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit Care Med 1996;24:392–397. [DOI] [PubMed] [Google Scholar]
- 157. Fowler AA, Syed AA, Knowlson S, Sculthorpe R, Farthing D, DeWilde C, Farthing CA, Larus TL, Martin E, Brophy DF, Gupta S, Fisher BJ, Natarajan R; Medical Respiratory Intensive Care Unit Nursing. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J Transl Med 2014;12:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV.. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 2002;236:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J.. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: a retrospective before-after study. Chest 2017;151:1229–1238. [DOI] [PubMed] [Google Scholar]
- 160. Fowler AA III, Truwit JD, Hite RD, Morris PE, DeWilde C, Priday A, Fisher B, Thacker LR II, Natarajan R, Brophy DF, Sculthorpe R, Nanchal R, Syed A, Sturgill J, Martin GS, Sevransky J, Kashiouris M, Hamman S, Egan KF, Hastings A, Spencer W, Tench S, Mehkri O, Bindas J, Duggal A, Graf J, Zellner S, Yanny L, McPolin C, Hollrith T, Kramer D, Ojielo C, Damm T, Cassity E, Wieliczko A, Halquist M.. Effect of vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: the CITRIS-ALI randomized clinical trial. JAMA 2019;322:1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Stefanec T. Endothelial apoptosis: could it have a role in the pathogenesis and treatment of disease? Chest 2000;117:841–854. [DOI] [PubMed] [Google Scholar]
- 162. Pohlman TH, Harlan JM.. Human endothelial cell response to lipopolysaccharide, interleukin-1, and tumor necrosis factor is regulated by protein synthesis. Cell Immunol 1989;119:41–52. [DOI] [PubMed] [Google Scholar]
- 163. Bombeli T, Karsan A, Tait JF, Harlan JM.. Apoptotic vascular endothelial cells become procoagulant. Blood 1997;89:2429–2442. [PubMed] [Google Scholar]
- 164. Mitra D, Jaffe EA, Weksler B, Hajjar KA, Soderland C, Laurence J.. Thrombotic thrombocytopenic purpura and sporadic hemolytic-uremic syndrome plasmas induce apoptosis in restricted lineages of human microvascular endothelial cells. Blood 1997;89:1224–1234. [PubMed] [Google Scholar]
- 165. Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, Benevolenskaya EV, Rehman J, Malik AB.. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun 2019;10:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zahran AM, Elsayh KI, Mohamad IL, Hassan GM, Abdou MA.. Circulating endothelial cells and endothelial progenitor cells in pediatric sepsis. Pediatr Emerg Care 2016;32:163–167. [DOI] [PubMed] [Google Scholar]
- 167. Friedman RC, Farh KK, Burge CB, Bartel DP.. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2008;19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Gao N, Dong L.. MicroRNA-146 regulates the inflammatory cytokines expression in vascular endothelial cells during sepsis. Pharmazie 2017;72:700–704. [DOI] [PubMed] [Google Scholar]
- 169. Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE.. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med 2013;5:1017–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW.. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J Clin Invest 2012;122:1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Wu XY, Fan WD, Fang R, Wu GF.. Regulation of microRNA-155 in endothelial inflammation by targeting nuclear factor (NF)-kappaB P65. J Cell Biochem 2014;115:1928–1936. [DOI] [PubMed] [Google Scholar]
- 172. Wang P, Zhang X, Li F, Yuan K, Li M, Zhang J, Li B, Liang W.. MiR-130b attenuates vascular inflammation via negatively regulating tumor progression locus 2 (Tpl2) expression. Int Immunopharmacol 2017;51:9–16. [DOI] [PubMed] [Google Scholar]
- 173. Rajput C, Tauseef M, Farazuddin M, Yazbeck P, Amin MR, Avin Br V, Sharma T, Mehta D.. MicroRNA-150 suppression of angiopoetin-2 generation and signaling is crucial for resolving vascular injury. Arterioscler Thromb Vasc Biol 2016;36:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Chatterjee V, Beard RS Jr, Reynolds JJ, Haines R, Guo M, Rubin M, Guido J, Wu MH, Yuan SY.. MicroRNA-147b regulates vascular endothelial barrier function by targeting ADAM15 expression. PLoS One 2014;9:e110286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Li X, Gao Y, Meng Z, Zhang C, Qi Q.. Regulatory role of microRNA-30b and plasminogen activator inhibitor-1 in the pathogenesis of cognitive impairment. Exp Ther Med 2016;11:1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Chen YS, Shen L, Mai RQ, Wang Y.. Levels of microRNA-181b and plasminogen activator inhibitor-1 are associated with hypertensive disorders complicating pregnancy. Exp Ther Med 2014;8:1523–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Villadsen SB, Bramsen JB, Ostenfeld MS, Wiklund ED, Fristrup N, Gao S, Hansen TB, Jensen TI, Borre M, Orntoft TF, Dyrskjot L, Kjems J.. The miR-143/-145 cluster regulates plasminogen activator inhibitor-1 in bladder cancer. Br J Cancer 2012;106:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang Y, Chen M, Zhang Y, Peng P, Li J, Xin X.. miR-96 and miR-330 overexpressed and targeted AQP5 in lipopolysaccharide-induced rat lung damage of disseminated intravascular coagulation. Blood Coagul Fibrinolysis 2014;25:731–737. [DOI] [PubMed] [Google Scholar]
- 179. Zhuang Y, Peng H, Mastej V, Chen W.. MicroRNA regulation of endothelial junction proteins and clinical consequence. Mediators Inflamm 2016;2016:1–6. 5078627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Madoiwa S, Nunomiya S, Ono T, Shintani Y, Ohmori T, Mimuro J, Sakata Y.. Plasminogen activator inhibitor 1 promotes a poor prognosis in sepsis-induced disseminated intravascular coagulation. Int J Hematol 2006;84:398–405. [DOI] [PubMed] [Google Scholar]
- 181. Shapiro NI, Schuetz P, Yano K, Sorasaki M, Parikh SM, Jones AE, Trzeciak S, Ngo L, Aird WC.. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care 2010;14:R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM.. Microparticles: biomarkers and beyond. Clin Sci 2013;124:423–441. [DOI] [PubMed] [Google Scholar]
- 183. Piccin A, Murphy WG, Smith OP.. Circulating microparticles: pathophysiology and clinical implications. Blood Rev 2007;21:157–171. [DOI] [PubMed] [Google Scholar]
- 184. Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M.. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008;112:2512–2519. [DOI] [PubMed] [Google Scholar]
- 185. Ghosh A, Li W, Febbraio M, Espinola RG, McCrae KR, Cockrell E, Silverstein RL.. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest 2008;118:1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sadallah S, Eken C, Martin PJ, Schifferli JA.. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J Immunol 2011;186:6543–6552. [DOI] [PubMed] [Google Scholar]
- 187. Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, Sturk A.. Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost 2001;85:810–820. [PubMed] [Google Scholar]
- 188. Mortaza S, Martinez MC, Baron-Menguy C, Burban M, de la Bourdonnaye M, Fizanne L, Pierrot M, Cales P, Henrion D, Andriantsitohaina R, Mercat A, Asfar P, Meziani F.. Detrimental hemodynamic and inflammatory effects of microparticles originating from septic rats. Crit Care Med 2009;37:2045–2050. [DOI] [PubMed] [Google Scholar]
- 189. Delabranche X, Boisrame-Helms J, Asfar P, Berger A, Mootien Y, Lavigne T, Grunebaum L, Lanza F, Gachet C, Freyssinet JM, Toti F, Meziani F.. Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med 2013;39:1695–1703. [DOI] [PubMed] [Google Scholar]