

Abstract
Among the many brain abnormalities in schizophrenia are those related to mitochondrial functions such as oxidative stress, energy metabolism and synaptic efficacy. The aim of this paper is to provide a brief review of mitochondrial structure and function and then to present abnormalities in mitochondria in postmortem brain in schizophrenia with a focus on anatomy. Deficits in expression of various mitochondrial genes have been found in multiple schizophrenia cohorts. Decreased activity of complexes I and IV are prominent as well as abnormal levels of individual subunits that comprise the complexes of the electron transport chain. Ultrastructural studies have shown layer, input and cell specific decreases in mitochondria. In cortex, there are fewer mitochondria in axon terminals, neuronal somata of pyramidal neurons and oligodendrocytes in both grey and white matter. In the caudate and putamen mitochondrial number is linked with symptoms and symptom severity. While there is a decrease in the number of mitochondria in astrocytes, mitochondria are smaller in oligodendrocytes. In the nucleus accumbens and substantia nigra, mitochondria are similar in density, size and structural integrity in schizophrenia compared to controls. Mitochondrial production of ATP and calcium buffering are essential in maintaining synaptic strength and abnormalities in these processes could lead to decreased metabolism and defective synaptic activity. Abnormalities in mitochondria in oligodendrocytes might contribute to myelin pathology and underlie dysconnectivity in the brain. In schizophrenia, mitochondria are affected differentially depending on the brain region, cell type in which they reside, subcellular location, treatment status, treatment response and predominant symptoms.
Keywords: psychosis, electron microscopy, neuropathology, cytochrome oxidase
The aim of this paper is to provide a brief introduction to schizophrenia, to review mitochondrial structure as it relates to function and then to present abnormalities in mitochondria that have been identified in postmortem brains of schizophrenia patients with a focus on anatomy and electron transport chain abnormalities.
Schizophrenia
Schizophrenia (SZ) is a biologically complex disease with several risk factors, a developmental and genetic basis, and neuropathology throughout the brain involving several transmitter systems. Briefly, SZ is a devastating mental illness that affects 1% of the world’s population (DSM). In spite of decades of research the causes, prevention and effective treatments remain elusive. SZ typically manifests itself in early adulthood with hallucinations, delusions and disorganized thought and behavior. In addition, most patients suffer from cognitive impairments and a subset present enduring negative symptoms (for example poverty of thought and speech, loss of motivation and affect). Cognitive and negative symptoms usually precede the first floridly psychotic episode and have no effective treatments, with the exception of clozapine. Antipsychotic drugs (APDs) are used to treat psychotic symptoms, but are not effective in approximately one third of patients; in treatment responders there is a gradient of response (Meltzer, 1997; Sheitman and Lieberman, 1998). Pharmacological evidence indicates that the efficacy of APDs is directly related to their ability to block dopamine D2 receptors, which are primarily located in the striatum (Creese 1976; Seeman et al., 1976). A preponderance of evidence shows that psychosis arises from an over-abundance of DA in the striatum, while cognitive and/or negative symptoms arise from an under-abundance of DA in the cortex (reviewed in Howes et al., 2012). Evidence from in vivo imaging, postmortem studies and animal models of schizophrenia implicate the glutamatergic system in schizophrenia as well (Coyle, 2006; Goff and Coyle, 2001; Javitt, 2004; Krystal, 2008), particularly in treatment resistant schizophrenia (Demjaha et al., 2014). The GABAergic system is impaired in schizophrenia, particularly in cortical and hippocampal interneurons (Lewis, 2014; Heckers and Konradi, 2015). Mitochondria are also affected in the illness, and mitochondrial defects will be discussed after a short introduction on normal mitochondrial function and anatomy.
Mitochondrial function
Mitochondria produce 95% of cellular ATP through oxidative phosphorylation, a process performed by complexes I through IV of the electron transport chain (ETC) (Wong-Riley, 1989; Huttemann et al., 2008). Mitochondria are also crucial for cellular functions such as calcium buffering (Gunter et al., 1994; Babcock and Hille, 1998; Duchen et al., 2008), modulation of synaptic activity (Li et al., 2004; Miller and Sheetz, 2004; Duchen et al., 2008; Sheng and Cai, 2012), regulation of apoptosis (Susin et al., 1999), and production of reactive oxygen species (Chang and Reynolds, 2006). Mitochondria are dynamic organelles that change intracellular location in response to energy demands (Ligon and Steward, 2000; O’Toole et al., 2008; Niescier et al., 2016). They are essential for normal formation of dendritic cytoarchitecture and dendritic spines (Li et al., 2004; Sheng and Cai, 2012), and are in part regulated by DISC1 for this particular function (Norkett et al., 2016). At the synaptic level, mitochondria provide the vast majority of energy for ionic homeostasis in axon terminals, synaptogenesis, synaptic transmission, synaptic vesicle recycling, and long-term potentiation (Mjaatvedt and Wong-Riley, 1988; Li et al., 2004; Change et al., 2006; Vos et al., 2010; Sheng and Cai, 2012; Pathak et la., 2015). The production of ATP and calcium buffering are essential in maintaining synaptic strength and abnormalities in these processes could lead to decreased metabolism and defective synaptic activity (Ben-Shachar and Laifenfeld, 2004; Chang and Reynolds, 2006; Duchen et al., 2008).
Cellular function and proper energy generation requires the tricarboxylic acid (TCA) cycle, which is located in the mitochondrial matrix. The enzymes of the TCA cycle (also known as the citric acid cycle or the Krebs cycle) produce the reducing equivalents NADH and FADH2, which in turn deliver electrons to complexes of electron transport chain (ETC), which drives ATP production. Optimal cellular function requires proper functioning of the ETC, which is comprised of four enzymes located within the inner mitochondrial membrane. These enzymes create a proton gradient used to power the enzyme ATP synthase (sometimes referred to as Complex V), which produces ATP. Each complex of the ETC is comprised of several subunits encoded either by nuclear or mitochondrial DNA (70 and 13 subunits, respectively). Abnormalities in a single enzyme of the electron transport chain are sufficient to cause disruption of cellular metabolism. Complexes I, II/III and IV of the electron transport chain can be measured to assess mitochondrial function (Wong-Riley, 1989). The evidence that Complex IV (cytochrome c oxidase, COX) is coupled to neuronal energy demands is derived from studies in which changes in COX activity can be induced by experimental interventions that alter neuronal activity.
Mitochondrial function declines in the aging brain (Bornstein et al., 2020), due in part to the accumulation of oxidative damage (Shigenaga et al., 1994). In the aging nervous system, there are reports of fewer mitochondria, but they are larger in size (Shigenaga et al., 1994; Martinelli et al., 2006; Soghomonian et al., 2010). Functionally, bigger mitochondria are able to meet short energy demands, but sustained energy demands are not met (Shigenaga et. al., 1994; Soghomonian et al., 2010; Martinelli et al., 2006).
Mitochondrial structure
Mitochondria are structurally complex, dynamic organelles that fuse, divide, change shape and move around the cell (Isaacs et al., 1992; Hollenbeck, 1996; Legros et al., 2002; Hollenbeck and Saxton, 2005; MacAskill et al., 2010; Otera et al., 2010; Loson et al., 2013; Bertholet et al., 2016; Ploumi et al., 2017) (Figure 1A). Mitochondria can assume different shapes (Picard and McEwen, 2014), which in most cases have functional implications (Youle and van der Bliek, 2012; Ahmad et al., 2013) (Figure 1B). For example, round and rod shapes reflect healthy mitochondria, while blob and donut shapes indicate diseased states (Liu and Hajnóczky, 2011; Ahmad et al., 2013; Picard and McEwen, 2014; Hara et al., 2014). Moreover, there is a relationship between the shape of mitochondria and the production of reactive oxygen species. In cell culture, mitochondrial stressors can induce the sequential conversion of mitochondria from rod-shaped to donut-shaped, to blob-shaped (Liu and Hajnóczky, 2011; Ahmad et al., 2013) (Figure 1C). Blob-shaped mitochondria generate the highest levels of reactive oxygen species, followed by donut shaped compared to straight mitochondria (Liu and Hajnóczky, 2011; Ahmad et al., 2013). While donut-shaped mitochondria can revert to the straight configuration, blob-shaped mitochondria are unable to revert to healthier configurations. In axon terminals in the dorsolateral prefrontal cortex of non-human primates, donut-shaped mitochondria are associated with shorter synapses, fewer docked vesicles and are correlated with poor delayed response memory (Hara et al., 2014). This finding could be relevant for schizophrenia pathophysiology as there is a robust decline in prefrontal cortical cognitive abilities in the illness (Goldman-Rakic, 1999).
Figure 1.
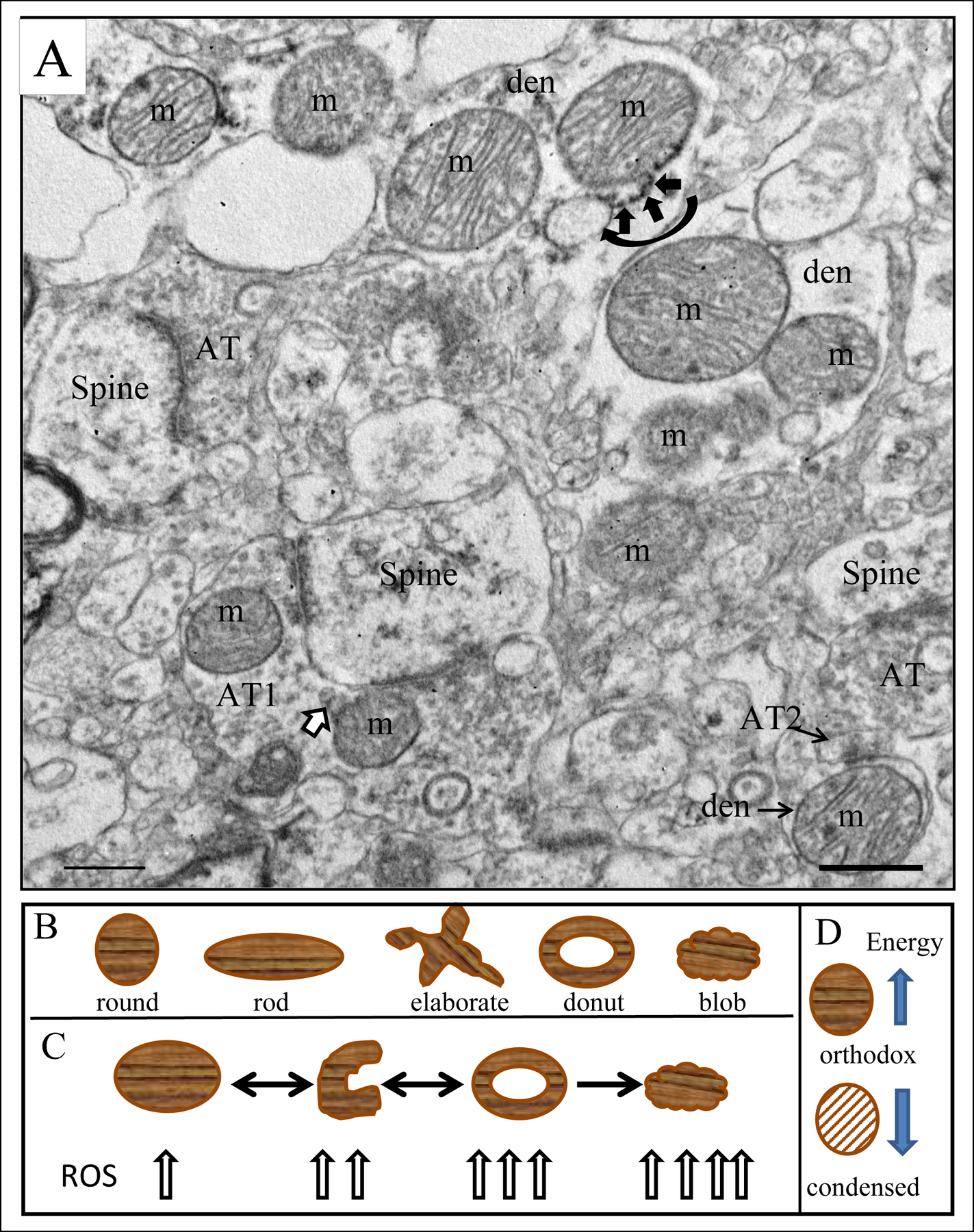
A) Electron micrograph of human striatum. Mitochondria (m) are indicated in various subcellular locations. In the dendrite (den) at the top of the field, a mitochondrial associated ER (MAM) is shown (curved black arrow) with ER (short black arrows) connecting to the adjacent mitochondrion. Axon terminal (AT1) forms an excitatory synapse on a spine in the lower part of the field; mitochondrial derived vesicles (MDVs) are shown (white arrow with black outline) budding off of a mitochondrion in the terminal. Axon terminal AT2 forms an inhibitory synapse on the dendrite (den). Scale bar = 0.5 µm. Figure is modified from Figure 2a in Somerville et al., 2011b and Figure 1 in Roberts, 2017). B) Drawings of different shaped mitochondria. C) Transformation of a mitochondrion from round/rod to curved to donut to blob shape and the corresponding amount of reactive oxygen species each produces. Arrows are bidirectional between round and curved and donut shaped mitochondria indicating the ability to change shape in either direction. Once a mitochondrion has assumed a blob shape, it cannot recover healthier configurations, thus the unidirectional arrow. D) Depiction of the orthodox and condensed form of mitochondria. Orthodox configuration is high energy producing, while condensed configuration is low energy producing, indicated by the directionality of the arrows.
The morphology of the cristae, matrix and inner mitochondrial membrane correspond to the activity of the electron transport chain (Hackenbrock, 1968). The orthodox configuration of mitochondria, which is typically illustrated in electron micrographs (Figure 1A), corresponds to higher energy producing states (Hackenbrock, 1968). The condensed configuration corresponds to low energy producing states. The morphological features of the condensed configuration include a small and dense matrix, an irregularly organized inner membrane with few cristae, and an enlarged space between inner and outer membranes (Figure 1D). Thus, examining the size and shape of mitochondria can reveal important information about their functionality.
Mitochondrial abnormalities in schizophrenia
Among the many brain abnormalities in schizophrenia are those related to mitochondrial functions such as oxidative stress, energy metabolism and synaptic efficacy (see reviews by Shao et al., 2008; Clay et al., 2011; Martins-de-Souza et al., 2011; Anglin et al., 2012; Manji et al., 2012; Hjelm et al., 2015; Ni and Chung, 2020). Indeed, mitochondrial pathology is a frequent finding in schizophrenia, as shown by various techniques in patients, postmortem samples, cell lines and animal models. That said there are many non-replications in the literature, which is a common plague in schizophrenia research. Part of the problem in reconciling the literature on mitochondria in schizophrenia are the differences between studies in techniques, brain areas, and patient characteristics. In addition, it is difficult to compare many findings because different things were being studied, such as different subunits of a given complex. The present review will concentrate on findings, particularly anatomical, derived from postmortem studies (Tables II–IV).
Table II:
Genetics
| finding | Brain area | comments | reference |
|---|---|---|---|
| ↓ in four mitochondrial rRNA three encode parts of the 16s rRNA | PFC | Mulcrone, et al., 1995 Whatley, et al., 1996 | |
| ↓ in malate shuttle & TCA genes | PFC | Middleton et al., 2002 | |
| ↓ mitochondrial genes related to energy metabolism and oxidative stress | DLPFC | ↓ transcript levels of pyruvate dehydrogenase APD effects ruled out | Prabakaran et al., 2004 |
| Δ expression of 11% of mitochondria-related genes 82% of those were ↓ | DLPFC | Findings held up when controlled for pH, might be APD effect | Iwamoto et al., 2005 |
| synonymous base pair substitutions in the coding regions of the mtDNA genome was 22% higher in SZ | DLPFC | pH sensitive, PMI independent | Rollins et al, 2009 |
| Genetic polymorphisms in molecules associated with proline metabolism | BA10 & superior temporal gyrus | Genetic data showing abnormal metabolism of proline. | Nagaoka et al., 2020 |
| ↓ in mitochondrial genes mitochondrial oxidative energy metabolism (isocitrate, lactate, malate, NADH, complexes II, IV, ATP synthase) | Hippocampus, laser captured granule cells | Data derived from multiple cohorts | Altar et al., 2005 |
| Review cites 57 mitochondrial genes changed in SZ in at least 2 studies | data support that SZ has many dysregulated mitochondrial genes | Hjelm et al., 2015 | |
| ↓ in mitochondrial genes with QPCR confirmation | DLPFC | Layer 3 pyramidal neurons, controlled for APD effects | Arion et al., 2015 |
| No Δ mtDNA common deletion | Frontal cortex | Cavelier et al.,, 1995; Kakiuchi et al., 2005; Sabunciyan et al., 2007; Fuke et al., 2008; Shao et al.,2008 | |
| No Δ mtDNA common deletion in schizophrenia ↑mtDNA common deletion with age, not diagnosis mtDNA common deletion very variable across brain areas |
DLPFC ACC, OFC, amygdala hippocampus caudate, NAcc putamen, SN, thalamus, cerebellum | ↑mtDNA common deletion in striatum, NAcc and amygdala with age ↑mtDNA common deletion SN> putamen, NAcc & Caudate vs other brain areas |
Sequeria et al., 2012 |
| ↓ brain mtDNA common deletion in DA rich areas when corrected for age, sex, PMI and pH | DLPFC, ACC, OFC, amygdala hippocampus caudate, NAcc putamen, SN, thalamus | Common deletion has genes encoding sub-units of COX, NADH-d and ATP synthase, affecting mitochondrial function in DA areas. | Mamdani et al., 2014 |
The table is organized by the brain area from cortex to subcortical regions. Abbreviations are in Table I.
Table IV:
Mitochondrial number, structure, and localization
| finding | brain area | comments | reference |
|---|---|---|---|
| ↓ # mitochondria in axon terminals | ACC | ↓ synaptic efficacy | Aganova & Uranova 1992 |
| Layer III: ↓#mitochondria at excitatory synapses Layer V/VI: ↓#mitochondria at inhibitory synapses and in soma. Structure & size normal. | ACC | ↓ synaptic efficacy of thalamic inputs ↓ synaptic efficacy of DA input and/or interneuron connections ↓ energy capacity in projection neurons |
Roberts et al., 2015 |
| ↓ # and size of mitochondria in oligodendrocytes | Prefrontal cortex | Abnormalities in oligodendrocyte energy might disturb myelin, axonal integrity and thus circuitry. | Uranova et al., 2007 |
| ↓ # and size of mitochondria in oligodendrocytes | Frontal cortex white matter | Vikhreva et al., 2016 | |
| ↓ # and size of mitochondria in oligodendrocytes adjacent to microglia | Frontal cortex white matter | oligodendrocyte dystrophy is not associated with microglial activation | Uranova et al., 2018 |
| ↓ # and size of mitochondria in oligodendrocytes adjacent to microglia | Frontal cortex grey matter | Microglial dystrophy might contribute to oligodendrocyte dystrophy in SZ during relapse of positive symptoms | Uranova et al., 2020 |
| ↓ # and area of mitochondria in astrocytes in SZ with DOI >20 years vs NCs and SZ with DOI<20yrs | hippocampus | Decreased energy of astrocytes with DOI | Kolomeets et al., 2010 |
| ↓ # mitochondria in astrocytes | caudate | Decreased energy available in astrocytes | Uranova et al., 1996 |
| ↓ size mitochondria in oligodendrocytes | caudate | Compromised function in oligodendrocytes | Uranova et al., 2001 |
| No Δ in mitochondrial size in the neuropil | caudate putamen | No obvious morphological abnormalities | Kung & Roberts 1999 |
| ↓ # mitochondria in neuropil | caudate putamen | Treatment responders and paranoid SZ may be able to decrease number of mitochondria and thus lower energy capabilities in the hyperactive glutamate system. This could translate into better outcomes than in treatment resistant or CUT SZ. | Somerville et al., 2012a |
| ↓ # mitochondria in axon terminals in treatment responders, but not treatment resistant SZ | caudate putamen | Somerville et al., 2011 | |
| ↓ # mitochondria in axon terminals in chronic paranoid SZ, but not chronic undifferentiated SZ | putamen | Somerville et al., 2012b | |
| mitochondria appearance, density, and size are normal in dopaminergic axon terminals | nucleus accumbens | Normal number and morphology of mitochondria in dopaminergic inputs to the nucleus accumbens. | McCollum et al., 2015 |
| Hyperplasia of mitochondria in axon terminals synapsing onto dopamine neurons | substantia nigra | Unmet energy requirements in terminals synapsing onto dopamine neurons | Kolomeets et al., 1999 |
| = density, size & structure of mitochondria in dopamine neurons | substantia nigra | Normal number and morphology of mitochondria in dopaminergic neurons and in axon terminals in the substantia nigra | Walker et al., 2018 |
| = density, size & structure of mitochondria in axon terminals | substantia nigra | Mabry et al., 2019 |
The table is organized by the brain area from cortex to subcortical regions. Abbreviations are in Table I.
Genetics
Deficits in expression of various mitochondrial genes have been found in multiple schizophrenia cohorts (Table II). Most of the brain regions studied have been cortical regions (Mulcrone et al., 1995; Whatley et al., 1996; Middleton et al., 2002; Prabakaran et al., 2004; Iwamoto et al., 2005; Rollins et al., 2009; Nagaoka et al., 2020) or the hippocampus (Altar et al., 2005). While not all studies of the same brain region identify similar genes, it is clear that mitochondrial genes are affected in the illness. A recent review (Hjelm et al., 2015) identified 57 mitochondrial genes that were found to be dysregulated (mostly downregulated) in at least two independent studies. Reductions in expression in genes include those involved in proline metabolism (Nagaoka et al., 2020), the mitochondrial malate shuttle system, the tricarboxylic acid cycle and the electron transport chain (Middleton et al., 2002; Altar et al., 2005). Proteomics studies showed decreases in gene expression involved in energy metabolism and oxidative stress in 90% of the schizophrenia cohort examined (Prabakaran et al., 2004). Mitochondrial gene expression is affected by pH, with more genes affected in subjects with prolonged agonal status and low pH (Iwamoto et al., 2005; Vawter et al., 2006). Therefore, pH and agonal status are important considerations when evaluating or planning studies in schizophrenia, and inconsistencies in attention to these details may account for different reports in the literature.
The mtDNA common deletion is a somatic 4,977 base pair deletion of the mitochondrial genome (Soong et al., 1992). Several findings regarding the common deletion are very well replicated in normal brains. The common deletion is found in adult but not fetal tissue suggesting that it accumulates with age. The amount of the common deletion varies greatly depending on the brain region. Levels o f the deletion are highest in dopamine containing nuclei and projection sites (Soong et al., 1992). There are several reports on the levels of the mtDNA common deletion in schizophrenia, but most of the results show no change. Sequeria et al., (2012) found an increase in the common deletion with age especially in the dopamine rich areas, such as the SN and dorsal striatum, but no change in schizophrenia. In addition, no changes were detected in the common deletion in several cortical areas, striatum, limbic system and thalamus (Sequeria et al., 2012). Others have also shown no changes in the common deletion in the frontal cortex or caudate nucleus (Cavelier et al., 1995; Kakiuchi et al., 2005; Sabunciyan et al., 2007; Fuke et al., 2008; Shao et al., 2008). In contrast, Mamdani et al., (2014) reported a decrease in the common deletion in schizophrenia with the largest abnormalities in dopaminergic regions including the ventral midbrain. The common deletion contains genes encoding subunits of cytochrome oxidase, NADH dehydrogenase and ATP synthase (Samuels et al., 2004; Verge et al., 2011). Oxidative stress mechanisms related to dopamine metabolism might be involved in the accumulation of the common deletion suggesting that mitochondrial function is impaired in dopaminergic nuclei and projection sites. While these are key areas affected in schizophrenia, if the common deletion plays a role in the pathology of schizophrenia, one would expect an increase in accumulation of the common deletion in key brain areas already in late teens and young adulthood when the disease first manifests itself. Since there is no evidence that this happens, it appears that the common deletion does not play a role in the pathogenesis of schizophrenia.
Activity of the electron transport chain
Some of the most thoroughly studied metabolic abnormalities in schizophrenia indicate disruptions in oxidative phosphorylation in various cortical regions and the basal ganglia (Table III). The results in cortex are mixed. Some have found decreased activity of complex I (Cavelier et al., 1995; Maurer et al., 2001) and a decreased protein levels in complex I subunits (Holper et al., 2019), while Andreazza et al., (2010) found no change in activity of complex I or of complexes III and IV. In addition, decreases in COX subunit II mRNA were found (Whatley et al., 1996; Clark et al 1999; Maurer et al., 2001; Andreazza et al., 2010). Several studies have shown mitochondrial abnormalities in the striatum in subjects with schizophrenia such as decreases in complex I and III and IV activity, protein and/or mRNA levels (Cavelier et al., 1995; Prince et al., 1999, 2000; Maurer et al., 2001; Ben-Shachar and Karry, 2008; Ben-Shachar, 2017). The activity of the complexes do not necessarily change in the same direction in all nuclei. For instance, there is a decrease in COX (complex IV) activity in the caudate and an increase in COX and succinate dehydrogenase (complex II) in the putamen and nucleus accumbens in postmortem tissue from schizophrenia patients (Prince, et al., 1999). Interestingly, COX and complex II have been shown to correlate with the severity of symptoms in the putamen (Prince et al., 2000), linking symptoms with mitochondrial dysfunction. Most of the changes to complex I appear to be caused by antipsychotic drugs (APDs) (Burkhardt et. al., 1993; Maurer and Moller, 1997; Prince et al., 1997; Balijepalli et al., 1999, 2001; Karry et al., 2004; Streck et al., 2007; Rosenfeld et al., 2011), while COX appears to be less affected (Whatley et al., 1996).
Table III:
Mitochondrial enzymes and proteins
| finding | Brain area | comments | reference |
|---|---|---|---|
| ↓ COX subunit II mRNA expression but no Δ in COX activity | Frontal cortex | This can happen without any changes in COX activity. | Whatley et al., 1996; Clark et al., 1999 |
| ↓ complex IV activity | frontal & temporal cortex, basal ganglia | Deficit in OxPhos in cortex may contribute to deficits in energy generation | Maurer et al., 2001 |
| ↓ complex I, III activity | temporal cortex, basal ganglia | ||
| No Δ in levels of NDUFS7, a subunit of complex I, & complex I activity | prefrontal cortex | Complex I in PFC is unaffected | Andreazza et al., 2010 |
| Δ in enzymes of TCA cycle | DLPFC | Abnormalities in energy metabolism could contribute to brain pathology in SZ. | Bubber et al., 2011 |
| ↑ ALDH4A1 detected with IHC Genetic polymorphisms in molecules associated with proline metabolism. ALDH4A1 is step in the metabolism of proline to glutamate, which occurs in mitochondria. | frontal cortex (BA10) & superior temporal gyrus | Genetic and anatomical data shows abnormal metabolism of proline, which may affect glutamate neurotransmission. | Nagaoka et al., 2020 |
| ↑ levels of a marker of oxidative stress | anterior cingulate cortex | Oxidative damage may contribute in part to brain pathology in SZ. | Wang et al., 2009 |
| No Δ in protein levels of mitofusin2 | anterior cingulate cortex | No difference in SZ, no effect of treatment, or treatment response. | Barksdale et al., 2014 |
| ↓ in mitochodrial proteins in synaptosomes | Primary auditory cortex | MacDonald et al., 2019 | |
| ↓ COX activity | frontal cortex caudate, | Could lead to abnormalities in energy metabolism | Cavelier et al., 1995 |
| Meta analysis: variable Δ in complex I subunits; ↓ COX activity in cortex, but ↑ in striatum | frontal cortex, striatum | Holper et al., 2019 | |
| ↓ protein levels of pyruvate dehydrogenase | frontal cortex | shift away from the TCA cycle toward glycolysis | Prabakaran et al., 2004 |
| ↓ β subunit of pyruvate dehydrogenase; ↑ pyruvate, glucose, lactate | striatum | Impaired glucose metabolism; shift away from the TCA cycle toward glycolysis | Dean et al., 2016 |
| ↓ COX activity | caudate | ↓ COX activity can lead to ↑ susceptibility to apoptosis | Prince et al., 1999 |
| ↑ COX complex II activity | putamen | Negative correlation with the severity of symptoms | Prince et al, 1999 |
| ↓ protein levels of ARF1, a mitochondrial protein, in synaptosomes | ventromedial caudate | ↓ ARF1 especially in glutamatergic synapses may compromise excitatory synaptic function | Ramos-Miguel et al., 2019 |
| No Δ in COX activity ↓protein levels of COX subunits II and IV-I | rostral substantia nigra ventral tegmental area | Faulty assembly of COX enzyme could lead to greater vulnerability to metabolic insult. | Rice et al., 2014 |
| No Δ in complex I, III or IV | cerebellum | Cerebellum is not affected. | Maurer et al., 2001 |
The table is organized by the brain area from cortex to subcortical regions. Abbreviations are in Table I.
In one of our previous studies, COX activity and the protein expression of key subunits for its assembly were measured in postmortem substantia nigra/ventral tegmental area (SN/VTA) (Rice et al., 2014). While overall COX activity was similar between schizophrenia patients and controls, there were decreases in the protein expression of two of the COX subunits (II and IV-I) in schizophrenia in samples containing rostral regions of the SN/VTA. These changes in the schizophrenia group probably were not caused by medication because samples containing only the middle to caudal portions of the SN/VTA were unaffected as were the SN/VTA from rats chronically treated with antipsychotic drugs (Rice et al., 2014).
Subunit IV of the COX enzyme is crucial for the proper functioning of the COX complex as a whole (Nijtmans et al., 1998; Clark et al., 1999; Rahman et al., 1999). COX subunit II is responsible for the binding of cytochrome c and the subsequent electron transfer to subunit I of the COX enzyme (Taanman, 1997). Interestingly, decreases in complex IV-II mRNA expression in the frontal cortex in schizophrenia have not resulted in significant changes in overall COX activity (Clark et al., 1999), suggesting that there might be some compensatory mechanisms involved that restores overall COX activity to normal in spite of a deficit in COX-II. However, suppression of subunit IV has been linked to a reduced function in overall COX activity and an increased susceptibility to apoptosis (Huttemann et al., 2001; Li et al., 2006). Thus, deficits in COX-IV subunit protein expression may lead to a faulty assembly of the COX enzyme and a greater vulnerability to metabolic insult in a region specific manner in the SN/VTA.
Tricarboxylic acid cycle
A thorough discussion of all of the intricacies of the TCA cycle as it related to schizophrenia is outside of the scope of this review. Briefly, two recent meta-analyses of postmortem and imaging studies strongly suggest that schizophrenia is associated with increased lactate and decreased pH in the brain (Hagihara et al., 2018; Pruett and Meador-Woodruff, 2020). Pruett and Meador-Woodruff (2020) discuss that the consequence of this could lead to a shift away from the TCA cycle and oxidative phosphorylation toward increased glycolysis for energy production. Pyruvate dehydrogenase (PDH), the enzyme that converts pyruvate to acetyl-CoA in order for it to enter the TCA cycle, is downregulated in the brain of schizophrenia patients (Dean et al., 2016; Prabakaran et al., 2004), supporting this hypothesis. Functionally, this indicates decreased energy production, increased lactate, and decreased pH, which is linked to cognitive and emotional impairments (Rae et al., 1996; Shioiri et al., 1997; Rowland et al., 2016).
Anatomy
Interestingly, much of the anatomical studies on mitochondria in schizophrenia have been performed at the electron microscopic level by my laboratory and Uranova’s group (Table IV). This is striking because ultrastructural studies, especially quantitative ultrastructural studies with or without combined immunohistochemistry, are rare in postmortem schizophrenia research due to practical issues such as needing brains with very short postmortem intervals. We have both published extensively on ultrastructural differences in multiple brain regions and for the most part our results are compatible when we have studied the same cell type or brain region.
Intracellular Abnormalities:
Mitochondria have structural appendages called mitochondria derived vesicles (MDVs) and mitochondria-associated endoplasmic reticulum membranes (MAMs) (Hayashi et al., 2009). MDVs are structures that bud off mitochondria and transport damaged cargo to peroxisomes or lysosomes (Neuspiel et al., 2080) (Figure 1A). MDVs are stimulated by various forms of stress, and the vesicles incorporate cargo, whose composition depends upon the type of stress (Soubannier et al., 2012). MDVs have not been studied in schizophrenia but could be a fertile field of study considering their function.
Mitochondria are connected to the endoplasmic reticulum via MAMs (Hayashi et al., 2009) (Figure 1A). MAMs are enriched in cholesterol, anionic phospholipids, (Hayashi and Fujimoto, 2010) and proteins related to the control of mitochondrial division (Friedman et al., 2011) and dynamics (Schon and Area-Gomez, 2013). MAMs are involved in a number of key metabolic functions, including phospholipid and cholesterol metabolism (Hayashi et al., 2009). Mitofusin 2 tethers mitochondria to the endoplasmic reticulum (de Brito and Scorrano, 2008). Mitochondria move within neurons along microtubules via kinesin and adaptors for anterograde transport and via dynein and adaptors for retrograde transport; they also can be anchored via actin and neurofilaments (reviewed by Lin and Sheng, 2015).
Disrupted in Schizophrenia 1 (DISC1) is a scaffold protein that is involved in intracellular functions and abnormalities in DISC1 are linked to cognitive and emotional deficits in schizophrenia (see review by Roberts, 2007 and references therein). DISC1 is predominantly localized to mitochondria (James et al., 2004) and in particular to MAMs (Park et al., 2017). At the MAM, DISC1 modulates the transfer of calcium from endoplasmic reticulum to the mitochondria. Disrupted DISC1 causes increased calcium transfer leading to increased calcium accumulation in mitochondria following oxidative stress, which impairs mitochondrial functions. (Park et al., 2017).
Cortex:
The anterior cingulate cortex, a structurally and functionally diverse region, is one of several brain regions that is abnormal in schizophrenia (Fornito et al., 2009). Mitofusion-2 is a mitochondrial fusion protein (Koshiba et al., 2004) that is also necessary for the maintenance and operation of the mitochondrial network (Bach et al., 2003) and transporting mitochondria to their proper location in axons and dendrites (Misko et al., 2010; Sheng and Cai, 2012). Protein levels of mitofusin-2 were normal in schizophrenia cases; moreover, there were no effects of antipsychotic treatment or treatment response (Barksdale et al., 2014). Normal protein levels suggest that mitochondrial fusion, maintenance and operation of the mitochondrial network may be intact. However, while protein levels of mitofusin 2 are unaffected, there are many other mitochondrial proteins involved in these functions that might be abnormal.
In an ultrastructural study, the numbers of mitochondria per neuronal somata and per axon terminal were decreased in a layer and input specific manner (Aganova and Uranova, 1992; Roberts et al., 2015). Excitatory synapses in superficial layers, likely arising from the medial dorsal thalamus and contralateral cortex (see Hoftman et al., 2016), had fewer mitochondria per axon terminal (Aganova and Uranova, 1992; Roberts et al., 2015). Synapses characteristically made by inhibitory interneurons and/or dopaminergic inputs (Kubota et al., 2016) had fewer mitochondria per axon terminal in deep layers (Roberts et al., 2015). Fewer mitochondria in axon terminals suggest a decrease in efficacy of synaptic transmission (Brodin et al., 1999; Verstreken et al., 2005; Hall et al., 2012), and in the case of the anterior cingulate cortex this abnormality affects both excitatory and inhibitory connections. Pyramidal neurons in the deep layers, which project to the striatum, brainstem, or thalamus (Goldman and Nauta, 1977), had fewer mitochondria per soma, suggesting compromised metabolism in one or more of those pathways (Roberts et al., 2015). There were no structural differences and no obvious blob or donut shaped mitochondria. This is somewhat surprising considering the observed increase in reactive oxygen species in schizophrenia (Wang et al., 2009; Madireddy and Madireddy, 2020) that are produced at a higher rates in mitochondria with those shapes (Liu and Hajnóczky, 2011; Ahmad et al., 2013). However, it is possible that upstream biochemical pathways, such as the pentose phosphate shuttle, may be responsible for the increase in reactive oxygen species (Koo et al., 2018). The layer specific location of the mitochondrial abnormalities suggests multiple connections are affected that might impact the cortex as well as several downstream pathways. Madireddy
Fewer mitochondria may be a primary deficit of the disease, or mitochondria may die as an epiphenomenon of the disease. Alternatively, mitochondria may be sequestered in neuronal somata located either extrinsic to the region studied. An inability of mitochondria to move into axon terminals or dendrites could account for a decreased number of mitochondria in these structures. Since mitochondria move around the neuron along microtubules between the soma and processes, damage to cytoskeletal elements might lead to a failure of proper mitochondrial movement. In the cingulum bundle, but not the arcuate fasciculus or the corpus callosum, we found abnormally high protein levels of αlpha-tubulin, a component of microtubules, in off drug schizophrenia, which was normalized by APD treatment (Schoonover et al., 2018). Moreover, in the schizophrenia cohort correlations between alpha-tubulin and other markers of white matter integrity (neurofilament heavy, myelin basic protein, and the autophagosome marker LC3) were opposite to controls in the cingulum bundle. These data suggest there is a dysregulation of the relationship between α-tubulin and the other markers of white matter integrity in the cingulum bundle in schizophrenia. Taken together, cytoskeletal abnormalities that could lead to faulty transport of mitochondria in a regionally specific manner.
Striatum:
Previous ultrastructural studies of striatal neuropil in schizophrenia have shown similar numbers (Somerville et al., 2011a) and size (Kung and Roberts, 1999) of mitochondria. However, decreases in the number of mitochondria per synapse were detected in both the caudate and putamen in schizophrenia. Since the majority of mitochondria are in dendrites, fewer mitochondria in axon terminals might have been overlooked in overall neuropil counts. Further analysis showed that subjects divided by treatment status into off drug, atypical APD or typical APD, all showed significant decreases in the putamen compared to controls (Somerville et al., 2011a). Since the patients on APD had similar decreases in mitochondrial number compared to the off-drug subjects, this result may not be an APD effect. Moreover, it seems unlikely that APDs would affect the putamen but not the caudate. However, haloperidol, a typical APD, does reduce the number of mitochondria in striatal neuropil of chronically treated rats (Roberts et al., 1995).
Mitochondrial pathology is linked with symptoms and symptom severity.
Symptoms of schizophrenia can vary markedly between patients, and similar symptoms may be related to shared pathophysiology. For example, a relationship between symptoms and mitochondrial pathology is evident in blood. Lymphocytes analyzed from paranoid schizophrenia patients showed less mitochondrial volume than in controls (Uranova et al., 2007). Moreover, the severity of the mitochondrial deficit was positively correlated with symptom severity, linking the severity of paranoid symptoms with mitochondrial impairment, albeit in blood (Uranova et al., 2007). Also, lower levels of COX and complex II activity correlate with increased severity of emotional and cognitive impairment in the putamen, but not other basal ganglia regions (Prince et al., 2000), again linking symptoms with mitochondrial dysfunction in a region specific way.
In an ultrastructural study of the striatum, decreases in the density of mitochondria were observed in the neuropil in chronic paranoid subjects compared to both controls and the chronic undifferentiated group (Somerville et al., 2012). In addition, the number of mitochondria in axon terminals in the putamen was decreased selectively in chronic paranoid subjects compared to controls and chronic undifferentiated patients. The number of mitochondria per synapse showed similar decreases compared to controls in both subgroups in the putamen, and a similar albeit insignificant pattern in the caudate. These deficits were found only in the matrix compartment, and not in the striosomes; cognition and memory are processed through the matrix, while limbic information is processed through the patches (Graybiel and Ragsdale, 1978; Flaherty and Graybiel, 1993; Eblen and Graybiel, 1995; Goldman-Rakic, 1999). Thus, it could be expected that decreased numbers of mitochondria in the striatal matrix in schizophrenia could impact cognitive skills.
Given the period of sample collection, the schizophrenia subjects in that study were diagnosed in accordance with the DSM-III and DSM-IV criteria, which define prominent symptomology at the time of death (Deep-Soboslay et al., 2005). In contrast, the DSM-V recognizes that predominant symptoms may fluctuate over the course of the illness, and that the diagnosis initially given may not reflect symptomology at the time of death. Importantly, in this study, all paranoid schizophrenia subjects were given a lifetime diagnosis of chronic paranoid schizophrenia with no history of formal thought disorder. Taken together, fewer mitochondria than normal in blood and the striatum could be associated with the symptoms of paranoia and/or could represent a protective mechanism against some of the symptoms that are less pronounced in this subtype than in the undifferentiated subgroup, such as formal though disorder.
One third of patients with schizophrenia do not respond to medication and remain psychotic (Meltzer, 1997). The patients that do respond, do so on a continuum, but only respond to positive symptoms. Cognitive and negative symptoms are poorly treated in all people with schizophrenia. Although treatment response and resistance have a biological basis (Sheitman and Lieberman, 1998), all studies conducted outside of our own work have been imaging live people. In a cohort of subjects rated for treatment response or resistance, treatment-responsive schizophrenia subjects had a large decrease in the number of mitochondria per synapse in the caudate nucleus and putamen compared to controls. In the putamen, treatment-responsive subjects also had decreases in this measure compared to treatment-resistant subjects (Somerville et al., 2011b). These results provide further support for a biological distinction between treatment response and treatment resistance in schizophrenia. Because treatment-resistant subjects had normal levels of mitochondria per synapse, but treatment responders had fewer mitochondria per synapse than controls, fewer mitochondria per synapse may be related to treatment response.
Fewer mitochondria per synapse were observed in a combined cohort of subjects and does not appear to be caused by APDs. This change was confined to treatment responders, and was not observed in treatment resistant subjects. A decrease in mitochondrial density in the neuropil distinguishes the paranoid from the undifferentiated schizophrenia subgroup. Fewer mitochondria may contribute to the pathophysiology of the illness, may be a medication effect, or an adaptive response to normalize overactive neurotransmission that may occur from the higher than normal number of excitatory striatal synapses previously found (Roberts et al., 2005a,b, 2008, 2012).
Nucleus Accumbens:
Given the report of increased COX and succinate dehydrogenase activity in the nucleus accumbens in schizophrenia compared to controls (Prince, et al., 1999), the observation that the structural integrity and general appearance of mitochondria were normal in the schizophrenia group in this same structure was surprising (McCollum et al., 2015). Moreover, the density of mitochondria in the neuropil, the average diameter, and the number of calcium deposits per mitochondrion were similar between controls and schizophrenia in both the core and shell. Taken together, alterations in mitochondrial function may not be detected with morphology.
Substantia Nigra:
In spite of the fact that the substantia nigra (SN) and ventral tegmental area (VTA) house the largest proportion of dopamine neurons in the brain and that antipsychotic medication works by blocking dopamine receptors (Creese et al., 1976), there have been very few studies of the SN/VTA in schizophrenia. At the ultrastructural level, mitochondrial hyperplasia has been observed within axon terminals that synapse onto dopamine neurons in a qualitative study of a small cohort of schizophrenia subjects (Kolomeets and Uranova, 1999); however, there were no comments on the number of mitochondria per terminal. In one of our recent studies, we quantified the number of mitochondria per terminal and found no difference in the number of mitochondria in all terminals or in subsets of excitatory or inhibitory terminals (Mabry et al., 2019); qualitatively, we did not observe any differences in the size of the mitochondria, so we did not measure them. Moreover, we found that mitochondria in dopamine neuronal somata were similar in size, density and structural integrity between schizophrenia patients and controls (Walker et al., 2018). From a structural standpoint, mitochondria do not appear to be affected in the substantia nigra in schizophrenia.
Medication effects:
The effects of antipsychotic drugs (APDs) on mitochondrial function is a vast topic beyond the scope of this review. However, an excellent recent review (Chan et al., 2020) addresses this complex issue. The authors conclude a complicated relationship between APDs and mitochondrial function with several scenarios: 1) mitochondrial damage precedes the onset of schizophrenia, 2) can be reversed by APDs, but 3) can also be caused by APD treatment. The general consensus is that antipsychotic drugs can alter mitochondrial function, number and size (for reviews see Carboni and Domenici, 2016; Roberts 2017; Chan et al., 2020). Antipsychotic drugs have differential effects on mitochondrial structure and function depending on brain location, type of antipsychotic drug, length of use, length of withdrawal period, dose and route of administration (for some examples see Takeichi and Sato, 1987; Uranova et al., 1991; Roberts et al., 1995; Prince et al., 1999; Streck et al., 2007). For instance, there are more striatal mitochondria after 3 weeks of haloperidol treatment (Uranova et al., 1991), but less after 6 months (Roberts et al., 1995). The majority of evidence is in agreement that complex I and succinate dehydrogenase appear to be adversely affected by antipsychotic drugs, however COX may not be as vulnerable (Burkhardt, et al., 1993; Balijepalli et al., 1999, 2001; Karry et al., 2004; Rosenfeld et al., 2011; Maurer and Moller, 1997; Prince et al., 1997; Streck et al., 2007).
Mitochondrial abnormalities in glial cells:
Mitochondrial abnormalities have been observed in glial cells in various brain regions. In the cortex, Uranova and colleagues have found fewer and smaller mitochondria in oligodendrocytes in both gray and white matter (Uranova et al., 2007; Vikhreva et al., 2016), suggesting that oligodendrocytes have less available energy, which may have an impact on proper myelination. In recent studies, Uranova and colleagues found a decrease in the number and size of mitochondria in oligodendrocytes adjacent to microglia in both grey (Uranova et al., 2020) and white matter (Uranova et al., 2018) in the prefrontal cortex. They concluded that oligodendrocyte dystrophy is not associated with microglial activation in white matter, but that microglial dystrophy might contribute to oligodendrocyte dystrophy in grey matter.
In the caudate nucleus, there are fewer mitochondria in astrocytes (Uranova et. al. 1996), and mitochondria are smaller in oligodendrocytes (Uranova et al., 2001). Either of these results could compromise the function of these cell types as discussed above.
Conclusions:
In the brains of subjects with schizophrenia, mitochondria are differentially affected depending on the brain region, cell type, and subcellular location in which they are located. Moreover, mitochondrial abnormalities differ depending on treatment status, treatment response and symptoms. While certain morphological configurations definitely correspond to energy capacity and other functions, it appears that mitochondria can appear intact, while being functionally compromised. Decreases in functional measures may be reflected by decreased number of mitochondria rather than decreased size or structural configuration.
Table I.
List of abbreviations
| Abbreviations and aliases | |
|---|---|
| ALDH4A1 | aldehyde dehydrogenase 4 family, member A1 |
| APD | antipsychotic drugs |
| ATP | adenosine triphosphate |
| Complex I | NADH-CoQ oxidoreductase |
| Complex II | succinate dehydrogenase |
| Complex III | cytochrome bc complex |
| Complex IV | cytochrome c oxidase (COX) |
| Complex V | ATP synthase |
| DLPFC | dorsal lateral prefrontal cortex |
| DOI | duration of illness |
| ETC | electron transport chain |
| mtDNA | mitochondrial DNA |
| NAcc | nucleus accumbens |
| NADH | nicotinamide adenine dinucleotide |
| NDUFS7 | a subunit of complex I |
| OFC | olfactory cortex |
| OxPhos | oxidative phosphorylation |
| PFC | prefrontal cortex |
| ROS | reactive oxygen species |
| SN | substantia nigra |
| TCA cycle | tricarboxylic acid cycle |
| VTA | ventral tegmental area |
Acknowledgements
I would like to thank the past and present members of my lab whose work I cite in this review, especially Keri A. Barksdale, Samuel J. Mabry, Lesley A. McCollum, Matthew W. Rice, Joy K. Roche, Kirsten E. Schoonover, Shahza M. Somerville and Courtney K. Walker.
This research reviewed in this manuscript was supported in part by R01MH66123 & R01 MH60744 to RCR, MH073461 to SMS, F31MH098566 to LAM, and the President’s Summer Research Scholarship (SJM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aganova EA, Uranova NA., 1992. Morphometric analysis of synaptic contacts in the anterior limbic cortex in the endogenous psychoses. Neurosci. Behav. Physiol 22(1):59–65. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, et al. , 2013. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis 4:e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, Jurata LW, Charles V, Lemire A, Liu P, et al. , 2005. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry 58:85–96. [DOI] [PubMed] [Google Scholar]
- Andreazza AC, Shao L, Wang JF, Young LT, 2010. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch. Gen. Psychiatry 67(4):360–8. [DOI] [PubMed] [Google Scholar]
- Anglin RE, Mazurek MF, Tarnopolsky MA, Rosebush PI, 2012. The mitochondrial genome and psychiatric illness. Am. J. Med. Genetics Part B 159B (7):749–759. Review [DOI] [PubMed] [Google Scholar]
- Arion D, Corradi JP, Tang S, Datta D, Boothe R, et al. 2015. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry 20(11):1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Hille B, 1998. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol 8:398–404. [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, et al. , 2003. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism, a novel regulatory mechanism altered in obesity. J. Biol. Chem 278:17190–17197. [DOI] [PubMed] [Google Scholar]
- Balijepalli S, Boyd MR, Ravindranath V, 1999. Inhibition of mitochondrial complex I by haloperidol: the role of thiol oxidation. Neuropharm 38 (4):567–577. [DOI] [PubMed] [Google Scholar]
- Balijepalli S, Kenchappa RS, Boyd MR, Ravindranath V, 2001. Protein thiol oxidation by haloperidol results in inhibition of mitochondrial complex I in brain regions: comparison with atypical antipsychotics. Neurochem. Int 38 (5):425–435. [DOI] [PubMed] [Google Scholar]
- Barksdale K, Lahti AC, Roberts RC, 2014. Synaptic proteins in the postmortem anterior cingulate cortex in schizophrenia: Relationship to treatment and treatment response. Neuropsychopharmacology 39 (9):2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, 2017. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr. Res 187:3–10. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Laifenfeld D, 2004. Mitochondria, synaptic plasticity, and schizophrenia. Int. Rev. Neurobiol 59:273–296. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Karry R, 2008. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS One 3 (11): e3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, et al. , 2016. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis 90: 3–19. [DOI] [PubMed] [Google Scholar]
- Brodin L, Bakeeva L, Shupliakov O, 1999. Presynaptic mitochondria and the temporal pattern of neurotransmitter release. Philos. Trans. R. Soc. Lond. B. Biol. Sci 354:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubber P, Hartounian V, Gibson GE, Blass JP, 2011. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. European Neuropsychopharm 21:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt C, Kelly JP, Lim Y-H, Filley CM, Parker WD Jr., 1993. Neuroleptic medications inhibit complex I of the electron transport chain. Ann. Neurol 33:512–517. [DOI] [PubMed] [Google Scholar]
- Carboni L, Domenici E., 2016. Proteome effects of antipsychotic drugs: Learning from preclinical models. Proteomics Clin. Appl 10 (4):430–41. Review. [DOI] [PubMed] [Google Scholar]
- Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, et al. , 1995. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics 29:217–224. [DOI] [PubMed] [Google Scholar]
- Chan ST, McCarthy MJ, Vawter MP, 2020. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res 217:136–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ, 2006. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci 26:7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DTW, Reynolds IJ, 2006. Mitochondrial trafficking and morphology in healthy and injured neurons. Progress in Neurobiol 80:241–268. [DOI] [PubMed] [Google Scholar]
- Clark KM, Taylor RW, Johnson MA, Chinnery PF, Chrzanowska-Lightowlers ZM, et al. , 1999. An mtDNA mutation in the initiation codon of the cytochrome C oxidase subunit II gene results in lower levels of the protein and a mitochondrial encephalomyopathy. Am. J. Hum. Genet 64:1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Sillivan S, Konradi C, 2011. Mitochrondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci 29 (3):311–324. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, 2006. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26(4–6):365–384. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH, 1976. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192(4238):481–483. [DOI] [PubMed] [Google Scholar]
- Davis AF, Clayton DA, 1996. In situ localization of mitochondrial DNA replication in intact mammalian cells. J. Cell. Biol 135 (4):883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L, 2008. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 4:456(7222):605–610. [DOI] [PubMed] [Google Scholar]
- Dean B, Thomas N, Scarr E, Udawela M, 2016. Evidence for impaired glucose metabolism in the striatum, obtained postmortem, from some subjects with schizophrenia. Transl. Psychiatry 6 (11), e949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deep-Soboslay A, Akil M, Martin CE, Bigelow LB, Herman MM, et al. , 2005. Reliability of psychiatric diagnosis in postmortem research. Biol. Psychiatry 57(1):96–101. [DOI] [PubMed] [Google Scholar]
- Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, McGuire PK, 2014. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psych 75(5): e11–3. [DOI] [PubMed] [Google Scholar]
- DSM Diagnostic and Statistical Manual of Mental Disorders (DSM), (2013) published by the American Psychiatric Association (APA). [Google Scholar]
- Duchen MR, Verkhratsky A, Muallem S, 2008. Mitochondria and calcium in health and disease. Cell Calcium 44 (1):1–5. [DOI] [PubMed] [Google Scholar]
- Eblen F, Graybiel AM, 1995. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci 15 (9):5999–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty AW, Graybiel AM, 1993. Two input systems for body representations in the primate striatal matrix: experimental evidence in the squirrel monkey. J. Neurosci 13 (3):1120–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Yücel M, Dean B, Wood SJ, Pantelis C, 2009. Anatomical abnormalities of the anterior cingulate cortex in schizophrenia: bridging the gap between neuroimaging and neuropathology. Schizophr. Bull 35 (5):973–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK, 2011. ER tubules mark sites of mitochondrial division. Science 334 (6054):358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuke S, Kametani M, Kato T, 2008. Quantitative analysis of the 4977-bp common deletion of mitochondrial DNA in postmortem frontal cortex from patients with bipolar disorder and schizophrenia. Neurosci. Lett 439(2):173–7. [DOI] [PubMed] [Google Scholar]
- Goff DC, Coyle JT, 2001. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158(9):1367–1377. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, 1999. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol. Psychiatry 46(5):650–661. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Nauta WJ, 1977. An intricately patterned prefronto-caudate projection in the rhesus monkey. J. Comp. Neurol 72 (3):369–386. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE, 1994. Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol 267 (2 Pt 1):C313–339. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW Jr., 1978. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl. Acad. Sci 75 (11):5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR, 1968. Ultrastructural bases for metabolically linked mechanical activity in mitochondria; II Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol 37 (2):345–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara H, Catts VS, Katayama Y, Shoji H, Takagi T, Huang FL, Nakao A, Mori Y, Huang KP, Ishii S, Graef IA, Nakayama KI, Shannon Weickert C, Miyakawa T,2018. Decreased brain pH as a shared endophenotype of psychiatric disorders. Neuropsychopharmacology 43 (3), 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, et al. , 2001Genome-wide expression analysis reveals dysregulation of myelination related genes in chronic schizophrenia. Proc. Natl. Acad. Sci 98:4746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Klein-Flugge MC, Howarth C, Attwell D, 2012. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci 32:8940–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH, 2014. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc. Natl. Acad. Sci 111 (1):486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Fujimoto M, 2010. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmocol 77 (4):517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP, 2009. MAM: more than just a housekeeper. Trends Cell Biol 19:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C, 2015. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res 2015 Sep;167(1–3):4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelm BE, Rollins B, Mamdani F, Lauterborn JC, Kirov,et al. , 2015. Evidence of Mitochondrial Dysfunction within the Complex Genetic Etiology of Schizophrenia. Mol. Neuropsychiatry 1(4):201–19. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftman GD, Datta D, Lewis DA, 2016. Layer 3 Excitatory and Inhibitory Circuitry in the Prefrontal Cortex: Developmental Trajectories and Alterations in Schizophrenia. Biol. Psychiatry pii: S0006-3223(16) 32427-1. [DOI] [PMC free article] [PubMed]
- Hollenbeck PJ, 1996. The pattern and mechanism of mitochondrial transport in axons. Front Biosci 1:d91–102. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM, 2005. The axonal transport of mitochondria. J. Cell Sci 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holper L, Ben-Shachar D, Mann JJ, 2016. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 44(5):837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes O, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S 2012. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry, 69(8): 776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW, 2008. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr 40:445–456. [DOI] [PubMed] [Google Scholar]
- Huttemann M, Kadenbach B, Grossman LI, 2001. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 267:111–123. [DOI] [PubMed] [Google Scholar]
- Isaacs KR, Anderson BJ, Alcantara AA, Black JE, Greenough WT, 1992. Exercise and the brain: angiogenesis in the adult rat cerebellum after vigorous physical activity and motor skill learning. J. Cereb. Blood Flow Metab 12 (1):110–119. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Kato T, 2005. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet 14(2):241–253. [DOI] [PubMed] [Google Scholar]
- James R, Adams RR, Christie S, Buchanan SR, Porteous DJ, et al. Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol Cell Neurosci 26(1):112–22 [DOI] [PubMed] [Google Scholar]
- Javitt DC, 2004. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry 9(11):984–997. [DOI] [PubMed] [Google Scholar]
- Kakiuchi C, Ishiwata M, Kametani M, Nelson C, Iwamoto K, Kato T 2005. Quantitative analysis of Neuropsychopharmacol 8(4):515–22. [DOI] [PubMed] [Google Scholar]
- Karry R, Klein E, Ben Shachar D, 2004. Mitochondrial complex I subunits expression is altered in schizophrenia: a postmortem study. mitochondrial DNA deletions in the brains of patients with bipolar disorder and schizophrenia. Int J Biol Psychiatry 55 (7):676–684. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Uranova NA, 1999. Synaptic contacts in schizophrenia: studies using immunocytochemical identification of dopaminergic neurons. Neurosci. Behav. Physiol 2: 217–221. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Uranova N, 2010. Ultrastructural abnormalities of astrocytes in the hippocampus in schizophrenia and duration of illness: a postortem (sic) morphometric study. World J Biol Psychiatry 11(2 [DOI] [PubMed] [Google Scholar]
- Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ 2018. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front Immunol 9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC, 2004. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305 (5685):858–862. [DOI] [PubMed] [Google Scholar]
- Krystal JH, 2008. Capitalizing on extrasynaptic glutamate neurotransmission to treat antipsychotic-resistant symptoms in schizophrenia. Biol Psychiatry 64(5):358–360. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Karube F, Nomura M, Kawaguchi Y, 2016. The Diversity of Cortical Inhibitory Synapses. Front Neural Circuits. 25 (10): 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung L, Roberts RC, 1999. Mitochondrial pathology in human schizophrenic striatum: a postmortem ultrastructural study. Synapse 3:67–75. [DOI] [PubMed] [Google Scholar]
- Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA, 2008. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol. Psychiatry 13:685–696. [DOI] [PubMed] [Google Scholar]
- Legros F, Lombès A, Frachon P, Rojo M, 2002. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusions. Mol. Biol. Cell 13 (12):4343–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, 2014. Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr Opin Neurobiol 26:22–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Park JS, Deng JH, Bai Y, 2006. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr 38: 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto KI, Hayashi Y, Sheng M, 2004. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O, 2000. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol 427 (3):351–361. [DOI] [PubMed] [Google Scholar]
- Lin MY, Sheng ZH, 2015. Regulation of mitochondrial transport in neurons. Exp. Cell Res 334 (1):35–44. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hajnóczky G, 2011. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ 18 (10):1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, Chan DC, 2013. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24 (5):659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabry SJ, McCollum LA, Farmer CB, Bloom ES, Roberts RC, 2019. Evidence for altered excitatory and inhibitory tone in the post-mortem substantia nigra in schizophrenia. World J. Biol. Psych 4:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Kittle JT, 2010. Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20 (2):102–112. [DOI] [PubMed] [Google Scholar]
- MacDonald ML, Garver M, Newman J, Sun Z, Kannarkat J, et al. , 2019. Synaptic proteome alterations in the primary auditory cortex of individuals with schizophrenia. JAMA Psychiatry 23;77(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madireddy S, Madireddy S, 2020. Regulation of reactive oxygen species-mediated damage in the pathogenesis of schizophrenia Brain Sci 10(10):E742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamdani F, Rollins B, Morgan L, Sequeira PA, Vawter MP, 2014. The somatic common deletion in mitochondrial DNA is decreased in schizophrenia. Schizophr. Res 159 (2–3): 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G, 2012. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci 13:293–307. Review [DOI] [PubMed] [Google Scholar]
- Martinelli C, Sartori P, Ledda M, Pannese E, 2006. A study of mitochondria in spinal ganglion neurons during life: quantitative changes from youth to extremely advanced age. Tissue Cell 38 (2) 93–8. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D, Harris LW, Guest PC, Bahn S, 2011. The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxidants Redox Signal 15 (7):2067–2079. Review [DOI] [PubMed] [Google Scholar]
- Martorell L, Segues T, Folch G, Valero J, Joven J, et al. , 2006. New variants in the mitochondrial genomes of schizophrenic patients. Eur. J. Hum. Genet 14:520–528. [DOI] [PubMed] [Google Scholar]
- Maurer I, Moller HJ, 1997. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol. Cell Biochem 174:255–259. [PubMed] [Google Scholar]
- Maurer I, Zierz S, Moller H, 2001. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr. Res 48:125–136. [DOI] [PubMed] [Google Scholar]
- McCollum LA, Walker CK, Roche JK, Roberts RC, 2015. Elevated excitatory input to the nucleus accumbens in schizophrenia: a postmortem ultrastructural study. Schizophr. Bull 41 (5):1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY, 1997. Treatment-resistant schizophrenia--the role of clozapine. Curr. Med. Res. Opin 14 (1):1–20. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P., 2002. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J. Neurosci 22(7):2718–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP, 2004. Axonal mitochondrial transport and potential are correlated. J. Cell Sci 117 (13):2791–2804. [DOI] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH, 2010. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci 30 (12):4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjaatvedt AE, Wong-Riley MT, 1988. Relationship between synaptogenesis and cytochrome oxidase activity in Purkinje cells of the developing rat cerebellum. J. Comp. Neurol 277 (2):155–182. [DOI] [PubMed] [Google Scholar]
- Mulcrone J, Whatley SA, Ferrier IN, Marchbanks RM. Schizophr Res. 1995. A study of altered gene expression in frontal cortex from schizophrenic patients using differential screening 14(3):203–13. [DOI] [PubMed] [Google Scholar]
- Nagaoka A, Kunii Y, Hino M, Izumi R, Nagashima C, et al. , 2020. ALDH4A1 expression levels are elevated in postmortem brains of patients with schizophrenia and are associated with genetic variants in enzymes related to proline metabolism. J. Psychiatr. Res 123:119–127. [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, et al. , 2008. Cargo-selected transport from the mitochondria to peroxisomes ismediated by vesicular carriers. Curr. Biol 18 (2), 102–108. [DOI] [PubMed] [Google Scholar]
- Ni P, Chung S, 2020. Mitochondrial Dysfunction in Schizophrenia. Bioessays 42(6):e1900202 Review [DOI] [PubMed] [Google Scholar]
- Niescier RF, Kwak SK, Joo SH, Chang KT, Min KT, 2016. Dynamics of Mitochondrial Transport in Axons. Front. Cell Neurosci (10):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans LG, Taanman JW, Muijsers AO, Speijer D, Van den Bogert C, 1998. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem 254:389–394. [DOI] [PubMed] [Google Scholar]
- Norkett R, Modi S, Birsa N, Atkin TA, Ivankovic D, et al. , 2016. DISC1-dependent Regulation of Mitochondrial Dynamics Controls the Morphogenesis of Complex Neuronal Dendrites. J. Biol. Chem 291(2):613–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole M, Latham R, Baqri RM, Miller KE, 2008. Modeling mitochondrial dynamics during in vivo axonal elongation. J. Theor. Biol 255:369–377. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, et al. , 2010. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol 191:1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee SB, Suh Y, Kim SJ, Lee N, et al. , 2017. DISC1 Modulates Neuronal Stress Responses by Gate-Keeping ER-Mitochondria Ca (2+) Transfer through the MAM. Cell Rep 21(10):2748–2759. [DOI] [PubMed] [Google Scholar]
- Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, et al. , 2015. The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem 290:22325–22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McEwen BS, 2014. Mitochondria impact brain function and cognition. Proc. Natl. Acad. Sci 111 (1):7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploumi C, Daskalaki I, Tavernarakis N, 2017. Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284(2):183–195. Review [DOI] [PubMed] [Google Scholar]
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, et al. , 2004. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 9:684–97. [DOI] [PubMed] [Google Scholar]
- Prince JA, Blennow K, Gottfries CG, Karlsson I, Oreland L, 1999. Mitochondrial function is differentially altered in the basal ganglia of chronic schizophrenics. Neuropsychopharmacology 21:372–379. [DOI] [PubMed] [Google Scholar]
- Prince JA, Harro J, Blennow K, Gottfries CG, Oreland L, 2000. Putamen mitochondrial energy metabolism is highly correlated to emotional and intellectual impairment in schizophrenics. Neuropsychopharmacology 22 (3):284–292. [DOI] [PubMed] [Google Scholar]
- Prince JA, Yassin MS, Oreland L, 1997. Neuroleptic-induced mitochondrial enzyme alterations in the rat brain. J. Pharmacol. Exp. Ther 280:261–267. [PubMed] [Google Scholar]
- Pruett BS, and Meador-Woodruff JH, 2020. Evidence for altered energy metabolism, increased lactate, and decreased pH in schizophrenia brain: A focused review and meta-analysis of human postmortem and magnetic resonance spectroscopy studies. Schizophr Res Sep 18;S0920-9964(20)30459-X. Review [DOI] [PubMed] [Google Scholar]
- Rae C, Scott RB, Thompson CH, Kemp GJ, Dumughn I, Styles P, Tracey I, Radda GK, 1996. Is pH a biochemical marker of IQ? Proc. Biol. Sci 263 (1373), 1061–1064. [DOI] [PubMed] [Google Scholar]
- Rahman S, Taanman JW, Cooper JM, Nelson I, Hargreaves I, et al. , 1999. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am. J. Hum. Genet 65:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Miguel A, Barakauskas V, Alamri J, Miyauchi M, Barr AM, et al. , 2019. The SNAP25 Interactome in Ventromedial Caudate in Schizophrenia Includes the Mitochondrial Protein ARF1. Neuroscience 420:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice MW, Smith KL, Roberts RC, Perez-Costas E, Melendez-Ferro M, 2014. Assessment of cytochrome C oxidase dysfunction in the substantia nigra/ventral tegmental area in schizophrenia. PLOS1 9 (6) e100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, 2007. Schizophrenia in translation: disrupted in schizophrenia (DISC1): integrating clinical and basic findings. Schizophr Bull 33(1):11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, 2017. Postmortem studies on mitochondria in schizophrenia. Schizophr. Res 187:17–25. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, Barksdale KA, Roche JK, Lahti AC, 2015. Decreased synaptic and mitochondrial density in the postmortem anterior cingulate cortex in schizophrenia. Schizophr. Res 168 (1–2):543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, Gaither LA, Gao XM, Kashyap SM, Tamminga CA, 1995. Ultrastructural correlates of haloperidol-induced oral dyskinesias in rat striatum. Synapse 20 (3):234–243. [DOI] [PubMed] [Google Scholar]
- Roberts RC, McCollum LA, Schoonover KE, Mabry SJ, Roche JK, Lahti AC. 2020. Ultrastructural evidence for glutamatergic dysregulation in schizophrenia. Schizophr Res January 31 pii: S0920–9964(20)30032–3. [DOI] [PMC free article] [PubMed]
- Roberts RC, Roche JK, Conley R, 2005a. Synaptic differences in the postmortem striatum of subjects with schizophrenia: a stereological ultrastructural analysis. Synapse 56 (4): 185–197. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley R, 2005b. Synaptic differences in the patch matrix compartments of the striatum of subjects with schizophrenia: a postmortem ultrastructural analysis. Neurobiol. Dis 20:324–335. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR, 2008. Differential synaptic changes in the striatum of subjects with undifferentiated versus paranoid schizophrenia. Synapse 62 (8): 616–627. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Somerville SM, Conley RR, 2012. Ultrastructural Distinctions Between Treatment Responders and Non-Responders in Schizophrenia: Postmortem Studies of the Striatum, in: Labate L (Ed.), Mental Illnesses - Evaluation, Treatments and Implications, InTech, Croatia, 261–286. [Google Scholar]
- Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan, et al. , 2009. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One 4: e4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M, Brenner-Lavie H, Ari SG-B, Kavushansky A, Ben-Shachar D 2011. Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol. Psychiatry 69:980–988. [DOI] [PubMed] [Google Scholar]
- Rowland LM, Pradhan S, Korenic S, Wijtenburg SA, Hong LE, Edden RA, Barker PB, 2016. Elevated brain lactate in schizophrenia: a 7 Tmagnetic resonance spectroscopy study. Transl. Psychiatry 6 (11), e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabunciyan S 2019. Gene expression profiles associated with brain aging are altered in schizophrenia Sci. Rep 9(1):5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabunciyan S, Kirches E, Krause G, Bogerts B, Mawrin C, Llenos IC, Weis S 2007. Quantification of total mitochondrial DNA and mitochondrial common deletion in the frontal cortex of patients with schizophrenia and bipolar disorder. J Neural Transm (Vienna) 114(5):665–74. [DOI] [PubMed] [Google Scholar]
- Samuels DC, Schon EA, Chinnery PF, 2004. Two direct repeats cause most human mtDNA deletions. Trends Genet 20 (9): 393–398. [DOI] [PubMed] [Google Scholar]
- Seeman P, Lee T, Chau-Wong M, Wong K 1976. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature, 261(5562): 717–19.. [DOI] [PubMed] [Google Scholar]
- Sequeira A, Martin MV, Rollins B, Moon EA, Bunney WE, et al. , 2012. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet 3:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Area-Gomez E, 2013. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci 55:26–36. [DOI] [PubMed] [Google Scholar]
- Schulmann A, Ryu E, Goncalves V, Rollins B, Christiansen M, Frye MA, Biernacka J, Vawter MP, 2019. ‘Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Mol. Neuropsychiatry 5(1):13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, et al. , 2008. Mitochondrial involvement in psychiatric disorders. Ann. Med 40:281–295. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheitman BB, Lieberman JA, 1998. The natural history and pathophysiology of treatment resistant schizophrenia. J. Psychiatry Res. 32:143–150. [DOI] [PubMed] [Google Scholar]
- Sheng Z-H, Cai Q, 2012. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci 13 (2):77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN, 1994. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci 91:10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioiri T, Someya T, Murashita J, Kato T, Hamakawa H, Fujii K, Inubushi T, 1997. Multiple regression analysis of relationship between frontal lobe phosphorus metabolism and clinical symptoms in patients with schizophrenia. Psychiatry Res 76 (2–3), 113–122. [DOI] [PubMed] [Google Scholar]
- Smirnova L, Griparic L, Shurland DL, van der Bliek AM, 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soghomonian JJ, Sethares C, Peters A, 2010. Effects of age on axon terminals forming axosomatic and axodendritic inhibitory synapses in prefrontal cortex. Neuroscience 168 (1):74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville SM, Conley RR, Roberts RC 2011a. Mitochondria in the striatum of subjects with schizophrenia. World J. Biol. Psychiatry 12 (1):48–56. [DOI] [PubMed] [Google Scholar]
- Somerville SM, Conley RR, Roberts RC, 2012. Striatal mitochondria in subjects with chronic undifferentiated vs. chronic paranoid schizophrenia. Synapse 66, 29–41. [DOI] [PubMed] [Google Scholar]
- Somerville SM, Lahti AC, Conley RR, Roberts RC, 2011b. Mitochondria in the striatum of subjects with schizophrenia: relationship to treatment response. Synapse 65:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong NW, Hinton DR, Cortopassi G, Arnheim N, 1992. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet 2(4):318–23. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM, 2012. Reconstitution of mitochondira derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7 (12):e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streck EL, Rezin GT, Barbosa LM, Assis LC, Grandi E, Quevedo J, 2007. Effect of antipsychotics on succinate dehydrogenase and cytochrome oxidase activities in rat brain. Naunyn Schmiedebergs Arch. Pharmacol 376(1–2):127–33. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, et al. , 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397 (6718):441–446. [DOI] [PubMed] [Google Scholar]
- Taanman JW, 1997. Human cytochrome c oxidase: structure, function, and deficiency. J. Bioenerg. Biomembr 29, 151–163. [DOI] [PubMed] [Google Scholar]
- Takeichi M and Sato T, 1987. Quantitative electron microscopic investigation on changes of mitochondria in long-term CPZ administration in rat brain, liver and heart. The Japanese J. Psych. Neurol 41:749–753. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Casanova MF, DeVaughn NM, Orlovskaya DD, Denisov DV, 1996. Ultrastructural alterations of synaptic contacts and astrocytes in postmortem caudate nucleus of schizophrenic patients. Schizophr. Res 22 (1):81–3. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Orlovskaya DD, Apel K, Klintsova AJ, Haselhorst U, Schenk H., 1991. Morphometric study of synaptic patterns in the rat caudate nucleus and hippocampus under haloperidol treatment. Synapse (4):253–259. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V, 2001. Electron microscopy of oligodendroglia in severe mental illness. Brain Res. Bull 55:597–610. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rakhmanova VI, Orlovskaya DD, 2018. Ultrastructural pathology of oligodendrocytes adjacent to microglia in prefrontal white matter in schizophrenia. NPJ Schizophr 4(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rakhmanova VI, Orlovskaya DD, 2020. Dystrophy of Oligodendrocytes and Adjacent Microglia in Prefrontal Gray Matter in Schizophrenia. Front. Psychiatry 11:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya DD, 2007. The role of oligodendrocyte pathology in schizophrenia. Int. J. Neuropsychopharmacol 10 (4): 537–545 [DOI] [PubMed] [Google Scholar]
- Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood WH III, et al. , 2001. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res. Bull 55(5):641–650. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, et al. , 2002. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr. Res 58(1):11–20. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Tomita H, Meng F, Bolstad B, Li J, et al. , 2006. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry 11(7):615, 663–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge B, Alonso Y, Valero J, Miralles C, Vilella E, Martorell L, 2011. Mitochondrial DNA (mtDNA) and schizophrenia. Eur. Psychiatry 26 (1):45–56. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJT, Koh TW, Zhou Y, Bellen HJ, 2005. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47:365–378. [DOI] [PubMed] [Google Scholar]
- Vikhreva OV, Rakhmanova VI, Orlovskaya DD, Uranova NA, 2016. Ultrastructural alterations of oligodendrocytes in prefrontal white matter in schizophrenia: A post-mortem morphometric study. Schizophr. Res 177(1–3):28–36. [DOI] [PubMed] [Google Scholar]
- Vos M, Lauwers E, Verstreken P. 2010. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci 2:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CK, Roche JK, Sinha V, Roberts RC, 2018. Substantia nigra ultrastructural pathology in schizophrenia. Schizophr. Res 197: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Shao L, Sun X, Young LT, 2009. Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disorders 11 (5):523–529. [DOI] [PubMed] [Google Scholar]
- Whatley SA, Curti D, Marchbanks RM, 1996. Mitochondrial involvement in schizophrenia and other functional psychoses. Neurochem Res 21(9):995–1004. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, 1989. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12 (3):94–101. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]