

A new method for detecting N-glycosylation reveals a role for the TRAP complex and a mechanism for cell state–specific regulation.
Abstract
Asparagine (N)–linked glycosylation is required for endoplasmic reticulum (ER) homeostasis, but how this co- and posttranslational modification is maintained during ER stress is unknown. Here, we introduce a fluorescence-based strategy to detect aberrant N-glycosylation in individual cells and identify a regulatory role for the heterotetrameric translocon-associated protein (TRAP) complex. Unexpectedly, cells with knockout of SSR3 or SSR4 subunits restore N-glycosylation over time concurrent with a diminished ER stress transcriptional signature. Activation of ER stress or silencing of the ER chaperone BiP exacerbates or rescues the glycosylation defects, respectively, indicating that SSR3 and SSR4 enable N-glycosylation during ER stress. Protein levels of the SSR3 subunit are ER stress and UBE2J1 dependent, revealing a mechanism that coordinates upstream N-glycosylation proficiency with downstream ER-associated degradation and proteostasis. The fidelity of N-glycosylation is not static in both nontransformed and tumor cells, and the TRAP complex regulates ER glycoprotein quality control under conditions of stress.
INTRODUCTION
The endoplasmic reticulum (ER) is a specialized eukaryotic organelle that enables the synthesis of proteins that partially or completely reside in secretory compartment or plasma membrane microenvironments. These microenvironments differ greatly from the cytosol with respect to pH, redox potential, and the presence of specialized enzymes or structural proteins. A chief function of the ER, therefore, is to facilitate protein folding and conformations that function in these distinct cellular destinations. A critical factor for achieving precise protein folding in the ER lumen is N-linked glycosylation, a co- and posttranslational modification encoded by a tripeptide amino acid consensus sequence (NXT/S/C, where X cannot be P). N-glycosylation contributes to protein solubility, stability, and trafficking, as well as to calnexin/calreticulin cycle entry and diversion to ER-associated degradation (ERAD) pathways for those proteins that do not fold properly (1). The ER also coordinates the unfolded protein response (UPR), convergent signaling pathways that restore ER homeostasis and protein production following ER stress (2). Although protein N-glycosylation is required for ER homeostasis, the mechanisms by which this modification is maintained during ER stress are not understood.
N-glycan precursor synthesis and protein glycosylation have long been considered constitutive biosynthetic processes coordinated by essential enzymes that act in series. This framework precludes the idea that the N-glycosylation pathway is actively regulated and contains specific molecular mechanisms to ensure glycosylation under diverse cellular conditions. An exception, however, is the oligosaccharyltransferase (OST), an ER lumen embedded and multimeric enzymatic complex of eight nonidentical subunits. Yeast synthetic lethal screening initially led to the discovery of both essential and nonessential OST subunits and identified a mechanism for improved glycosylation of cysteine containing nascent polypeptides via inclusion of the auxiliary OST3p and OST6p oxidoreductase subunits (MAGT1 and TUSC3 in mammals) (3–5). In parallel, the discovery of the yeast OST catalytic subunit paralogs in mammalian cells, STT3A and STT3B, as well as the DC2 and KCP2 subunits, which enable STT3A docking with the translocon, has expanded our understanding of how N-glycosylation can be adjusted and calibrated in eukaryotic cells (6–9).
The OST is associated with the ER translocation machinery, which includes the membrane bound ribosome, the heterotrimeric Sec61p translocation channel complex, the translocating chain–associated membrane (TRAM) protein, and the heterotetrameric TRAP complex. While the ribosome and Sec61p complex are indispensable for ER translation, TRAM, TRAP, and the OST are required for translation efficiency and maturation of secretory proteins. TRAM and TRAP have been implicated in controlling insertion of integral membrane proteins into the lipid bilayer during translocation and facilitating translocation of proteins with specific ER signal peptides, respectively (10–13). A role for either TRAM or TRAP in regulating N-glycosylation has not been demonstrated by genetic screens or biochemical analysis. However, patients with an aberrant glycosylation phenotype that harbor germline TRAP subunit mutations have recently been identified (14–16), although the role of TRAP or other components of the translocon in mediating glycosylation has yet to be elucidated.
A fundamental obstacle for advancing studies of N-glycosylation is the lack of rapid methods for detection of glycosylation changes at the single-cell level. To address this obstacle, we sought to establish a new molecular imaging platform to quantify the fidelity of this biosynthetic process and to discriminate between cells with or without N-glycan site occupancy deficits. Ideally, this platform would not only provide analytical value but also facilitate broader investigations into the mechanisms that regulate this process. Here, we report a methodology for fluorescent detection of reduced N-glycan site occupancy in human cells and its successful implementation for a pooled genetic CRISPR-Cas9 screen. This work identifies the network of genetic components that support the glycosylation machinery and identifies the translocon-associated protein (TRAP) complex as a critical regulator for maintaining this protein modification under conditions of cellular stress.
RESULTS
Fluorescent detection and selection of cells with abnormal N-linked glycosylation
We sought to engineer a fluorescence-based method for detecting alterations in N-glycosylation with the goal of broadly detecting changes in this protein modification at the single-cell level. We reasoned that, similar to our drug screening efforts with a modified luciferase (17, 18), a fluorescent signal dependent on enzymatic activity would also be susceptible to changes in glycosylation and identified the HaloTag (subsequently referred to as Halo) as a candidate for generating a molecular tool to measure N-glycan site occupancy in intact cells. Halo is a modified dehalogenase, and the two-step enzymatic mechanism has been well described (19). Although Halo is not an inherently fluorescent protein, substrates with fluorogenic moieties can be used to covalently label Halo for detection. We therefore introduced N-glycan sequons into Halo to identify sites that could prevent interactions with its ligand (Fig. 1A). Halo mutants fused to the epidermal growth factor receptor (EGFR) signal peptide were transiently expressed in HEK-293T cells and scored for glycosylation and compartmental localization by Western blot and fluorescence microscopy (fig. S1, A and B). The NPNSS mutant was efficiently glycosylated and displayed a unique increase in ER localization after treatment with tunicamycin or the OST inhibitor, NGI-1, suggesting that inhibition of N-glycosylation also stabilized the protein in the ER. A cell line expressing the NPNSS mutant was therefore generated and demonstrated virtually no Halo ligand binding at baseline, but rapid induction of fluorescence by inhibition of N-glycosylation with either tunicamycin or NGI-1 treatment (Fig. 1B). Western blots from these samples demonstrated that increased fluorescence was associated with both increased protein levels and loss of glycosylation (Fig. 1C), suggesting that rapid induction of fluorescence by reduced N-glycosylation is due to two effects: stabilization of Halo ligand interactions and stabilization of the Halo protein itself. Time course experiments for either induction or decay of fluorescence after NGI-1 treatment or washout (Fig. 1, D to G) demonstrate reversible signal changes and coupled with dose-response experiments (Fig. 1, H and I), validate the NPNSS mutant (named Halo1N) as a reliable indicator of N-linked glycan site occupancy. Last, mixing experiments with a ratio of 1:1 between NGI-1–treated and untreated cells demonstrate the ability to discriminate populations with impaired N-glycosylation by flow cytometry (Fig. 1, J and K). Together, these results show that Halo1N expression coupled with fluorescence-activated cell sorting (FACS) can be used to select individual cells with aberrant glycosylation.
Fig. 1. Fluorescent detection of N-linked glycan site occupancy.
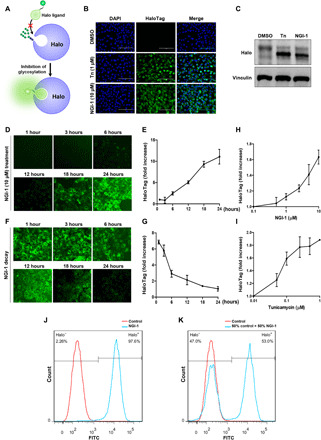
(A) Conceptual overview for generating the glycosylated Halo reporter that interacts with ligand and fluoresces only in cellular states of hypoglycosylation. (B) Fluorescence of A549 cells with stable expression of ER-Halo1N after Halo ligand exposure (470/525 nm for excitation/emission spectra) following 24-hour treatment with dimethyl sulfoxide (DMSO), Tn, or NGI-1. 4′,6-diamidino-2-phenylindole (DAPI) costaining of DNA was used to show the uniformity of cell numbers across conditions. (C) Western blots of Halo protein corresponding to treatments in (B). (D and E) Time course for induction of Halo ligand fluorescence with live cell microscopy or quantitation of fluorescent signal (at 525 nm) after treatment with 10 μM NGI-1. (F and G) Time course and quantitation of fluorescent signal decay after washout of NGI-1. (H and I) Dose-dependent Halo1N fluorescence in A549-Halo1N cells after 24-hour treatment with NGI-1 or tunicamycin. (J and K) Fluorescence of A549-Halo1N cells treated with (J) 10 μM NGI-1 after 24 hours or (K) a mixing experiment with 50% untreated control or 50% NGI-1–treated cells. Scale bars, 100 μm. FITC, fluorescein isothiocyanate.
CRISPR-Cas9 screening to identify regulators of N-linked glycosylation
Halo1N provides the ability to measure changes in N-glycosylation in the ER compartment of individual cells under diverse cellular conditions. To identify genes that regulate N-glycosylation, we performed a whole-genome CRISPR-Cas9 screen using the A549 human lung adenocarcinoma cell line. We chose this cell line because partial inhibition of N-glycosylation with NGI-1 does not affect viability (18), and we that thought this would be advantageous for the selection of live cells with impaired N-glycosylation. A549-Halo1N-Cas9 cells were screened using the human genome-scale CRISPR knock-out (hGeCKO) guide RNA (gRNA) library [containing 123,411 individual gRNAs targeting 19,050 genes; (20)]. The FACS strategy for identifying gRNAs that cause abnormal glycosylation is shown in Fig. 2A. In each of three biologically independent replicates, fluorescence was detected in ≤1% of the cell population. These cells were sorted, expanded, and harvested for DNA isolation (fig. S1C). gRNA enrichment was determined by model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) analysis of the next-generation sequencing results (21), with >80% of gRNAs detected in controls (fig. S1D). MAGeCK identified 24 genes with P < 1 × 10−4 and false discovery rate–corrected p value <0.07 (Fig. 2B and table S1). Twenty of these genes have an established role in lipid-linked oligosaccharide (LLO) biosynthesis or glycan transfer and also include the glucose transporter slc2a1/GLUT1 (Fig. 2, B and C, red text). Together, these genes can be ordered to trace the import of glucose, its conversion to mannose and GlcNAc precursors, LLO incorporation of nucleotide sugars in the cytosol, import of dolichol-linked sugar substrates, ER elongation of the LLO, transfer of the glycan by the OST, and sregeneration of dolichol phosphate (Fig. 2C). These results validate the ability to identify genetic regulators of N-glycosylation with this phenotypic screening methodology. Two additional genes, ube4a and ubxn4, are false positives and are explained by defined roles in the cytosolic component of ERAD (fig. S3). In addition, 15 genes known to be required for glycosylation showed enriched gRNAs but were not significant by the MAGeCK analysis criteria (Fig. 2, B and C, blue text; and table S2). The two remaining genes, ssr1 and ssr3 (table S3), are members of the translocon associated protein (TRAP) complex, which resides at the interface of the translocon (22) and has an undescribed role in mediating protein N-glycosylation.
Fig. 2. CRISPR-Cas9 pooled screening to identify genetic regulation of N-linked glycosylation.
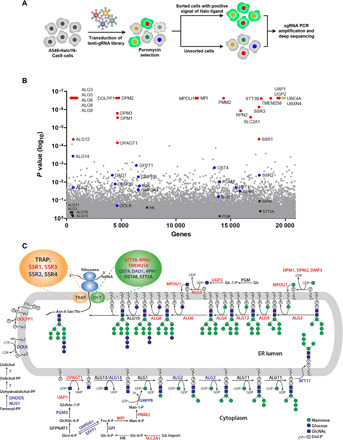
(A) Strategic overview to screen and sort gRNAs that cause inhibition of N-linked glycosylation and induction of fluorescence by enhancing Halo ligand interactions with Halo1N. (B) Scatter plot showing genes corresponding to gRNAs that were significantly enriched in the fluorescent cell populations of three independent replicates using model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK). (C) Assignment of gene functions for biosynthesis and transfer of N-linked glycans. Genes in red were significant by MAGECK analysis, genes in blue had gRNA enrichment but were not significant, genes in black showed no enrichment, and genes in green were significantly enriched and involved in the ERAD pathway. Genes identified for the TRAP complex (orange oval) and oligosaccharyltransferase (green oval) are highlighted.
The TRAP complex regulates N-linked glycosylation in mammalian cells
The TRAP complex is composed of four subunits [signal sequence receptor (SSR)1, SSR2, SSR3, and SSR4], and we therefore independently knocked out each TRAP subunit using CRISPR-Cas9 to determine the effects of each on N-glycosylation. Using FACS (Fig. 3A), we isolated both fluorescent and nonfluorescent cell populations from TRAP subunit knockout (KO) cultures (Fig. 3B) and found, unexpectedly, that both subpopulations demonstrated complete subunit KO or, in the case of SSR1, similar levels of protein loss (Fig. 3C). We also found that KO of each subunit reduced Halo glycosylation, and consistent with prior experiments, only the fluorescent cells showed changes in Halo1N glycosylation (Fig. 3D). These data show that TRAP regulates N-glycosylation in a discrete subset of cells, a finding that may explain the difficulty in identifying this complex as a factor required for normal cellular glycosylation.
Fig. 3. Validation of TRAP subunits as regulators of N-linked glycosylation.
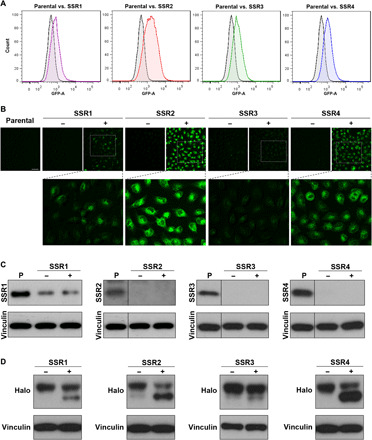
(A) Flow cytometry results for pooled cultures of A549-Halo1N-Cas9 cells after lentiviral infection and puromycin selection with single gRNAs for either SSR1, SSR2, SSR3, or SSR4 compared to parental cells. (B) Fluorescence microscopy results for FACS unsorted (−) or sorted (+) populations (scale bar, 100 μm). (C) Western blots for control or sorted populations demonstrating effective reduction or KO for SSR1, SSR2, SSR3, and SSR4. (D) Effects of TRAP subunit KO on Halo1N glycosylation indicated by reduced molecular weight and increased protein mobility on Western blot for FACS-sorted cells.
TRAP regulates glycosylation for a subset of ER-translated proteins
To identify proteins that are abnormally glycosylated in the setting of TRAP dysfunction, we profiled cell surface glycoprotein expression in SSR4 KO cells compared to controls using selective exo-enzymatic labeling (SEEL) coupled with mass spectrometry as previously reported (23). Spectra for the majority of glycoproteins detected in two independent experiments were reduced in SSR4 KO, and 24 of 87 (28%) were reduced by ≥40% (Fig. 4A and table S4). Receptor tyrosine kinases (RTKs), a class of highly N-glycosylated proteins whose functions are sensitive to aberrant glycosylation (24–26), were identified in this screening dataset, and we examined the glycosylation of receptors that had either reduced (EGFR and MET) or borderline to unaffected (IGF-1R and INSR) cell surface expression (Fig. 4B). Compared to parental cells, the molecular weight of EGFR and MET was found to decrease, similar to the effects of OST inhibition with NGI-1 (Fig. 4C). The size of INSR and IGF-1R, however, was not changed in SSR4 KO cells. We also generated HEK-293 cells with SSR4 KO and observed the same differential effects on glycosylation in this second cell type for all four receptors. The functional consequences of SSR4 KO on these RTKs were also investigated in both A549 and HEK-293 cells. SSR4 KO reduced ligand-induced EGFR phosphorylation in A549 (Fig. 4D) and HEK-293 (Fig. 4E) cells and impaired downstream activation of mitogen-activated protein kinase (MAPK), and AKT was also observed (fig. S3). In contrast, the activation of IGF-1R by IGF-1 was not different between the parental and SSR4 KO cells (Fig. 4, F and G). Together, these results indicate that TRAP regulates glycosylation for a subset of glycoprotein clients.
Fig. 4. Differential effects of SSR4 knockout on RTK glycosylation and signaling.
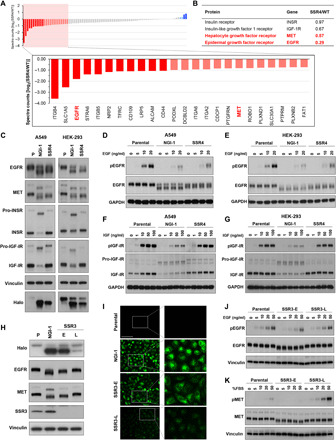
(A) Proteins that require TRAP for N-glycosylation as identified by SEEL and mass spectrometry; results represent the 87 proteins found in two independent experiments. (B) Fold difference of identified receptor tyrosine kinases (RTKs) between parental wild type (WT) and SSR4 KO cells. (C) Western blots of EGFR, MET, INSR, and IGF-1R in parental cells with or without NGI-1 compared to SSR4 KO in A549 and HEK-293 cells. (D and E) EGF-stimulated EGFR phosphorylation differences in A549 or HEK-293 SSR4 KO cells, respectively. Cells were treated with indicated concentrations of EGF for 10 min. (F and G) IGF-stimulated IGF-1R phosphorylation differences in A549 or HEK-293 SSR4 KO cells, respectively. Cells were treated with indicated concentrations of IGF for 10 min. NGI-1 treatment (10 μM) for 24 hours was used as a control, and the results are representative of three independent experiments. (H) Western blots demonstrating gel mobility changes in SSR3 KO cells after early (E) isolation compared to cells kept in culture for 3 months (late; L). (I) Loss of Halo ligand-Halo1N fluorescence in SSR3-L cells; scale bar, 100 μm. NGI-1 treatment (10 μM) for 24 hours was used as a positive control. (J) Comparison of EGFR and (K) MET protein size or activation by Western blot analysis in SSR3-E or SSR3-L cells. Cells were treated with indicated concentrations of EGF or fetal bovine serum (FBS) for 10 min or 1 hour, respectively.
Cells with TRAP subunit KO regain N-linked glycosylation
In parallel, we sought to investigate the effects of SSR3 KO on RTK function in A549 cells but found that over several weeks, the glycosylation-deficient phenotype was lost. To confirm this finding, individual clones were grown in culture for up to 3 months [SSR3-Late (L)] and compared to initial clone isolates [SSR3-Early (E)]. The amount of glycosylated Halo was significantly increased in SSR3-L compared to SSR3-E cells indicated by both Western blot and fluorescence microscopy (Fig. 4, H and I), signifying that N-glycosylation had been largely restored. Moreover, the glycosylation status of EGFR and MET was also rescued with molecular weights greater than SSR3-E and similar to parental controls, despite having loss of the SSR3 protein. We further tested the activation states of EGFR (Fig. 4J) and MET (Fig. 4K) in SSR3-E and SSR3-L cells and found that although RTK activation by EGF or fetal bovine serum (FBS) treatment was reduced in SSR3-E cells, ligand-induced activation was restored in SSR3-L cells. These data indicate that A549 cells undergo a cellular adaptation after SSR3 KO that restores N-glycosylation and glycoprotein function. In addition, these findings imply that SSR3 is required to enable N-glycosylation under a distinct cellular state.
RNA-seq reveals an ER stress signature associated with TRAP KO adaptation
To understand the cellular mechanisms that underlie adaptation and rescue of N-glycosylation in SSR3 KO cells, we performed RNA sequencing (RNA-seq) comparing triplicate samples of parental, NGI-1–treated, SSR3-E, and SSR3-L cells. Total RNA was extracted and analyzed using Illumina NovaSeq and DESEq2 and grouped using PANTHER14.1 (Fig. 5A). These results broadly demonstrated that transcription of ER-associated genes was up-regulated in SSR3-E cells, similar to NGI-1 treatment, but returned to basal levels in SSR3-L cells. The expression of TRAP subunits between early and late SSR3 KO cells, however, was not different at the RNA or protein levels (Fig. 5B), indicating that up-regulation of other TRAP subunits was not the cause for restored glycosylation. ERdj6, ERO1B, GRP94 (HSP90B1), BiP (HSPA5), PDI (P4HB), and SYVN1 transcripts were increased in early SSR3-E KO cells but were found to be reduced to control levels in late SSR3-L cells (Fig. 5C). These findings are consistent with activation of an ER stress cellular program in SSR3-E and a return to basal levels in SSR3-L cells. Together, the RNA-seq data suggest that SSR3 is critical for proficient N-glycosylation under conditions of ER stress.
Fig. 5. Homeostasis of N-linked glycosylation via modulating ER stress response.
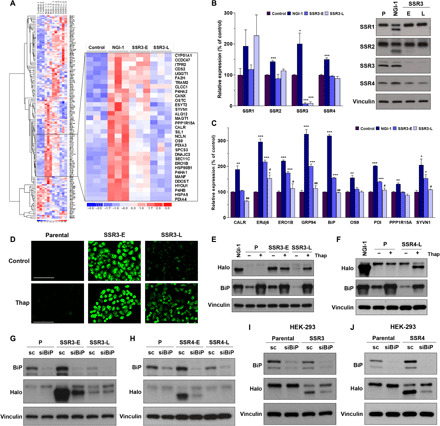
(A) RNA-seq results from three independent samples of A549 control cells compared to 10 μM NGI-1 treatment for 24 hours, SSR3-E cells, and SSR3-L cells. The inset identifies ER genes with enhanced expression in SSR3-E and reduced expression in SSR3-L. (B) The expression of TRAP subunits was compared between SSR3-E and SSR3-L cells. Parental and NGI-1–treated cells were used as the control cells. (C) RNA-seq results of genes involved in ER stress and the unfolded protein response pathways. *P < 0.05, **P < 0.01, and ***P < 0.001 (compared with control); #P < 0.05 and ##P < 0.01 (SSR3-E versus SSR3-L). (D) Effects of thapsigargin (Thap) treatment (100 nM, 24 hours) on N-linked glycosylation in SSR3-L KO cells as measured by induction of fluorescence in A549 Halo-1N cells (scale bars, 100 μm) and (E) hypoglycosylation of Halo1N by Western blot. (F) Effects of thapsigargin on glycosylation in SSR4-L cells by Western blot. BiP was used as the indicator for ER stress induction. (G and H) Effects of BiP knockdown with siRNA on Halo1N glycosylation in SSR3-E and SSR4-E, scramble (sc) siRNA was used as the control. (I and J) BiP knockdown improves N-linked glycosylation in both HEK-293 SSR3 KO and SSR4 KO cells. The Western blots are representative figures from at least two independent experiments.
To directly test whether the TRAP complex regulates N-glycosylation during ER stress, we treated SSR3-E and SSR3-L cells with 100 nM thapsigargin, a pharmacologic inducer of ER stress. Thapsigargin induced fluorescence in SSR3-L cells consistent with a glycosylation deficiency, but had no effect in control cells (Fig. 5D). Western blots of thapsigargin-treated cells confirmed that ER stress causes loss of Halo1N glycosylation in SSR3-L cells (Fig. 5E). We next sought to determine whether this effect was unique to SSR3 KO and established long-term cultures of A549 SSR4 KO cells by serial passaging over 3 months. We found that SSR4 KOs also restored glycosylation over time and that induction of ER stress with thapsigargin again reduced Halo1N glycosylation in adapted cells but not in controls (Fig. 5F). SSR3 and SSR4 KO cells also showed increased XBP-1 splicing at early time points and reduced splicing at later time points consistent with adaptation, while eIF2α phosphorylation remained unchanged (fig. S4, A and B).
To determine whether inverse manipulations of the ER stress response affected glycosylation in cells with TRAP subunit KO, we used small interfering RNA (siRNA) to knockdown BiP, an ER stress–induced chaperone. In both SSR3-E (Fig. 5G) and SSR4-E cells (Fig. 5H), BiP knockdown increased Halo1N glycosylation. In contrast, BiP knockdown had no effect on Halo1N glycosylation in the SSR3-L, SSR4-L, or control cells. Knockdown of BiP in HEK-293 cells also showed improved glycosylation for both SSR3 and SSR4 KOs without affecting glycosylation in control HEK-293 cells (Fig. 5, I and J). Together, both pharmacologic activation of ER stress and genetic manipulation of the ER stress effector BiP demonstrate that SSR3 and SSR4 are required for normal glycosylation during cellular conditions of ER stress.
Unique role of SSR2 in TRAP complex stability
With the finding that cells can adapt and N-glycosylate in the absence of SSR3 and SSR4, we reexamined a recent model proposing that loss of any subunit leads to TRAP complex dissolution and degradation of all subunits (27, 28). Comparison of SSR1, SSR2, SSR3, and SSR4 protein levels in three individual clones of SSR1–4 KOs was consistent and revealed that complete or significant reduction of SSR1, SSR3, or SSR4 protein levels did not eliminate protein levels of the other subunits (Fig. 6A). To verify this result, similar KO experiments were performed in HEK-293–Halo1N-Cas9 cells, where two independent, FACS-derived KO cultures for each subunit were tested and showed identical findings (Fig. 6, B and C). In contrast, KO of SSR2 was sufficient to affect protein levels for all members of the TRAP complex in both A549 and HEK-293 cells. These results show that loss of different subunits is not equivalent and assign a complex-stabilizing role for SSR2. To test this hypothesis, SSR2 KOs were treated with the proteasome inhibitor, bortezomib (Fig. 6D), or the cysteine peptidase inhibitor, E-64 (Fig. 6E), and results showed that inhibition of protein degradation increased the levels of SSR1, SSR3, and SSR4, consistent with a role for SSR2 in stabilizing these proteins. Together, these results show that SSR2 provides a scaffold function for the TRAP complex and is the chief regulator of TRAP subunit protein levels.
Fig. 6. Sustainability of protein glycosylation by the TRAP complex during ER stress.
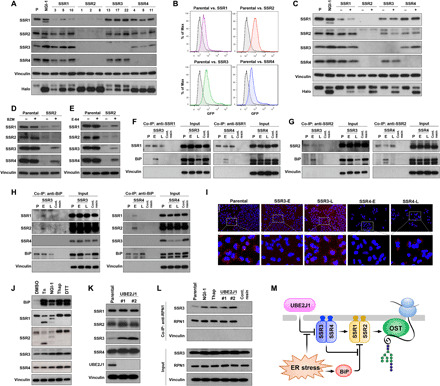
(A) TRAP subunit protein levels for independent A549 clones with SSR1, SSR2, SSR3, or SSR4 KO. (B) FACS and (C) Western blots of HEK-293–Halo1N-Cas9 with individual subunit KO. (D) TRAP subunit protein levels in parental or SSR2 KO cells treated with the proteasome inhibitor bortezomib (BZM; 100 nM) for 24 hours or (E) the cysteine peptidase inhibitor, E-64 (10 μM for 24 hours). (F to H) Co-immunoprecipitation (Co-IP) of BiP in A549-SSR3 KO or SSR4 KO cells using (F) anti-SSR1, (G) anti-SSR2, and (H) anti-BiP. (I) SSR1 and SSR2 interactions determined by the proximity ligation assay in parental, SSR3, and SSR4 early (E) or late (L) cells (scale bar, 100 μm). (J) TRAP subunit levels after ER stress activation by [tunicamycin (Tn), NGI-1, thapsigargin (Thap), and dithiothreitol (DTT)]. (K) TRAP subunit expression in UBE2J1 KO clones compared to the control cells. (L) Co-IP of SSR3 with RPN1 in Tn, NGI-1, Thap, and UBE2J1 KO cells. Control resin is used as the nonspecific binding control and vinculin is used as the negative interaction control. (M) Systems model of the functional roles for TRAP subunits in regulating N-glycosylation during ER stress.
SSR1 and SSR2 associate following either SSR3 or SSR4 KO
Because KO of neither SSR3 nor SSR4 causes loss of other TRAP subunits, we hypothesized that individual subunits can associate to restore N-glycosylation. A specialized role for SSR3 and SSR4 is also consistent with their absence from lower eukaryotes, which express only SSR1 and SSR2 orthologs (28). Because BiP knockdown in SSR3 and SSR4 KOs improves glycosylation (Fig. 5, G to J), we asked whether BiP is the ER stress effector that disrupts SSR1 and SSR2 function. Immunoprecipitation experiments with SSR1 or SSR2 specific antibodies show that in the setting of either early SSR3 or SSR4 KO, BiP coprecipitates with both SSR1 (Fig. 6F) and SSR2 (Fig. 6G). However, in SSR3-L or SSR4-L cells, where ER stress is reduced, BiP no longer associates with either TRAP subunit. Reciprocal coprecipitation experiments using an antibody specific for BiP confirmed an association with SSR1 or SSR2 subunits only under conditions of ER stress, but no interaction with either SSR3 or SSR4 (Fig. 6H). To rule out the possibility that BiP indirectly associates with SSR1 or SSR2 through a Sec61 interaction, we also evaluated SSR1-, SSR2-, and BiP-immunoprecipitation experiments for Sec61A1 and did not observe association with either SSR1 or SSR2 (fig. S5, A to C), while an interaction between Sec61A1 and BiP was demonstrated (fig. S5D). In addition, the interactions between TRAP subunits (SSR1 and SSR2) and BiP were not a general consequence of ER stress as coprecipitation could not be demonstrated in wild-type cells treated with thapsigargin (fig. S5, F to H). Together, these results suggest that BiP associates with and prevents SSR1 and SSR2 function under conditions of ER stress.
We also analyzed SSR1 and SSR2 interactions using the proximity ligation assay (PLA) in SSR3 and SSR4 KO cells. The fluorescent PLA signal was observed in controls cells but absent from either SSR3-E or SSR4-E cells (Fig. 6I). Upon resolution of ER stress, the fluorescent PLA signal was restored in both SSR3-L and SSR4-L cells, indicating that restoration of N-glycosylation occurs with a reassociation of SSR1 and SSR2. We also found that the coprecipitation of the S6 ribosomal protein (RPS6) with SSR1 or SSR2 is lost in SSR4-E cells but restored in SSR4-L cells (fig. S5, A and B). These results therefore suggest that SSR1 and SSR2 are capable of forming functional super-complexes in the absence of SSR3 or SSR4.
ER stress reduces SSR3 degradation and increases interactions with the OST
Our results suggest that SSR3 and SSR4 have chaperone roles that enable N-glycosylation during ER stress. Because BiP levels are induced by ER stress and could compete with SSR3 and SSR4, we asked whether ER stress also induces TRAP subunit expression or stability to ensure normal N-glycosylation. Partial inhibition of glycosylation with NGI-1 induces ER stress (29) and causes a small but significant enhancement of transcription for each SSR gene (Fig. 5B), consistent with this notion. At the protein level, we found that induction of ER stress by inhibition of either glycosylation (Tn or NGI-1), calcium homeostasis (thapsigargin), or disulfide bond formation (dithiothreitol) also increased TRAP subunit protein levels to varying degrees (Fig. 6J). Each pharmacologic inhibitor increased SSR3, suggesting that a specific mechanism governs SSR3 levels in the setting of ER stress. To our knowledge, only the ubiquitin-conjugating enzyme UBE2J1 (UBC6e in yeast) is known to regulate a limited number of specific client proteins during the UPR and ERAD (30). We therefore knocked out UBE2J1 and observed an increase in SSR3 protein levels (Fig. 6K), but not those of SSR1, SSR2, and SSR4, demonstrating that SSR3 is UBE2J1 dependent. SSR3 participates with the other translocon subunits in localizing TRAP to the ER membrane (28, 31) and has been reported to coprecipitate with the OST subunit RPN1 [BioGRID 3.5; (32)]. We therefore tested whether enhanced SSR3 protein levels increase association with the OST and found that both pharmacologic (NGI-1 and thapsigargin) and genetic manipulation of SSR3 levels (UBE2J1 KO) enhanced SSR3 association with RPN1 (Fig. 6L) but not with Sec61A1 (fig. S5E). Together, these results suggest that ER stress exerts an additional level of homeostatic control on N-glycosylation by increasing SSR3 protein levels and its association with the OST.
DISCUSSION
Understanding the dynamic regulation of N-glycosylation in eukaryotic cells has been a challenge for the field of glycobiology. While the majority of essential and nonessential genes required for glycan precursor biosynthesis and transfer have been identified, factors that alter the fidelity of N-glycosylation under varied cellular conditions have been more difficult to isolate. This is due to the high levels of N-glycan site occupancy found in the baseline state of most eukaryotic cells (33) and the technical obstacles for measuring protein glycosylation at the single-cell level. Here, we have designed and implemented a fluorescent methodology to determine the efficiency of N-linked glycosylation under physiologic conditions in single cells. Cellular expression of the Halo1N provides a new approach for rapidly determining the functional status of the N-linked glycosylation apparatus and can be used in diverse model systems to understand genetic, pharmacologic, and environmental effects on protein N-glycosylation. In this work, we use Halo1N expression to successfully complete a pooled genome CRISPR-Cas9 screening campaign, to comprehensively identify the genetic factors that regulate N-glycosylation, and to facilitate the discovery of the TRAP complex’s role in regulating N-glycosylation during ER stress.
The results of this phenotypic genetic screen clearly demonstrate the reliability of Halo1N fluorescence as a marker for abnormal N-glycosylation. Of the 48 known genes required for transforming glucose monomers into the mature 14 carbohydrate LLO precursor and transferring them to newly synthesized proteins, gRNAs for 38 were found to be enriched following deep sequencing of the fluorescent cell population. Twenty-four of these genes were significant by the MaGECK analysis including uap1, ugp2, and slc2a1, which can logically be placed in the pathway for glycan precursor biosynthesis. Although mutations in these genes have not previously been associated with aberrant glycosylation phenotypes in humans, the results from this screen imply that inactivating mutations should be considered in patients with otherwise uncharacterized disorders of N-glycosylation. How slc2a1 affects glycosylation is of particular interest as it may be required for external glucose in the synthesis of precursors needed for N-glycosylation as well as for optimal glucose uptake for energy production to support N-glycan biosynthesis. The screen was most effective in identifying genes involved in elongation of the Man5GlcNAc2 glycan precursor in the ER lumen, known to be nonessential in yeast, and only the alg10a/alg10b paralogs were absent from the screen because of inefficient KO of both genes simultaneously. Several essential genes required for both glycan precursor biosynthesis and transfer were enriched, reflecting the ability of CRISPR gene editing to introduce hypomorphic mutations that reduce but do not eliminate enzymatic activity. Other essential genes such as stt3a, alg1, and alg11 were not identified, possibly because of a loss of cell fitness that prevented positive selection of live cells. The screen also did not select for genes required for de novo dolichol phosphate synthesis or for the LLO scramblases that flip glycan precursors into the ER lumen, and these incompletely described mechanisms will require further investigation.
The significant enrichment of gRNAs for subunits of the TRAP complex led us to pursue the role of this heterotetramer in N-glycosylation. Recent reports of patients with either an SSR4 (14, 15) or SSR3 (16) mutation provided additional rationale to elucidate the functional role for this complex, and we therefore established individual SSR1–4 subunit KO cells. These models unambiguously demonstrate that TRAP regulates N-glycosylation. This finding was confirmed through glycoproteomic analyses of SSR4 WT and KO cells, which identified reduced N-glycosylation and cell surface expression of specific RTKs such as EGFR and MET. In contrast, neither the IGF-1R nor IR RTKs were affected by TRAP subunit KO, which is of interest as dysfunction of these RTKs could lower cytoplasmic glucose levels and exacerbate an N-glycosylation defect. TRAP’s role in regulating the fidelity of N-glycosylation is unexpected because unlike the fine-tuning demonstrated for subunits of the OST (5, 6), TRAP has no enzymatic activity. In addition, the observation that SSR3 and SSR4 provide a discrete mechanism for cell state–specific regulation of N-glycosylation is unprecedented and demonstrates that our understanding of the factors required for glycosylation continues to evolve.
Our ability to rapidly detect and select cells with changes in N-glycosylation led to the discovery that TRAP is only necessary for N-glycosylation in discrete cellular states. The ability of cells to adapt in cell culture over time may explain why the role of the TRAP complex in regulating N-glycosylation has not previously been defined. In addition, early genetic studies on N-glycosylation were performed in yeast, which do not have a TRAP complex, and further underscore the value of organism agnostic platforms to detect changes in N-glycosylation. Our results with pharmacologic and genetic manipulations provide clear evidence that a significant function of the TRAP complex is to preserve N-glycosylation under conditions of ER stress. Recently, using an RNA interference approach, TRAP was suggested to be involved with efficient translocation and synthesis of both glycosylated and nonglycosylated proteins with signal peptides that have high glycine-plus-proline content and/or low hydrophobicity (13). However, reduced translocation was not a factor in regulating N-glycosylation, and a reduction of protein levels either with (EGFR and MET) or without (IGF-1R and IR) alterations in N-glycosylation was not observed.
We found that SSR2 is absolutely essential for TRAP complex assembly and stability of the other subunits. SSR2 KO reduced protein levels of the other subunits through lysosome or proteasome degradation without affecting gene transcription. These findings emphasize a unique role for SSR2 in the assembly of the TRAP complex and also raised the possibility that TRAP’s role in N-glycosylation was not completely eliminated by loss of other subunits. Subsequently, we showed that loss of SSR1, SRR3, or SSR4 does not eliminate cellular protein levels of other subunits. Coupled with the finding that SSR3 and SSR4 KOs underwent a cellular adaptation characterized by reduced expression of ER stress–dependent genes and loss of the aberrant glycosylation phenotype, we then demonstrated that adaptation is coincident with SSR1 and SSR2’s disassociation with BiP and reassociation with each other. These findings strongly suggest that SSR3 and SSR4 prevent BiP interactions with SSR1 and SSR2, although are not essential for regulating N-glycosylation. The appearance of SSR3 and SSR4 in higher eukaryotes thus suggests the evolution of specialized functions for the heterotetrameric TRAP complex that are advantageous for responding to cellular stress, including coordination of ER stress responses and regulation of N-glycosylation.
In agreement with this role, another unexpected finding is the posttranslational regulation of SSR3 by UBE2J1. This ubiquitin-conjugating enzyme was identified in yeast to mediate constitutive degradation of EDEM1, OS-9, and SEL1L, fine-tuning glycoproteostasis through up-regulation of these ERAD enhancers (30). Our discovery that SSR3 is also regulated by UBE2J1 suggests a more integrated pathway where UBE2J1 not only enhances glycoprotein ERAD but also facilitates proper N-glycosylation at the same time through up-regulation of the membrane-localizing TRAP subunit, SSR3. In support of this interpretation, we demonstrate that UBE2J1 KO or pharmacologic induction of ER stress increases SSR3 and its association with the OST.
A mechanism for preventing abnormal N-glycosylation during ER stress has not previously been reported, although resolution of ER stress through signaling (ATF6, IRE-1, and PERK), the UPR, and ERAD are known to be essential pathways for maintaining ER homeostasis and progression along paths of either cell death or survival (34–37). How cells ensure that N-glycosylation remains intact under conditions of transient ER dysfunction or overload remains an important question. Here, we present a model for the regulation of N-glycosylation by the TRAP complex (Fig. 6M) where SSR1 and SSR2 are sufficient for enabling N-glycosylation. SSR3 and SSR4 are not absolutely required and instead prevent association of SSR1 and SSR2 with BiP to maintain N-glycosylation under ER stress. ER stress also disrupts active degradation of SSR3 by UBE2J1, increases SSR3 protein levels and OST association, and ensure that newly synthesized proteins are appropriately N-glycosylated.
MATERIALS AND METHODS
Cell culture and treatments
The A549, HEK-293, and HEK-293T cell lines were purchased from the American Type Culture Collection (Rockville, MD). The A549 cells were cultured in RPMI (Gibco, Life Technologies, Grand Island, NY), and HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FBS (Gibco) and penicillin and streptomycin (Gibco) in a humidified incubator with 5% CO2. Cells were kept in culture for no more than 6 months after resuscitation from the original stocks. The A549 and HEK-293 Cas9-expressing cells were generated via lentiviral transduction using virus produced from the lentiCas9-Blast (Addgene, 52962), psPAX2 packaging, and pMD2.GVG envelope plasmids. Tunicamycin (Tn; 1 μM) and NGI-1 (10 μM) were used to inhibit N-linked glycosylation in cell culture, and dimethyl sulfoxide treatment was used as a vehicle control. Thapsigargin (100 nM; Selleckchem, Houston, TX) was used to induce ER stress. The proteasome and protease inhibitors, bortezomib (BZM; 100 nM, Selleckchem) and E64 (10 μM, Selleckchem), were used to inhibit cellular proteolytic activity. EGFR and IGF-1R activity was assessed by treatment with either EGF (R&D Systems, Minneapolis, MN) or IGF-1 (R&D Systems) ligands, respectively.
Crispr-Cas9 screening and FACS
The hGeCKO library (hGeCKOa and hGeCKOb; Addgene, 1000000049) was generated in the same way as described above. For each replicate of the pooled screen, a total of 1 × 108 cells that constitutively express Cas9 were infected with lentiGuide-Puro from the hGeCKO library at a multiplicity of infection of 0.3 and selected with puromycin at 2 μg/ml for 10 days. After puromycin selection, pooled cells were treated with trypsin, stained with HaloTag ligand (1:1000; Promega, G2801) for 1 hour at 37°C, and then sorted by flow cytometry after washing twice in phosphate-buffered saline (PBS) to remove unbound ligand. Unsorted cells were treated in the same manner and used as a control. Individual cell line KOs were constructed using lentiviral infection with a specific single gRNA as shown in table S3. KO was confirmed by sequencing for SSR2, SSR3, and SSR4. All SSR1 clones displayed loss of one allele and an in-frame mutation of a second allele, consistent with a hypomorphic mutation.
Genomic DNA sequencing and analysis
Genomic DNA isolation from both sorted and unsorted cell pellets was performed using QIAamp DNA columns (Qiagen, Hilden, Germany), and gRNA sequences were amplified in a two-step polymerase chain reaction (PCR): For the first PCR, the amount of input genomic DNA for each sample was calculated to achieve 160× coverage of the hGeCKOa and hGeCKOb libraries; a second PCR was performed to attach Illumina adapters and barcodes for next-generation sequencing. All PCRs were performed using Phusion Flash High Fidelity Master Mix (Thermo Fisher Scientific, F548L). Sequencing was performed with Illumina HiSeq and 75–base pair (bp) single-end reads at the Yale Center for Genome Analysis. Primers and barcode sequences are listed in table S5. Reads were aligned to index sequences using the Bowtie aligner, and a maximum of one mismatch was allowed in the 20-bp gRNA sequence. The number of uniquely aligned reads for each library sequence was calculated after alignment for each of three biologically independent replicates.
siRNA transfection
The expression of HSPA5 (BiP) was suppressed using siRNA specific to HSPA5 (siGENOME human HSPA5-SMART pool, Dharmacon, Lafayette, CO). The cells (3 × 105 cells per well) were cultured in a six-well plate for 24 hours and then transfected with 100 pmol of si-HSPA5 using Lipofectamine 2000 (2 μg/ml) (Invitrogen) according to the recommendations from the manufacturer. Excess transfection complex was removed after 6 hours, and cells continued to be cultured in 10% FBS in RPMI for the subsequent experiments. Control experiments were performed using cells treated with negative control siRNA (OriGene, Rockville, MD, SR30004).
Whole transcriptome sequencing (RNA-seq)
RNA was extracted from whole cells with the RNeasy Mini kit (Qiagen) according to the manufacturer’s protocol. RNA quality and integrity were determined with an Agilent Bioanalyzer gel with RNA integrity number score higher than 7. RNA-seq libraries were prepared from total RNA with the Kapa mRNA HyperPrep kit (Kapa Biosystems), and library size distributions were determined with the LabChip GX or Agilent Bioanalyzer. Sequencing was performed with Illumina NovaSeq using 100-bp paired-end sequencing RNA-seq at the Yale Center for Genome Analysis. A positive control (prepared bacteriophage PhiX library) provided by Illumina was spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time. Primary analysis, sample demultiplexing and alignment to the human genome, was performed using Illumina’s CASAVA 1.8.2 software suite. The reads were trimmed for quality using custom scripts. Minimum length accepted was 45 bases. The trimmed reads were then aligned to the mm10 reference genome using Gencode annotation, HISAT2 for alignment, and StringTie for transcript abundance estimation (38, 39). The generated counts were processed with DESeq2 (40) in R to determine statistically significantly expressed genes (q < 0.05).
Semiquantitative reverse transcription PCR
RNA from the cells was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The complementary DNA (cDNA) was synthesized using the ProtoScript First Strand cDNA Synthesis Kit (New England Biolabs, Ipswich, MA). Semiquantitative PCR was performed using Taq DNA polymerase (Thermo Fisher Scientific) with the following primers: XBP1s (forward, 5′-GGTCTGCTGAGTCCGCAGCAGG-3′; reverse, 5′-GGGGCTTGGTATATATGTGG-3′); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-GGATGATGTTCTGGAGAGCC-3′; reverse, 5′-CATCACCATCTTCCAGGAGC-3′) (29).
One-step SEEL labeling and proteomics
Briefly, cell surface glycoproteins were labeled with sialic acid-C5-triazole-biotin by ST6Gal1 at 37°C for 2 hours as previously described (23). Labeled cells were collected using radioimmunoprecipitation assay lysis buffer, and 1 mg of lysate was immunoprecipitated using an anti-biotin antibody. The precipitated proteins were then separated using SDS–polyacrylamide gel electrophoresis (PAGE), stained by silver staining, and proceeded to the in-gel digestion. The peptides were purified and subjected to Orbitrap Fusion Tribrid Mass Spectrometry (Thermo Fisher Scientific, Waltham, MA). The raw spectra for proteins 100 to 250 kDa in size were analyzed using SEQUEST (Proteome Discoverer 1.4, Thermo Fisher Scientific) based on the human protein database (UniProt, October 2014). Quantification was performed by comparison of normalized spectral counts generated by ProteoIQ (v2.7, Premier Biosoft).
SDS-PAGE and Western blot
Following cell lysis, proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The following primary antibodies were used: anti-HaloTag (1:3000; Promega, Madison, WI, G9281), anti-SSR1 (1:500; Proteintech, Rosemont, IL, 10583-1-AP), anti-SSR2 (1:500; Proteintech, 10278-1-AP), anti-SSR3 (1:500; Sigma-Aldrich, St. Louis, MO, HPA014906), anti-SSR4 (1:500; Proteintech, 11655-2-AP), anti-INSR (1:1000; Cell Signaling Technology, Danvers, MA, 4B8, 3025), anti–IGF-1Rβ (1:1000; Cell Signaling Technology, D23H3, 9750), anti–pIGF-1Rβ (1:1000; Cell Signaling Technology, Y1135, DA7A8, 3918), anti-MET (1:1000; Cell Signaling Technology, 4560), anti-pMET (1:500; Cell Signaling Technology, Y1234/1235, D26, 3077), anti-EGFR (1:1000; Cell Signaling Technology, D38B1, 4267), anti-pEGFR (1:1000; Cell Signaling Technology, Y1068, D7A5, 3777), anti-BiP (1:1000; Cell Signaling Technology, C50B12, 3177), anti-UBE2J1 (1:800; Santa Cruz Biotechnology, Dallas, TX, B-6, sc-377002), anti-RPN1 (1:500; Proteintech, 12894-1-AP), anti-pAkt (1:1000; Cell Signaling Technology, S473, D9E, 4060), anti-Akt (1:1000; Cell Signaling Technology, 9272), anti-pErk1/2 (1:1000; Cell Signaling Technology, D13.14.4E, 4370), anti-Erk1/2 (1:1000; Cell Signaling Technology, 137F5, 4695), anti–p-eIF2α (1:1000; Cell Signaling Technology, S51, 119A11), anti-eIF2α (1:1000; Cell Signaling Technology, D7D3, 5324), anti-Sec61A1 (1:1000; Cell Signaling Technology, D7Q6V, 14868), anti-S6 ribosomal protein (1:1000; Cell Signaling Technology, 54D2, 2317), and anti-Vinculin (1:2000; Cell Signaling Technology, E1E9V, 13901). The immunoreactivity was detected using the ECL Western Blotting Detection Reagents (GE Healthcare).
HaloTag ligand detection
To measure the loss of glycosylation on Halo protein, the HaloTag Oregon Green Ligand (Promega, Madison, WI) was used to detect the deglycosylated Halo. The cells were incubated with 1:1000 HaloTag ligand in culture media at 37°C for 15 min. The unbound ligands were washed twice with PBS and then washed with completed media at 37°C for 30 min. The signal of HaloTag ligand was observed under the fluorescent microscope with ×200 magnification.
Co-immunoprecipitation
Protein interactions were determined using Pierce Co-Immunoprecipitation (Co-IP) Kit (Pierce Biotechnology) according to the manufacturer’s protocol. Briefly, 2 to 4 μg of antibodies were immobilized with AminoLink Plus Coupling Resin, and the total 1500 μg of whole-cell lysate was incubated overnight with the resin-antibody complex at 4°C. Protein-bound antibody was eluted and solubilized in Lane Marker Sample Buffer before being subjected to SDS-PAGE. The lysate incubated with antibody-control agarose resin complex was used as the negative control.
Proximity ligation assay
The in situ interaction between SSR1 and SSR2 was demonstrated by PLA according to the manufacturer’s instructions. Briefly, anti-SSR1 and anti-SSR2 were conjugated with PLA oligonucleotides using Duolink PLA Multicolor Probemaker Kit-Red (Sigma-Aldrich, DUO96010-1KT). Fixed cells were permeabilized using 0.1% Triton X-100 for 10 min at room temperature and then blocked nonspecific binding using Duolink Blocking Solution for 1 hour at 37°C. The cells were incubated with oligo-conjugated primary antibodies at 4°C, overnight, followed by ligation, amplification, and detection reactions using Duolink PLA Multicolor Reagent Pack (Sigma-Aldrich, DUO96000). The single antibody condition was used as the negative control of the PLA reaction. After washing, cells were then mounted using Duolink In Situ Mounting Media with DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, DUO82040). The PLA signal was observed under the fluorescent microscope.
Statistical analysis
Statistical analysis not described above was determined using Student’s t test (GraphPad Prism 8.0 software; GraphPad software Inc., La Jolla, CA), and P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the U.S. NIH through R01GM127383 (J.N.C.), R01GM086524 (R.S.), and R01GM130915 (L.W.). Author contributions: C.P., W.C., T.J.H., L.W., R.S., and J.N.C. conceived and designed the experiments. C.P., W.C., T.J.H., S.-H.Y., and P.Z. performed the experiments and analyzed the data. C.P. and W.C. prepared the figures and tables. C.P., W.C., and J.N.C. wrote the manuscript. All authors participated in the interpretation of the studies and reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabc6364/DC1
REFERENCES AND NOTES
- 1.Helenius A., Aebi M., Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Hebert D. N., Lamriben L., Powers E. T., Kelly J. W., The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat. Chem. Biol. 10, 902–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karaoglu D., Kelleher D. J., Gilmore R., Functional characterization of Ost3p. Loss of the 34-kD subunit of the Saccharomyces cerevisiae oligosaccharyltransferase results in biased underglycosylation of acceptor substrates. J. Cell Biol. 130, 567–577 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz B. L., Aebi M., Analysis of glycosylation site occupancy reveals a role for Ost3p and Ost6p in site-specific N-glycosylation efficiency. Mol. Cell. Proteomics 8, 357–364 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Schulz B. L., Stirnimann C. U., Grimshaw J. P., Brozzo M. S., Fritsch F., Mohorko E., Capitani G., Glockshuber R., Grütter M. G., Aebi M., Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc. Natl. Acad. Sci. U.S.A. 106, 11061–11066 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelleher D. J., Karaoglu D., Mandon E. C., Gilmore R., Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell 12, 101–111 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Ruiz-Canada C., Kelleher D. J., Gilmore R., Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roboti P., High S., The oligosaccharyltransferase subunits OST48, DAD1 and KCP2 function as ubiquitous and selective modulators of mammalian N-glycosylation. J. Cell Sci. 125, 3474–3484 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Braunger K., Pfeffer S., Shrimal S., Gilmore R., Berninghausen O., Mandon E. C., Becker T., Förster F., Beckmann R., Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conti B. J., Devaraneni P. K., Yang Z., David L. L., Skach W. R., Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 58, 269–283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snapp E. L., Reinhart G. A., Bogert B. A., Lippincott-Schwartz J., Hegde R. S., The organization of engaged and quiescent translocons in the endoplasmic reticulum of mammalian cells. J. Cell Biol. 164, 997–1007 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fons R. D., Bogert B. A., Hegde R. S., Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 160, 529–539 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen D., Stutz R., Schorr S., Lang S., Pfeffer S., Freeze H. H., Forster F., Helms V., Dudek J., Zimmermann R., Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 9, 3765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losfeld M. E., Ng B. G., Kircher M., Buckingham K. J., Turner E. H., Eroshkin A., Smith J. D., Shendure J., Nickerson D. A., Bamshad M. J.; University of Washington Center for Mendelian Genomics, Freeze H. H., A new congenital disorder of glycosylation caused by a mutation in SSR4, the signal sequence receptor 4 protein of the TRAP complex. Hum. Mol. Genet. 23, 1602–1605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng B. G., Raymond K., Kircher M., Buckingham K. J., Wood T., Shendure J., Nickerson D. A., Bamshad M. J.; University of Washington Center for Mendelian Genomics, Wong J. T., Monteiro F. P., Graham B. H., Jackson S., Sparkes R., Scheuerle A. E., Cathey S., Kok F., Gibson J. B., Freeze H. H., Expanding the Molecular and Clinical Phenotype of SSR4-CDG. Hum. Mutat. 36, 1048–1051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng B. G., Lourenco C. M., Losfeld M. E., Buckingham K. J., Kircher M., Nickerson D. A., Shendure J., Bamshad M. J.; University of Washington Center for Mendelian Genomics, Freeze H. H., Mutations in the translocon-associated protein complex subunit SSR3 cause a novel congenital disorder of glycosylation. J. Inherit. Metab. Dis. 42, 993–997 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contessa J. N., Bhojani M. S., Freeze H. H., Ross B. D., Rehemtulla A., Lawrence T. S., Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res. 16, 3205–3214 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Sambrooks C., Shrimal S., Khodier C., Flaherty D. P., Rinis N., Charest J. C., Gao N., Zhao P., Wells L., Lewis T. A., Lehrman M. A., Gilmore R., Golden J. E., Contessa J. N., Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol. 12, 1023–1030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., Zhu J., Darzins A., Klaubert D. H., Bulleit R. F., Wood K. V., HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W., Xu H., Xiao T., Cong L., Love M. I., Zhang F., Irizarry R. A., Liu J. S., Brown M., Liu X. S., MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeffer S., Brandt F., Hrabe T., Lang S., Eibauer M., Zimmermann R., Förster F., Structure and 3D arrangement of endoplasmic reticulum membrane-associated ribosomes. Structure 20, 1508–1518 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Klaver E., Zhao P., May M., Flanagan-Steet H., Freeze H. H., Gilmore R., Wells L., Contessa J., Steet R., Selective inhibition of N-linked glycosylation impairs receptor tyrosine kinase processing. Dis. Model Mech. 12, dmm039602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez Sambrooks C., Baro M., Quijano A., Narayan A., Cui W., Greninger P., Egan R., Patel A., Benes C. H., Saltzman W. M., Contessa J. N., Oligosaccharyltransferase inhibition overcomes therapeutic resistance to EGFR tyrosine kinase inhibitors. Cancer Res. 78, 5094–5106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baro M., Lopez Sambrooks C., Quijano A., Saltzman W. M., Contessa J., Oligosaccharyltransferase inhibition reduces receptor tyrosine kinase activation and enhances glioma radiosensitivity. Clin. Cancer Res. 25, 784–795 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croci D. O., Cerliani J. P., Dalotto-Moreno T., Méndez-Huergo S. P., Mascanfroni I. D., Dergan-Dylon S., Toscano M. A., Caramelo J. J., García-Vallejo J. J., Ouyang J., Mesri E. A., Junttila M. R., Bais C., Shipp M. A., Salatino M., Rabinovich G. A., Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., Forster F., Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Pfeffer S., Dudek J., Schaffer M., Ng B. G., Albert S., Plitzko J. M., Baumeister W., Zimmermann R., Freeze H. H., Engel B. D., Forster F., Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 8, 14516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinis N., Golden J. E., Marceau C. D., Carette J. E., Van Zandt M. C., Gilmore R., Contessa J. N., Editing N-glycan site occupancy with small-molecule oligosaccharyltransferase inhibitors. Cell Chem Biol. 25, 1231–1241.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagiwara M., Ling J., Koenig P. A., Ploegh H. L., Posttranscriptional regulation of glycoprotein quality control in the endoplasmic reticulum is controlled by the E2 Ub-conjugating enzyme UBC6e. Mol. Cell 63, 753–767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartmann E., Gorlich D., Kostka S., Otto A., Kraft R., Knespel S., Burger E., Rapoport T. A., Prehn S., A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur. J. Biochem. 214, 375–381 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Oughtred R., Stark C., Breitkreutz B.-J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R., Zhang F., Dolma S., Willems A., Coulombe-Huntington J., Chatr-aryamontri A., Dolinski K., Tyers M., The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zielinska D. F., Gnad F., Wisniewski J. R., Mann M., Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., Kaufman R. J., ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong F., Liu B., Wu B. X., Morreall J., Roth B., Davies C., Sun S., Diehl J. A., Li Z., CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat. Struct. Mol. Biol. 24, 834–839 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J., Wang Y., Song L., Zeng L., Yi W., Liu T., Chen H., Wang M., Ju Z., Cong Y. S., A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1α stability. Nat. Commun. 8, 14186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinkaew D., Chattopadhyay A., King M. D., Chunhacha P., Liu Z., Stevenson H. L., Chen Y., Sinthujaroen P., McDougal O. M., Fujise K., Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death. Nat. Commun. 8, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frankish A., Diekhans M., Ferreira A. M., Johnson R., Jungreis I., Loveland J., Mudge J. M., Sisu C., Wright J., Armstrong J., Barnes I., Berry A., Bignell A., Carbonell Sala S., Chrast J., Cunningham F., Di Domenico T., Donaldson S., Fiddes I. T., García Girón C., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Hunt T., Izuogu O. G., Lagarde J., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Xu J., Yates A., Zerbino D., Zhang Y., Aken B., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Reymond A., Tress M. L., Flicek P., GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabc6364/DC1