

A hominoid-specific gene promotes cortical expansion via regulating histone modification.
Abstract
Genomic changes during human linage evolution contribute to the expansion of the cerebral cortex to allow more advanced thought processes. The hominoid-specific gene TBC1D3 displays robust capacity of promoting the generation and proliferation of neural progenitors (NPs), which are thought to contribute to cortical expansion. However, the underlying mechanisms remain unclear. Here, we found that TBC1D3 interacts with G9a, a euchromatic histone lysine N-methyltransferase, which mediates dimethylation of histone 3 in lysine 9 (H3K9me2), a suppressive mark for gene expression. TBC1D3 displayed an inhibitory role in G9a’s histone methyltransferase activity. Treatment with G9a inhibitor markedly increased NP proliferation and promoted human cerebral organoid expansion, mimicking the effects caused by TBC1D3 up-regulation. By contrast, blockade of TBC1D3/G9a interaction to disinhibit G9a caused up-regulation of H3K9me2, suppressed NP proliferation, and impaired organoid development. Together, this study has demonstrated a mechanism underlying the role of a hominoid-specific gene in promoting cortical expansion.
INTRODUCTION
The expansion of the cerebral cortex during primate evolution is assumed to be associated with the acquisition of higher intelligence especially in the human species (1, 2). This process involves increased proliferative ability of cortical neural progenitors (NPs), including the ventricular radial glial cells (vRGs), the intermediate progenitors, and basal or outer RGs (bRGs or oRGs) (3, 4), which give rise to neurons or glia cells directly or indirectly. By contrast, disruption of the proliferative capacity of RGs has been shown to cause malformations of the cortex, which usually leads to intellectual disability (5). It has been shown that the epigenetic mechanisms, especially modifications of chromatin, play a critical role in regulating transcriptional programs that govern the stemness of NPs (6). However, the role of epigenetic factors in the process of cortical expansion during human evolution remains to be explored.
The epigenetic regulation at the level of chromatin is tightly controlled by posttranslational histone modification (7). The mono- or dimethylation of lysine 9 (K9) site at histone 3 (H3) (H3K9me1 and H3K9me2) mediated by histone methyltransferases G9a [also known as EHMT2 (euchromatic histone-lysine N-methyltransferase 2)] and G9a-like protein (GLP, also known as EHMT1) marks transcriptionally repressive genomic loci, which symbolize gene silencing in mammals (8). Notably, genetic ablation of G9a or GLP in the forebrain of adult mice has been shown to reactivate NP genes, leading to defects in cognitive and adaptive behaviors (9). Deletions or mutations of G9a/GLP genes are a cause of Kleefstra syndrome, a rare genetic disorder characterized by intellectual disability, autistic-like features, childhood hypotonia, and distinctive facial features (10). In addition, the differentiation of retinal progenitor cells requires G9a-mediated silencing of genes that sustain a proliferative state (11). It would be of interest to determine how the G9a activity is tightly controlled in neural stem cells during proliferation, in particular in human cortical NPs, which exhibit increased numbers of mitotic cell division compared with mice.
The cross-species analyses of epigenetic modifications between primate species and rodents have revealed major phenotypic changes during mammalian evolution (12, 13). Notably, comparative epigenetic analysis of human, rhesus macaque, and mouse genomes has identified the gained activity of promoters and enhancers in humans, which are substantially enriched in modules crucial for neural proliferation (14). Nevertheless, the factors responsible for these epigenetic differences underlying human brain development remain unclear.
Recently, we have found that the expression of the hominoid-specific gene TBC1D3 promotes production of cortical NPs, leading to expansion and folding of the cortex in mice (15). Here, we report a regulatory role of TBC1D3 for G9a-mediated H3K9me2 modification in human cortical NPs. We found that TBC1D3 physically interacted with G9a and inhibited G9a activity. Down-regulation of G9a promoted the proliferation of human NPs, resulting in expansion of human cerebral organoids. Disruption of TBC1D3/G9a interaction up-regulated the level of H3K9me2 and suppressed the expansion of human cerebral organoid. These results indicate that the inhibition of G9a by TBC1D3 ensures enhanced proliferation of human NPs and the expansion of human cerebral cortex.
RESULTS
TBC1D3 facilitates the proliferation of NPs and expansion in human cerebral organoids
In line with the hypothesis that duplication of specific genes in human might contribute to brain evolution, we found that expression of TBC1D3 gene, which is duplicated to form multiple paralogs in human genome and present in the chimpanzee genome as a single copy (16, 17), promoted cortex expansion and folding in mice (15). To assess whether the copy number correlated with the expression level of TBC1D3 in the chimpanzee and human, we analyzed accessible online datasets of RNA-sequencing (RNA-seq) information in NPs and differentiated neurons derived from induced pluripotent stem cells (18). Notably, the expression of TBC1D3 in chimpanzee cells was barely detectable, while it exhibited a substantial level in human cells, both in NPs and neurons (fig. S1A). To further determine the role of TBC1D3 in human cellular contexts, we generated human cerebral organoids by using guided differentiation of the H9 human embryonic stem cells (hESCs) into neuroectoderm with the addition of inhibitors for transforming growth factor–β (TGF-β) or bone morphogenetic protein (BMP) pathway (19), as well as matrix embedding to promote neuroepithelium formation and organoid assembling (20). The quantitative polymerase chain reaction (PCR) analysis of organoids at different cultured stages revealed the expression of TBC1D3 since cultured day 16 (D16) as well as the appearance of forebrain marker FOXG1 (fig. S1, B and C). Then, we analyzed the effect of TBC1D3 up-regulation on organoid development by transducing hESCs with lentivirus encoding TBC1D3 (rLV-TBC1D3) or vehicle alone (rLV-Ctrl) (Fig. 1A). As shown in fig. S1D, the level of TBC1D3 mRNA in hESCs transduced with rLV-TBC1D3 was significantly higher than that in control cells. Intriguingly, the organoids with TBC1D3 up-regulation were markedly larger in size compared with control organoids at either the neuroectodermal stage (D12) or 6 days after induction for neuronal differentiation (D18) (Fig. 1B). Further analysis showed that the percentage of cells positive for KI67, a marker for proliferative cells, or phospho-vimentin (P-VIM), which marks mitotic radial glia cells, as well as PAX6, a typical marker for cortical NPs, among total DAPI+ (4′,6-diamidino-2-phenylindole–positive) cells markedly increased in TBC1D3 organoids (Fig. 1C and fig. S1E). Thus, TBC1D3 up-regulation promotes the proliferation of NPs in human cerebral organoids.
Fig. 1. Overexpression of TBC1D3 induces human cerebral organoid expansion.
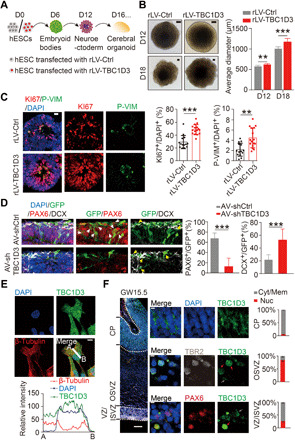
(A) Schematic diagram for human cerebral organoid culture. (B) Analysis for the size of organoids (Ctrl, 6 organoids in D12 and 15 in D18; TBC1D3, 11 organoids in D12 and 17 in D18). Scale bars, 100 μm. (C) Analysis for the percentage of KI67+ or P-VIM+ cells in D18 organoids (14 neuroepithelial rosettes from 6 control organoids; 16 rosettes from 8 TBC1D3 organoids). Scale bar, 20 μm. (D) Analysis for the percentage of PAX6+ (white arrowheads) or DCX+ (yellow arrowheads) cells among GFP+ cells in D40 organoids infected with adenovirus (AV) expressing shTBC1D3 or shCtrl. Scale bar, 20 μm. shCtrl, 23 rosettes from 15 organoids; shTBC1D3, 19 rosettes from 13 organoids. (E) TBC1D3 distribution in ReN cells. Scale bar, 10 μm. (F) Immunostaining for PAX6, TBR2, and TBC1D3 in GW15.5 fetal human cortex. Scale bars, 200 μm (left) and 5 μm (magnified). Histograms show the percentage of cells with TBC1D3 enriched in cytoplasm/membrane (Cyt/Mem; gray arrows) or nucleus (Nuc; red arrows). Five regions for cortical plate (CP) or OSVZ; six regions for VZ/ISVZ from three slices. Data are presented as means ± SD, unpaired Student’s t test. **P < 0.01; ***P < 0.001.
To determine TBC1D3’s loss-of-function effects, organoids at D37 were infected by adenovirus encoding small hairpin RNA sequence against TBC1D3 (AV-shTBC1D3) or scrambled control sequence (AV-shCtrl) (15), with vector-encoded green fluorescent protein (GFP) marking infected cells. As shown in Fig. 1D, the shTBC1D3 organoids at D40 exhibited marked decrease in the percentage of PAX6+ cells among GFP-marked infected cells, whereas the percentage of newborn differentiated neurons positively labeled by doublecortin (DCX) increased significantly (Fig. 1D). Furthermore, the shTBC1D3 organoids at D60 also exhibited a marked reduction in the percentage of cells labeled by HOPX (fig. S1F), a marker for oRGs (21). Thus, down-regulation of TBC1D3 impeded NP proliferation and caused precocious neuronal differentiation in human cerebral organoids.
TBC1D3 is expressed in the nuclei of NPs and interacts with G9a
Previous studies have shown the cytoplasmic or membrane localization of TBC1D3 in several non-neuronal cell types (22–24). To reveal the molecular mechanism of TBC1D3 in human cortical development, we determined the subcellular localization of TBC1D3 in human neural cells. First, immunostaining of cultured human neural stem cell ReN showed that albeit TBC1D3 was widely distributed in the whole cell, it was enriched in the nuclei, which were marked by DAPI and devoid of β-tubulin (Fig. 1E). Next, we determined the expression of TBC1D3 in human fetal brain at gestation week 15.5 (GW15.5) and found that TBC1D3 was expressed widely in PAX6- or TBR2-labeled NPs in ventricular zone (VZ)/inner subventricular zone (ISVZ) or outer subventricular zone (OSVZ) regions (fig. S1G). Intriguingly, while the signal of TBC1D3 was distributed dominantly in cytoplasmic and membrane regions in cells located in the cortical plate, it was enriched in the nuclei of a majority of TBR2+ cells in the OSVZ and a fraction of PAX6+ cells in VZ/ISVZ (Fig. 1F). The nuclear localization of TBC1D3 was also confirmed by immunoblot analysis for the biochemical fractions of human fetal brain tissues and ReN cells (fig. S1H). Substantial TBC1D3 signals were also observed in the nuclei of cells in cultured human cerebral organoids at D16 or D24, and the cells in organoids at D40 exhibited much less TBC1D3 signals in the nuclei (fig. S1I). The immunofluorescence signals for TBC1D3 were specific because cells in organoids infected by AV-shTBC1D3 exhibited markedly decreased TBC1D3 signals compared with uninfected cells, while AV-shCtrl had no effect (fig. S1J). These results suggest that the subcellular localization of TBC1D3 is dynamic, and its nuclear distribution in human NPs suggests a mechanism underlying its role in regulating NP proliferation.
To gain further insights into molecular mechanisms by which TBC1D3 executes its functions in human cortex development, we searched for proteins that directly interact with TBC1D3 using the yeast two-hybrid (Y2H) system (Fig. 2A). A screen of human fetal brain complementary DNA (cDNA) library using as bait the full length of TBC1D3 led to the identification of around 20 hits. Among them, G9a was the only candidate that has been shown to control transcriptional regulation in nervous system (9). Three cDNA clones in Y2H encoded fragments of G9a, with the sequences covering a part of ankyrin repeats (ANK) and the entire SET domain, which has the methyltransferase activity (Fig. 2B) (8, 25). To determine whether TBC1D3 and G9a interact in mammalian cells, hemagglutinin (HA)–tagged G9a and Myc-TBC1D3 were cotransfected into human embryonic kidney (HEK) 293 cells, and cell lysates were subjected to immunoprecipitation (IP). We found that IP of HA-G9a caused co-IP of Myc-TBC1D3, and vice versa (Fig. 2C). Furthermore, we observed the interaction between endogenous TBC1D3 and G9a, as IP with G9a antibody caused co-IP of TBC1D3 in homogenates of the GW15 human cortical tissue (Fig. 2D). In the human fetal brain, the TBC1D3 signals were colocalized with that of G9a in the nucleus of cells in the OSVZ (fig. S2A). The direct interaction between TBC1D3 and G9a was further verified using a pull-down assay. HEK293T cells were transfected with a construct encoding Myc-tagged TBC1D3 (Myc-TBC1D3), and then the cell lysates were incubated with beads containing glutathione S-transferase (GST) protein or GST-tagged recombinant fragment of 649 to 1210 amino acids of G9a, which contained C-terminal ANK and SET domains and thus was shortened as G9a-CF (CF represents C-terminal fragment). As shown in Fig. 2E, G9a-CF, but not GST alone, interacted with TBC1D3. The truncated CF containing the catalytic SET domain (879 to 1210 amino acids), but not the ANK domain (649 to 879 amino acids), was able to bind TBC1D3 directly (fig. S2B). These results indicate that TBC1D3 directly interacts with G9a in the developing human cortex.
Fig. 2. Interaction between TBC1D3 and G9a inhibits H3K9me2 modification.
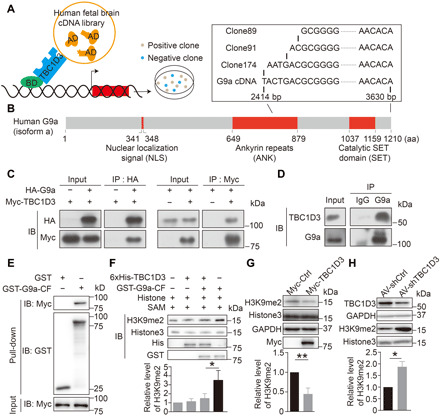
(A) Schematic diagram for Y2H screening assay. (B) Domain structure of human G9a protein and sequences of positive clones. (C) IB analysis of reciprocal co-immunoprecipitation (co-IP) results in HEK293T cells transfected with HA-G9a or HA-G9a plus Myc-TBC1D3. (D) Homogenates of GW15 human fetal cortical tissues were subjected to IP with anti-G9a antibody with immunoglobulin G (IgG) as a control and IB with antibodies against TBC1D3 or G9a. Shown is an example of two independent experiments with similar results. (E) Homogenates of HEK293cells transfected with Myc-TBC1D3 were subjected to pull-down with beads coupled with GST or GST-G9a-CF [649 to 1210 amino acids (aa)], followed by IB with anti-Myc antibody. (F) Addition of 6xHis-TBC1D3 attenuates the Histone3 methylation activity of G9a. Relative levels of H3K9me2 with respect to that of Histone3 from three independent experiments were quantified. (G) Levels of H3K9me2 in ReN cells transfected with control (Myc-Ctrl) or Myc-TBC1D3 plasmid (six independent experiments). (H) Levels of H3K9me2 in human cerebral organoids infected with AV-shCtrl or AV-shTBC1D3 (three independent experiments). The quantified data are presented as means ± SD by unpaired Student’s t test. *P < 0.05; **P < 0.01. bp, base pair.
TBC1D3 inhibits histone methylation activity of G9a
Having shown the interaction between TBC1D3 and G9a, we next investigated whether this interaction regulates the methyltransferase activity of G9a. To this end, we used the in vitro histone methylation system. Because the full length of G9a was difficult to be purified, we generated G9a-CF as the catalytic enzyme instead. As shown in fig. S2C, in the presence of methyl group donor SAM (S-adenosyl-l-methionine), purified G9a-CF was capable of mediating H3K9me2 modification. Addition of TBC1D3 to the histone methylation system significantly decreased the level of H3K9me2 (Fig. 2F, lanes 3 and 4), suggesting the inhibitory effect of TBC1D3 on G9a activity. TBC1D3 itself had no effect on H3K9me2 modification (Fig. 2F, lanes 1 and 2).
We then determined whether TBC1D3 regulates histone methylation in human neural stem cells. We found that transfection with Myc-TBC1D3 in ReN cells caused a marked decrease in the level of H3K9me2 but had no effect on some other histone modifications such as H3K9me3, H3K27me2, and H3K36me2, compared with the vehicle control group (Fig. 2G and fig. S2D). In addition, H9 hESCs transduced with rLV-TBC1D3 as well as later induced cerebral organoids also showed markedly decreased H3K9me2 compared with vehicle control group (fig. S2, E and F). By contrast, down-regulation of TBC1D3 by small interference RNA in D40 human cerebral organoids caused a marked increase in the level of H3K9me2 (Fig. 2H). Thus, the level of TBC1D3 is reversely correlated with that of H3K9me2, supporting the hypothesis that TBC1D3 suppresses the activity of G9a.
Our previous study has demonstrated that the TBC1D3 transgenic (TG) mice show increased cortical expansion and folding (15). We determined levels of H3K9me2 in wild-type and TG mice and found that TG mice exhibited a decreased level of H3K9me2 at embryonic days 14.5 (E14.5) and 17.5 (E17.5) (fig. S2, G and H). These results suggest a correlation between the states of H3K9me2 modification and cortex expansion during evolution.
G9a inhibition promotes expansion of cultured human cerebral organoids
Next, we determined whether inhibition of G9a had any effect on cortex development by using compound UNC0638, a specific and competitive inhibitor of G9a with high efficiency and low cytotoxicity (26). First, we treated ReN cells with different concentrations of UNC0638 for 24 hours and found that these treatments reduced the level of H3K9me2 in a dose-dependent manner (fig. S3A). Then, an optimized concentration of UNC0638 (1 μM) was added into the medium during organoid induction (D12) with dimethyl sulfoxide (DMSO) as control, with drug-containing medium renewed every other day. Again, UNC0638 treatment resulted in notable reduction in the level of H3K9me2, as measured by either immunoblotting (IB) (Fig. 3A) or immunostaining (fig. S3B). UNC0638-treated organoids exhibited a marked increase in size at different culture stages as exampled in D30 and D40 (Fig. 3B). Then, different molecular markers were used for detailed immunochemistry analysis for NPs and differentiated neurons. We found that the percentage of cells labeled by KI67 or the expression of PAX6 increased significantly in UNC0638-treated organoids at D18 (fig. S3, C and D). The augmentation of NPs in UNC0638-treated organoids persisted until later stages at D30, as reflected from increased percentage of cells labeled by PAX6, P-VIM, or KI67 (Fig. 3, C, D, and E to G). The cortical identity of cultured organoids was further confirmed by the appearance of TBR2-marked intermediate progenitors and DCX-labeled differentiated neurons, which were also increased in UNC0638-treated samples (Fig. 3, C, D, F, and I). We believe that G9a inhibition might have promoted replenishment of NPs, subsequently leading to enhanced neurogenesis.
Fig. 3. Effects of G9a inhibition on cortical expansion in human cerebral organoids.
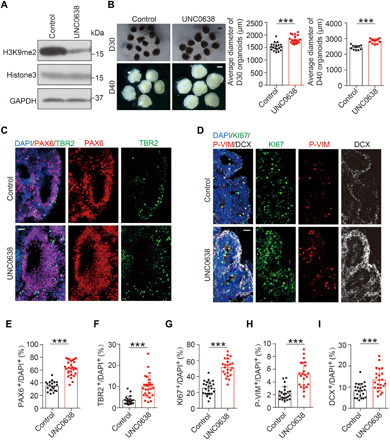
(A) Levels of H3K9me2 in D16 cerebral organoids treated with G9a inhibitor UNC0638 or DMSO control, determined by IB. (B) Representative images of D30 and D40 cerebral organoids treated with UNC0638 or vehicle control and quantification for the average diameter. Numbers of organoids analyzed: D30, 19 organoids for control and 20 organoids for UNC0638 group; D40, 11 organoids for control and 12 organoids for UNC0638 group. Scale bar, 1000 μm. (C) Immunostaining for signals of PAX6, TBR2, and DAPI in D30 organoids treated with UNC0638 or DMSO. Scale bar, 50 μm. (D) Immunostaining for signals of KI67, P-VIM, and DCX in D30 organoids treated with UNC0638 or DMSO control, with DAPI marking cell nucleus. Scale bar, 50 μm. (E and F) Quantification for the percentage of PAX6+ cells (E) or TBR2+ cells (F) among DAPI+ cells (20 rosettes from 14 control organoids; 31 rosettes from 17 UNC0638-treated organoids). (G to I) Quantification for the percentage of KI67+ (G), P-VIM+ (H), or DCX+ cells (I). Twenty-three rosettes from 15 organoids were analyzed in each group. Data are presented as means ± SD, unpaired Student’s t test. ***P < 0.001.
To investigate the proliferation and neuronal competency of NPs, D30 organoids were infected with GFP-expressing retrovirus, followed by culture for an additional 3 days in virus-free medium, to label dividing cells and the daughter progeny (fig. S3E). We found that the percentage of GFP+ cells among DAPI+ cells increased in UNC0638-treated organoids (fig. S3F), while the percentage of KI67+ GFP+ or DCX+ GFP+ among total GFP+ cells had no difference (fig. S3, G and H). These results suggest that G9a inhibition expanded the pool of dividing NPs without altering cellular lineage composition and neuronal competency. The effect induced by UNC0638 is reminiscent of the expansion of the human cerebral organoids with overexpression of TBC1D3 as well as a mutation of PTEN (27).
Domain mapping of TBC1D3 for its interaction with G9a
To precisely manipulate the interaction between TBC1D3 and G9a and seek the functional relevance, we mapped the TBC1D3 region that is essential for the interaction. We generated a battery of truncated forms of TBC1D3 tagged with Myc at the N terminus (Fig. 4A) and cotransfected each of them with HA-G9a into HEK293 cells. Then, the cell lysates were subjected to IP with anti-HA antibody. In the first round of domain mapping, we generated mutated forms of TBC1D3 with sequential deletion of a quarter of full length and found that the mutant with the deletion of C terminus (TBC1D3Δ413–549) failed to be associated with G9a, whereas other mutants maintained the binding activity (Fig. 4B). This result suggests that the C terminus of TBC1D3 is essential for the interaction with G9a. Based on this, we did other rounds of narrowing down to pinpoint the regions essential for the interaction (Fig. 4, C to E). Last, the minimal region was mapped to 465 to 481 amino acids, because TBC1D3Δ465–481 did not interact with G9a (Fig. 4E), but further partitioning had no effect (Fig. 4F).
Fig. 4. The 465– to 481–amino acid segment in TBC1D3 is essential for its interaction with G9a.
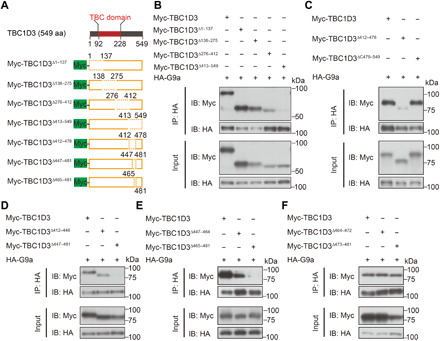
(A) Schematic representation for full length of TBC1D3 protein and mutated forms with indicated fragment deletions. (B to F) HEK293 cells were cotransfected with constructs encoding HA-tagged G9a and Myc-tagged full-length or mutated forms of TBC1D3. Cell homogenates were subjected to IP with antibody against HA, followed by IB with antibody against Myc or HA. Data shown are blots of representative experiments performed for at least three times with similar results.
TBC1D3 interaction with G9a promotes the proliferation of cortical NPs
Because the 465– to 481–amino acid segment of TBC1D3 was essential for the interaction with G9a, we asked whether the peptide covering this sequence is able to interfere with TBC1D3-G9a interaction. We synthesized the peptide composed of the cell-penetrating TAT sequence derived from the trans-activator of transcription of human immunodeficiency virus (28) and the 465 to 481 amino acids of TBC1D3 (shortened as T-T) or scrambled sequence (shortened as T-S) (Fig. 5A) and tested their effect on TBC1D3/G9a interaction. As shown in fig. S4A, the association between Myc-TBC1D3 and HA-G9a was attenuated in transfected HEK293 cells treated with T-T, but not T-S. Furthermore, the efficiency of GST-G9a-CF in pulling down Myc-TBC1D3 was markedly decreased in reactions with the presence of T-T, but not T-S (fig. S4B). Next, we asked whether the blockade of TBC1D3/G9a interaction changed the level of H3K9me2 or proliferation of NPs in human cerebral organoids. For this purpose, the organoids at D12 were treated with T-T or T-S for 4 days, followed by immunostaining with various antibodies. Notably, we found that the volume of human cerebral organoids reduced significantly in the T-T group (Fig. 5A). Moreover, the level of H3K9me2 was markedly increased, whereas the percentage of KI67+ cells or the intensity of PAX6+ signals was markedly decreased in T-T–treated organoids (Fig. 5, B to D). These results were unlikely caused by a direct effect of peptides on G9a, because addition of T-T to the in vitro histone methylation assay did not change the level of H3K9me2 (fig. S4C). In addition, treatment of mouse neural stem cell N2A, which does not harbor TBC1D3, with T-T had no effect on H3K9me2 modification (fig. S4D). These results were in line with the idea that TBC1D3 interaction with G9a represses its histone dimethylation activity and thus maintains H3K9me2 at a low level, which may ensure high proliferative potency of human cortical NPs.
Fig. 5. Blockade of TBC1D3 interaction with G9a attenuates NP proliferation.
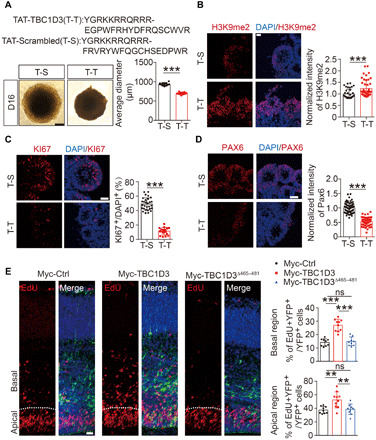
(A) Blockade of TBC1D3/G9a interaction with peptides suppresses organoid growth. At least 14 organoids were analyzed in each group. Scale bar, 200 μm. (B) H3K9me2 signals in D16 organoids. Data are represented as means ± SD of 25 rosettes from 9 T-S organoids and 33 rosettes from 12 T-T organoids, with average value of T-S group normalized as 1. Scale bar, 20 μm. (C) Analysis for the percentage of KI67+ in D16 organoids (29 rosettes from 7 T-S organoids; 16 rosettes from 6 T-T organoids). Scale bar, 40 μm. (D) PAX6 signals in D16 organoids. Normalized intensity of PAX6 was quantified with the value of T-S group set as 1 (79 rosettes from 6 T-S organoids; 49 rosettes from 6 T-T organoids). Scale bar, 40 μm. (E) Proliferative cells marked by EdU in mouse cortex transfected with indicated plasmids and yellow fluorescent protein (YFP). Histograms indicate percentage of EdU+ cells among YFP+ cells (9 embryos in the Myc-Ctrl or Myc-TBC1D3Δ465–481 group; 10 embryos in the Myc-TBC1D3 group). Scale bar, 20 μm. Data are presented as means ± SD, unpaired Student’s t test. **P < 0.01; ***P < 0.001; ns, no significant difference.
We have shown previously that TBC1D3 expression promoted the generation and proliferation of basal cortical progenitors, leading to cortex expansion in mice (15). We wondered whether these effects were attributable to the TBC1D3 regulation of G9a. The fetal mice at E13.5 were subjected to in utero electroporation (IUE) to introduce various constructs into NPs in the VZ, followed by analysis of cell proliferation at E15.5. We first examined the effects of full-length (Myc-TBC1D3) and the mutated form of TBC1D3 with the deletion of 465 to 481 amino acids (Myc-TBC1D3Δ465–481) on NP proliferation by calculating the proportion of cells in the S phase determined by incorporation of pyrimidine analog 5-ethynyl-2′-deoxyuridine (EdU) (Fig. 5E). Consistent with previous observation (15), we found that the Myc-TBC1D3 IUE mice exhibited an increase in EdU+ proliferating cells, as compared with control mice subjected to IUE with vesicle plasmid, while IUE with Myc-TBC1D3Δ465–481 had no effect on NP proliferation (Fig. 5E). These effects were evident in both apical and basal regions (Fig. 5E, right). Unlike the full length of TBC1D3, forced expression of TBC1D3Δ465–481 in ReN cells did not reduce the level of H3K9me2 (fig. S4E). These results suggest that the suppression of G9a-mediated histone dimethylation by TBC1D3 underlies its role in promoting NP proliferation.
TBC1D3/G9a interaction regulates pathways involved in proliferation of NPs
As H3K9me2 modification is considered to be a typical repressive transcription mark (7), we analyzed the gene expression networks regulated by TBC1D3/G9a interaction. First, we compared global transcriptome profiles between human cerebral organoids (D16) treated with T-S or T-T using RNA-seq information. We conducted two replicates in each group to validate experimental consistency in terms of peptide treatment and sequencing process. The hierarchical clustering and Pearson correlation analysis revealed similar patterns in duplicates of either T-S (T-S-1 and T-S-2) or T-T (T-T-1 and T-T-2) duplicates (fig. S5, A and B). Among the differentially expressed genes (DEGs), the significantly changed ones were selected (fig. S5C) for further analysis. Gene ontology (GO) analysis showed that the down-regulated genes in T-T organoids were enriched in functional forebrain development or neuronal differentiation, while the up-regulated genes were enriched in apoptotic signaling pathways (fig. S5D). Further analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database showed that DEGs were enriched in pathways involved in cell proliferation, such as AKT, WNT, or MAPK (mitogen-activated protein kinase) signaling, as well as pluripotency of stem cells (Fig. 6A). Among down-regulated genes, we found several genes that encode WNT ligands and receptor FZD10, FGF receptors, or PAX6 (Fig. 6B). Quantitative gene expression analysis using real-time reverse transcription PCR (RT-PCR) also validated the down-regulation of these representative genes in T-T–treated organoids (Fig. 6C). These results support the conclusion that TBC1D3 interaction with G9a down-regulates the level of H3K9me2 and, hence, promotes the expression of genes involved in the proliferation of NPs.
Fig. 6. Blockade of TBC1D3 interaction with G9a down-regulates expression of genes associated with NP proliferation.
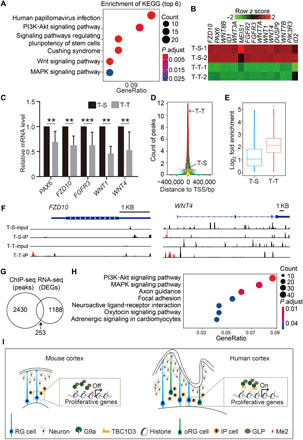
(A) Top six enriched KEGG pathways by clustering analysis of down-regulated genes in T-T groups compared with T-S groups. (B) Heatmap showing representative down-regulated genes in T-T–treated organoids. (C) Relative mRNA levels of indicated genes to GAPDH measured by quantitative PCR analysis for D16 organoids treated with T-T or T-S peptides. Data are presented as means ± SD of at least five experiments in each group with values from T-S group normalized as 1. Unpaired Student’s t test. **P < 0.01; ***P < 0.001. (D) ChIP-seq showing the count and distribution of peaks around TSSs in T-S– or T-T–treated organoids at D16. (E) H3K9me2-binding peaks increase in T-T–treated organoids. (F) Genomic tracks showing differential H3K9me2 enrichment regions near TSS of FZD10 and WNT4. (G and H) Venn diagrams (G) and GO biological enrichment analysis (H) of genes corresponding to ChIP-seq peaks and DEGs identified by RNA-seq. (I) A proposed model showing a role of TBC1D3 in promoting NP proliferation and cortex expansion through down-regulating the level of H3K9me2. PI3K, phosphatidylinositol 3-kinase.
To identify the genomic locus associated with H3K9me2, we performed the genome-wide H3K9me2 chromatin immunoprecipitation sequencing (ChIP-seq) in human cerebral organoids treated with T-S or T-T peptides. The peak reads of H3K9me2 binding on transcriptional start sites (TSSs) were markedly increased in D16 organoids treated with T-T (Fig. 6, D and E, and fig. S5E). For example, many differential peaks appeared to be enriched in promoter regions of regulated genes, such as FZD10 or WNT4 (Fig. 6F, see the red peaks). Moreover, the comprehensive analysis showed that 253 genes were overlapped between the DEGs of RNA-seq and differential peaks observed in ChIP-seq (Fig. 6G). Further GO analysis indicated that the overlapped genes were mainly enriched in proliferative pathways, such as PI3K-Akt and MAPK signaling (Fig. 6H). These results support the conclusion that blockade of the TBC1D3/G9a interaction reactivates G9a activity, leading to increased level of H3K9me2, which marks suppressive gene expression. Together, the inhibitory role of TBC1D3 in G9a activity through direct interaction may maintain H3K9me2 at a low level, which allows expression of genes involved in NP proliferation and hominoid cortical expansion (Fig. 6I).
DISCUSSION
The expansion in human cerebral cortex is believed to facilitate emergence of higher cognitive skills (1). Prolonged duration of cortical neurogenesis may contribute to cortical expansion and folding, and this process involves markedly increased proliferation capacity of cortical NPs. Recently, several human-specific genes have been shown to promote cortical progenitor proliferation and expansion, and the underlying mechanisms varied from cell cycle transition (29) to glutaminolysis regulation (30). In this study, we found that the level of H3K9me2 modification is reversely correlated with the proliferation capacity of cortical NPs. The hominoid-specific protein TBC1D3 inhibits G9a-mediated H3K9me2 modification, and this regulation underlies TBC1D’s role in promoting the proliferation of cortical NPs. The down-regulation of H3K9me2 caused by TBC1D3 interaction with G9a may derepress the expression of genes involved in the proliferation of NPs, which, in turn, resulted in cortical expansion. This study shows an epigenetic mechanism underlying enhanced stemness of NPs during the evolution of neocortex.
Compared with rodents, the human cortex exhibits increased radial and tangential expansion and more abundant cortical progenitors, which have sustained capability of multiple rounds of division and prolonged neurogenic period (4, 31). Interspecies comparisons have led to identification of specific genomic changes on the human linage, including individual nucleotide variation, insertion-deletions, gene duplications, and a few purely de novo human-specific genes (32). Besides coding regions, many forms of variants in regulatory regions or epigenetic elements, such as human-specific microRNAs (33) and differential histone methylation compared with other nonhuman primates, have been identified (34). Nevertheless, connecting these changes to functions in human brain development has been challenging due to the limitation of ethical issues and the lack of appropriate experimental approaches. In this study, we have established a link between TBC1D3 and H3K9me2 modification in cultured human cerebral organoid system.
Previous studies have shown that the level of H3K9me2 is dynamically regulated in the contexts of memory formation, addiction, and stress (35–37). G9a-deficient mice display severe growth retardation and early lethality, and H3-K9 methylation is decreased markedly in G9a-deficient embryos (38). In humans, haploinsufficiency or disruption of the GLP gene has been shown to be associated with congenital intellectual disability, including Kleefstra syndrome and autism spectrum disorder (10). In mice, heterozygous ablation of GLP gene caused developmental delay and abnormal behavior (39). It would be of interest to determine whether TBC1D3 is involved in any intellectual disability or neural developmental disorders. Given that many mammals without TBC1D3 also have cortical expansion compared with rodents, this study does not preclude other mechanisms governed by multiple genetic elements underlying cortex expansion during evolution.
TBC1D3 has been shown to be involved in RAB guanosine triphosphatase signaling, vesicle trafficking, and tissue repair (16, 22–24). All these functions seem to rely on its cytosolic and/or membrane localization. Notably, TBC1D3 can also shuttle between cytoplasm and nucleus, and its cytoplasmic retention needs microtubule network (40). In this study, we unraveled a role of TBC1D3 in the nucleus, especially in human NPs. Intriguingly, TBC1D3 was expressed in almost all PAX6+ or TBR2+ cells, and notably, most of those in the OSVZ and a fraction of vRGs had TBC1D3 enriched in the nucleus. This heterogeneous subcellular localization may reflect different states of vRGs. As shown in our previous study, the expression of TBC1D3 in vRGs caused destabilization of Cdh2 mRNA, leading to down-regulation of N-cadherin and delamination of vRGs in mice (15). How the dynamic spatial localization of TBC1D3 is determined in NPs at various states and/or positions warrants further study. Furthermore, its dominant distribution in the cytosol of differentiated neurons suggests multifaceted functions.
MATERIALS AND METHODS
Mice maintenance
C57BL6/J mice and TBC1D3-TG mouse line maintained in C57BL6/J background were used for the IUE experiments. All animal manipulations including mouse housing, breeding, and surgical procedures were executed in compliance with the ethical guidelines of the Institutional Animal Care and Use Committee of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, and ShanghaiTech University. All mice were housed under a 12-hour light-dark cycle in the institutional animal care facility. The TBC1D3-TG mice were generated as described previously (15). Mice at E13 to E17 were used for experimental processing without discrimination of the sex of embryos.
Human fetal tissue samples
The human fetal cortical tissue samples were obtained from medical pregnancy termination. The collection and usage of the human fetuses were conducted in strict observance of the ethical guidelines approved by the ethics committee in Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (approval identifier number: ER-SIBS-221506). After release from clinical autopsy procedure, the brain tissues were transported in ice-cold Leibowitz-15 medium (Gibco, 21083027) and stored in liquid nitrogen for protein extraction or embedded in optimal cutting temperature (OCT) compound (Tissue-Tek Sakura, catalog no. 4583) for further frozen sectioning and immunostaining.
Cell culture and transfection
N2A and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, 11966-025) supplemented with 10% fetal bovine serum (FBS; Gibco, 10099-141) in a 37°C incubator with 5% CO2. HEK293T cells were transfected with plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019). ReN cells derived from human mesencephalon were grown in DMEM/F12 medium (Gibco, H330057) supplemented with B27 (Gibco, 17504044), heparin (10 U/ml; Sigma-Aldrich, H3393), epidermal growth factor (20 ng/ml; Stem cell, 78073), and fibroblast growth factor (10 ng/ml; Stem cell, 78003). The cultured dishes for ReN cells were pretreated with 0.5% laminin (Sigma-Aldrich, L2020) dissolved in DMEM medium for at least 4 hours at 37°C. ReN cells were transfected with plasmids using Nucleofector (Lonza Nucleofector II, 2B). H9 hESCs (NEST) were grown on Matrigel (BD Biosciences, 354277)–pretreated dishes and cultured in mTeSR medium (STEMCELL Technologies, 5850). Clones of H9 hESCs were passaged using the ReLeSR kit (STEMCELL Technologies, 5872) according to the protocol when the size of clones reached 1 to 2 mm in diameter.
Constructs
Constructs encoding mutated forms of TBC1D3 with various deletions were generated using site-directed mutagenesis with a PrimeSTAR GXL DNA polymerase kit (Takara, R050A) according to standard protocol with Myc-TBC1D3 plasmid (15) as template and primers listed in table S1. TBC1D3 was also subcloned into PGB plasmid as bait for Y2H screening, into lentiviral vector rLV-EF1a-2A-EGFP-T2A-puro-WPRE for transduction of ES clones, and into PET-28a plasmid to produce 6xHis-tagged recombinant proteins. The full-length coding sequence of G9a or 649– to 1210–amino acid fragment was amplified by PCR and subcloned into pGEX-2T-GST or pKH3-HA vector (see table S1 for the list of plasmids and sequences of primers used in PCR amplification).
Y2H screening
Y190 yeast cells were cotransfected with PGB-TBC1D3 plasmid and the human fetal brain cDNA library (Clontech, catalog no. HL4028AH). The hits in the positive yeast clones were amplified and sequenced to obtain the gene information. False-positive clones were excluded from the following analysis.
Purification of recombinant proteins and pull-down assay
The plasmids encoding GST-tagged G9a truncated fragments or 6xHis-tagged TBC1D3 were transformed into Rosetta Escherichia coli BL21 strain. After 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) induction (16°C for 20 hours) for protein expression, cells were collected and lysed in phosphate-buffered saline (PBS) buffer supplemented with dithiothreitol (DTT) and phenylmethylsulfonyl fluoride (PMSF) using ultrasonication. For GST-fusion protein, the precleared supernatants were collected and incubated with Glutathione Sepharose 4B (GE Healthcare, 17-0756-01) beads, followed by washes in PBS and elution with glutathione (5 mg/ml). For 6xHis-tagged proteins, the lysis buffer was changed to phosphate buffer containing 10 mM imidazole, 300 mM sodium chloride, 50 mM sodium phosphate buffer, 10% glycerol, and 0.5% Tween, and recombinant proteins were purified with Ni column using elution buffer (30 mM sodium phosphate buffer, 300 mM imidazole, 300 mM sodium chloride, and 10% glycerol).
The cell lysates (1 mg/ml protein) of HEK293T cells transfected with Myc-TBC1D3 or various mutants were incubated with GST-tagged G9a fragments coupled with glutathione agarose beads at 4°C with gentle rotation. For TAT blockade experiments, the peptide of T-S or T-T (100 μM) was added into the mixture of Myc-TBC1D3 and GST-G9a-CF before following incubation. The beads were then washed in cell lysis buffer and subjected to IB analysis with corresponding antibody.
Co-immunoprecipitation, subcellular fractionation, and IB analysis
HEK293T cells transfected with various plasmids were lysed in modified radioimmunoprecipitation assay (RIPA) buffer containing 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, 1 mM EDTA, and protease inhibitor cocktail. After centrifugation (12,000 rpm for 15 min), supernatants were collected and incubated with primary antibodies at 4°C overnight and then incubated with Protein-G or Protein-A beads at 4°C for 4 hours. After five washes with lysis buffer, the beads were boiled in 30 to 50 μl of 1× SDS loading buffer and subjected to IB analysis. Nuclear and cytoplasmic fractions were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, 78833) following the manufacturer’s instructions. For IB analysis, protein samples were loaded and separated in SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membranes. After blocking in 5% milk in TBS-T (tris buffered saline-Tween) for 1 hour at room temperature, the membranes were probed with primary antibodies and visualized with horseradish peroxidase (HRP)–conjugated secondary antibodies. Antibodies for IB analysis were as follows: TBC1D3 (rabbit, Abcam, Ab139034), H3K9me2 (Cell Signaling Technology, 4658s), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Proteintech, 60004-1), Histone3 (Cell Signaling Technology, 4499s), Myc (rabbit, Sigma-Aldrich, C3956; mouse, Millipore, 05-419), HA (rabbit, Cell Signaling Technology, 3724s; mouse, Sigma-Aldrich, H3663), H3K9me3 (Abcam, Ab8898), H3K27me2 (Cell Signaling Technology, 9728T), H3K36me2 (Cell Signaling Technology, 2901T), HRP anti-mouse (Abcam, Ab64259), and HRP anti-rabbit (Abcam, Ab64261).
In vitro methylation assay
Histone proteins were extracted and purified from cultured ReN cells following the protocol as described previously (41). Briefly, 5 × 106 cells were collected and lysed in hypotonic lysis buffer containing 10 mM tris-HCl (pH 8.0), 1 mM KCl, 1.5 mM MgCl2, 1 mM DTT, and protease inhibitor cocktail (Selleck, B14001) to release the intact nuclei. The nuclei pellets were then resuspended in 0.4 N H2SO4 and incubated on a rotator for at least 30 min or overnight at 4°C. Histone proteins were precipitated in 33% trichloroacetic acid (TCA) (Sigma-Aldrich, T4885), washed with ice-cold acetone, air dried for 20 min at room temperature, and, lastly, dissolved in appropriate volume of ddH2O.
The purified histone proteins (about 30 ng/μl) were mixed with GST-G9a (649 to 1210 amino acids) (30 ng/μl) in 30 μl of reaction buffer containing 0.5 mM SAM (New England Biolabs) as the methyl group donor, 50 mM tris-HCl (pH 8.0), 2 mM MgCl2, 0.01% Trion X-100 (Takara), 1 mM tris (2-carboxyethyl) phosphine (TCEP) (Hampton Research), and protease inhibitor cocktail, and incubated for 12 hours at room temperature, without or with 6xHis-TBC1D3 (30 ng/μl). The SDS-PAGE sample buffer was added to stop the reaction, and the products were subjected to IB analysis.
Human cerebral organoid culture, drug treatments, and virus infection
Human cerebral organoids were derived from H9 ES cells following the protocol introduced previously (20) with some modifications. First, after culturing for 48 hours, the ES clones were digested to single cells and passaged to new dishes. Then, the clones were digested into single cells using 1 ml of Accutase at 37°C for 5 min, washed in mTeSR medium, centrifuged at 800 rpm for 90 s, and resuspended in 2 ml of mTeSR medium. The cell suspension was seeded into low-attachment V-bottom 96-well plates with about 7000 cells per well in 180 μl of mTeSR medium supplemented with 10 μM Y27632 (Stem cell, 72304). After 2 days, the medium was changed to the hES medium containing 80% DMEM/F12, 20% KSR (KnockOutTM Serum Replacement) (Gibco, 10828028), 1% GlutaMAX (Thermo Fisher Scientific, 35050061), 1% MEM_NEAA (Invitrogen, 11140050), 0.0004% 2-mercaptoethanol, 2.5 μM dorsomorphine (Tocris, Oct-93), and 2 μM A83-01 (Tocris, Oct-39) and cultured for 4 days. At D6 to D12, half of the hES medium was replaced with neural induction medium: DMEM/F12 supplemented with 1% N2 supplement (Thermo Fisher Scientific, 17502048), 1% GlutaMAX, 1% MEM_NEAA, heparin (1 μg/ml; Sigma-Aldrich, H3393), 200 nM LDN-193189 (Selleck, S7507), and 2 μM SB431542 (Selleck, S1067). At D12, the organoids were embedded in Matrigel and cultured in neural differentiation medium containing 50% DMEM/F12, 50% Neurobasal medium (Life Technology, 12348-017), 0.5% N2 supplement, 1% GlutaMAX, 1% MEM_NEAA, 1% B27, 0.0004% 2-mercaptoethanol, and 0.025% insulin (Sigma-Aldrich, I9278) for 4 days, without or with the addition of small molecular inhibitors UNC0638 (1 μM; Selleck, S8071), DMSO vehicle control, or 30 μM peptides (T-T: YGRKKRRQRRR-EGPWFRHYDFRQSCWVR; T-S: YGRKKRRQRRR-FRVRYWFQGCHSEDPWR). The drug-containing medium was renewed every other day. At D16, B27 supplemented with vitamin A was used in differentiation medium, and the culture condition was maintained for the following days. For lentivirus-transduced cerebral organoids, H9 ES clones (1 to 2 mm in diameter) were infected with control lentivirus (rLV-EF1a-2A-EGFP-T2A-puro-WPRE, shortened as rLV-Ctrl) or TBC1D3 expressing lentivirus (rLV-EF1a-TBC1D3-2A-EGFP-T2A-puro-WPRE, shortened as rLV-TBC1D3). Two days later, puromycin (1/1000; Sigma-Aldrich, P8833) was added to select transfected cells with GFP as selection marker. The clones grown from single GFP+ cells were subjected to quantitative real-time PCR to determine the RNA level of TBC1D3 and used in the following analyses. The nucleotide sequence of small interference RNA for TBC1D3 (target sequence: 5′-GCCTCTATGAAGAAACTAA-3′) or control (target sequence: 5′-TTCTCCGAACGTGTCACGT-3′) was inserted into adenovirus vector to generate the pDKD-CMV-eGFP-U6-shTBC1D3 construct. The packaged adenovirus was added into organoid culture medium at D37 and maintained for 3 days. For retrovirus infection, virus was added into organoid culture medium at D26 and maintained for 24 hours. Then, the medium with virus was removed, and organoids were washed immediately with fresh medium for at least three times. After another 3-day culture, organoids were fixed in 4% paraformaldehyde (PFA) and subjected to immunostaining.
Quantitative PCR for mRNA expression
The total RNA from brain tissue or human brain organoids was extracted with TRIzol reagent (Life Technology, 15596018) following the manufacturer’s instructions. RNA samples were subjected to reverse transcription and quantitative real-time PCR using SYBR Green (Selleck, B21702) on QuantStudio 7 Flex System (Life Technologies). The QuantStudio Real-Time PCR Software v1.3 was used for data analysis. The primers used were as follows: TBC1D3, 5′-AGGTTCAGCAGAAGCGCCTCA-3′ (forward), 5′-GCCTGGATGCCGACGACCCTT-3′ (reverse); human GAPDH, 5′-GACCTGCCGTCTAGAAAAACCT-3′ (forward), 5′-CTGTTGCTGTAGCCAAATTCGT-3′ (reverse); mouse GAPDH, 5′-GGGTCATCATCTCCGCCCC-3′ (forward), 5′-TTGGCAGCACCAGTGGATGCA-3′ (reverse); PAX6, 5′-TGCATTTGCATGTTGCGGAG-3′ (forward), 5′-TTAGCGAAGCCTGACCTCTG-3′ (reverse); FZD10, 5′-CAAACCTCGAAACAGCTGCC-3′ (forward), 5′-AACAATACCGGGAAGCGAGG-3′ (reverse); FGFR3, 5′-AGGAGCTCTTCAAGCTGCTG-3′ (forward), 5′-ACAGGTCCAGGTACTCGTCG-3′ (reverse); WNT1, 5′-CAAGATCGTCAACCGAGGCT-3′ (forward), 5′-AAGGTTCATGAGGAAGCGCA-3′ (reverse); WNT4, 5′-CGTGCCTGCGTTCGCT-3′ (forward), 5′-GGCAAGGAGTCGAGTGTGG-3′ (reverse); FOXG1, 5′-CCCTCCCATTTCTGTACGTTT (forward), 5′-CTGGCGGCTCTTAGAGAT (reverse).
RNA sequencing
High-throughput sequencing of total RNA isolated from human cerebral organoids was performed on Illumina NovaSeq 6000 system with average length of 150 nucleotides for every read of paired end. Raw data were filtered by FASTX-Toolkit to generate clean reads and then mapped to human GRCh38. The level of a specific transcript was expressed as FRKM (fragments per kilobase of transcript per million fragments mapped) measured using StringTie software with statistical criterion set as P < 0.05. GO analysis was performed using EdgeR software with false discovery rate <0.05 and log2 (fold change) >1 or <−1.
ChIP sequencing
The samples for ChIP analysis were extracted using SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, 91820s). Briefly, tissues were fixed in 1% formaldehyde to cross-link proteins to DNA for 10 to 20 min at room temperature, and the reaction was stopped by addition of glycine. Then, cells were lysed to release nucleus, and Micrococcal Nuclease was added to digest chromatin into protein-associated DNA fragments, followed by sonication to break nuclear membranes and generate chromatin fragments of appropriate size. The samples were incubated with antibody against H3K9me2 (Cell Signaling Technology, 4658s) at 4°C overnight and then incubated with ChIP-grade Protein G magnetic beads at 4°C for 2 hours, followed by DNA elution and purification. DNA samples in input and IP groups were pair-end sequenced on HiSeq 2500 (XTen) platform. The sequence reads were trimmed for adaptor sequence using FASTP software (version 0.19.11), and peak calling was conducted using MACS2 (version 2.1.0). Peaks were mapped to genome using BWA software (version 0.7.12-r1039), and GO analysis was conducted using Goseq and Bioconductor (version 4.10.2). The enrichGO function in the clusterProfiler (v3.13) R package was used for overrepresentation analysis of GO biological processes overlapped in ChIP-seq and RNA-seq. Venn diagram is plotted by VennDiagram R package.
In utero electroporation
Pregnant mice with embryos at D13.5 or D14.5 were anesthetized with a mixture of pentobarbital sodium (2.5 g/kg body weight) and ketamine (50 mg/ml of solution) and subjected to IUE. The uterus was exposed under sterile conditions, and plasmid solutions containing DNA (1 to 2 μg/μl) mixed with fast green (0.1 mg/ml; Sigma-Aldrich, F7252) were manually injected into the lateral ventricles with a beveled glass micropipette (VWR International, 53432-921). Two tweezer electrodes connected to an electroporator (BTX830) were used in the electroporation procedure to deliver five 50-ms pulses of 30-V voltage with 950-ms interval. The mice were surgically sutured and placed on warm electric blanket until recovery.
Immunostaining and EdU labeling
Cultured cells on coverslips were fixed in 4% PFA for 10 min at room temperature. Embryonic mouse brains were dissected out and postfixed in 4% PFA overnight at 4°C. Cultured human brain organoids were soaked in 4% PFA for 2 to 4 hours at 4°C. The fixed tissues were dehydrated in 20% sucrose in PBS at 4°C and then sectioned at 30-μm (mouse brain) or 20-μm thickness (organoids) using a freezing microtome (Leica, CM1950). Sections of control and experimental groups were pasted on the same slide to maintain uniform conditions during staining and image collection processes. For immunohistochemistry, fixed cells were washed with PBS for three times and permeabilized in 0.1% Triton X-100 in PBS for 10 min. Tissue slices were subjected to antigen retrieval by citrate and then permeabilized in 0.3% Triton X-100 in PBS for 30 min. After blocking with 10% FBS for 50 min, the cells or slices were incubated with various primary antibodies at 4°C overnight, washed with PBS for three times, incubated with secondary antibodies for 2 hours at room temperature in the dark, and mounted with mounting reagent (DAKO, S3023) for observation.
For EdU labeling, pregnant mice that recovered from the IUE surge were injected intraperitoneally with EdU (50 mg/kg body weight) (Thermo Fisher Scientific, C10640). EdU staining was performed immediately after the secondary antibody incubation using a Click-iT Plus EdU Imaging Kit (Thermo Fisher Scientific, C10640). Antibodies for immunostaining were as follows: DAPI (Beyotime, C1002), TBC1D3 (rabbit, Abcam, Ab139034), TBC1D3 (mouse, Santa Cruz, sc-376073), KI67 (Abcam, Ab66155), PAX6 (Covance, PRB-278P), H3K9me2 (Cell Signaling Technology, 4658s), G9a (Abcam, Ab185050), β-tubulin (Cell Signaling Technology, 2128s), TBR2 (Invitrogen, 14-4877-82), DCX (Santa Cruz, sc-8066), HOPX (Sigma-Aldrich, HPA030180), Alexa Fluor 488 (Jackson, 703-546-155), Alexa Fluor 555 (Invitrogen, A31572), and Alexa Fluor 647 (Invitrogen, A31571).
Image processing and quantification
Images of immunostaining were collected using confocal microscopy with Nikon TiE, Nikon A1R, Leica P8, or Olympus FV3000 and processed with ImageJ software. The shooting parameters were kept the same between each control and experimental group. Statistical tests were performed using GraphPad Prism software, and data were presented as means ± SD. Data satisfied to Gaussian distribution test were quantified with Student’s t test, while others were quantified with unpaired Student’s t test. The statistical significance was indicated by *P < 0.05; **P < 0.01; ***P < 0.001; ns, no significant difference.
Supplementary Material
Acknowledgments
We are grateful to A. L. Sheng for the assistance with brain organoid culture, L. Du for suggestions on ChIP-seq analysis, Y. Jin for sharing ReN cells, and J. P. Ding for the modified PET-28a vector. Funding: This study was partially supported by grants from the National Natural Science Foundation of China (31490591 to Z.-G.L. and 31871034 to X.-C.J.), the National Key R&D Program of China (2017YFA0700500), the Frontier Key Project of the Chinese Academy of Sciences (QYZDJ-SSW-SMC025), and Shanghai Municipal Science and Technology Projects (2018SHZDZX05 and 201409001700). Author contributions: Q.-Q.H. designed the experiments, conducted data collection and analysis, and wrote the original draft. Q.X. and X.-C.J. participated in RNA-seq and ChIP- seq data analysis. X.-Y.S. participated in human cerebral organoid culture. Z.-G.L. conceived the project and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: RNA-seq data and ChIP-seq data were deposited in the Gene Expression Omnibus (GEO) with accession number GSE136283. The dataset for evaluating TBC1D3 expression in human and chimpanzee neural cells was deposited in the GEO with accession number GSE83638. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eaba8053/DC1
REFERENCES AND NOTES
- 1.Rakic P., Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun T., Hevner R. F., Growth and folding of the mammalian cerebral cortex: From molecules to malformations. Nat. Rev. Neurosci. 15, 217–232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fietz S. A., Huttner W. B., Cortical progenitor expansion, self-renewal and neurogenesis—A polarized perspective. Curr. Opin. Neurobiol. 21, 23–35 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Lui J. H., Hansen D. V., Kriegstein A. R., Development and evolution of the human neocortex. Cell 146, 18–36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrini R., Dobyns W. B., Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 13, 710–726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirabayashi Y., Gotoh Y., Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 11, 377–388 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Dambacher S., Hahn M., Schotta G., Epigenetic regulation of development by histone lysine methylation. Heredity (Edinb) 105, 24–37 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y., Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer A., Sampath S. C., Intrator A., Min A., Gertler T. S., Surmeier D. J., Tarakhovsky A., Greengard P., Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleefstra T., Smidt M., Banning M. J., Oudakker A. R., van Esch H., de Brouwer A. P., Nillesen W., Sistermans E. A., Hamel B. C., de Bruijn D., Fryns J. P., Yntema H. G., Brunner H. G., de Vries B. B., van Bokhoven H., Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 42, 299–306 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katoh K., Yamazaki R., Onishi A., Sanuki R., Furukawa T., G9a histone methyltransferase activity in retinal progenitors is essential for proper differentiation and survival of mouse retinal cells. J. Neurosci. 32, 17658–17670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubinstein M., de Souza F. S. J., Evolution of transcriptional enhancers and animal diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vermunt M. W., Tan S. C., Castelijns B., Geeven G., Reinink P., de Bruijn E., Kondova I., Persengiev S., Bank N. B., Bontrop R., Cuppen E., de Laat W., Creyghton M. P., Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat. Neurosci. 19, 494–503 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Reilly S. K., Yin J., Ayoub A. E., Emera D., Leng J., Cotney J., Sarro R., Rakic P., Noonan J. P., Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ju X. C., Hou Q. Q., Sheng A. L., Wu K. Y., Zhou Y., Jin Y., Wen T., Yang Z., Wang X., Luo Z. G., The hominoid-specific gene TBC1D3 promotes generation of basal neural progenitors and induces cortical folding in mice. eLife 5, e18197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodzic D., Kong C., Wainszelbaum M. J., Charron A. J., Su X., Stahl P. D., TBC1D3, a hominoid oncoprotein, is encoded by a cluster of paralogues located on chromosome 17q12. Genomics 88, 731–736 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Perry G. H., Yang F., Marques-Bonet T., Murphy C., Fitzgerald T., Lee A. S., Hyland C., Stone A. C., Hurles M. E., Tyler-Smith C., Eichler E. E., Carter N. P., Lee C., Redon R., Copy number variation and evolution in humans and chimpanzees. Genome Res. 18, 1698–1710 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nuttle X., Giannuzzi G., Duyzend M. H., Schraiber J. G., Narvaiza I., Sudmant P. H., Penn O., Chiatante G., Malig M., Huddleston J., Benner C., Camponeschi F., Ciofi-Baffoni S., Stessman H. A. F., Marchetto M. C. N., Denman L., Harshman L., Baker C., Raja A., Penewit K., Janke N., Joyce Tang W., Ventura M., Banci L., Antonacci F., Akey J. M., Amemiya C. T., Gage F. H., Reymond A., Eichler E. E., Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 536, 205–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadoshima T., Sakaguchi H., Nakano T., Soen M., Ando S., Eiraku M., Sasai Y., Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. U.S.A. 110, 20284–20289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lancaster M. A., Knoblich J. A., Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollen A. A., Nowakowski T. J., Chen J., Retallack H., Sandoval-Espinosa C., Nicholas C. R., Shuga J., Liu S. J., Oldham M. C., Diaz A., Lim D. A., Leyrat A. A., West J. A., Kriegstein A. R., Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin S., Dorschner R. A., Masini I., Lavoie-Gagne O., Stahl P. D., Costantini T. W., Baird A., Eliceiri B. P., TBC1D3 regulates the payload and biological activity of extracellular vesicles that mediate tissue repair. FASEB J. 33, 6129–6139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frittoli E., Palamidessi A., Pizzigoni A., Lanzetti L., Garrè M., Troglio F., Troilo A., Fukuda M., di Fiore P. P., Scita G., Confalonieri S., The primate-specific protein TBC1D3 is required for optimal macropinocytosis in a novel ARF6-dependent pathway. Mol. Biol. Cell 19, 1304–1316 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wainszelbaum M. J., Charron A. J., Kong C., Kirkpatrick D. S., Srikanth P., Barbieri M. A., Gygi S. P., Stahl P. D., The hominoid-specific oncogene TBC1D3 activates Ras and modulates epidermal growth factor receptor signaling and trafficking. J. Biol. Chem. 283, 13233–13242 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milner C. M., Campbell R. D., The G9a gene in the human major histocompatibility complex encodes a novel protein containing ankyrin-like repeats. J. Biol. Chem. 290, 811–818 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T. J., DiMaggio P. A., Wasney G. A., Siarheyeva A., Dong A., Tempel W., Wang S.-C., Chen X., Chau I., Mangano T. J., Huang X. P., Simpson C. D., Pattenden S. G., Norris J. L., Kireev D. B., Tripathy A., Edwards A., Roth B. L., Janzen W. P., Garcia B. A., Petronis A., Ellis J., Brown P. J., Frye S. V., Arrowsmith C. H., Jin J., A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Muffat J., Omer A., Bosch I., Lancaster M. A., Sur M., Gehrke L., Knoblich J. A., Jaenisch R., Induction of Expansion and folding in human cerebral organoids. Cell Stem Cell 20, 385–396.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagahara H., Vocero-Akbani A. M., Snyder E. L., Ho A., Latham D. G., Lissy N. A., Becker-Hapak M., Ezhevsky S. A., Dowdy S. F., Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4, 1449–1452 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Liu J., Liu W., Yang L., Wu Q., Zhang H., Fang A., Li L., Xu X., Sun L., Zhang J., Tang F., Wang X., The primate-specific gene TMEM14B marks outer radial glia cells and promotes cortical expansion and folding. Cell Stem Cell 21, 635–649.e8 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Namba T., Dóczi J., Pinson A., Xing L., Kalebic N., Wilsch-Bräuninger M., Long K. R., Vaid S., Lauer J., Bogdanova A., Borgonovo B., Shevchenko A., Keller P., Drechsel D., Kurzchalia T., Wimberger P., Chinopoulos C., Huttner W. B., Human-specific ARHGAP11B acts in mitochondria to expand neocortical progenitors by glutaminolysis. Neuron 105, 867–881.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Chenn A., Walsh C. A., Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369 (2002). [DOI] [PubMed] [Google Scholar]
- 32.O’Bleness M., Searles V. B., Varki A., Gagneux P., Sikela J. M., Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 13, 853–866 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu H. Y., Guo S., Xi J., Yan Z., Fu N., Zhang X., Menzel C., Liang H., Yang H., Zhao M., Zeng R., Chen W., Pääbo S., Khaitovich P., MicroRNA expression and regulation in human, chimpanzee, and macaque brains. PLOS Genet. 7, e1002327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shulha H. P., Crisci J. L., Reshetov D., Tushir J. S., Cheung I., Bharadwaj R., Chou H.-J., Houston I. B., Peter C. J., Mitchell A. C., Yao W.-D., Myers R. H., Chen J.-F., Preuss T. M., Rogaev E. I., Jensen J. D., Weng Z., Akbarian S., Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLOS Biol. 10, e1001427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benevento M., van de Molengraft M., van Westen R., van Bokhoven H., Kasri N. N., The role of chromatin repressive marks in cognition and disease: A focus on the repressive complex GLP/G9a. Neurobiol. Learn. Mem. 124, 88–96 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Maze I., Covington H. E. III, Dietz D. M., LaPlant Q., Renthal W., Russo S. J., Mechanic M., Mouzon E., Neve R. L., Haggarty S. J., Ren Y., Sampath S. C., Hurd Y. L., Greengard P., Tarakhovsky A., Schaefer A., Nestler E. J., Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta-Agarwal S., Franklin A. V., DeRamus T., Wheelock M., Davis R. L., McMahon L. L., Lubin F. D., G9a/GLP Histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 32, 5440–5453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., Shinkai Y., G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balemans M. C. M., Ansar M., Oudakker A. R., van Caam A. P. M., Bakker B., Vitters E. L., van der Kraan P. M., de Bruijn D. R. H., Janssen S. M., Kuipers A. J., Huibers M. M. H., Maliepaard E. M., Walboomers X. F., Benevento M., Nadif Kasri N., Kleefstra T., Zhou H., van der Zee C. E. E. M., van Bokhoven H., Reduced Euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev. Biol. 386, 395–407 (2014). [DOI] [PubMed] [Google Scholar]
- 40.He Z., Tian T., Guo D., Wu H., Chen Y., Zhang Y., Wan Q., Zhao H., Wang C., Shen H., Zhao L., Bu X., Wan M., Shen C., Cytoplasmic retention of a nucleocytoplasmic protein TBC1D3 by microtubule network is required for enhanced EGFR signaling. PLOS ONE 9, e94134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shechter D., Dormann H. L., Allis C. D., Hake S. B., Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eaba8053/DC1