

Abstract
Resource limitation underlies competition in the living world, even between intracellular populations of mitochondria. A new study shows that reducing the availability of an essential cellular resource, namely the enzyme that replicates mitochondrial DNA (mtDNA), can alter the selective advantage of one mtDNA type over another.
In his publication On the Origin of Species, Charles Darwin remarked that competition over limited resources underlies natural selection. Through a division of labor, collaborating for mutual benefit allows for efficient utilization of resources. Hence, competition for survival and reproduction can select for mutualistic symbiotic relationships. Among the most successful symbiotic relationships in the history of life was the endosymbiosis that led to the formation and evolution of eukaryotic organisms. The success of this relationship is predicated on cooperation for mutual benefit: mitochondria supply energy to the host, and in return the host supplies the protein machinery and building blocks for replicating mitochondria.
Owing to their heritage as previously autonomous organisms, mitochondria retain their own genomes, which themselves can undergo natural selection, particularly when variation in mitochondrial DNA (mtDNA) sequences accounts for differences in mitochondrial function and replication. However, mtDNA mutations need not enhance cellular respiration in order to give a selective advantage to the mutated mtDNA. On the contrary, deleterious mtDNA mutations can proliferate within a host despite compromising host fitness [1], hence their categorization as ‘selfish mtDNA’. Accordingly, the field of mitochondrial genetics garners widespread interest among evolutionary biologists and biomedical scientists alike, as disease-causing mtDNA mutations have been estimated to affect approximately 1 in 5,000 individuals [2].
Because their reproductive fitnesses are intertwined, the nuclear and mitochondrial genomes have evolved in tandem. For example, nuclear genes serve an adaptive benefit for the host if they limit the spread of deleterious mtDNA mutations. Their coevolution has thus selected for nuclear-encoded quality control mechanisms that function within the female germline, where mtDNA is transmitted to the next generation [3,4]. So why does the nucleus not always protect the organism from selfish mtDNA? A crucial detail of natural selection that Darwin sought to communicate is the conditional nature of fitness. Indeed, mounting evidence suggests that the competitive advantage of one mtDNA variant over another is highly context dependent. One recent study, for example, showed that the balance between mitochondrial fusion and fission modulates the proliferation of mutant mtDNA [5]. Specifically, mitochondrial fission enables the host to fragment its mitochondria, selectively promoting the sequestration and degradation of selfish mtDNA, thereby suppressing its proliferation. Mitochondrial fusion, on the other hand, promotes the congregation of mtDNA variants into more contiguous organelle networks, thereby shielding the mutant genomes from degradation.
Proteins encoded by the nucleus supply vital resources for mtDNA replication. Their abundance therefore represents another condition governing the competition between selfish and cooperative mtDNA molecules. A paper in this issue of Current Biology features the example of POLG — the enzyme that catalyzes the replication of mtDNA. Using fruit flies, Chiang et al. [6] sought to identify nuclear-encoded genes responsible for determining the selective advantage between selfish versus metabolically competent mtDNA copies. They take advantage of a fly stock that contains a mixture of metabolically competent and selfish mtDNAs. The selfish genome has a transmission advantage, allowing it to persist at approximately 95% frequency, whereas the metabolically competent mtDNA is maintained at approximately 5% frequency. They screened a collection of chromosomal deletions within the nucleus, looking for those that alter the balance of the mtDNA types. In particular, one region of the nuclear genome, located on chromosome 2, was observed to enable the high levels of the selfish mtDNA. Deleting one of the two copies of this nuclear region allowed the competent mtDNA to quickly outcompete the selfish mtDNA, rising from 5% to 100% in five generations.
Several genes located within this nuclear region encode mitochondrial proteins, including those involved in mtDNA replication and protein synthesis, thus providing a list of candidate genes for the research group to further interrogate for their potential roles in modulating mtDNA competition. Using more precise mutations that disrupt these genes one at a time, Chiang et al. [6] found that tam, the gene encoding the catalytic subunit of POLG, is responsible for modulating the competition between the two mitochondrial genomes. By deleting and then replacing one copy of tam, they were able to shift the selection advantage from favoring the selfish to the competent mtDNA, and back again. Furthermore, these trends were likewise observed among different pairings of selfish with functionally competent mtDNA, and among different mutations in tam. These findings suggest that the enzyme activity of POLG is not only important for mtDNA replication in general, but that an abundance of POLG permits the selective amplification of selfish mtDNA (Figure 1).
Figure 1. Deleterious mutant mtDNA levels depend on POLG levels.
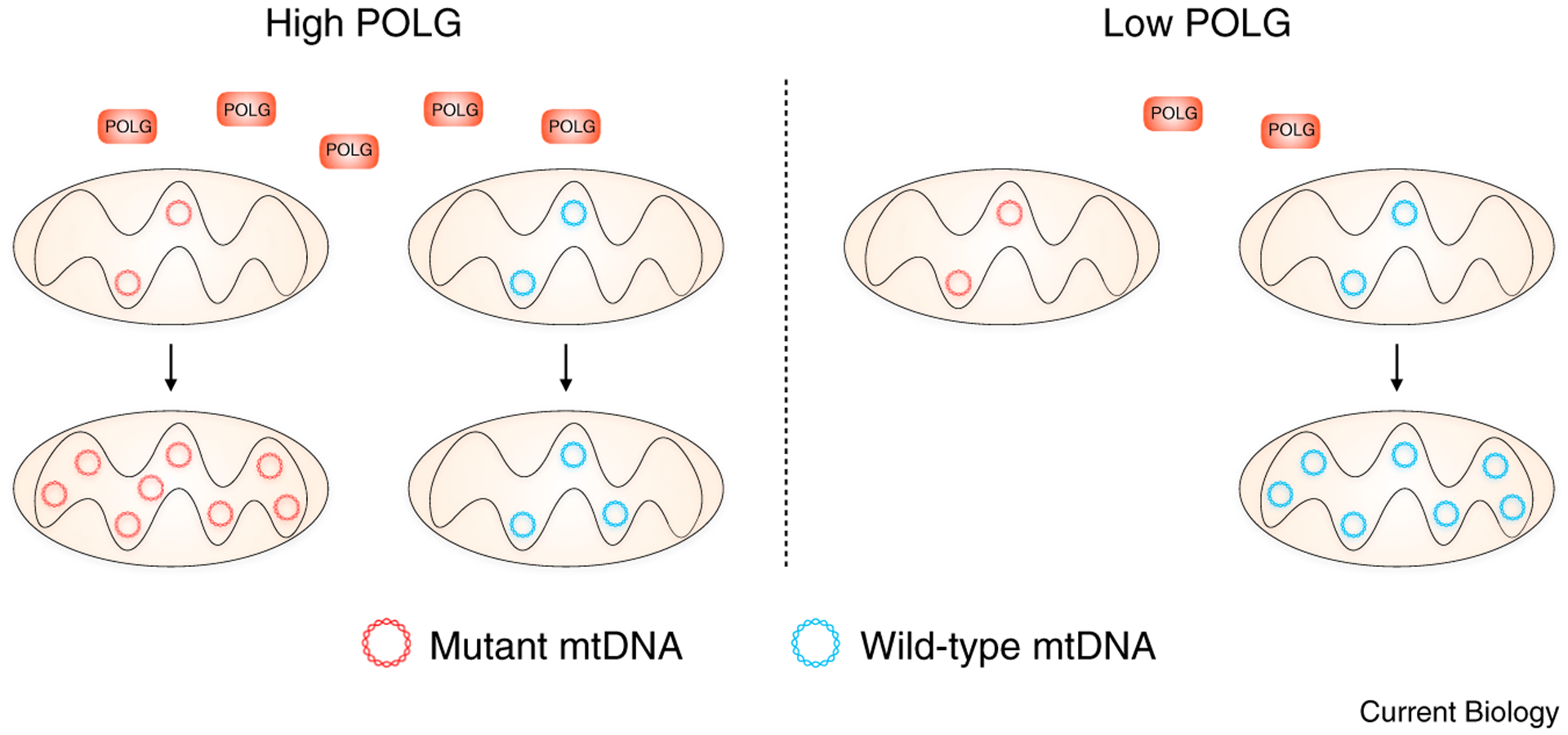
Under normal conditions, a selfish genome can experience a selective advantage, comprising the majority of mtDNA copies within a cell. However, when the levels of POLG are reduced, the selective advantage is flipped, from favoring selfish mtDNA propagation to favoring the amplification of the wild-type, or functionally competent, genome.
How might POLG affect the selection between competing mtDNA? One possible answer involves the regulation of protein synthesis on the outer mitochondrial membrane, shown to be important for mtDNA replication [7]. Earlier this year, the same group found that the protein PINK1 accumulates on the outer membrane of dysfunctional mitochondria, inhibiting protein synthesis and corresponding to lower POLG levels and lower deleterious mtDNA frequency [8]. However, it remained unresolved whether POLG levels directly influenced the competition between mtDNA variants, or whether these observations were merely correlated. The new work now sheds light on this question by demonstrating that reduced availability of POLG itself is sufficient to inhibit selfish mtDNA proliferation (Figure 1).
These findings showcase the exciting discovery that the availability of a vital resource needed for mtDNA replication not only determines overall mtDNA copy number within a cell, but also the relative fitness of different mtDNA variants. Moreover, given that the expression of the mtDNA polymerase gene was previously shown to be important for the proliferation of a selfish mtDNA in nematodes [9], these findings raise the interesting possibility that the underlying mechanism is widely conserved between distantly related species. As the field of mitochondrial genetics moves forward, it will likely prove illuminating to more fully explore the ways in which the availability of vital resources can be modified to alter the competitiveness of selfish genomes. These studies promise to provide insights into both evolution and mitochondrial diseases.
REFERENCES
- 1.Ma H, and O’Farrell PH (2016). Selfish drive can trump function when animal mitochondrial genomes compete. Nat. Genet 48, 798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, et al. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol 77, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, and Wallace DC (2008). A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, and Larsson NG (2008). Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieber T, Jeedigunta SP, Palozzi JM, Lehmann R, and Hurd TR (2019). Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature 570, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang AC-Y, McCartney E, O’Farrell PH, and Ma H (2019). A genome-wide screen reveals that reducing mitochondrial DNA polymerase can promote elimination of deleterious mitochondrial mutations. Curr. Biol 29, 4330–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Chen Y, Gucek M, and Xu H (2016). The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J. 35, 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Wang ZH, Liu Y, Chen Y, Sun N, Gucek M, Zhang F, and Xu H (2019). PINK1 inhibits local protein synthesis to limit transmission of deleterious mitochondrial DNA mutations. Mol. Cell 73, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, and Haynes CM (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]