

Abstract
The metabolic syndrome (MetS) is associated with dementia, but it is unclear whether MetS is related to Alzheimer’s disease (AD). We investigated the association of MetS with brain amyloid, a key AD feature, and neurodegeneration. A community-based sample of 350 middle-aged Hispanics in New York City had cerebral amyloid β (Aβ) burden ascertained with 18F-Florbetaben PET. Neurodegeneration was ascertained as cortical thickness in AD signature regions from 3T brain MRI. MetS and its components (glucose, blood pressure, triglycerides, high-density lipoprotein, adiposity) were defined using the National Institutes of Health criteria. Neither MetS presence nor MetS score were associated with Aβ or neurodegeneration. Among the MetS components, elevated glucose was associated with lower Aβ burden, and this association was not explained by diabetes treatment. Glucose and triglycerides were related to smaller cortical thickness. Our findings suggest that the MetS as an arbitrary measure of aggregate metabolic and vascular risk does not capture the risk of AD neuropathology in late middle-age, and that other approaches to measure aggregate risk should be examined.
Keywords: Alzheimer’s disease, neuroimaging, epidemiology, metabolic syndrome
1. Introduction
The metabolic syndrome is defined as a cluster of cardiometabolic conditions, including elevated blood pressure, elevated blood glucose, excess central adiposity, high levels of triglycerides, and abnormal high density lipoprotein cholesterol (Grundy et al., 2005). Prior studies estimate the prevalence of the metabolic syndrome to be nearly 34–39% among U.S. adults, with estimates varying due to differences in adiposity thresholds applied across studies (Alberti et al., 2009; Moore et al., 2017). The highest estimates are among racial/ethnic minority groups, including Hispanics and non-Hispanic Blacks. Data from the National Health and Nutrition Examination Survey (NHANES), showed that Mexican Americans had the highest prevalence of metabolic syndrome compared to non-Hispanic Whites and African Americans (Ford et al., 2002).
Metabolic syndrome is a known risk factor for diabetes, cardiovascular disease, and both cardiovascular-related and all-cause mortality (Katzmarzyk et al., 2005). Metabolic syndrome is also associated with cognitive impairment (Exalto et al., 2015), vascular dementia (Raffaitin et al., 2009), and dementia due to Alzheimer’s disease (AD) (Vanhanen et al., 2006). However, it is unclear if the metabolic syndrome is related to AD neuropathology. Current understanding of the natural history leading to dementia due to AD can be summarized as follows (Jack et al., 2013): the 2 main proteinopathies underlying AD, amyloid and tau, are separate processes, but amyloid deposition is the main driver of AD and accelerates tau deposition; amyloid and tau deposition precede and cause neurodegeneration, which leads to the clinical syndromes of amnestic mild cognitive impairment (MCI) and dementia. The constructs of amyloid, tau, and neurodegeneration feature prominently in the recent National Institute on Aging (NIA)/Alzheimer’s Association (AA) 2018 research framework (Jack, C.R., Jr. et al., 2018). This framework proposes to conduct research in which biomarkers of amyloid, tau, and neurodegeneration are used as outcomes in research for the purpose of better understanding the mechanisms and sequence of neuropathology. Our primary objective was to examine whether the metabolic syndrome and its components are related to brain amyloid β (Aβ) burden, the main pathological hallmark of AD. Our secondary objective was to examine the relation of the metabolic syndrome and its components with neurodegeneration. We hypothesized that the metabolic syndrome and its components would be related to higher brain Aβ burden and neurodegeneration in late middle-age.
2. Materials and Methods
2.1. Study Population and Design
This was a cross-sectional analysis of 350 participants of a study focusing on the relation of metabolic conditions and brain amyloid in middle-aged Hispanics conducted at Columbia University Irving Medical Center (CUIMC) in New York City, recruited between 03/01/2016 and 07/31/2019. We targeted Hispanics because they are the most common ethnic group in the community surrounding CUIMC (Luchsinger et al., 2015) and because there is a paucity of AD biomarkers studies in Non-Whites (Jack et al., 2013). In addition, Hispanics have a higher prevalence of the metabolic syndrome and a higher risk of cognitive impairment compared with Non-Hispanic Whites (Evans et al.; Gurland et al.; Haerer et al., 1987; Perkins et al.; Prineas et al.; Tang et al.; Tang et al.; Whitmer et al., 2014; Yaffe et al., 2013). Participants were recruited from the community through various outreach activities, including presentations at churches and senior centers, posters at CUIMC and around the community, health fairs, and newspaper ads. Study research staff collaborated with the Community Engagement Core Resource of the Irving Institute for Clinical and Translational Research, the Clinical Translational Science Award at CUIMC, to conduct outreach in the community and promote the study. Recruitment was exclusively community-based and did not include recruitment from clinics or using electronic medical records. Inclusion criteria were: aged 55 to 69 years, self-reported Hispanic men and women, willing and able to undergo phlebotomy, clinical and neuropsychological assessments, 3T brain magnetic resonance imaging (MRI), and positron emission tomography (PET) with injection of the Aβ radioligand 18F-Florbetaben. All MRI scans were processed with FreeSurfer software to derive regions of interest for PET analyses. Exclusion criteria were: diagnosis of dementia, cancer other than non-melanoma skin cancer, and MRI contraindications (eFigure 1). We screened 659 potential participants; 114 (17.30%) declined to participate, 178 (27.01%) were ineligible, 16 (2.43%) did not complete study procedures after being deemed eligible, and 1 participant (0.15%) was recruited into the study but did not complete APOE-ε4 genotyping. Our final analytic sample included 350 middle-aged Hispanic participants. The interval between Aβ PET and MRI in this sample was 15.79 ± 33.41 days. This study was approved by the Institutional Review Board and the Joint Radiation Safety Commission at Columbia University. All study participants provided written informed consent.
2.2. Exposure: Metabolic Syndrome
Presence or absence of the metabolic syndrome and its components were the primary exposures. We defined the metabolic syndrome and its components using the definition from the National Institutes of Health (eTable 1, (Alberti et al., 2009)). The metabolic syndrome includes the following components and definitions: elevated blood pressure (systolic blood pressure ≥130 and/ or diastolic blood pressure ≥ 85 mmHg or use of anti-hypertensive medication use), high fasting glucose (≥100 mg/dL or diabetes medication use), high waist circumference (women: ≥35 inches or ≥ 89 centimeters; men: ≥40 inches or ≥102 centimeters), high triglycerides level (≥150 mg/dL (1.7 mmol/L) or triglyceride medication use), and abnormal high density lipoprotein ([HDL]; women: <50 mg/dL (1.3 mmol/L); men: <40 mg/dL (1.04 mmol/L) or low HDL medication use). HDL and triglycerides were measured on an automated immunochemistry analyzer, Integra 400 plus (Roche Diagnostics, Indianapolis, IN) using an enzymatic colorimetric assay with a lower limit of quantitation of 3.09 mg/dL for HDL and 0.1mmol/L for triglycerides. Glucose is measured in plasma with NaF preservative using an enzymatic reference method on the automated analyzer, Integra 400 plus (Roche Diagnostics, Indianapolis, IN). All reagents, calibrators and controls were purchased from Roche. The lower limit of detection of the assay was 0.24mmol/L. The intra-assay precision was 0.7% and inter-assay precision was 1.3%.
The presence of the metabolic syndrome was defined by the presence of 3 out of the 5 components. We also examined the metabolic syndrome score (range: 0–5) and each component separately (defined categorically and continuously) as exposures.
2.3. Outcomes
2.3.1. Brain Amyloid Burden
Our primary outcome was brain Aβ burden ascertained as global standardized uptake value ratio (SUVR) measured with 18F-Florbetaben PET.
The dose of 18F-Florbetaben was 300 MBq (8.1 mCi), maximum 30 mcg mass dose, administered as a single slow intravenous bolus. Images were acquired over 20 minutes starting 90 minutes after injection. Dynamic PET frames (4 scans) were aligned to the first frame using rigid-body registration to correct for potential head motion during imaging session (6 degrees of freedom [Dof]). A single static PET image was then obtained by averaging the four registered frames (voxelwise operation). A computerized tomography scan (CT) was used for attenuation correction for the PET and was only used when the registration between MRI and PET fails. The static PET and CT scans are coregistered using a rigid-body intermodal registration (6 DoF, normalized mutual information) and merged to generate a composite image in PET static space. Next, each participant’s structural T1-weighted scan was reconstructed in FreeSurfer space (v6.0 (http://surfer.nmr.mgh.harvard.edu/) and was then registered to the participant’s merged image using rigid-body registration (6 DoF, normalized mutual information) to obtain a second transformation matrix. A combination of the 2 transformation matrices is used to transfer the regional masks (ROIs) from Freesurfer space to static PET image space. Additional information on the Aβ PET scan processing protocol has been previously published (Tahmi et al., 2019). The standardized uptake value (SUV), defined as the decay-corrected brain radioactivity concentration normalized for injected dose and body weight, was calculated in all FreeSurfer regions. The SUV in each region as well as each voxel was also normalized to the SUV in cerebellar gray matter to derive the regional and voxel-wise ratio (SUVR). Analyses incorporated voxel-based, individual region of interests (ROI) (lateral temporal cortex, parietal cortex, cingulate cortex, and frontal cortex), and an overall mean SUVR value of Aβ burden across all ROIs was derived.
Our primary analysis examined Aβ SUVR as a continuous outcome. The 2018 National Institute on Aging/Alzheimer’s Association research framework recommends categorizing AD biomarkers as positive or negative (or high or low), but there is uncertainty about the right approach for categorization (Jack, C.R. et al., 2018). Brain Aβ SUVR has a bimodal distribution and Aβ positivity is typically examined using cutoffs that vary by study (Jansen, W. J. et al., 2015), but usually represent the point of separation in the bimodal distribution (eFigure 2). In a secondary analysis, we categorized Aβ positivity using an SUVR threshold of 1.34, determined using the K-means clustering method (Tahmi et al., 2020 ), which identifies the partition between the 2 peaks in the Aβ SUVR distribution (eFigure 2).
2.3.2. Neurodegeneration
The measure of neurodegeneration was cortical thickness obtained by averaging values from AD-related regions as specific patterns of cortical thinning are found in AD.(Dickerson et al., 2009) Regional patterns were derived with FreeSurfer v6.0 (http://surfer.nmr.mgh.harvard.edu/). These regions included entorhinal cortex, parahippocampus, inferior parietal lobule, pars opercularis, pars orbitalis, pars triangularis, inferior temporal pole, supramarginal gyrus, superior parietal lobe, and superior frontal lobe. Cortical thickness was analyzed as a continuous outcome.
2.4. Covariates
The following covariates were considered: age, sex, education in years, Hispanic subgroup, and APOE-ε4 genotype, the strongest risk factor for sporadic dementia due to AD (Atti et al., 2019), and also the strongest determinant of amyloid burden (Jansen, Willemijn J. et al., 2015). We also compared medications for diabetes, hypertension, and dyslipidemia between persons with and without the metabolic syndrome. Hispanic subgroup was classified following the format of the 2010 Census by country or region of origin (e.g. Mexican, Puerto Rican, Cuban, Dominican, (Humes et al., 2011)). APOE-ε4 genotyping was conducted by LGC genomics (Beverly, MA) using single nucleotide polymorphisms rs429358 and rs7412. Participants were classified as APOE-ε4 carriers if they were homozygous or heterozygous for APOE-ε4. Medications for diabetes included metformin, sulfonylureas, dipeptidyl peptidase 4 (DPP-4) inhibitors, peroxisome proliferator-activator γ (PPAR-γ) agonists, Glucagon Like Peptide 1 (GLP1) agonists, and sodium glucose transport 2 (SGLT2) inhibitors; none of the study participants reported using insulin. Blood pressure medications included betablockers, Angiotensin Converting Enzyme (ACE) inhibitors, Angiotensin Receptor Blockers (ARB), diuretics, calcium channel blockers, and vasodilators. Lipid medications included HMG-CoA reductase inhibitors (statins), bile acids, ezetimibe, and fibrates.
2.5. Statistical Analysis
Descriptive analysis used chi-squared and ANOVA tests to examine differences in demographic and clinical characteristics across participants with and without the metabolic syndrome. For all analyses, comparisons were made between the presence and absence of metabolic syndrome, total metabolic syndrome score, and the presence and absence of each of the individual metabolic syndrome components. We used standardized linear regression models to estimate the association of the metabolic syndrome and its components with continuous measures of global amyloid SUVR and cortical thickness in the AD signature region. Continuous values of global Aβ SUVR and cortical thickness were standardized to a mean of zero and standard deviation of 1 to allow for comparisons across the outcomes. Logistic regression models estimated the odds ratio of amyloid positivity by metabolic syndrome, total metabolic syndrome score, and the metabolic syndrome components. All models were adjusted for age, sex, and APOE-ε4. Multiple comparisons were adjusted using a Bonferroni correction. All analyses were performed using SAS version 9.4m5 and R version 3.6.0.
3. Results
No differences in sex, Hispanic subgroup, APOE-ε4, or education were observed between participants with and without the metabolic syndrome (Table 1). Persons with the metabolic syndrome were approximately one year older than those without the metabolic syndrome, and this difference was close to statistical significance. As expected, participants with the metabolic syndrome were more often taking medications for diabetes, particularly metformin, anti-hypertensive medications, and lipid-lowering medications. As expected, APOE-ε4 genotype was a strong predictor of Aβ burden. Global Aβ SUVR examined as a continuous outcome was higher in APOE-ε4 carriers and APOE-ε4 non-carriers (age and sex adjusted means, 1.21 ± 0.01 vs. 1.13 ± 0.09; p<0.0001, data not shown), respectively. The unadjusted scatterplots of continuous global brain Aβ SUVR and cortical thickness in the AD signature region with each metabolic syndrome component, measured continuously, are provided in eFigure 3.
Table 1.
Participant characteristics, overall and by metabolic syndrome status (n=350)
| Characteristic | Total sample n = 350 | No Metabolic Syndrome n = 182 | Metabolic Syndrome n = 168 | p-value |
|---|---|---|---|---|
| Age, mean (SD), year | 64.2 ± 3.3 | 63.6 ± 3.3 | 64.4 ± 3.6 | 0.051 |
| Women, n (%) | 252 (72.0%) | 128 (70.3%) | 124 (73.8%) | 0.469 |
| Ethnicity, n (%) | ||||
| Dominican | 300 (85.7%) | 157 (86.3%) | 143 (85.1%) | 0.875 |
| Other Caribbean Hispanic | 21 (6.0%) | 12 (6.6%) | 9 (5.4%) | |
| South American | 18 (5.1%) | 11 (6.0%) | 7 (4.2%) | |
| Unspecified Hispanic | 7 (2.0%) | 4 (2.2%) | 3 (1.8%) | |
| Central American | 4 (1.1%) | 2 (1.1%) | 2 (1.2%) | |
| Education, mean (SD), y | 10.5 ± 3.9 | 10.7 ± 3.6 | 10.1 ± 3.8 | 0.19 |
| Medication Use, n (%) | ||||
| Diabetes | 85 (24.3%) | 14 (7.7%) | 71 (42.3%) | <0.0001 |
| Metformin | 73 (20.9%) | 14 (7.7%) | 59 (35.1%) | <0.0001 |
| Sulfonylurea | 20 (5.7%) | 2 (1.1%) | 18 (10.7%) | |
| DPP-4i | 16 (4.6%) | 1 (0.5%) | 15 (8.9%) | |
| PPAR | 6 (1.7%) | 2 (1.1%) | 4 (2.4%) | |
| GLP-1a | 4 (1.1%) | 0 (0.0%) | 4 (2.4%) | |
| SGLT2i | 2 (0.6%) | 1 (0.5%) | 1 (0.6%) | |
| Blood pressure | 151 (43.1%) | 57 (31.3%) | 94 (56.0%) | <0.0001 |
| Lipid-lowering | 131 (37.4%) | 57 (31.3%) | 74 (44.0%) | 0.014 |
| APOE-ε4, n (%) | 124 (35.4%) | 71 (39.0%) | 53 (31.5%) | 0.144 |
| Fasting glucose, mean (SD), mmol/L | 6.1 ± 2.3 | 5.3 ± 1.3 | 6.8 ± 2.8 | <0.0001 |
| Waist circumference, mean (SD), cm | 96.4 ± 11.3 | 91.7 ± 9.5 | 101.3 ± 9.6 | <0.0001 |
| Triglycerides, mean (SD), mmol/L | 6.5 ± 3.4 | 5.2 ± 2.6 | 7.8 ± 3.5 | <0.0001 |
| High density lipoprotein, mean (SD), mmol/L | 3.0 ± 0.8 | 3.4 ± 0.8 | 2.6 ± 0.6 | <0.0001 |
| Low density lipoprotein, mean (SD), mmol/L | 6.1 ± 1.9 | 6.3 ± 1.8 | 6.0 ± 2.1 | 0.212 |
| Systolic blood pressure, mean (SD), mm Hg | 134.2 ± 17.8 | 133.4 ± 18.3 | 136.7 ± 17.1 | 0.139 |
| Diastolic blood pressure, mean (SD), mm Hg | 82.7 ± 10.2 | 83.3 ± 11.2 | 83.5 ± 9.4 | 0.907 |
Note: SD = Standard deviation
Metabolic syndrome presence or a continuous metabolic syndrome score were not associated with global brain Aβ SUVR examined continuously (Table 2). Among metabolic syndrome components examined individually (Table 3), compared to normal glucose, high glucose was associated with lower global brain Aβ SUVR (Beta = −0.22, 95% CI: −0.42, −0.02). This result was not significant after adjustment for multiple comparisons. None of the other components of the metabolic syndrome, waist circumference, triglycerides, blood pressure, or HDL were associated with global brain Aβ SUVR.
Table 2.
Coefficients and 95% confidence intervals from linear regression models relating the metabolic syndrome to continuous global brain amyloid standardized uptake value ratio (SUVR) and cortical thickness in the AD signature regions (n=350)
| Exposure Group | n | Model 1 | Model 2 |
|---|---|---|---|
| β | β | ||
| (95% Confidence Interval) | (95% Confidence Interval) | ||
| Global Brain Amyloid SUVR | |||
| Metabolic Syndrome Score | 350 | −0.02 (−0.10, 0.06) | −0.01 (−0.09, 0.07) |
| No Metabolic Syndrome Present | 182 | Reference | Reference |
| Metabolic Syndrome Present | 168 | −0.01 (−0.22, 0.20) | 0.01 (−0.19, 0.21) |
| AD Signature Region Cortical Thickness | |||
| Metabolic Syndrome Score | 350 | −0.06 (−0.14, 0.02) | −0.06 (−0.14, 0.02) |
| No Metabolic Syndrome Present | 182 | Reference | Reference |
| Metabolic Syndrome Present | 168 | −0.07 (−0.28, 0.14) | −0.06 (−0.27, 0.15) |
Note: Model 1: Unadjusted
Model 2: Adjusted for age, sex, and APOE-ε4
p <0.05
Table 3.
Adjusted association of dichotomized metabolic syndrome components with standardized continuous measures of global brain amyloid standardized uptake value ratio (SUVR) and cortical thickness in AD signature regions (n=350)
| Exposure Group | n | Global Brain Amyloid SUVR | AD Signature Region Cortical Thickness |
|---|---|---|---|
| Glucose | |||
| Normal | 191 | Reference | Reference |
| High | 159 | −0.22 (−0.42, −0.02)* | −0.15 (−0.35, 0.06) |
| Waist Circumference | |||
| Healthy | 133 | Reference | Reference |
| High | 217 | 0.02 (−0.20, 0.24) | −0.10 (−0.32, 0.12) |
| Triglycerides | |||
| Healthy | 274 | Reference | Reference |
| High | 76 | −0.04 (−0.28, 0.20) | −0.14 (−0.39, 0.11) |
| HDL Cholesterol | |||
| Healthy | 224 | Reference | Reference |
| Low | 126 | 0.12 (−0.08, 0.33) | 0.02 (−0.19, 0.23) |
| Systolic Blood Pressure | |||
| Healthy | 99 | Reference | Reference |
| High | 251 | 0.01 (−0.22, 0.24) | −0.06 (−0.29, 0.17) |
| Diastolic Blood Pressure | |||
| Healthy | 119 | Reference | Reference |
| High | 231 | −0.01 (−0.22, 0.21) | −0.03 (−0.25, 0.19) |
Note: Estimates are βetas (95% Confidence Interval)
Models adjusted for age, sex, and APOE-ε4
Indicates significant result after adjustment for multiple comparisons (p<0.0083 by Bonferroni correction (6 tests))
p<0.05 before accounting for multiple comparisons
Neither the presence of metabolic syndrome nor a continuous metabolic syndrome score was associated with continuous measures of cortical thickness (Table 2). None of the metabolic syndrome components were associated with cortical thickness in adjusted models (Table 3). In analyses of metabolic syndrome components defined continuously in relation to neurodegeneration (Figure 1) using linear regression adjusted for age, sex, and APOE-ε4, a one-standard deviation (SD) higher glucose level was associated with 0.15 mm smaller cortical thickness (95% CI: −0.27, −0.03), and a one-standard deviation higher triglyceride level was also associated with a smaller cortical thickness (Beta=−0.13, 95% CI: −0.23, −0.02). These results were not significant after adjustment for multiple comparisons.
Figure 1. Adjusted Association of Continuous Metabolic Syndrome Components with Standardized Continuous Measures of Global Brain Amyloid Standardized Uptake Value Ratio (SUVR) and Cortical Thickness in AD signature regions (n=350).
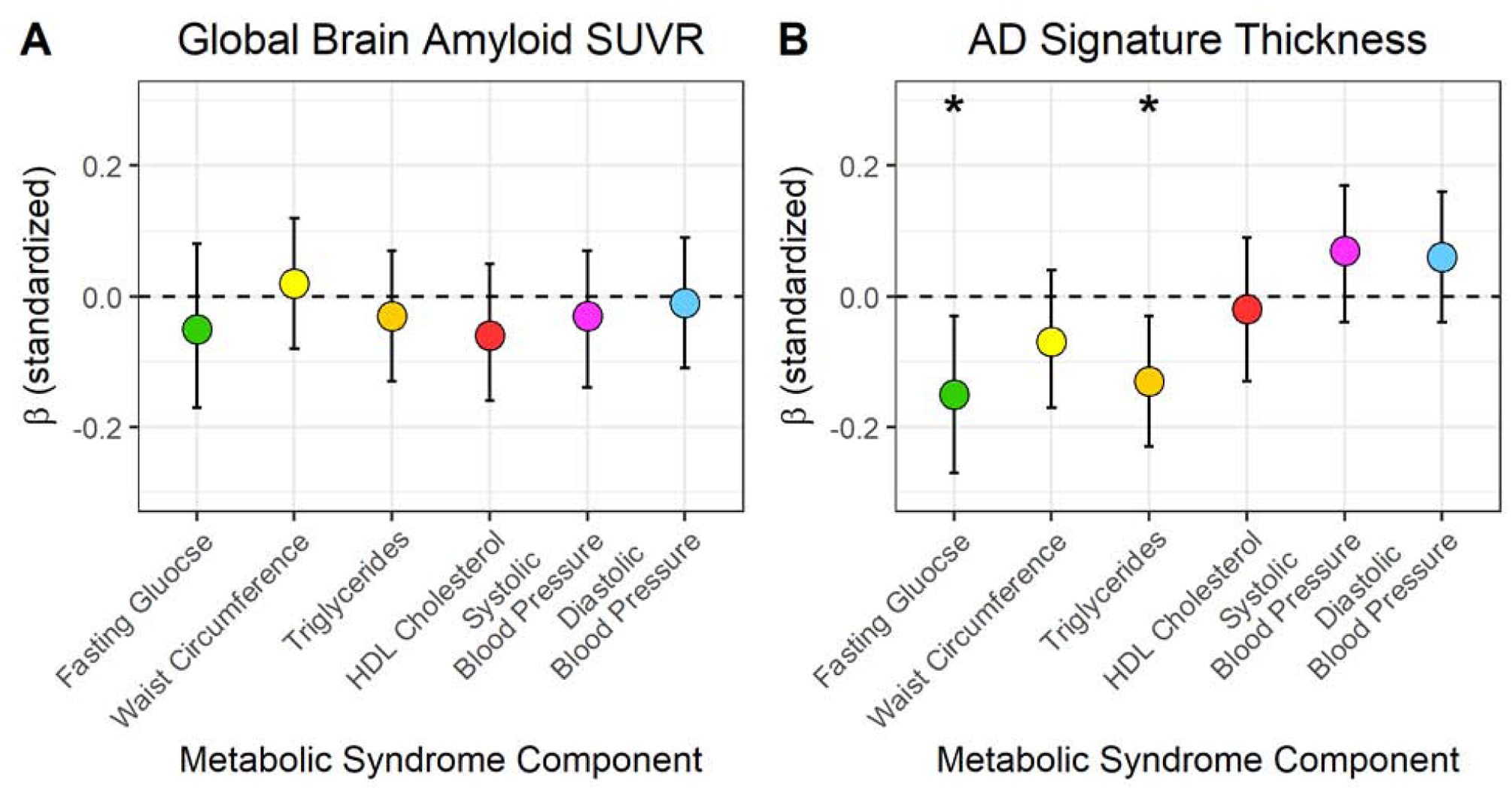
Note: 1 standard deviation= Fasting glucose: 41.0 mg/dL; Waist circumference: 11.3 cm; Triglycerides: 60.9, mg/dL; HDL cholesterol=14.7 mg/dL; Systolic blood pressure: 17.8 mmHg; Diastolic blood pressure: 10.2 mmHg; Global brain amyloid SUVR: 0.14; Cortical thickness in AD signature regions: 0.10 mm. *p<0.05 before accounting for multiple comparisons.
Neither the presence of metabolic syndrome nor the total metabolic syndrome score was associated with Aβ positivity (Table 4). Using Aβ negativity as the reference, high glucose compared to normal glucose was associated with a lower odds of Aβ positivity (Odds Ratio (OR): 0.28, 95% confidence interval (CI): 0.11, 0.75), after adjustment for age, sex, and APOE-ε. No other component of the metabolic syndrome was associated with Aβ positivity after adjustment.
Table 4.
Unadjusted and adjusted odds ratios from logistic regression models relating the metabolic syndrome and its components, defined by the National Institutes of Health criteria, to Global Brain Amyloid Positivity, n=350
| Exposure Group | n | Model 1 | Model 2 |
|---|---|---|---|
| Odds Ratio | Odds Ratio | ||
| (95% Confidence Interval) | (95% Confidence Interval) | ||
| Presence/Absence of Metabolic Syndrome | |||
| No Metabolic Syndrome Present | 182 | Reference | Reference |
| Metabolic Syndrome Present | 168 | 0.68 (0.31, 1.50) | 0.65 (0.28, 1.50) |
| Total Metabolic Syndrome Score | |||
| Metabolic Syndrome Score | 350 | 0.83 (0.61, 1.12) | 0.82 (0.60, 1.12) |
| Metabolic Syndrome Components | |||
| Glucose | |||
| Normal | 191 | Reference | Reference |
| High | 159 | 0.30 (0.12, 0.76)* | 0.28 (0.11, 0.75)* |
| Waist Circumference | |||
| Healthy | 133 | Reference | Reference |
| High | 217 | 0.94 (0.43, 2.08) | 0.85 (0.35, 2.03) |
| Triglycerides | |||
| Healthy | 274 | Reference | Reference |
| High | 76 | 0.58 (0.19, 1.72) | 0.60 (0.20, 1.87) |
| HDL Cholesterol | |||
| Healthy | 224 | Reference | Reference |
| Low | 126 | 1.37 (0.63, 2.99) | 1.28 (0.56, 2.90) |
| Systolic Blood Pressure | |||
| Healthy | 99 | Reference | Reference |
| High | 251 | 0.99 (0.42, 2.32) | 0.90 (0.36, 2.22) |
| Diastolic Blood Pressure | |||
| Healthy | 119 | Reference | Reference |
| High | 231 | 0.78 (0.35, 1.72) | 0.77 (0.33, 1.76) |
Model 1: Unadjusted
Model 2: Adjusted for age, sex, and APOE-ε4
Indicates significant result after adjustment for multiple comparisons (p<0.0083 by Bonferroni correction (6 tests))
p<0.05 before accounting for multiple comparisons
The finding relating blood glucose to lower Aβ was contrary to our hypothesis. Thus, we conducted a post-hoc examination relating diabetes treatment to Aβ to examine whether diabetes treatment could account for this finding. Among the 159 participants classified as high glucose by the metabolic syndrome criteria, 148 participants had elevated glucose only (glucose >100 mg/dL), of which 74 reported taking diabetes medications and 74 reported not taking diabetes medications. Those taking diabetes medications had similar global brain Aβ SUVR compared to those not taking medications (means adjusted for age, sex, APOE-ε4 : 1.14 ± 0.011 vs 1.14 ± 0.011; p =0.74). Lastly, glucose defined continuously was not related to Aβ SUVR (Figure 1).
4. Discussion
We found that both the presence of the metabolic syndrome by the NIH definition and the metabolic syndrome score were not associated with our primary outcome, brain amyloid, the primary driver of AD pathology. The metabolic syndrome and the metabolic syndrome score were also not related to neurodegeneration. Although the metabolic syndrome, a measure of aggregate metabolic and vascular risk, was not related to AD neuropathology, we found that individual metabolic syndrome components showed relationships with our outcomes. First, unexpectedly, higher glucose was related to lower amyloid burden, and this finding did not seem to be explained by diabetes medications. No other metabolic syndrome component was related to brain amyloid. Higher glucose and higher triglycerides were related to neurodegeneration in our sample.
There are conflicting reports on the association of the metabolic syndrome with AD dementia. A recent systematic review and meta-analysis of clinical and population-based studies included 9 longitudinal studies examining the association between metabolic syndrome and all-cause, Alzheimer’s disease, and vascular dementias (Atti et al., 2019). In a total sample of 18,313 participants with an average of 9.41 years of follow-up, it was concluded that there is no statistically significant association between metabolic syndrome and incident dementia or Alzheimer’s disease (Atti et al., 2019). Metabolic syndrome was, however, associated with an increased incidence of vascular dementia. The lack of an association was attributed to large proportions of drop-out and non-participation from the included studies, which may have introduced a survival bias impacting the results towards the null (Atti et al., 2019). Furthermore, high statistical heterogeneity was reported in the meta-analysis.
Few studies have examined the association of the metabolic syndrome with AD neuropathology. A recent analysis of 165 cognitively normal participants with a mean age of 76.4 years from the Baltimore Longitudinal Study of Aging (BLSA), metabolic syndrome was not associated with amyloid positivity; however, this study reported accelerated beta amyloid accumulation over an average of 2.6 years among participants who were amyloid positive and had metabolic syndrome (Gomez et al., 2018). This study also found that impaired fasting glucose and high blood pressure showed associations with higher amyloid. In a sample of 73 nondemented subjects 65 years and older who received Florbetapir PET imaging, impaired glycemia was associated with cerebral amyloid deposition (Morris et al., 2016); however, in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), fasting glucose was not associated with amyloid burden among 99 participants (mean age: 75 years) who received PiB-PET imaging (Toledo et al., 2012). In general, our study is consistent with the existing literature in finding no association of the metabolic syndrome as an aggregate measure of metabolic and vascular risk with AD neuropathology. Contrary to previous studies, we found an inverse relationship between glucose and amyloid burden in a larger sample at an earlier period of the lifespan. A recent analysis of data from 900 participants in ADNI who underwent lumbar puncture with a mean age of 73.54 years (McIntosh and Nation, 2019) showed that persons with untreated diabetes had higher biomarkers of AD in the cerebrospinal fluid (phosphorylated-tau, total-tau, and phosphorylated -tau/ Aβ 1–42 levels) than those with treated diabetes groups, suggesting that diabetes treatment lowers AD neuropathology. However, we found that diabetes treatment was not related to amyloid burden, and we cannot address the tau in our analyses. It is possible that our observation that higher glucose was associated with lower amyloid burden could be due to chance. However, we observed that APOE-ε4 is clearly related to higher Aβ burden as expected using our operationalization of this measure, providing face validity for our Aβ measure and supporting our finding. Also, glucose defined continuously was related to neurodegeneration consistent with other studies (Reitz et al., 2017).
Other components of the metabolic syndrome, waist circumference, blood pressure, triglycerides, and HDL, were not associated with amyloid burden or neurodegeneration, and only high triglycerides examined continuously were related to neurodegeneration. There are few studies examining the relation of these measures with amyloid burden and neurodegeneration. A study of 942 individuals in the Mayo Clinic Study of Aging found among individuals 70 to 79 years of age that obesity was not related to amyloid burden on PET or neurodegeneration on MRI (Vemuri et al., 2017). An analysis from two subsamples of BLSA, in which 191 participants underwent autopsy and neuropathological assessment, and 75 non-demented individuals underwent brain amyloid imaging, examined the association between midlife adiposity ascertained as BMI at 50 years of age and AD neuropathology (Willette et al., 2015). Higher midlife BMI was associated with greater Braak neurofibrillary, but not CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) neuritic plaque scores at autopsy overall. Higher midlife BMI was also associated with greater fibrillar amyloid on PET, but without statistical significance. Among 5,362 participants from the Insight 46 birth cohort study, continuous blood pressure measures were not associated with positive versus negative Aβ status determined based on an SUVR cutoff=0.6104 measured with Florbetapir amyloid-PET imaging (Lane et al., 2019). In the Atherosclerosis Risk in Communities (ARIC) Study, midlife hypertension, defined as systolic blood pressure ≥ 140 mmHg or diastolic blood pressure > 90 mmHg or participant report of anti-hypertensive medication use, was also not associated with elevated late-life brain amyloid, defined according to the median SUVR value >1.2, and measured with Florbetapir PET imaging (Gottesman et al., 2017). There is a dearth of studies examining the association of HDL and triglycerides with AD biomarkers. A Swedish study in 318 elderly persons with cerebrospinal fluid (CSF) biomarkers found that triglycerides in early midlife were related to higher amyloid burden measured 2 decades later on CSF, and amyloid PET in a subsample of 134 participants (Nägga et al., 2018). Our study could not replicate this findings with concurrent measures of lipids and in-vivo amyloid pathology. In the ARIC Study, neither midlife total cholesterol, LDL cholesterol, HDL cholesterol, or triglycerides were significantly associated with amyloid burden measured in late-life (Bennett et al., 2020).
Our study has several limitations that deserve consideration. First, the metabolic syndrome is an arbitrary concept that seeks to capture aggregate vascular and metabolic risk using arbitrary cutoffs for its components. In this study, we used the NIH definition of the metabolic syndrome, and some of our results, for example, for glucose and triglycerides, suggest that it may be better to examine the components continuously rather than with arbitrary categories. Second, this study’s main limitation is the cross-sectional design, and therefore we cannot infer causality. However compared with other studies, we have concurrent measures of the metabolic syndrome and in-vivo AD biomarkers. Third, this is a study of urban Hispanics in New York City who are able to undergo brain imaging, and the results may not be generalizable to other populations. However, our results relating APOE-ε4 with higher brain amyloid as observed in non-Hispanic populations (Jansen, Willemijn J. et al., 2015) suggest that our results are generalizable. Lastly, we did not report on tau, another important hallmark of AD along with Aβ and neurodegeneration (Jack, C.R. et al., 2018), which has been related to type 2 diabetes (McIntosh and Nation, 2019; Moran et al., 2015) in biomarker studies. We are currently implementing tau PET in the same participants (NCT03389100) and will report results once we achieve a sample size comparable to the present study. Despite these limitations, the main strength of our study is the availability of state-of-the-art biomarkers of amyloid in a community-based sample of middle-aged Hispanics, in which there is a paucity of information on in-vivo AD neuropathology (Jack, C.R. et al., 2018). Our sample size of 350 subjects is larger than most other studies examining AD biomarkers in middle age.
4.1. Conclusions
In conclusion, the metabolic syndrome using the NIH definition is not related to brain amyloid or neurodegeneration. Contrary to our hypothesis, glucose was found to be inversely related to amyloid SUVR. This finding does not seem to be related to diabetes treatment, requires replication in other samples, and deserves further study. Glucose and triglycerides examined continuously are related to neurodegeneration. Our findings suggest that the metabolic syndrome as an arbitrary measure of aggregate metabolic and vascular risk does not capture the risk of AD neuropathology in late middle age, and that other approaches to capture aggregate risk should be examined.
Supplementary Material
Highlights.
The metabolic syndrome is associated with dementia, but it is unclear whether metabolic syndrome is related to Alzheimer’s disease neuropathology
Neither metabolic syndrome presence nor metabolic syndrome score were associated with brain amyloid or neurodegeneration
Elevated glucose was associated with lower brain amyloid burden, but this association was not explained by diabetes treatment
Glucose and triglycerides were related to smaller cortical thickness
Funding
This work was supported by United States National Institutes of Health grants R01AG050440 and RF1AG051556. Partial support was also provided by grants K24AG045334, P30AG059303, ULT1TR001873. Dr. Palta is in part supported by grant R00AG052830 from the National Institute on Aging. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institute of Aging; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest/Disclosures
JA Luchsinger receives a stipend from Wolters Kluwer, N.V. as Editor in Chief of the journal Alzheimer’s Disease and Associated Disorders, and has served as a paid consultant to vTv therapeutics, Inc. and Recruitment Partners. AM Brickman is a paid consultant for Regeneron Pharmaceuticals and Cognition Therapeutics, Inc and owns equity in Mars Holding Limited. The other authors have no other conflicts of interests to declare.
References
- Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr., International Diabetes Federation Task Force on, E., Prevention, Hational Heart, L., Blood, I., American Heart, A., World Heart, F., International Atherosclerosis, S., International Association for the Study of, O., 2009. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120(16), 1640–1645. [DOI] [PubMed] [Google Scholar]
- Atti AR, Valente S, Iodice A, Caramella I, Ferrari B, Albert U, Mandelli L, De Ronchi D, 2019. Metabolic Syndrome, Mild Cognitive Impairment, and Dementia: A Meta-Analysis of Longitudinal Studies. Am J Geriatr Psychiatry 27(6), 625–637. [DOI] [PubMed] [Google Scholar]
- Bennett EE, Gianattasio KZ, Hughes TM, Mosley TH, Wong DF, Gottesman RF, Power MC, 2020. The Association Between Midlife Lipid Levels and Late-Life Brain Amyloid Deposition. Neurobiol Aging In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL, 2009. The Cortical Signature of Alzheimer’s Disease: Regionally Specific Cortical Thinning Relates to Symptom Severity in Very Mild to Mild AD Dementia and is Detectable in Asymptomatic Amyloid-Positive Individuals. Cerebral Cortex 19(3), 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DA, Bennett DA, Wilson RS, Bienias JL, Morris MC, Scherr PA, Hebert LE, Aggarwal N, Beckett LA, Joglekar R, Berry-Kravis E, Schneider J, 2003. Incidence of Alzheimer disease in a biracial urban community: relation to apolipoprotein E allele status. Archives of Neurology 60, 185–189. [DOI] [PubMed] [Google Scholar]
- Exalto LG, van der Flier WM, van Boheemen CJM, Kappelle LJ, Vrenken H, Teunissen C, Koene T, Scheltens P, Biessels GJ, 2015. The metabolic syndrome in a memory clinic population: relation with clinical profile and prognosis. J Neurol Sci 351(1–2), 18–23. [DOI] [PubMed] [Google Scholar]
- Ford ES, Giles WH, Dietz WH, 2002. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287(3), 356–359. [DOI] [PubMed] [Google Scholar]
- Gomez G, Beason-Held LL, Bilgel M, An Y, Wong DF, Studenski S, Ferrucci L, Resnick SM, 2018. Metabolic Syndrome and Amyloid Accumulation in the Aging Brain. J Alzheimers Dis 65(2), 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, Schneider AL, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH, 2017. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. Jama 317(14), 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr., Spertus JA, Costa F, 2005. Diagnosis and Management of the Metabolic Syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112(17), 2735–2752. [DOI] [PubMed] [Google Scholar]
- Gurland BJ, Wilder D, Lantigua R, Stern Y, Chen J, Killeffer EHP, Mayeux R, 1998. Rates of dementia in three ethnoracial groups. [PubMed]
- Haerer AF, Anderson DW, Schoenberg BS, 1987. Survey of major neurologic disorders in a biracial United States population: the Copiah County Study. Southern Medical Journal 80, 339–343. [DOI] [PubMed] [Google Scholar]
- Humes KR, Jones NA, Ramirez RR, 2011. Overview of Race adn Hispanic Origin: 2010, in: Bureau, U.C. (Ed.) 2010 Census Briefs. U.S. Department of Commerce, Washington, DC. [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14(4), 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, et al. , 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14(4), 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ, 2013. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12(2), 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, et al. , 2015. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313(19), 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, Visser PJ, and the Amyloid Biomarker Study, G., 2015. Prevalence of Cerebral Amyloid Pathology in Persons Without Dementia: A Meta-analysisPrevalence of Cerebral Amyloid Pathology in Persons Without DementiaPrevalence of Cerebral Amyloid Pathology in Persons Without Dementia. JAMA 313(19), 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmarzyk PT, Church TS, Janssen I, Ross R, Blair SN, 2005. Metabolic syndrome, obesity, and mortality: impact of cardiorespiratory fitness. Diabetes Care 28(2), 391–397. [DOI] [PubMed] [Google Scholar]
- Lane CA, Barnes J, Nicholas JM, Sudre CH, Cash DM, Parker TD, Malone IB, Lu K, James SN, Keshavan A, Murray-Smith H, Wong A, Buchanan SM, Keuss SE, Gordon E, Coath W, Barnes A, Dickson J, Modat M, Thomas D, Crutch SJ, Hardy R, Richards M, Fox NC, Schott JM, 2019. Associations between blood pressure across adulthood and late-life brain structure and pathology in the neuroscience substudy of the 1946 British birth cohort (Insight 46): an epidemiological study. Lancet Neurol 18(10), 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Cabral R, Eimicke JP, Manly JJ, Teresi J, 2015. Glycemia, Diabetes Status, and Cognition in Hispanic Adults Aged 55–64 Years. Psychosomatic medicine 77(6), 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh EC, Nation DA, 2019. Importance of Treatment Status in Links Between Type 2 Diabetes and Alzheimer’s Disease. Diabetes Care 42(5), 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JX, Chaudhary N, Akinyemiju T, 2017. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev Chronic Dis 14, E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran C, Beare R, Phan TG, Bruce DG, Callisaya ML, Srikanth V, Alzheimer’s Disease Neuroimaging I, 2015. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 85(13), 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Vidoni ED, Wilkins HM, Archer AE, Burns NC, Karcher RT, Graves RS, Swerdlow RH, Thyfault JP, Burns JM, 2016. Impaired fasting glucose is associated with increased regional cerebral amyloid. Neurobiol Aging 44, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägga K, Gustavsson A-M, Stomrud E, Lindqvist D, van Westen D, Blennow K, Zetterberg H, Melander O, Hansson O, 2018. Increased midlife triglycerides predict brain β-amyloid and tau pathology 20 years later. Neurology 90(1), e73–e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins P, Annegers JF, Doody RS, Cooke N, Aday L, Vernon SW, 1997. Incidence and prevalence of dementia in a multiethnic cohort of municipal retirees. Neurology 49, 44–50. [DOI] [PubMed] [Google Scholar]
- Prineas RJ, Demirovic J, Bean JA, Duara R, Gomez-Marin O, Loewenstein DA, Sevush S, Stitt F, Szapocznik J, 1995. South Florida Program on Aging and Health. Assessing the prevalence of Alzheimer’s disease in three ethnic groups. Journal of the Florida Medical Association 82, 805–810. [PubMed] [Google Scholar]
- Raffaitin C, Gin H, Empana JP, Helmer C, Berr C, Tzourio C, Portet F, Dartigues JF, Alperovitch A, Barberger-Gateau P, 2009. Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: the Three-City Study. Diabetes Care 32(1), 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Guzman VA, Narkhede A, DeCarli C, Brickman AM, Luchsinger JA, 2017. Relation of Dysglycemia to Structural Brain Changes in a Multiethnic Elderly Cohort. J Am Geriatr Soc 65(2), 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmi M, Bou-Zeid W, Razlighi QR, 2019. A fully automatic technique for precise localization and quantification of Amyloid-β PET scans. Journal of Nuclear Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmi M, Rippon B, Palta P, Soto L, Ceballos F, Pardo M, Sherwood G, Hernandez G, Arevalo R, He H, Sedaghat A, Arabshani S, Teresi J, Moreno H, Brickman AM, Razlighi Q, Luchsinger JA, 2020. Brain amyloid burden and resting-state functional connectivity in late middle-aged Hispanics. Front Neurol In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MX, Cross P, Andrews H, Jacobs DM, Small S, Bell K, Merchant C, Lantigua R, Costa R, Stern Y, Mayeux R, 2001. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology 56, 49–56. [DOI] [PubMed] [Google Scholar]
- Tang MX, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R, 1998. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics [see comments]. Jama 279(10), 751–755. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Toledo E, Weiner MW, Jack CR Jr., Jagust W, Lee VM, Shaw LM, Trojanowski JQ, Alzheimer’s Disease Neuroimaging I, 2012. Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 8(6), 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhanen M, Koivisto K, Moilanen L, Helkala EL, Hanninen T, Soininen H, Kervinen K, Kesaniemi YA, Laakso M, Kuusisto J, 2006. Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology 67(5), 843–847. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Knopman DS, Lesnick TG, Przybelski SA, Mielke MM, Graff-Radford J, Murray ME, Roberts RO, Vassilaki M, Lowe VJ, Machulda MM, Jones DT, Petersen RC, Jack CR Jr., 2017. Evaluation of Amyloid Protective Factors and Alzheimer Disease Neurodegeneration Protective Factors in Elderly Individuals. JAMA Neurol 74(6), 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer RA, Mayeda ER, Quesenberry CP, Lu W, Glymour M, 2014. Ethnic and racial disparities in ten-year cumulative prevalence of dementia and Alzheimer’s disease. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association 10(4), P152. [Google Scholar]
- Willette AA, Bendlin BB, Starks EJ, Birdsill AC, Johnson SC, Christian BT, Okonkwo OC, La Rue A, Hermann BP, Koscik RL, Jonaitis EM, Sager MA, Asthana S, 2015. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol 72(9), 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Falvey C, Harris TB, Newman A, Satterfield S, Koster A, Ayonayon H, Simonsick E, 2013. Effect of socioeconomic disparities on incidence of dementia among biracial older adults: prospective study. BMJ 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.