

Abstract
Both basal and glucose-stimulated insulin release occur primarily by insulin secretory granule exocytosis from pancreatic β cells, and both are needed to maintain normoglycemia. Loss of insulin-secreting β cells, accompanied by abnormal glucose tolerance, may involve simple exhaustion of insulin reserves (which, by immunostaining, appears as a loss of β cell identity), or β cell dedifferentiation, or β cell death. While various sensing and signaling defects can result in diminished insulin secretion, somewhat less attention has been paid to diabetes risk caused by insufficiency in the biosynthetic generation and maintenance of the total insulin granule storage pool. This Review offers an overview of insulin biosynthesis, beginning with the preproinsulin mRNA (translation and translocation into the ER), proinsulin folding and export from the ER, and delivery via the Golgi complex to secretory granules for conversion to insulin and ultimate hormone storage. All of these steps are needed for generation and maintenance of the total insulin granule pool, and defects in any of these steps may, weakly or strongly, perturb glycemic control. The foregoing considerations have obvious potential relevance to the pathogenesis of type 2 diabetes and some forms of monogenic diabetes; conceivably, several of these concepts might also have implications for β cell failure in type 1 diabetes.
Brief description of insulin mRNA quantity and quality
Many factors may contribute to the control of insulin mRNA levels (1, 2), including transcriptional networks that control β cell function (3) as well as mRNA stability (4). While a detailed analysis of the transcriptional regulation of β cell development, differentiation, and dedifferentiation is beyond the scope of this Review, it is certainly clear that PDX1 and NEUROD1 can act together to directly stimulate transcription from the INS promoter (5). Moreover, PDX1, in combination with NKX2.2 (downstream of NEUROG3; ref. 6) and FOXA2, stimulates the expression of the β cell–specific activator MafA (7); in turn, MafA homodimers (or MafA/MafB heterodimers) help support β cell INS gene transcription (8) (two alleles in humans, four alleles in mice and rats), along with additional upstream factors (9). A number of human diabetes–related genes associated with INS mRNA expression are summarized in Table 1.
Table 1. Human diabetes–related genes associated with INS gene transcriptionA.

Classic studies of the islets of insulin-resistant C57BL/KsJ db/db mice, a genetic model of type 2 diabetes (T2D), have shown that at 5 weeks of age (when glycemia is not yet significantly perturbed and pancreatic insulin content is still similar to that in +/db controls), there is an elevation of Ins mRNA that is likely to be disproportional to β cell mass, which helps to provide the insulin needed to mount a hyperinsulinemic response in these animals (10). The db/db mice then progress to diabetes, and with this, the abundance of Ins mRNA is not sustained (although interventions to limit obesity and diabetes with diet or medications that provide β cell rest tend to preserve some of the elevation of Ins mRNA levels, as well as insulin secretory capacity; refs. 10, 11). Thus, regulating INS mRNA level is at least part of the response by which β cells compensate for insulin resistance, although this may not be maintained as hyperglycemia progresses.Accumulated clinical and basic research data suggest that declining INS mRNA levels may be just one feature of a larger phenomenon of β cell dedifferentiation, and this may be a relatively early signature of β cell failure in the natural progression of T2D in humans and animal models (12–15). Not surprisingly, many genes associated with β cell differentiation (including INS mRNA expression) also appear as (loss-of-function) GWAS candidates for T2D in humans (Tables 1, 2, 3, and 4).
Table 2. Human diabetes–related genes linked to preproinsulin translation and/or translocationA.

Table 3. Human diabetes–related genes involved in proinsulin folding or ER homeostasisA.

Table 4. Human diabetes–related genes associated with insulin granule packaging, processing, or secretionA.

There are also murine models in which hyperglycemia (from experimentally induced partial insulin deficiency) appears to lead directly to decreased β cell transcription factors (PDX1, Nkx6.1, HNF1A, and MafA — all of which contribute to differentiated function) in pancreatic islets, along with downregulation of β cell–enriched genes (Glut2 and Gck) and accompanied by increased endocrine progenitor markers (Ngn3) and β cell–disallowed genes (Ldha and G6pc). The effects of hyperglycemia have also been observed in residual islets after incomplete pancreatectomy (16), as well as in transgenic mice expressing a mutated form of the ATP channel (Kir6.2-V59M, which impairs insulin secretion in β cells, causing hyperglycemia; ref. 17). Additionally, even without known genetically predisposing factors, INS mRNA has been shown to be dramatically decreased in human islet grafts exposed in vivo to hyperglycemic conditions (i.e., after transplantation into NSG mice), again suggesting that chronic hyperglycemia — along with other factors (18) — is detrimental to the maintenance of β cell INS mRNA as well as other key transcriptional machinery needed for the differentiated β cell phenotype (19, 20).
The INS mRNA may also be subject to abnormal splicing, including formation of the INS-IGF2 chimera, which — although it may occur at only a low level (21) — could offer an INS-specific sequence contribution limited to the signal peptide, B chain, cleavage site, and a short portion of the C-peptide (22). Another errant splice donor-acceptor combination occurs in patients with early-onset autosomal dominant diabetes (MIDY, discussed in greater detail below) caused by a mutation within the INS gene intron 2, creating a preferential splice acceptor site, thus terminating the normal preproinsulin sequence in the same place as in INS-IGF2, but thereafter leading to a frameshifted protein replacing the remaining C-peptide and A chain with a novel peptide sequence (23).
Preproinsulin translation and translocation into the ER
Insulin biogenesis (Figure 1) begins with preproinsulin translation and translocation across the endoplasmic reticulum (ER) membrane, to synthesize proinsulin. Translation from the INS mRNA is acutely regulated by multiple drugs, hormones, and nutrients, especially glucose and its metabolites (24–27). Glucose-dependent preproinsulin translation is, to a considerable extent, dependent on factors that associate with untranslated regions (UTRs) of the INS mRNA (4, 28, 29). In isolated rat pancreatic islets, total islet protein synthesis increases about 2-fold within ≤1 hour of high glucose exposure, but much of this overall increase comprises the biosynthesis of preproinsulin along with copackaged secretory granule content proteins (30), with a much higher fold change during the same period (31, 32). Most Ins mRNA is associated with the ER, and a key step of glucose-dependent preproinsulin biosynthesis includes ribosome recruitment to initiate translation (33).
Figure 1. An overview of insulin biosynthesis in β cells.
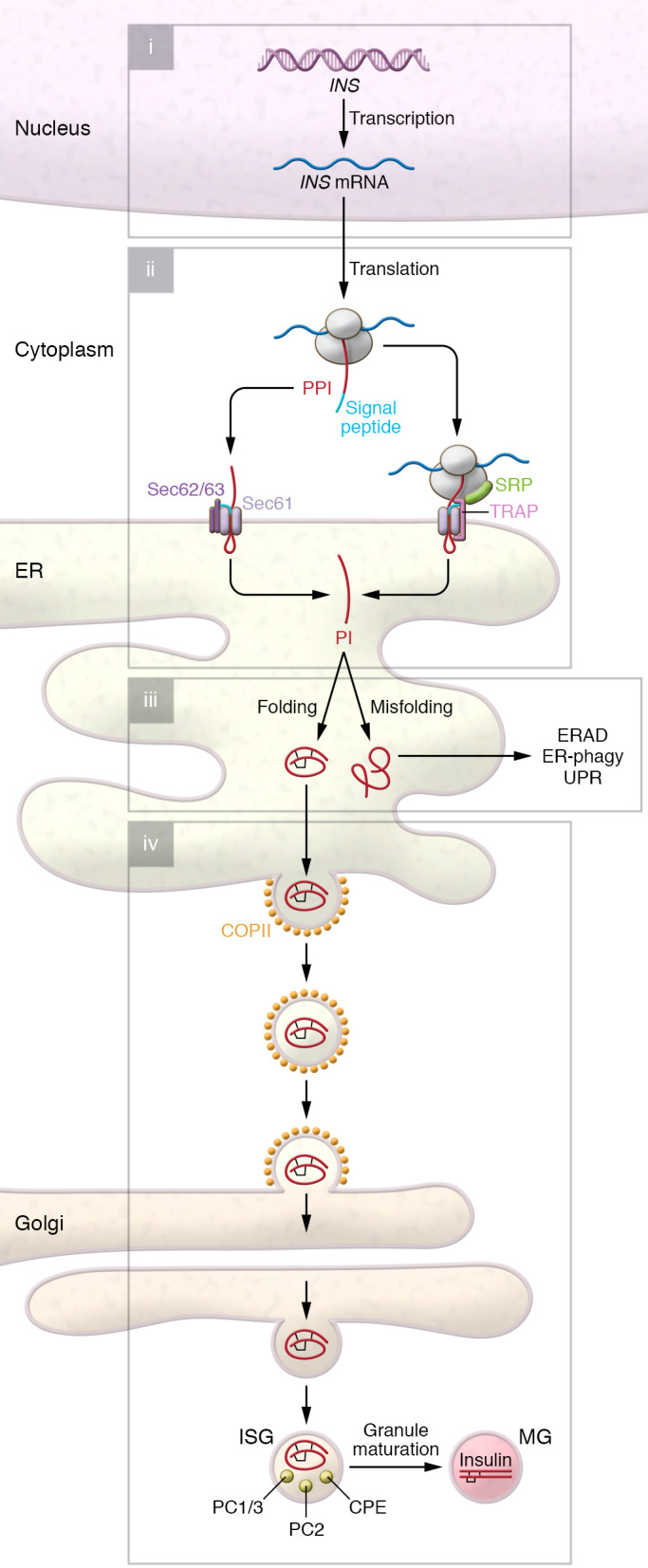
(i) Expression of INS gene is controlled quantitatively and qualitatively by transcriptional networks including PDX1, NGN3, NEUROD1, NKX2.2, FOXA2, and MAFA. (ii) Newly synthesized preproinsulin (PPI) undergoes cotranslational (SRP- and Sec61-dependent) and posttranslational (Sec62/63-dependent) translocation across the ER membrane, in which TRAP plays an important yet poorly defined role. (iii) Upon delivery to the ER lumen, the preproinsulin signal peptide is rapidly excised, forming proinsulin (PI) that undergoes rapid oxidative folding — ultimately forming three highly conserved disulfide bonds. ER folding machinery (BiP, ERO1, PDI, ERp46, GRP94, etc.) acts in concert to promote folding, and may recognize unfolded/misfolded proinsulin for degradation through ERAD and ER-phagy. Misfolded proinsulin can also activate the unfolded protein response (UPR; including three classic arms involving PERK, IRE1, and ATF6). (iv) Well-folded proinsulin is exported from the ER via COPII vesicles for delivery to the Golgi complex, where proinsulin forms hexamers in the presence of Zn2+. Upon proinsulin delivery to (more acidic, Ca2+-rich) immature secretory granules (ISGs), PC1/3, PC2, and CPE act to convert proinsulin to insulin, which is ultimately stored in mature granules (MGs).
The preproinsulin mRNA yields a polypeptide that can be synthesized via cap-dependent or -independent translation (4), encoding a protein from N- to C-terminus comprising the signal peptide to direct translocation into the ER, the B chain, the connecting (C)-peptide (that is removed within immature secretory granules), and concluding with the A chain. Mature, optimally bioactive insulin requires a two-chain structure with two disulfide bonds that link the B and A chains plus one internal disulfide bond within the A chain.
Problems with preproinsulin translation may affect either the quantity or the quality of the translation product. Interestingly, based on rodent studies (that will require further validation in humans as well as animal models), it has been suggested that there is a steady decline of preproinsulin translation as a function of age (34); this might correlate with the age-related increase in the incidence of T2D (35). As for quality, aberrant preproinsulin translation has been linked to defects in Cdkal1 (a tRNA methylthiotransferase required for the fidelity of reading AAA and AAG), and its deficiency can result in the misreading of Lys codons (36). Thus, in addition to effects on insulin target tissues, Cdkal1 deficiency can adversely affect the quality of the proinsulin translation product (triggering ER stress; ref. 37) as well as subsequent proinsulin-to-insulin conversion (38). Further, it was recently demonstrated that proper β cell iron status, mediated by iron-responsive element binding protein 2 (IRP2), is required for the ongoing function of Cdkal1, and IRP2 deficiency also results in Lys codon misreading that leads to impaired proinsulin folding with diminished insulin biosynthesis (39). (Alternatively, diabetogenic SNPs in the CDKAL1 locus may reflect alteration in the expression of SRY-box transcription factor 4 [SOX4], as reviewed in ref. 40.) Moreover, translational initiation at an improper AUG start site could potentially lead either to the production of a preproinsulin bearing an N-terminally foreshortened signal peptide, or to a novel defective ribosomal initiation product that shares no sequence identity whatsoever with preproinsulin (41). Along with aberrant splicing noted above, these mistranslated products are fascinating proteins, potentially generating neoantigens that may be recognized by the immune system to trigger autoimmune response (42, 43).
A critical step for preproinsulin entry into the secretory pathway involves its translocation across the ER membrane. There are at least two modes of entry: cotranslational translocation that is signal recognition particle–dependent (SRP-dependent), and posttranslational translocation that does not require SRP (44–46). Cotranslational translocation is the main ER entry pathway for most secretory proteins; however, posttranslational translocation may be important especially for small secretory proteins and those with suboptimal SRP recognition/binding sequences (47, 48). Preproinsulin had long been thought to enter the ER exclusively through cotranslational translocation (i.e., formation of the ribosome-preproinsulin-SRP complex for delivery to the Sec61 translocon via binding to the SRP receptor on the ER membrane; ref. 49). However, recent experimental evidence from human islets (as well as non-β cells transfected to express wild-type preproinsulin) indicates that up to 15%–20% of full-length preproinsulin (110 residues) can be detected immediately after its biosynthesis. As cleavage of the signal peptide is not itself thought to be rate-limiting, these data suggest that completion of the full-length translation product is too rapid to fully engage SRP-dependent cotranslational translocation (50, 51). This subset of fully translated molecules can nevertheless undergo subsequent conversion to proinsulin, strongly suggesting posttranslational translocation into the ER (47, 48). This may be an important “backup” mechanism to enhance newly synthesized preproinsulin translocation efficiency (44, 47). A positive charge within the N-terminal portion of the preproinsulin signal peptide appears to be an important factor in determining translocation efficiency for preproinsulin and other small secretory proteins (47, 50, 52).
Importantly, recent studies suggest that translocation into the ER is regulated, and decreased translocation efficiency may be a cellular response to overload of the ER (53, 54). This has been termed “preemptive quality control” accompanied by proteasomal disposal of untranslocated proteins, with the notion that decreased ER protein loading can alleviate ER stress (53, 55, 56). This preemptive quality control has been observed primarily for small secretory proteins and/or those with a suboptimal SRP recognition sequence (44, 57, 58). However, it is worth noting that although this mechanism may reduce ER stress, it may also result in cytosolic accumulation of untranslocated preproteins. Indeed, failure to translocate prion protein has been linked to neurodegenerative diseases (56). Thus, in order to avoid toxic accumulation in cells, untranslocated preproteins need to be efficiently degraded. Components of the ER-associated protein degradation (ERAD) machinery, including Derlins, p97 (valosin-containing protein), Bag6, and AIRAPL (zinc finger AN1-type containing 2B), have all been suggested to play a role in the preemptive proteasomal clearance of untranslocated preproteins (54, 59). Given the fact that preproinsulin is a small protein that may be predisposed to inefficient translocation (44, 50), it will be important to know whether it is also subject to preemptive regulation under ER stress, and whether inefficiently translocated preproinsulin can contribute to β cell failure during the ER stress of type 1 diabetes (T1D), T2D, or selective forms of monogenic diabetes (60–65). Certainly, the pathophysiological significance of inefficient translocation for preproinsulin is highlighted by the recent discovery of INS gene–encoded signal peptide mutations that cause early-onset diabetes in humans (66–69). Further, even in the absence of such signal peptide mutations, defective IRE1α-mediated ER stress signaling is known to result in the accumulation of unprocessed preproinsulin (70), which normally is virtually undetectable in healthy β cells (71). Recently, translocon-associated protein-α (TRAPα), encoded by the T2D-associated gene SSR1 (signal sequence receptor 1), has also been shown to play an important role in the efficiency of preproinsulin translocation (71). These realizations open new avenues for further studies to evaluate the potential pathophysiological importance of defective preproinsulin translation and/or translocation during the development of diabetes, and the gene variants that may contribute to these defects (Table 2).
ER quality control in proinsulin folding/misfolding
In general, proteotoxic forms of proinsulin are selected against in evolution (72), as they seem to impair β cell mass in addition to their impairment of insulin biogenesis (73, 74) as well as general dysfunction of the secretory pathway (75). Nevertheless, misfolding of the INS gene product is not incompatible with life, as a large family of INS gene mutations underlie the autosomal dominant disorder known as MIDY (mutant INS gene–induced diabetes of youth) (76). MIDY does not represent all INS gene mutations, as some mutant alleles may merely decrease preproinsulin expression (77), or alter aspects of the biology of the INS gene product other than proinsulin folding (60). All MIDY patients thus far described are heterozygotes, still bearing one wild-type (WT) INS allele (78). It is clear that misfolded MIDY proinsulins are synthesized yet entrapped within the ER lumen (79), and this is the location where the mutant protein first engages (and blocks the trafficking of) WT bystander proinsulin (80). Indeed, MIDY mutant proinsulins can form aberrant, intermolecular disulfide bonds with WT proinsulin, propagating the defects of impaired folding and ER export onto the WT bystanders (51, 76, 81). These abnormal interactions appear to be an initiating event leading to insulin-deficient diabetes, well before decreased β cell mass (82–84).
How these aberrant interactions are initiated has not been clear, but recent evidence suggests that initial contact between mutant and WT molecules involves one or more residues that would normally be used for noncovalent proinsulin dimerization (85), which engages a relatively hydrophobic helical region spanning residues 8–29 of the insulin B chain from two apposed partners. Structural analysis indicates that Tyr-B16 contributes more surface area to the dimerization contact surface than any other single residue (85). Whereas substitution of Tyr-B16 with either Ala or Asp does not affect the folding and trafficking of WT proinsulin, introduction of these substitutions into a MIDY proinsulin (such as the Akita mutant) functions as an intragenic suppressor, substantially limiting cross-dimerization, decreasing abnormal intermolecular disulfide bonding between mutant and WT proinsulin, and increasing WT insulin production. These data support the idea that dominant-negative propagation of misfolding from mutant proinsulin onto bystander proinsulin is initiated by intermolecular contact involving residues of the natural proinsulin dimerization surface. Independently, misfolded proinsulin may also impair, in trans, other exportable proteins in the β cell secretory pathway, such as the precursor insulin receptor (ProIR), which can inhibit ProIR maturation and insulin signaling in β cells (86).
In addition to the foregoing interactions, proinsulin has the potential to interact with multiple resident proteins of the ER, and these interactions are likely to be altered under pathological conditions. Proinsulin has been reported to coprecipitate with ERp46, but the expression level of this ER oxidoreductase (and thus its probable interaction with proinsulin) decreases upon onset of diabetes (at least in C57BL/KsJ db/db mice) — although this may be partially rescued upon treatment with GLP-1 agonists (87). BiP is a well-established ER chaperone with preferential binding to misfolded proinsulin (88, 89). BiP function is potentially supported by the Sil1 nucleotide exchange factor (90). Additionally, GRP94 function is needed for proper post-ER processing of proinsulin, and deficiency of GRP94 increases the numbers of abnormally enlarged secretory granules bearing diminished mature insulin (91).
Further, proinsulin that is misfolded under altered redox conditions may present itself for interactions with ER-resident proteins that perhaps do not participate as importantly in the normal proinsulin folding pathway. New evidence indicates that PRDX4 may be an important proinsulin interactor in normal human islets — particularly under altered redox conditions (92). Further, the lectin-like chaperone calreticulin appears to interact with reduced proinsulin and facilitates its lysosomal degradation (93). Misfolded mutant proinsulin is also known to interact with PDIA1 (also known as PDI; encoded by P4HB) (94), which assists proinsulin under conditions of oxidative stress (95); misfolded proinsulin also interacts with GRP170 (encoded by HYOU1) (96). ERO1 is thought to function upstream of PDIA1 and other isomerases/oxidoreductases. Deletion or downregulation of ERO1 impairs proinsulin oxidative folding and decreases insulin production (97, 98). Conversely, overexpression of ERO1 in β cells appears to accelerate oxidative folding and enhances the ER export of WT proinsulin in the presence of MIDY mutants (51, 99), although excessive β cell–specific expression of ERO1β does not improve insulin production (100), suggesting that a fine-tuned oxidative state of the ER is needed to optimize insulin biosynthesis.
Misfolded proinsulin, at least in part, undergoes intracellular disposal (101) by ERAD and ER macroautophagy. ERAD has an important role to play in normal β cell ER homeostasis (102). The evolutionarily conserved ERAD core machinery includes Hrd1 (involved in retrotranslocation of the target protein) and Sel1L (the cofactor essential for Hrd1 protein stability; ref. 103); additional factors may contribute to the expression and/or stability of these core components (104). The basic steps involved in ERAD of proinsulin (105) and other secretory proteins involve (a) recognition of the (presumably misfolded) protein target via binding to various resident proteins within the ER that either positively or negatively facilitate the efficiency of the degradation process (96, 106); (b) retrotranslocation from the luminal side of the ER membrane to the cytosolic side (94); (c) ubiquitylation of the target protein (107) via Hrd1; (d) extraction of the ubiquitylated target protein via the p97 ATPase for the delivery to proteasomes (108); and (e) proteasomal degradation, which may involve the stress-inducible proteasome subunit β5i (109).
Deficiency of Sel1L expression (which causes instability of Hrd1 protein; ref. 110) has long been known to block glucose-stimulated insulin secretion from islet β cells (111). Hrd1 may act on proinsulin but also on other β cell degradation substrates (112). Indeed, new work has highlighted that Sel1L-Hrd1 activity in β cells is needed for the maintenance of full functionality and identity of β cells — especially including insulin biosynthesis — in particular by limiting the activity of the TGF-β signaling pathway (113). Thus, loss of β cell Sel1L appears to trigger hyperactivation of TGF-β signaling, which accompanies a dedifferentiated β cell phenotype.
Beyond ERAD, quality control of misfolded proinsulin results in active clearance by ER autophagy (ER-phagy); this occurs under all conditions but is especially true when ERAD of proinsulin is impaired (114), or in β cells bearing Ins mutations that cause proinsulin misfolding (115). In the latter case, mTOR activity appears diminished (73) and AMP kinase activity appears stimulated. While ER-phagy is likely to be beneficial in clearing misfolded proinsulin and thereby enhancing efficiency of the remaining proinsulin to be properly folded and to undergo successful intracellular transport (114), generalized hyperactivation of autophagy is not a cure-all for β cells, as this can stimulate enhanced autophagic turnover of transport-competent proinsulin (116) as well as turnover of mature insulin secretory granules, leading to a diminution of the insulin storage pool (117, 118). The regulation of lysosome-mediated proinsulin turnover is also not yet understood (119); however, if therapies can be designed that selectively activate β cell ER macroautophagy, such therapies may hold promise as potential treatments for diabetes linked to proinsulin misfolding (114).
ER stress and ER stress response signaling pathways in pancreatic β cells have been extensively reviewed elsewhere (120–123), including heterogeneity in responses that accompany the heterogeneity of proinsulin biosynthetic/folding activity among islet β cells (124). The biosynthesis and folding of proinsulin itself controls a set-point of ER stress activation within β cells (125). Additionally, β cell ER stress appears to be a hallmark of T2D in both humans and rodent models (126) — and failure to mount adaptive ER stress responses is known to make matters worse (127, 128).
The Ire1 limb of the ER stress response is of acute and profound benefit to insulin biosynthesis (129). In particular, Ire1-Xbp1 upregulates multiple ER factors promoting proinsulin biosynthesis, including preproinsulin translocation across the ER membrane (70, 130) (see previous section). Moreover, Ire1-Xbp1 upregulation of various ER oxidoreductases is thought to participate in the oxidative folding of proinsulin to the native state (including PDIA1, PDIA5 [also known as PdiR], PDIA6 [P5], ERp44 [PDIA10], and TXNDC5 [PDIA15, or ERp46]) — of these, PDIA1 has been demonstrated to contribute to the reduction of aberrant intermolecular proinsulin disulfide bonds that are formed upon redox stress (94, 95). Nevertheless, Ire1 activation must be fine-tuned, as its hyperactivation appears clearly detrimental to insulin biosynthesis and β cell survival (131–133).
The role of PERK in regulating insulin biogenesis is firmly established, but the proposed mechanism(s) have varied considerably between different groups and systems. Classic work described PERK as one of the four kinases involved in the phosphorylation of eIF2α (134), negatively regulating pancreatic (exocrine and endocrine) translation (135, 136). Consistent with this, conditions of insufficient PERK activity have been reported to result in hypersynthesis of proinsulin, albeit with an increase in the abundance of non-native folded forms (88, 89, 137) that may contribute to the β cell failure of Wolcott-Rallison syndrome (138, 139). However, other studies have not corroborated that PERK activity negatively regulates proinsulin biosynthesis (140), concluding instead that PERK regulates multiple other functions (141) including β cell proliferation (142, 143) — although this too has been challenged (144). Further, there is reason to suspect that PERK expression is needed for the maintenance of β cell differentiation as a result of its role in regulating multiple aspects of ER homeostasis (82, 145). Like the findings regarding Ire1, these findings may not necessarily be internally contradictory, as they may reflect the homeostatic importance of PERK in β cells in which both too little and too much activity can be detrimental (146). However, in studies of proinsulin translation and its regulation by PERK, we do wish to sound a strong note of caution about conclusions based largely on the INS-832/13 rat β cell line. These cells may indeed be useful for studies of proinsulin trafficking, conversion to insulin, and glucose-stimulated insulin secretion, as well as sensitivity to ROS and other β cell toxins (147). Nevertheless, this clone of INS-1 cells (expressing endogenous rat proinsulin-1 and -2) is uniquely complicated for studies of proinsulin biosynthesis, because the INS-832/13 cells also synthesize transfected human proinsulin expressed from a nonphysiological CMV promoter and bearing nonphysiological 5′- and 3′-UTR sequences (148) — thus its translation cannot be regulated in a physiological way.
PERK activity is also thought to support the expression of another ER stress sensor, ATF6 (149). As with Ire1 and PERK, different studies of ATF6 in β cells have reached somewhat differing conclusions regarding its impact on insulin production and release (150). In brief, physiological (i.e., mild) ER stress is thought to promote ATF6 expression in a manner that is favorable to β cell growth and survival (128, 151); on the other hand, transcription activated by ATF6 stimulates production of the proapoptotic transcription factor CHOP (152, 153). There is reason to think that inadequate ATF6 and Xbp1 expression may be linked to inadequate insulin production during the progression of T2D (154). In summary, each of the ER stress response signaling limbs requires fine tuning and has the potential to lead to both helpful and harmful effects on insulin biogenesis and β cell viability. A number of genes involved in ER homeostasis whose variants may be linked to human diabetes are listed in Table 3.
Delivery to granules with processing and insulin storage
A list of human diabetes–related genes associated with insulin granule packaging, processing, or secretion is presented in Table 4. Formation of insulin secretory granules, and maintenance of the granule compartment, is a broad topic that will be only briefly summarized here. It is important to note, however, that morphometric studies of human T2D islets have shown that the volume density of total mature insulin granules in β cells is significantly diminished, and this pool size correlates with the diminished magnitude of the glucose-stimulated insulin secretion response (155).
Recent proteomics studies have suggested that to support the upregulation and maintenance of insulin secretory granule biogenesis (in mice; ref. 156), there are differential posttranslational modifications of ER molecular chaperones and proteins involved in redox homeostasis (see previous section), as well as those involved in protein trafficking. This may be important, because ongoing ER-to-Golgi transport itself helps to maintain the ER environment and limit ER stress (157). Indeed, augmented proinsulin export is thought to be linked to enhanced insulin secretion (158).
Upon transport through the Golgi complex, proinsulin and its processing enzymes emerge in immature secretory granules (ISGs) budding from the trans-Golgi network (a process that appears to be microtubule-dependent; ref. 159). The formation and maturation of insulin granules involve a complex set of processes that coordinate ionic control of the organelle interior (160, 161), acquisition of competence for stimulus-dependent secretion (162, 163), enzymatic processing of proinsulin (164, 165), packaging (protein condensation) (166), and membrane trafficking and lipid modification (167–170), as well as stability of the organelle during its intracellular transport through the cytoplasm (171–173); many of these steps are interconnected, and they are ultimately transcriptionally orchestrated (174). For example, the peripheral membrane protein HID-1 is thought to support proinsulin-to-insulin conversion that is synchronized with other steps of insulin secretory granule maturation (175).
As for processing enzymes, ISG acidification and high Ca2+ are both required for optimal proinsulin conversion to insulin (176). Prohormone convertase-1 (PC1/3) is the dominant endoproteolytic activity required for proinsulin-to-insulin conversion, with a lesser contribution from prohormone convertase-2 (177). Additionally, the activity of carboxypeptidase E (CPE) is strongly linked to β cell insulin production and storage (178). These enzymes depend on the dibasic endoproteolytic cleavage sites of proinsulin; diabetes-related insulin deficiency is observed either when these cleavage sequences are not present, or when they are present but inefficiently acted upon. Indeed, deficiency of proinsulin conversion appears to correlate with insulin deficiency in diabetes (179–183).
With regard to intragranular packaging, chromogranins A and B have both been long suspected to play an important role (184), but their precise molecular role has remained somewhat elusive (185, 186), in part because it is difficult to distinguish direct and indirect effects of the expression of these molecules. Additionally, other proteins such as VGF or IA-2/ICA512 (islet cell autoantigen 512, an integral membrane protein with either a detached or an intact luminal domain) may also help to support luminal protein condensation in young granules (187, 188). While luminal condensation is occurring, there appears to be active modeling/remodeling of the ISG membrane, controlled in large part by the recruitment of cytosolic factors that interact with ISG membrane proteins to drive the remodeling (189), as well as dynamic membrane interchange with the endosomal system — in part regulated by the endosome-associated recycling protein (EARP) complex and the EARP-interacting protein EIPR1 (190).
Diabetes caused by defective β cell insulin storage
Absolute/relative insulin deficiency is a hallmark of both T1D and T2D. However, beginning with relatively healthy compensation including robust (or even increased) insulin biosynthesis, the onset and subsequent development of T2D leads β cells through a gradual progression of different phenotypes, from diminished insulin storage, to functionally exhausted cells that can no longer be identified as β cells owing to loss of immunostaining with anti-insulin antibodies, and ultimately to the loss of islet β cells that may result from dedifferentiation and eventual β cell death (Figure 2) (15, 36, 191–193). Importantly, accumulating evidence from both mouse (17) and human (14, 194, 195) studies supports the idea that at some points during the progression of T2D, β cell function can be partially or even fully restored in those favorably responding to weight loss; however, the ability to regain β cell function appears to be time-dependent (196). Although the rate of remission after extensive intervention can be as high as 50%–90% for patients with T2D of ≤1.5 years’ duration (194, 195), reversal essentially cannot be achieved in patients who have had T2D for more than 11 years (197). These data suggest progression beyond one or more transition points that determine recoverability across the β cell population (Figure 3).
Figure 2. β Cell phenotypes of defective insulin production and storage.
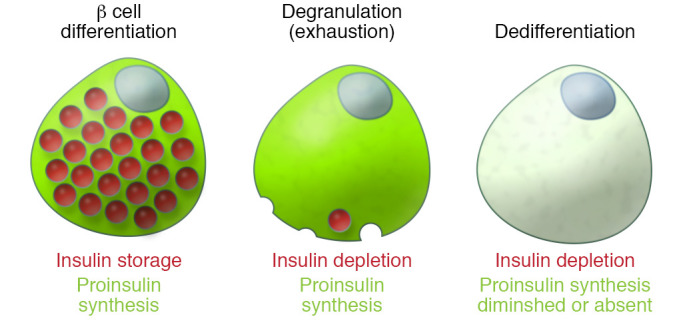
β Cell dysfunction may begin many years before diagnosis of diabetes, and progressive deterioration of β cell function represents the natural history of T2D. In healthy, well-differentiated and well-compensated β cells (left), the synthesis of proinsulin (green) and its folding and processing are homeostatically regulated to meet metabolic demand in order to maintain the biogenesis of insulin (red), which is stored in granules for insulin secretion. Well before end-stage β cell failure, proinsulin biosynthesis may be actively ongoing even in β cells that no longer maintain an insulin storage pool (middle). β Cell degranulation appears (by immunofluorescence analysis of insulin content) as a loss of β cell identity. As the disease progresses further (right), there is also a diminution of proinsulin biosynthesis in addition to the loss of insulin storage. This schematic figure shows individual β cells for clarity, but given the heterogeneity of islet β cells, these representations should be viewed not as a fixed stage for all β cells in the population, but rather as the phenotype for a prevailing portion of islet β cells across the pancreas during disease progression. β Cell death is not shown in the figure, but this is also a relevant consideration in many forms of diabetes.
Figure 3. Proinsulin and insulin during progression to β cell failure.

Prediabetes: β Cell function has a greater chance of recovery. β Cell insulin synthesis increases and insulin content is maintained, although intracellular proinsulin/Cpep or proinsulin/insulin ratio begins to rise, accompanied by defective GSIS and elevated proinsulin/Cpep or proinsulin/insulin ratio in the secretion. Early T2D: β Cell function still has a good chance of recovery. Insulin content declines and progresses toward depletion as demonstrated by immunostaining, making it more difficult to identify β cells (“loss of identity”), but (pre)proinsulin biosynthesis is still actively ongoing. Proinsulin/Cpep or proinsulin/insulin ratio in the islets and secretion is more markedly elevated. Later T2D: β Cell function may still be recoverable, but the chance of recovery is lower at this stage. Evidence of β cell dedifferentiation is detectable and (pre)proinsulin biosynthesis drops (though still detectable) with a decreased expression of pancreatic proinsulin. β Cell–specific transcription factors exhibit diminished expression; endocrine progenitor cell markers increase. End-stage T2D: β Cell function appears unrecoverable and β cell mass is diminished, consistent with some level of β cell demise. Cpep, C-peptide; GSIS, glucose-stimulated insulin secretion; PI, proinsulin.
Unfortunately, to date, there are no reliable biomarkers to identify/distinguish these stages in a living individual, or even in histological analysis of the pancreas from deceased individuals. Although absence of islet insulin immunostaining is perhaps the most widely used marker to define loss of β cell identity, depletion of insulin storage does not necessarily mean dedifferentiation or β cell death. For example, it has been shown that depleted insulin storage in the islets of C57BL/KsJ db/db diabetic mice can still be significantly restored when the islets are cultured ex vivo in 5.6 mmol/L glucose (198). It has also been found that in islets from Akita diabetic mice with profound insulin deficiency, (pre)proinsulin biosynthesis is nevertheless still ongoing, although this fails to result in the efficient formation of mature insulin (84). A generally similar phenotype can be discerned in the islets of young C57BL/KsJ db/db mice, in which many insulin-depleted β cells continue to abundantly express proinsulin (199). Such data indicate early depletion of the insulin storage pool prior to any diminution of translation of the INS mRNA.
Concluding questions and comments
Recent genetic and biological evidence suggests that early steps of insulin biosynthesis, including preproinsulin translocation into the ER, proinsulin folding, and ER export, are not as efficient as was once thought. Indeed, key questions in these areas remain to be addressed:
(a) Does the efficiency of co- and posttranslational translocation vary between healthy β cells and the islets of individuals with diabetes? To what extent do signaling pathways regulate the efficiency of preproinsulin translocation under different physiological and/or pathological conditions?
(b) Proinsulin dimerizes in the ER, and cross-dimerization between misfolded proinsulin and bystander WT proinsulin contributes to dominant-negative effects. If proinsulin dimerization in the ER is nonessential, could the dimerization contact surface be a druggable target to promote increased insulin biogenesis, comparable to pharmacochaperones being developed for other degenerative diseases caused by protein misfolding (200)?
(c) Correct disulfide pairing is a critical event both for proinsulin folding in the ER, and for the ultimate bioactivity of insulin. Does the oxidative environment of the β cell ER change in T2D, and if so, is this too a druggable target?
(d) Proinsulin folding is a kinetic process, during which we hypothesize that there are discrete folding intermediates. How do β cells differentiate “on-pathway” folding intermediates from “dead-end” misfolded proinsulin in order to distinguish the fates of not-yet-folded versus misfolded proinsulin molecules?
(e) If there really are transition zones during the development and progression of T2D at which β cell function can be fully (or even partially) restored, can β cell rest promote recovery of β cell function even after dedifferentiation has occurred, and could proinsulin status be used as a biomarker to determine this?
Accumulating evidence indicates that genetic or acquired defects in the Ins gene translation product are central to virtually all of these key questions. In the coming years, we would hope to see development of therapies designed to attenuate the detrimental effects of improperly translocated preproinsulin or misfolded proinsulin. These strategies may span approaches designed to increase the biological yield of properly folded proinsulin (and thus insulin) in β cells, or to enhance the degradative clearance of improperly folded products. Such approaches may well have broad translational significance for future developments in the management of β cells in diabetes.
Acknowledgments
The authors are supported by NIH R01-DK48280 and R01-DK111174, as well as R24-DK110973, R01-DK120047, and R01-DK113171; the Liu lab is supported by research grants 81620108004, 81830025, and 81700699 from the National Natural Science Foundation of China. We also acknowledge support from the Protein Folding Diseases Initiative of the University of Michigan as well as support from the National Key R&D Program of China (2019YFA0802502) and the Tianjin Municipal Science and Technology Commission (17ZXMFSY00150, 18JCQNJC82100).
Version 1. 01/19/2021
Electronic publication
Version 2. 01/29/2021
Modified figure 1
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(2):e142240.https://doi.org/10.1172/JCI142240.
Contributor Information
Ming Liu, Email: mingliu@tmu.edu.cn.
Yumeng Huang, Email: yumengh@umich.edu.
Xiaoxi Xu, Email: xiaoxix@umich.edu.
Xin Li, Email: lixin920616@163.com.
Maroof Alam, Email: mdal@umich.edu.
Anoop Arunagiri, Email: anooparu@med.umich.edu.
Leena Haataja, Email: leenah@med.umich.edu.
Li Ding, Email: dinglitml@163.com.
Shusen Wang, Email: shusen@vip.163.com.
Pamela Itkin-Ansari, Email: pitkin@sbpdiscovery.org.
Randal J. Kaufman, Email: rkaufman@sbpdiscovery.org.
Billy Tsai, Email: btsai@umich.edu.
Ling Qi, Email: lingq@med.umich.edu.
Peter Arvan, Email: parvan@umich.edu.
References
- 1.Carrano AC, Mulas F, Zeng C, Sander M. Interrogating islets in health and disease with single-cell technologies. Mol Metab. 2017;6(9):991–1001. doi: 10.1016/j.molmet.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu Y, Liu Q, Zhou Z, Ikeda Y. PDX1, Neurogenin-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther. 2017;8(1):240. doi: 10.1186/s13287-017-0694-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haliyur R, et al. Human islets expressing HNF1A variant have defective β cell transcriptional regulatory networks. J Clin Invest. 2019;129(1):246–251. doi: 10.1172/JCI121994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasiljević J, Torkko JM, Knoch KP, Solimena M. The making of insulin in health and disease. Diabetologia. 2020;63(10):1981–1989. doi: 10.1007/s00125-020-05192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glick E, Leshkowitz D, Walker MD. Transcription factor BETA2 acts cooperatively with E2A and PDX1 to activate the insulin gene promoter. J Biol Chem. 2000;275(3):2199–2204. doi: 10.1074/jbc.275.3.2199. [DOI] [PubMed] [Google Scholar]
- 6.Churchill AJ, Gutiérrez GD, Singer RA, Lorberbaum DS, Fischer KA, Sussel L. Genetic evidence that Nkx2.2 acts primarily downstream of Neurog3 in pancreatic endocrine lineage development. Elife. 2017;6:e20010. doi: 10.7554/eLife.20010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raum JC, et al. FoxA2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol Cell Biol. 2006;26(15):5735–5743. doi: 10.1128/MCB.00249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cyphert HA, et al. Examining how the MAFB transcription factor affects islet β-cell function postnatally. Diabetes. 2019;68(2):337–348. doi: 10.2337/db18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pappalardo Z, et al. A whole-genome RNA interference screen reveals a role for Spry2 in insulin transcription and the unfolded protein response. Diabetes. 2017;66(6):1703–1712. doi: 10.2337/db16-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orland MJ, Permutt MA. Quantitative analysis of pancreatic proinsulin mRNA in genetically diabetic (db/db) mice. Diabetes. 1987;36(3):341–347. doi: 10.2337/diab.36.3.341. [DOI] [PubMed] [Google Scholar]
- 11.Boland BB, et al. Pancreatic β-cell rest replenishes insulin secretory capacity and attenuates diabetes in an extreme model of obese type 2 diabetes. Diabetes. 2019;68(1):131–140. doi: 10.2337/db18-0304. [DOI] [PubMed] [Google Scholar]
- 12.Perreira M, et al. “Reversine” and its 2-substituted adenine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem. 2005;48(15):4910–4918. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, et al. β-Cell dedifferentiation in patients with T2D with adequate glucose control and nondiabetic chronic pancreatitis. J Clin Endocrinol Metab. 2019;104(1):83–94. doi: 10.1210/jc.2018-00968. [DOI] [PubMed] [Google Scholar]
- 15.Cinti F, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab. 2016;101(3):1044–1054. doi: 10.1210/jc.2015-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonas JC, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem. 1999;274(20):14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19(5):872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weir GC. Glucolipotoxicity, β-cells, and diabetes: the emperor has no clothes. Diabetes. 2020;69(3):273–278. doi: 10.2337/db19-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai C, et al. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J Clin Invest. 2016;126(5):1857–1870. doi: 10.1172/JCI83657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo S, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123(8):3305–3316. doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wernersson R, et al. Analysis artefacts of the INS-IGF2 fusion transcript. BMC Mol Biol. 2015;16:13. doi: 10.1186/s12867-015-0042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monk D, et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum Mol Genet. 2006;15(8):1259–1269. doi: 10.1093/hmg/ddl041. [DOI] [PubMed] [Google Scholar]
- 23.Garin I, Perez de Nanclares G, Gastaldo E, Harries LW, Rubio-Cabezas O, Castaño L. Permanent neonatal diabetes caused by creation of an ectopic splice site within the INS gene. PLoS One. 2012;7(1):e29205. doi: 10.1371/journal.pone.0029205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alarcon C, Wicksteed B, Prentki M, Corkey BE, Rhodes CJ. Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes. 2002;51(8):2496–2504. doi: 10.2337/diabetes.51.8.2496. [DOI] [PubMed] [Google Scholar]
- 25.Skelly RH, Bollheimer LC, Wicksteed BL, Corkey BE, Rhodes CJ. A distinct difference in the metabolic stimulus-response coupling pathways for regulating proinsulin biosynthesis and insulin secretion that lies at the level of a requirement for fatty acyl moieties. Biochem J. 1998;331(pt 2):553–561. doi: 10.1042/bj3310553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leibowitz G, et al. Mitochondrial regulation of insulin production in rat pancreatic islets. Diabetologia. 2005;48(8):1549–1559. doi: 10.1007/s00125-005-1811-6. [DOI] [PubMed] [Google Scholar]
- 27.Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature. 1980;283(5742):100–102. doi: 10.1038/283100a0. [DOI] [PubMed] [Google Scholar]
- 28.Wicksteed B, Uchizono Y, Alarcon C, McCuaig JF, Shalev A, Rhodes CJ. A cis-element in the 5’ untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab. 2007;5(3):221–227. doi: 10.1016/j.cmet.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Lee EK, et al. RNA-binding protein HuD controls insulin translation. Mol Cell. 2012;45(6):826–835. doi: 10.1016/j.molcel.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guest PC. 2D gel electrophoresis of insulin secretory granule proteins from biosynthetically labelled pancreatic islets. Adv Exp Med Biol. 2017;974:167–174. doi: 10.1007/978-3-319-52479-5_12. [DOI] [PubMed] [Google Scholar]
- 31.Wang SY, Rowe JW. Age-related impairment in the short term regulation of insulin biosynthesis by glucose in rat pancreatic islets. Endocrinology. 1988;123(2):1008–1013. doi: 10.1210/endo-123-2-1008. [DOI] [PubMed] [Google Scholar]
- 32.Wicksteed B, Alarcon C, Briaud I, Lingohr MK, Rhodes CJ. Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels in islet beta-cells but not regulated via a positive feedback of secreted insulin. J Biol Chem. 2003;278(43):42080–42090. doi: 10.1074/jbc.M303509200. [DOI] [PubMed] [Google Scholar]
- 33.Greenman IC, Gomez E, Moore CE, Herbert TP. The selective recruitment of mRNA to the ER and an increase in initiation are important for glucose-stimulated proinsulin synthesis in pancreatic beta-cells. Biochem J. 2005;391(pt 2):291–300. doi: 10.1042/BJ20050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang SY, Halban PA, Rowe JW. Effects of aging on insulin synthesis and secretion. Differential effects on preproinsulin messenger RNA levels, proinsulin biosynthesis, and secretion of newly made and preformed insulin in the rat. J Clin Invest. 1988;81(1):176–184. doi: 10.1172/JCI113291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halter JB, et al. Diabetes and cardiovascular disease in older adults: current status and future directions. Diabetes. 2014;63(8):2578–2589. doi: 10.2337/db14-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufman RJ. Beta-cell failure, stress, and type 2 diabetes. N Engl J Med. 2011;365(20):1931–1933. doi: 10.1056/NEJMcibr1109442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei FY, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest. 2011;121(9):3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirchhoff K, et al. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia. 2008;51(4):597–601. doi: 10.1007/s00125-008-0926-y. [DOI] [PubMed] [Google Scholar]
- 39.Santos MCFD, et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat Commun. 2020;11(1):296. doi: 10.1038/s41467-019-14004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krentz NAJ, Gloyn AL. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat Rev Endocrinol. 2020;16(4):202–212. doi: 10.1038/s41574-020-0325-0. [DOI] [PubMed] [Google Scholar]
- 41.Kracht MJ, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med. 2017;23(4):501–507. doi: 10.1038/nm.4289. [DOI] [PubMed] [Google Scholar]
- 42.Kanatsuna N, et al. Doubly reactive INS-IGF2 autoantibodies in children with newly diagnosed autoimmune (type 1) diabetes. Scand J Immunol. 2015;82(4):361–369. doi: 10.1111/sji.12325. [DOI] [PubMed] [Google Scholar]
- 43.Laban S, et al. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PLoS One. 2018;13(8):e0200818. doi: 10.1371/journal.pone.0200818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu M, et al. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab. 2018;20(suppl 2):28–50. doi: 10.1111/dom.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zimmermann R, Eyrisch S, Ahmad M, Helms V. Protein translocation across the ER membrane. Biochim Biophys Acta. 2011;1808(3):912–924. doi: 10.1016/j.bbamem.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 46.Rapoport TA. Protein transport across the endoplasmic reticulum membrane. FEBS J. 2008;275(18):4471–4478. doi: 10.1111/j.1742-4658.2008.06588.x. [DOI] [PubMed] [Google Scholar]
- 47.Guo H, et al. Positive charge in the n-region of the signal peptide contributes to efficient post-translational translocation of small secretory preproteins. J Biol Chem. 2018;293(6):1899–1907. doi: 10.1074/jbc.RA117.000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakkaraju AK, Thankappan R, Mary C, Garrison JL, Taunton J, Strub K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol Biol Cell. 2012;23(14):2712–2722. doi: 10.1091/mbc.e12-03-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okun MM, Shields D. Translocation of preproinsulin across the endoplasmic reticulum membrane. The relationship between nascent polypeptide size and extent of signal recognition particle-mediated inhibition of protein synthesis. J Biol Chem. 1992;267(16):11476–11482. doi: 10.1016/S0021-9258(19)49934-1. [DOI] [PubMed] [Google Scholar]
- 50.Guo H, et al. Inefficient translocation of preproinsulin contributes to pancreatic β cell failure and late-onset diabetes. J Biol Chem. 2014;289(23):16290–16302. doi: 10.1074/jbc.M114.562355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu M, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes. 2012;61(4):828–837. doi: 10.2337/db11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okamoto Y, Shikano S. Positive zip coding in small protein translocation. J Biol Chem. 2018;293(6):1908–1909. doi: 10.1074/jbc.H118.001415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127(5):999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kadowaki H, et al. Pre-emptive quality control protects the ER from protein overload via the proximity of ERAD components and SRP. Cell Rep. 2015;13(5):944–956. doi: 10.1016/j.celrep.2015.09.047. [DOI] [PubMed] [Google Scholar]
- 55.Liu M, Wright J, Guo H, Xiong Y, Arvan P. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitam Horm. 2014;95:35–62. doi: 10.1016/B978-0-12-800174-5.00002-8. [DOI] [PubMed] [Google Scholar]
- 56.Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell. 2008;15(3):359–370. doi: 10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson N, et al. The signal sequence influences post-translational ER translocation at distinct stages. PLoS One. 2013;8(10):e75394. doi: 10.1371/journal.pone.0075394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol. 2010;188(4):515–526. doi: 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braunstein I, Zach L, Allan S, Kalies KU, Stanhill A. Proteasomal degradation of preemptive quality control (pQC) substrates is mediated by an AIRAPL-p97 complex. Mol Biol Cell. 2015;26(21):3719–3727. doi: 10.1091/mbc.E15-02-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21(11):652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tersey SA, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61(4):818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papa FR. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb Perspect Med. 2012;2(9):a007666. doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Engin F. ER stress and development of type 1 diabetes. J Investig Med. 2016;64(1):2–6. doi: 10.1097/JIM.0000000000000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Franco E, et al. Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes. 2017;66(7):2044–2053. doi: 10.2337/db16-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arunagiri A, et al. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann N Y Acad Sci. 2018;1418(1):5–19. doi: 10.1111/nyas.13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hussain S, Mohd Ali J, Jalaludin MY, Harun F. Permanent neonatal diabetes due to a novel insulin signal peptide mutation. Pediatr Diabetes. 2013;14(4):299–303. doi: 10.1111/pedi.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meur G, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59(3):653–661. doi: 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu M, et al. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med. 2015;42:3–18. doi: 10.1016/j.mam.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan J, et al. Whole-exome sequencing identifies a novel INS mutation causative of maturity-onset diabetes of the young 10. J Mol Cell Biol. 2017;9(5):376–383. doi: 10.1093/jmcb/mjx039. [DOI] [PubMed] [Google Scholar]
- 70.Hassler JR, et al. The IRE1α/XBP1s pathway is essential for the glucose response and protection of β cells. PLoS Biol. 2015;13(10):e1002277. doi: 10.1371/journal.pbio.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li X, et al. Requirement for translocon-associated protein (TRAP) α in insulin biogenesis. Sci Adv. 2019;5(12):eaax0292. doi: 10.1126/sciadv.aax0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rege NK, et al. “Register-shift” insulin analogs uncover constraints of proteotoxicity in protein evolution. J Biol Chem. 2020;295(10):3080–3098. doi: 10.1074/jbc.RA119.011389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riahi Y, et al. Inhibition of mTORC1 by ER stress impairs neonatal β-cell expansion and predisposes to diabetes in the Akita mouse. Elife. 2018;7:e38472. doi: 10.7554/eLife.38472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balboa D, et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife. 2018;7:e38519. doi: 10.7554/eLife.38519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52(2):409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- 76.Liu M, et al. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS One. 2010;5(10):e13333. doi: 10.1371/journal.pone.0013333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garin I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A. 2010;107(7):3105–3110. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu M, et al. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab. 2018;20(suppl 2):28–50. doi: 10.1111/dom.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park SY, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391(3):1449–1454. doi: 10.1016/j.bbrc.2009.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haataja L, et al. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J Biol Chem. 2013;288(3):1896–1906. doi: 10.1074/jbc.M112.420018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wright J, et al. Dominant protein interactions that influence the pathogenesis of conformational diseases. J Clin Invest. 2013;123(7):3124–3134. doi: 10.1172/JCI67260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Renner S, et al. Permanent neonatal diabetes in INS(C94Y) transgenic pigs. Diabetes. 2013;62(5):1505–1511. doi: 10.2337/db12-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A. 2007;104(40):15841–15846. doi: 10.1073/pnas.0702697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun J, et al. Role of proinsulin self-association in mutant INS gene-induced diabetes of youth. Diabetes. 2020;69(5):954–964. doi: 10.2337/db19-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu S, et al. Misfolded proinsulin impairs processing of precursor of insulin receptor and insulin signaling in β cells. FASEB J. 2019;33(10):11338–11348. doi: 10.1096/fj.201900442R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lampropoulou E, Lymperopoulou A, Charonis A. Reduced expression of ERp46 under diabetic conditions in β-cells and the effect of liraglutide. Metabolism. 2016;65(1):7–15. doi: 10.1016/j.metabol.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 88.Scheuner D, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 89.Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem. 2005;280(14):13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ittner AA, Bertz J, Chan TY, van Eersel J, Polly P, Ittner LM. The nucleotide exchange factor SIL1 is required for glucose-stimulated insulin secretion from mouse pancreatic beta cells in vivo. Diabetologia. 2014;57(7):1410–1419. doi: 10.1007/s00125-014-3230-z. [DOI] [PubMed] [Google Scholar]
- 91.Ghiasi SM, et al. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes. 2019;68(4):747–760. doi: 10.2337/db18-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tran DT, et al. Unbiased profiling of the human proinsulin biosynthetic interaction network reveals a role for peroxiredoxin 4 in proinsulin folding. Diabetes. 2020;69(8):1723–1734. doi: 10.2337/db20-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikezaki M, et al. Calreticulin protects insulin against reductive stress in vitro and in MIN6 cells. Biochimie. 2020;171-172:1–11. doi: 10.1016/j.biochi.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 94.He K, Cunningham CN, Manickam N, Liu M, Arvan P, Tsai B. PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol Biol Cell. 2015;26(19):3413–3423. doi: 10.1091/mbc.E15-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jang I, et al. PDIA1/P4HB is required for efficient proinsulin maturation and β cell health in response to diet induced obesity. Elife. 2019;8:e44528. doi: 10.7554/eLife.44528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cunningham CN, et al. Chaperone-driven degradation of a misfolded proinsulin mutant in parallel with restoration of wild-type insulin secretion. Diabetes. 2017;66(3):741–753. doi: 10.2337/db16-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zito E, Chin KT, Blais J, Harding HP, Ron D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol. 2010;188(6):821–832. doi: 10.1083/jcb.200911086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khoo C, et al. Endoplasmic reticulum oxidoreductin-1-like β (ERO1lβ) regulates susceptibility to endoplasmic reticulum stress and is induced by insulin flux in β-cells. Endocrinology. 2011;152(7):2599–2608. doi: 10.1210/en.2010-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wright J, et al. Endoplasmic reticulum oxidoreductin-1α (Ero1α) improves folding and secretion of mutant proinsulin and limits mutant proinsulin-induced endoplasmic reticulum stress. J Biol Chem. 2013;288(43):31010–31018. doi: 10.1074/jbc.M113.510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Awazawa M, et al. Deregulation of pancreas-specific oxidoreductin ERO1β in the pathogenesis of diabetes mellitus. Mol Cell Biol. 2014;34(7):1290–1299. doi: 10.1128/MCB.01647-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang J, Chen Y, Yuan Q, Tang W, Zhang X, Osei K. Control of precursor maturation and disposal is an early regulative mechanism in the normal insulin production of pancreatic β-cells. PLoS One. 2011;6(4):e19446. doi: 10.1371/journal.pone.0019446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu Y, et al. Endoplasmic reticulum-associated degradation (ERAD) has a critical role in supporting glucose-stimulated insulin secretion in pancreatic β-cells. Diabetes. 2019;68(4):733–746. doi: 10.2337/db18-0624. [DOI] [PubMed] [Google Scholar]
- 103.Qi L, Tsai B, Arvan P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 2017;27(6):430–440. doi: 10.1016/j.tcb.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu B, et al. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep. 2018;24(1):181–196. doi: 10.1016/j.celrep.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sugawara T, Kano F, Murata M. Rab2A is a pivotal switch protein that promotes either secretion or ER-associated degradation of (pro)insulin in insulin-secreting cells. Sci Rep. 2014;4:6952. doi: 10.1038/srep06952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tiwari A, Schuiki I, Zhang L, Allister EM, Wheeler MB, Volchuk A. SDF2L1 interacts with the ER-associated degradation machinery and retards the degradation of mutant proinsulin in pancreatic β-cells. J Cell Sci. 2013;126(Pt 9):1962–1968. doi: 10.1242/jcs.117374. [DOI] [PubMed] [Google Scholar]
- 107.Allen JR, Nguyen LX, Sargent KE, Lipson KL, Hackett A, Urano F. High ER stress in beta-cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun. 2004;324(1):166–170. doi: 10.1016/j.bbrc.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 108.Hoelen H, et al. Proteasomal degradation of proinsulin requires Derlin-2, HRD1 and p97. PLoS One. 2015;10(6):e0128206. doi: 10.1371/journal.pone.0128206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khilji MS, et al. The inducible β5i proteasome subunit contributes to proinsulin degradation in GRP94-deficient β-cells and is overexpressed in type 2 diabetes pancreatic islets. Am J Physiol Endocrinol Metab. 2020;318(6):E892–E900. doi: 10.1152/ajpendo.00372.2019. [DOI] [PubMed] [Google Scholar]
- 110.Sun S, et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A. 2014;111(5):E582–E591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Diaferia GR, Cirulli V, Biunno I. SEL1L regulates adhesion, proliferation and secretion of insulin by affecting integrin signaling. PLoS One. 2013;8(11):e79458. doi: 10.1371/journal.pone.0079458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu T, et al. HRD1, an important player in pancreatic β-cell failure and therapeutic target for type 2 diabetic mice. Diabetes. 2020;69(5):940–953. doi: 10.2337/db19-1060. [DOI] [PubMed] [Google Scholar]
- 113.Shrestha N, et al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J Clin Invest. 2020;130(7):3499–3510. doi: 10.1172/JCI134874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P, Tsai B. Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol Cell. 2019;75(3):442–456.e4. doi: 10.1016/j.molcel.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bachar-Wikstrom E, et al. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 2013;62(4):1227–1237. doi: 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Riahi Y, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016;59(7):1480–1491. doi: 10.1007/s00125-016-3868-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamamoto S, et al. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 2018;23(11):3286–3299. doi: 10.1016/j.celrep.2018.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goginashvili A, et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science. 2015;347(6224):878–882. doi: 10.1126/science.aaa2628. [DOI] [PubMed] [Google Scholar]
- 119.Zhou Y, et al. RILP restricts insulin secretion through mediating lysosomal degradation of proinsulin. Diabetes. 2020;69(1):67–82. doi: 10.2337/db19-0086. [DOI] [PubMed] [Google Scholar]
- 120.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Herbert TP, Laybutt DR. A reevaluation of the role of the unfolded protein response in islet dysfunction: maladaptation or a failure to adapt? Diabetes. 2016;65(6):1472–1480. doi: 10.2337/db15-1633. [DOI] [PubMed] [Google Scholar]
- 122.Ghosh R, Colon-Negron K, Papa FR. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol Metab. 2019;27S:S60–S68. doi: 10.1016/j.molmet.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol. 2019;15(8):764–775. doi: 10.1038/s41589-019-0326-2. [DOI] [PubMed] [Google Scholar]
- 124.Dominguez-Gutierrez G, Xin Y, Gromada J. Heterogeneity of human pancreatic β-cells. Mol Metab. 2019;27S:S7–S14. doi: 10.1016/j.molmet.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Szabat M, et al. Reduced insulin production relieves endoplasmic reticulum stress and induces β cell proliferation. Cell Metab. 2016;23(1):179–193. doi: 10.1016/j.cmet.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 126.Huang H, et al. β-Cell compensation concomitant with adaptive endoplasmic reticulum stress and β-cell neogenesis in a diet-induced type 2 diabetes model. Appl Physiol Nutr Metab. 2019;44(12):1355–1366. doi: 10.1139/apnm-2019-0144. [DOI] [PubMed] [Google Scholar]
- 127.Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in β-cell gene expression and progression to diabetes. Diabetes. 2013;62(5):1557–1568. doi: 10.2337/db12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sharma RB, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831–3846. doi: 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lipson KL, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 130.Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A. 2011;108(21):8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS One. 2008;3(2):e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Han D, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Upton JP, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338(6108):818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 135.Harding HP, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7(6):1153–1163. doi: 10.1016/S1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 136.Scheuner D, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7(6):1165–1176. doi: 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 137.Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem. 2012;287(53):44338–44344. doi: 10.1074/jbc.M112.428987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(suppl 3):S455–S461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- 139.Back SH, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10(1):13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yamani L, Latreille M, Larose L. Interaction of Nck1 and PERK phosphorylated at Y561 negatively modulates PERK activity and PERK regulation of pancreatic β-cell proinsulin content. Mol Biol Cell. 2014;25(5):702–711. doi: 10.1091/mbc.e13-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang R, et al. Insulin secretion and Ca2+ dynamics in β-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem. 2013;288(47):33824–33836. doi: 10.1074/jbc.M113.503664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4(6):491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 143.Feng D, Wei J, Gupta S, McGrath BC, Cavener DR. Acute ablation of PERK results in ER dysfunctions followed by reduced insulin secretion and cell proliferation. BMC Cell Biol. 2009;10:61. doi: 10.1186/1471-2121-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gao Y, et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol. 2012;32(24):5129–5139. doi: 10.1128/MCB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sowers CR, et al. The protein kinase PERK/EIF2AK3 regulates proinsulin processing not via protein synthesis but by controlling endoplasmic reticulum chaperones. J Biol Chem. 2018;293(14):5134–5149. doi: 10.1074/jbc.M117.813790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kefalas G, Larose L. PERK leads a hub dictating pancreatic β cell homoeostasis. Biol Cell. 2018;110(2):27–32. doi: 10.1111/boc.201700059. [DOI] [PubMed] [Google Scholar]
- 147.Stancill JS, Happ JT, Broniowska KA, Hogg N, Corbett JA. Peroxiredoxin 1 plays a primary role in protecting pancreatic β-cells from hydrogen peroxide and peroxynitrite. Am J Physiol Regul Integr Comp Physiol. 2020;318(5):R1004–R1013. doi: 10.1152/ajpregu.00011.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49(3):424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 149.Teske BF, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22(22):4390–4405. doi: 10.1091/mbc.e11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Seo HY, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology. 2008;149(8):3832–3841. doi: 10.1210/en.2008-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Odisho T, Zhang L, Volchuk A. ATF6β regulates the Wfs1 gene and has a cell survival role in the ER stress response in pancreatic β-cells. Exp Cell Res. 2015;330(1):111–122. doi: 10.1016/j.yexcr.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 152.Yoshida H, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20(18):6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yang H, Niemeijer M, van de Water B, Beltman JB. ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience. 2020;23(2):100860. doi: 10.1016/j.isci.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Engin F, Nguyen T, Yermalovich A, Hotamisligil GS. Aberrant islet unfolded protein response in type 2 diabetes. Sci Rep. 2014;4:4054. doi: 10.1038/srep04054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Masini M, et al. Ultrastructural morphometric analysis of insulin secretory granules in human type 2 diabetes. Acta Diabetol. 2012;49(suppl 1):S247–S252. doi: 10.1007/s00592-012-0446-6. [DOI] [PubMed] [Google Scholar]
- 156.Kang T, et al. Characterization of signaling pathways associated with pancreatic β-cell adaptive flexibility in compensation of obesity-linked diabetes in db/db mice. Mol Cell Proteomics. 2020;19(6):971–993. doi: 10.1074/mcp.RA119.001882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhu R, et al. Defective endoplasmic reticulum export causes proinsulin misfolding in pancreatic β cells. Mol Cell Endocrinol. 2019;493:110470. doi: 10.1016/j.mce.2019.110470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Umeda M, Hiramoto M, Watanabe A, Tsunoda N, Imai T. Arginine-induced insulin secretion in endoplasmic reticulum. Biochem Biophys Res Commun. 2015;466(4):717–722. doi: 10.1016/j.bbrc.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 159.Trogden KP, Zhu X, Lee JS, Wright CVE, Gu G, Kaverina I. Regulation of glucose-dependent golgi-derived microtubules by cAMP/EPAC2 promotes secretory vesicle biogenesis in pancreatic β cells. Curr Biol. 2019;29(14):2339–2350.e5. doi: 10.1016/j.cub.2019.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deriy LV, et al. The granular chloride channel ClC-3 is permissive for insulin secretion. Cell Metab. 2009;10(4):316–323. doi: 10.1016/j.cmet.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wijesekara N, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. 2010;53(8):1656–1668. doi: 10.1007/s00125-010-1733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Matsunaga K, Taoka M, Isobe T, Izumi T. Rab2a and Rab27a cooperatively regulate the transition from granule maturation to exocytosis through the dual effector Noc2. J Cell Sci. 2017;130(3):541–550. doi: 10.1242/jcs.195479. [DOI] [PubMed] [Google Scholar]
- 163.Yau B, et al. A fluorescent timer reporter enables sorting of insulin secretory granules by age. J Biol Chem. 2020;295(27):8901–8911. doi: 10.1074/jbc.RA120.012432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Orci L, Ravazzola M, Amherdt M, Madsen O, Vassalli JD, Perrelet A. Direct identification of prohormone conversion site in insulin-secreting cells. Cell. 1985;42(2):671–681. doi: 10.1016/0092-8674(85)90124-2. [DOI] [PubMed] [Google Scholar]
- 165.Huang XF, Arvan P. Formation of the insulin-containing secretory granule core occurs within immature beta-granules. J Biol Chem. 1994;269(33):20838–20844. [PubMed] [Google Scholar]
- 166.Huang XF, Arvan P. Intracellular transport of proinsulin in pancreatic beta-cells. Structural maturation probed by disulfide accessibility. J Biol Chem. 1995;270(35):20417–20423. doi: 10.1074/jbc.270.35.20417. [DOI] [PubMed] [Google Scholar]
- 167.Gehart H, et al. The BAR domain protein Arfaptin-1 controls secretory granule biogenesis at the trans-Golgi network. Dev Cell. 2012;23(4):756–768. doi: 10.1016/j.devcel.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 168.Holst B, et al. PICK1 deficiency impairs secretory vesicle biogenesis and leads to growth retardation and decreased glucose tolerance. PLoS Biol. 2013;11(4):e1001542. doi: 10.1371/journal.pbio.1001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li H, et al. BIG3 inhibits insulin granule biogenesis and insulin secretion. EMBO Rep. 2014;15(6):714–722. doi: 10.1002/embr.201338181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carrat GR, et al. The type 2 diabetes gene product STARD10 is a phosphoinositide-binding protein that controls insulin secretory granule biogenesis. Mol Metab. 2020;40:101015. doi: 10.1016/j.molmet.2020.101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Müller A, Mziaut H, Neukam M, Knoch KP, Solimena M. A 4D view on insulin secretory granule turnover in the β-cell. Diabetes Obes Metab. 2017;19 Suppl 1:107–114. doi: 10.1111/dom.13015. [DOI] [PubMed] [Google Scholar]
- 172.Kebede MA, et al. SORCS1 is necessary for normal insulin secretory granule biogenesis in metabolically stressed β cells. J Clin Invest. 2014;124(10):4240–4256. doi: 10.1172/JCI74072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hussain SS, et al. Control of insulin granule formation and function by the ABC transporters ABCG1 and ABCA1 and by oxysterol binding protein OSBP. Mol Biol Cell. 2018;29(10):1238–1257. doi: 10.1091/mbc.E17-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Villamayor L, et al. GATA6 controls insulin biosynthesis and secretion in adult β-cells. Diabetes. 2018;67(3):448–460. doi: 10.2337/db17-0364. [DOI] [PubMed] [Google Scholar]
- 175.Hummer BH, et al. HID-1 controls formation of large dense core vesicles by influencing cargo sorting and trans-Golgi network acidification. Mol Biol Cell. 2017;28(26):3870–3880. doi: 10.1091/mbc.e17-08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hutton JC. Insulin secretory granule biogenesis and the proinsulin-processing endopeptidases. Diabetologia. 1994;37(suppl 2):S48–S56. doi: 10.1007/BF00400826. [DOI] [PubMed] [Google Scholar]
- 177.Ramzy A, Asadi A, Kieffer TJ. Revisiting proinsulin processing: evidence that human β-cells process proinsulin with prohormone convertase (PC) 1/3 but not PC2. Diabetes. 2020;69(7):1451–1462. doi: 10.2337/db19-0276. [DOI] [PubMed] [Google Scholar]
- 178.Jo S, Lockridge A, Alejandro EU. eIF4G1 and carboxypeptidase E axis dysregulation in O-GlcNAc transferase-deficient pancreatic β-cells contributes to hyperproinsulinemia in mice. J Biol Chem. 2019;294(35):13040–13050. doi: 10.1074/jbc.RA119.008670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46(11):1725–1732. doi: 10.2337/diab.46.11.1725. [DOI] [PubMed] [Google Scholar]
- 180.Nakamura A, et al. Proinsulin is sensitive to reflect glucose intolerance. J Diabetes Investig. 2020;11(1):75–79. doi: 10.1111/jdi.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rodriguez-Calvo T, et al. Increase in pancreatic proinsulin and preservation of β-cell mass in autoantibody-positive donors prior to type 1 diabetes onset. Diabetes. 2017;66(5):1334–1345. doi: 10.2337/db16-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wasserfall C, et al. Persistence of pancreatic insulin mRNA expression and proinsulin protein in type 1 diabetes pancreata. Cell Metab. 2017;26(3):568–575.e3. doi: 10.1016/j.cmet.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sims EK, et al. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl Res. 2019;213:90–99. doi: 10.1016/j.trsl.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chanat E, Huttner WB. Milieu-induced, selective aggregation of regulated secretory proteins in the trans-Golgi network. J Cell Biol. 1991;115(6):1505–1519. doi: 10.1083/jcb.115.6.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wollam J, et al. Chromogranin A regulates vesicle storage and mitochondrial dynamics to influence insulin secretion. Cell Tissue Res. 2017;368(3):487–501. doi: 10.1007/s00441-017-2580-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bearrows SC, Bauchle CJ, Becker M, Haldeman JM, Swaminathan S, Stephens SB. Chromogranin B regulates early-stage insulin granule trafficking from the Golgi in pancreatic islet β-cells. J Cell Sci. 2019;132(13):jcs231373. doi: 10.1242/jcs.231373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Toledo PL, et al. ICA512 RESP18 homology domain is a protein-condensing factor and insulin fibrillation inhibitor. J Biol Chem. 2019;294(21):8564–8576. doi: 10.1074/jbc.RA119.007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stephens SB, et al. The prohormone VGF regulates β cell function via insulin secretory granule biogenesis. Cell Rep. 2017;20(10):2480–2489. doi: 10.1016/j.celrep.2017.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Herlo R, et al. An amphipathic helix directs cellular membrane curvature sensing and function of the BAR domain protein PICK1. Cell Rep. 2018;23(7):2056–2069. doi: 10.1016/j.celrep.2018.04.074. [DOI] [PubMed] [Google Scholar]
- 190.Topalidou I, Cattin-Ortolá J, Hummer B, Asensio CS, Ailion M. EIPR1 controls dense-core vesicle cargo retention and EARP complex localization in insulin-secreting cells. Mol Biol Cell. 2020;31(1):59–79. doi: 10.1091/mbc.E18-07-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Moin ASM, Butler AE. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr Diab Rep. 2019;19(9):83. doi: 10.1007/s11892-019-1194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rhodes CJ. Type 2 diabetes—a matter of beta-cell life and death? Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 194.McInnes N, et al. Piloting a remission strategy in type 2 diabetes: results of a randomized controlled trial. J Clin Endocrinol Metab. 2017;102(5):1596–1605. doi: 10.1210/jc.2016-3373. [DOI] [PubMed] [Google Scholar]
- 195.Weng J, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet. 2008;371(9626):1753–1760. doi: 10.1016/S0140-6736(08)60762-X. [DOI] [PubMed] [Google Scholar]
- 196.[No authors listed] U.K. Prospective Diabetes Study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes. 1995;44(11):1249–1258. [PubMed] [Google Scholar]
- 197.Taylor R, Al-Mrabeh A, Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019;7(9):726–736. doi: 10.1016/S2213-8587(19)30076-2. [DOI] [PubMed] [Google Scholar]
- 198.Alarcon C, et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes. 2016;65(2):438–450. doi: 10.2337/db15-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Arunagiri A, et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife. 2019;8:e44532. doi: 10.7554/eLife.44532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Baranczak A, Kelly JW. A current pharmacologic agent versus the promise of next generation therapeutics to ameliorate protein misfolding and/or aggregation diseases. Curr Opin Chem Biol. 2016;32:10–21. doi: 10.1016/j.cbpa.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Törn C, et al. Role of type 1 diabetes-associated SNPs on risk of autoantibody positivity in the TEDDY study. Diabetes. 2015;64(5):1818–1829. doi: 10.2337/db14-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 203.Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59(9):2326–2331. doi: 10.2337/db10-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Malecki MT, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–328. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 205.Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002;99(10):6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Iacovazzo D, et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci U S A. 2018;115(5):1027–1032. doi: 10.1073/pnas.1712262115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Noso S, et al. Insulin transactivator MafA regulates intrathymic expression of insulin and affects susceptibility to type 1 diabetes. Diabetes. 2010;59(10):2579–2587. doi: 10.2337/db10-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11(13):1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- 209.Yasuda T, et al. PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes. 2002;51(1):224–230. doi: 10.2337/diabetes.51.1.224. [DOI] [PubMed] [Google Scholar]
- 210.ZeRuth GT, Takeda Y, Jetten AM. The Krüppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol Endocrinol. 2013;27(10):1692–1705. doi: 10.1210/me.2013-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dimitri P, et al. Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab. 2015;100(10):E1362–E1369. doi: 10.1210/jc.2015-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.De Franco E, et al. De novo mutations in EIF2B1 affecting eIF2 signaling cause neonatal/early-onset diabetes and transient hepatic dysfunction. Diabetes. 2020;69(3):477–483. doi: 10.2337/db19-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gregory LC, et al. Impaired EIF2S3 function associated with a novel phenotype of X-linked hypopituitarism with glucose dysregulation. EBioMedicine. 2019;42:470–480. doi: 10.1016/j.ebiom.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kasuga Y, et al. Association of common polymorphisms with gestational diabetes mellitus in Japanese women: a case-control study. Endocr J. 2017;64(4):463–475. doi: 10.1507/endocrj.EJ16-0431. [DOI] [PubMed] [Google Scholar]
- 215.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46(3):234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Donoviel DB, Bernstein A. SEL-1L maps to human chromosome 14, near the insulin-dependent diabetes mellitus locus 11. Genomics. 1999;56(2):232–233. doi: 10.1006/geno.1998.5534. [DOI] [PubMed] [Google Scholar]
- 217.Purcell AW, Sechi S, DiLorenzo TP. The evolving landscape of autoantigen discovery and characterization in type 1 diabetes. Diabetes. 2019;68(5):879–886. doi: 10.2337/dbi18-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25(4):406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 219.Acevedo-Calado MJ, et al. Autoantibodies directed toward a novel IA-2 variant protein enhance prediction of type 1 diabetes. Diabetes. 2019;68(9):1819–1829. doi: 10.2337/db18-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ndiaye FK, et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol Metab. 2017;6(6):459–470. doi: 10.1016/j.molmet.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58(2):221–232. doi: 10.1007/s00125-014-3451-1. [DOI] [PubMed] [Google Scholar]
- 222.Rutter GA, Chimienti F. SLC30A8 mutations in type 2 diabetes. Diabetologia. 2015;58(1):31–36. doi: 10.1007/s00125-014-3405-7. [DOI] [PubMed] [Google Scholar]
- 223.Shimomura K, Maejima Y. KATP channel mutations and neonatal diabetes. Intern Med. 2017;56(18):2387–2393. doi: 10.2169/internalmedicine.8454-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yamagata K, et al. Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 β-cell line. Biochem Biophys Res Commun. 2011;407(3):620–625. doi: 10.1016/j.bbrc.2011.03.083. [DOI] [PubMed] [Google Scholar]
- 225.Cho YS, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2011;44(1):67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yang JK, et al. From hyper- to hypoinsulinemia and diabetes: effect of KCNH6 on insulin secretion. Cell Rep. 2018;25(13):3800–3810.e6. doi: 10.1016/j.celrep.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 227.Marcelo KL, et al. Research Resource: Roles for calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in systems metabolism. Mol Endocrinol. 2016;30(5):557–572. doi: 10.1210/me.2016-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Peiris H, et al. Discovering human diabetes-risk gene function with genetics and physiological assays. Nat Commun. 2018;9(1):3855. doi: 10.1038/s41467-018-06249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Thomsen SK, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic β-cells. Nat Genet. 2018;50(8):1122–1131. doi: 10.1038/s41588-018-0173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Vlacich G, Nawijn MC, Webb GC, Steiner DF. Pim3 negatively regulates glucose-stimulated insulin secretion. Islets. 2010;2(5):308–317. doi: 10.4161/isl.2.5.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ishihara H, et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet. 2004;13(11):1159–1170. doi: 10.1093/hmg/ddh125. [DOI] [PubMed] [Google Scholar]
- 232.Rosengren AH, et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science. 2010;327(5962):217–220. doi: 10.1126/science.1176827. [DOI] [PubMed] [Google Scholar]
- 233.van de Bunt M, et al. Transcript expression data from human islets links regulatory signals from genome-wide association studies for type 2 diabetes and glycemic traits to their downstream effectors. PLoS Genet. 2015;11(12):e1005694. doi: 10.1371/journal.pgen.1005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Thomsen SK, et al. Systematic functional characterization of candidate causal genes for type 2 diabetes risk variants. Diabetes. 2016;65(12):3805–3811. doi: 10.2337/db16-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Li LC, et al. IG20/MADD plays a critical role in glucose-induced insulin secretion. Diabetes. 2014;63(5):1612–1623. doi: 10.2337/db13-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Carrat GR, et al. Decreased STARD10 expression is associated with defective insulin secretion in humans and mice. Am J Hum Genet. 2017;100(2):238–256. doi: 10.1016/j.ajhg.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Deisl C, et al. Sodium/hydrogen exchanger NHA2 is critical for insulin secretion in β-cells. Proc Natl Acad Sci U S A. 2013;110(24):10004–10009. doi: 10.1073/pnas.1220009110. [DOI] [PMC free article] [PubMed] [Google Scholar]