

Abstract
The phenotypic switch in tumor-associated macrophages (TAMs) mediates immunity escape of cancer. However, the underlying mechanisms in the TAM phenotypic switch have not been systematically elucidated. In this study, long noncoding RNA (lncRNA)-Xist, CCAAT/enhancer-binding protein (C/EBP)α, and Kruppel-like factor 6 (KLF6) were upregulated, whereas microRNA (miR)-101 was downregulated in M1 macrophages-type (M1). Knockdown of Xist or overexpression of miR-101 in M1 could induce M1-to-M2 macrophage-type (M2) conversion to promote cell proliferation and migration of breast and ovarian cancer by inhibiting C/EBPα and KLF6 expression. Furthermore, miR-101 could combine with both Xist and C/EBPα and KLF6 through the same microRNA response element (MRE) predicted by bioinformatics and verified by luciferase reporter assays. Moreover, we found that miR-101 knockdown restored the decreased M1 marker and the increased M2 marker expression and also reversed the promotion of proliferation and migration of human breast cancer cells (MCF-7) and human ovarian cancer (OV) cells caused by silencing Xist. Generally, the present study indicates that Xist could mediate macrophage polarization to affect cell proliferation and migration of breast and ovarian cancer by competing with miR-101 to regulate C/EBPα and KLF6 expression. The promotion of Xist expression in M1 macrophages and inhibition of miR-101 expression in M2 macrophages might play an important role in inhibiting breast and ovarian tumor proliferation and migration abilities.
Keywords: tumor-associated macrophages, polarization, lncRNA Xist, ceRNA, miR-101
Graphical Abstract
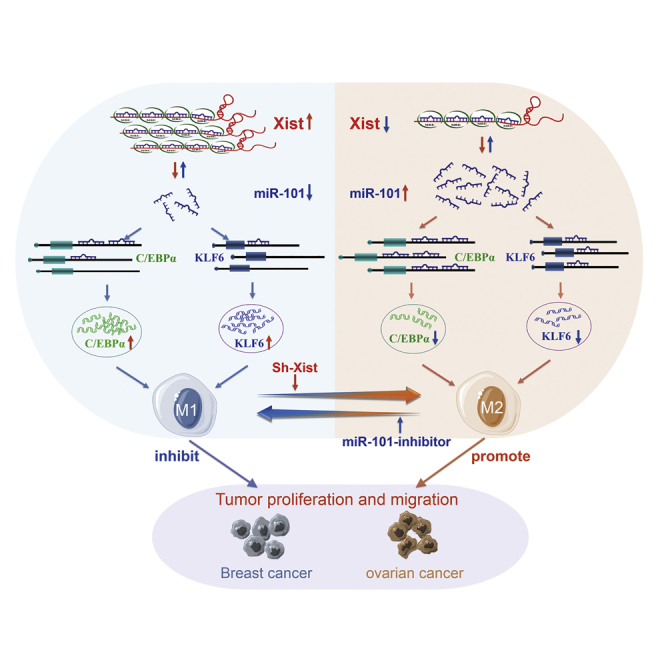
The phenotypic switch in tumor-associated macrophages (TAMs) mediates immunity escape of cancer. The present study indicates that Xist mediates macrophage polarization to affect cell proliferation and migration of breast and ovarian cancer by competing with miR-101 to regulate C/EBPα and KLF6 expression.
Introduction
Tumor-associated macrophages (TAMs) in the tumor microenvironment (TME)1 are closely associated with tumor proliferation, metastasis, invasion, and angiogenesis2, 3, 4 and have gradually become valuable therapeutic targets for cancers.5,6 TAMs are divided into M1 and M2 phenotypes based on their activation status and function. The former exert proinflammatory and anti-tumor properties, induced by lipopolysaccharide (LPS) and interferon-γ (IFN-γ), and highly express interleukin (IL)-1β, CD86, tumor necrosis factor-α (TNF-α), and so on.7, 8, 9 The latter, alternatively activated macrophages, play anti-inflammatory and protumorigenic roles, induced by IL-10, IL-4, and IL-13, and highly express CD206, CD163, and so on.10 In the early stage, a majority of macrophages are manifested as an M1 phenotype, which plays a role in inhibiting tumor growth. With the continuous progress of tumor cells, macrophages gradually tend to be M2 phenotype, promoting tumorigenesis and progression.11 Increasing evidence demonstrated that inducing macrophages from M1 to M2 phenotypes might boost cancer initiation and progression by promoting cell proliferation, metastasis, drug reliance, and immune evasion.12,13 Therefore, the discovery of mechanisms and targets that mediate TAM phenotypic transformation will provide a new approach for malignant tumor treatment.
Long noncoding RNA (lncRNA), as a type of endogenous nonprotein-coding RNA longer than 200 nt, could regulate occurrence and progression of many aspects of tumors at epigenetic modification, transcription, or post-transcription.14 Xist is an essential lncRNA for maintaining the stable inactivation of the X chromosome in female mammals.15 It is increasingly recognized that Xist plays an important role in the regulation of tumor progression.16 Many studies have shown that Xist was a pathogenic gene in colorectal, gastric, and non-small cell lung cancers.17, 18, 19 However, it was considered as a tumor suppressor in other cancers, including liver, breast, ovarian, and cervical cancers.20, 21, 22, 23 Recently, Li et al.24 found that Xist could promote proinflammatory M1 macrophages on osteoarthritis chondrocyte apoptosis by competing with Osteopontin (OPN) for microRNA (miR)-376-5p, suggesting that Xist could influence progression of the disease by modulating macrophage polarization. So far, it has not been reported whether Xist can affect tumorigenesis and progression through mediating macrophage phenotypic transformation.
In this study, we found that Xist was highly expressed in M1 macrophages, and knockdown of Xist could increase M1-to-M2 conversion, whereas miR-101 was highly expressed in M2 macrophages. Furthermore, CCAAT/enhancer-binding protein (C/EBPα) and Kruppel-like factor 6 (KLF6), critical factors regulating TAM phenotypic transformation, have the same microRNA (miRNA) response element (MRE) sequence binding to miR-101 as Xist. Therefore, the purpose of this study is to investigate whether Xist could regulate macrophage polarization and tumor progression and illustrate its function as a competing endogenous RNA (ceRNA) to regulate the expression of C/EBPα and KLF6 by competing for miR-101 binding.
Results
lncRNA Xist is highly expressed in M1 macrophages and participates in the maintenance of the macrophage phenotype
First, human monocyte cells (THP-1 cells) were differentiated to macrophages by treating with phorbol 12-myristate-13-acetate (PMA) for 24 h.25, 26, 27 Then, macrophages were activated to the polarized M1 phenotype by treating with LPS and IFN-γ and to the M2 phenotype by treating with IL-4 for 24 h. To confirm whether we successfully induced M1 and M2 phenotype macrophages, we tested the mRNA expressions of M1 macrophage markers IL-1β and TNF-α and M2 macrophage markers C-C motif chemokine 17 (CCL17), C-C motif chemokine 22 (CCL22), and CD163. The quantitative real-time PCR results showed the IL-1β and TNF-α expression was higher in M1 than in M2 macrophages, whereas CCL17, CCL22, and CD163 expression was higher in M2 than in M1 macrophages (Figure 1A). We also detected the levels of M1 surface marker CD86 and M2 surface marker CD206 by flow cytometry. We found the increased CD86 levels in M1 compared with M2 macrophages and the elevated CD206 levels in M2 compared with M1 macrophages (Figure 1B). These results suggested that we successfully induced M1 and M2 macrophages.
Figure 1.

lncRNA Xist is highly expressed in M1 macrophages
(A) Quantitative real-time PCR results showing the relative mRNA expression of IL-1β, TNF-α, CCL17, CCL22, CD163, and Xist in M1 and M2 macrophages. (B) The surface expression of CD86 and CD206 in M1 and M2 macrophages was measured by flow cytometry. (C) The relative mRNA expression of IL-1β, TNF-α, CCL17, CCL22, CD163, and Xist in macrophage and M1, M1-M2, M2, and M2-M1 macrophages by analyzed by quantitative real-time PCR. (D–F) The expression of (D) Xist, (E) C/EBPα, and (F) KLF6 in M1 and M2 macrophages was analyzed from GEO: GSE46903 data. (G) The relative mRNA expression of C/EBPα and KLF6 in M1 and M2 macrophages was analyzed by quantitative real-time PCR. (H) The protein levels of C/EBPα, KLF6, PU.1, and peroxisome proliferator-activated receptor (PPARγ) in M1 and M2 macrophages were tested by western blot. β-actin was used as an internal control. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3; mean ± SD.
Furthermore, we found that Xist was upregulated in M1 than M2 macrophages (Figure 1A). When IL-4 was added to M1 macrophages, the mRNA expression of M2 phenotypic markers CCL17, CCL22, and CD163 increased, whereas the mRNA expression of M1 phenotypic markers IL-1β and TNF-α decreased, suggesting that IL-4 could promote M1 to M2 macrophages. Simultaneously, we found the Xist expression also decreased. Conversely, when LPS and IFN-γ were added to M2 macrophages, the mRNA expression of M1 phenotypic markers IL-1β and TNF-α increased, and the mRNA expression of M2 phenotypic markers CCL17, CCL22, and CD163 decreased, showing that LPS and IFN-γ could promote M2 to M1 macrophages. Meanwhile, the Xist expression also increased (Figure 1C). Additionally, we analyzed the Xist expression in macrophages from chip GEO: GSE46903 data and found that Xist expression was higher in M1 than in M2 macrophages (Figure 1D). Taken together, these results suggested that Xist was highly expressed in M1 macrophages and participated in the maintenance of the macrophage phenotype.
Previous studies have shown that KLF6 is a novel important factor to regulate macrophage polarization, which could inhibit the formation of M2 macrophages by decreasing PPARγ expression.28 C/EBPα is an important transcription factor that regulates the polarization of M1 macrophages. C/EBPα and its target gene PU.1 take part in activating M1 macrophages induced by Toll-like receptor (TLR) ligands.29 To verify the function of C/EBPα and KLF6 on macrophage polarization, we first analyzed expression of C/EBPα and KLF6 in macrophages from GEO: GSE46903 data, and results showed that expression of C/EBPα and KLF6 was higher in M1 than M2 macrophages (Figures 1E and 1F). Next, we detected the expression levels of C/EBPα and KLF6 in macrophages. The quantitative real-time PCR results showed that the mRNA expression of C/EBPα and KLF6 was higher in M1 macrophages (Figure 1G). The western results revealed that the protein levels of C/EBPα, KLF6, and PU.1 were increased in M1 cells, whereas the PPARγ protein level was decreased in M1 compared with M2 macrophages (Figure 1H). Thus, the data showed the successfully induced M1 macrophages more highly expressed C/EBPα and KLF6 than M2 macrophages.
Knockdown of Xist promotes macrophage polarization to the M2 phenotype and inhibits the expression of C/EBPα and KLF6
To further assess the role of Xist on macrophage polarization, we transfected the silenced Xist plasmid (short hairpin [sh]-Xist-1, sh-Xist-2) into M1 macrophages for 24 h. The quantitative real-time PCR results showed that the expression of Xist and M1 markers IL-1β and TNF-α decreased, whereas M2 markers CCL17, CCL22, and CD163 increased in the M1 macrophages transfected with sh-Xist-1 and sh-Xist-2 plasmids, compared with the cells transfected with sh-negative control (NC) (Figure 2A). In addition, the flow cytometry results showed that the knockdown of Xist resulted in the decreased M1 surface marker CD86 and increased M2 surface marker CD206 in M1 macrophages (Figure 2B). These findings suggested that knockdown of Xist in M1 macrophages could promote macrophages to M2 phenotype.
Figure 2.

Knockdown of Xist reduces M1 macrophages by decreasing the expression of C/EBPα, KLF6, and their target genes
M1 macrophage was transfected with negative control (sh-NC) or sh-Xist-1 or sh-Xist-2 plasmids for 24 h. (A) The relative expression of Xist, IL-1β, TNF-α, CCL17, CCL22, and CD163 measured by quantitative real-time PCR analysis. (B) Flow cytometry results showing the surface expression of CD86 and CD206. (C) The correlation between Xist and C/EBPα and KLF6 expression in macrophage from GEO: GSE46903 data by linear regression analysis. (D) The relative mRNA expression of C/EBPα and KLF6 by quantitative real-time PCR analysis. (E) Western blot results showing the protein expression of C/EBPα, KLF6, PU.1, and PPARγ. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus sh-NC; n = 3; mean ± SD.
To further investigate whether the regulation of Xist on macrophage polarization is associated with C/EBPα, KLF6, and their target genes, we first analyzed the correlation of Xist with C/EBPα and KLF6 expression in macrophages from GEO: GSE46903 data. We found that Xist expression was significantly positively correlated with C/EBPα and KLF6 expression (Figure 2C). Then, we detected the mRNA and protein expression of C/EBPα, KLF6, PU.1, and PPARγ after inhibiting Xist expression in M1 macrophages. As shown in Figure 2D, the mRNA levels of C/EBPα and KLF6 were obviously reduced in the M1 macrophages transfected with sh-Xist-1 and sh-Xist-2 plasmids by quantitative real-time PCR analysis. Consistently, the western results showed that silencing Xist markedly decreased the protein level of C/EBPα, KLF6, and PU.1, whereas increased the protein expression of PPARγ (Figure 2E). Thus, the data elucidated that the suppression of Xist in M1 macrophages could regulate macrophages phenotypic transformation by inhibiting the expression of C/EBPα, KLF6, and their target genes.
Knockdown of Xist promotes cell proliferation and migration of breast and ovarian cancers
Several studies have shown that Xist has low expression and plays an anti-cancer role in gynecological diseases, such as breast cancer, ovarian cancer, and cervical cancer.21, 22, 23 We analyzed the differential expression of Xist among breast, ovarian cancer tissues, and their adjacent tissues in The Cancer Genome Atlas (TCGA) and GEO: GSE26712 datasets. The expression of Xist is higher in breast and ovarian cancer tissues, which is consistent with previous reports (Figures 3A and 3B). Furthermore, many studies confirmed M2 macrophages could promote tumor cell proliferation and migration.30, 31, 32 Therefore, to explore whether Xist could promote tumor proliferation and migration by regulating macrophage polarization, we cocultured human breast cancer cells (MCF-7) and human ovarian (OV) cancer cells with M1 conditional medium after transfection with sh-NC or sh-Xist-1 or sh-Xist-2 plasmids. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays showed that the cell viabilities of MCF-7 and OV cells in sh-Xist-1 and sh-Xist-2 groups were increased compared to the sh-NC group (Figure 3C). Further, we observed that the migration abilities of MCF-7 and OV cells in sh-Xist-1 and sh-Xist-2 groups were strengthened compared to the sh-NC group by the wound-healing and Transwell analysis (Figures 3D and 3E). Taken together, the suppression of Xist expression in M1 macrophages could improve the proliferation and migration abilities of MCF-7 and OV cells.
Figure 3.

Knockdown of Xist promotes tumor proliferation and migration
(A) The comparison of Xist expression between breast cancer tissues and adjacent tissues in TCGA dataset. (B) The comparison of Xist expression between ovarian cancer tissues and adjacent tissues in the GEO: GSE26712 dataset. MCF-7 and OV cells were cocultured with M1 conditional medium after transfected with sh-NC or sh-Xist-1 or sh-Xist-2 plasmids. (C) The cell viabilities of MCF-7 and OV cells were analyzed by MTT assays. (D) Wound-healing results showing the migration ability of MCF-7 and OV cells. (E) Transwell results showing the migration ability of MCF-7 and OV cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus sh-NC; n = 3; mean ± SD.
miR-101 induces transformation of M1 to M2 to promote tumor proliferation and migration by directly inhibiting C/EBPα and KLF6
miR-101 is predicted to bind to Xist, C/EBPα, and KLF6 with the same MRE through starBase 2.0, TargetScan, and miRDB databases (Figure 4A). To investigate whether Xist regulates macrophage polarization, and expression of C/EBPα and KLF6 is associated with miR-101, we first detected the targeted regulation of miR-101 on C/EBPα and KLF6. We, respectively, cloned the 3′ UTR of C/EBPα and 3′ UTR of KLF6 with wild-type (WT) and mutation (MT) binding sites into luciferase reporter. Then, we cotransfected WT/MT-C/EBPα or WT/MT-KLF6 luciferase reporter plasmids with miR-101-mimic or NC-mimic into 293T cells. We found that compared with NC groups, miR-101 downregulated luciferase activity of the reporter that contained 3′ UTR of C/EBPα or KLF6. Conversely, luciferase activity has little change in cells transfected with miR-101 and MT-C/EBPα or MT-KLF6 plasmids (Figure 4B). These results suggested that miR-101 could directly target C/EBPα and KLF6 3′ UTR regions.
Figure 4.

miR-101 inhibits the expression of C/EBPα and KLF6
(A) The binding sites of miR-101-3p to KLF6 and C/EBPα. (B) Dual luciferase reporter assays were measured in the 293T cells cotransfected with the miR-101-mimic and luciferase reporter plasmid that was inserted with the wild-type (WT)/mutation (MT)-C/EBPα 3′ UTR or WT/MT-KLF6 3′ UTR. Luc, firefly luciferase. (C) Quantitative real-time PCR results showing the relative expression of miR-101 in M1 and M2 macrophages. (D−F) miR-101-mimic or NC-mimic was transfected into M1 macrophages for 24 h. Quantitative real-time PCR results showing the relative expression of miR-101 (D), quantitative real-time PCR results showing the relative mRNA expression of C/EBPα and KLF6 (E), and western blot results showing the protein expression of C/EBPα, KLF6, PU.1, and PPARγ (F). (G−I) miR-101-inhibitor or NC-inhibitor was transfected in M2 macrophages for 48 h. Quantitative real-time PCR results showing the relative expression of miR-101 (G), quantitative real-time PCR results showing the relative mRNA expression of C/EBPα and KLF6 (H), and western blot results showing the protein expression of C/EBPα, KLF6, PU.1, and PPARγ (I). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3; mean ± SD.
To further explore whether miR-101 regulates the expression of C/EBPα and KLF6 in macrophages, we first detected the differential expression of miR-101 in M1 and M2 macrophages and found that miR-101 had a higher expression in M2 than in M1 macrophages (Figure 4C). Then, we transfected miR-101-mimic or NC-mimic into M1 macrophages to detect the expression of C/EBPα, KLF6, and their target genes. The overexpression of miR-101 (Figure 4D) markedly decreased mRNA expression of C/EBPα and KLF6 (Figure 4E), the protein levels of C/EBPα, KLF6, and PU.1 were increased, and PPARγ protein expression was decreased in miR-101-mimic-transfected M1 macrophages (Figure 4F). We also transfected miR-101-inhibitor or NC-inhibitor into M2 macrophages. The downregulation of miR-101 (Figure 4G) obviously increased the mRNA expression of C/EBPα and KLF6 (Figure 4H). The miR-101-inhibitor also notably elevated the protein expression of C/EBPα, KLF6, and PU.1 and reduced the PPARγ protein expression (Figure 4I). The data confirmed the targeted inhibitory function of miR-101 to C/EBPα and KLF6.
Furthermore, to examine whether miR-101 regulates macrophage polarization, we detected the expression of M1 and M2 markers after transfecting miR-101-mimic in M1 macrophages and transfecting miR-101-inhibitor in M2 macrophages. The quantitative real-time PCR results showed that enhanced miR-101 expression reduced the mRNA expression of IL-1β and TNF-α and improved the mRNA expression of CCL17, CCL22, and CD163 (Figure 5A), whereas knockdown of miR-101 increased the mRNA expression of IL-1β and TNF-α and decreased the mRNA expression of CCL17, CCL22, and CD163 (Figure 5B). These results suggested that miR-101 could promote macrophage polarization to M2 phenotype.
Figure 5.

miR-101 activates M2 macrophages to promote tumor proliferation and migration
(A and B) Quantitative real-time PCR was performed to detect the mRNA expression of M1 markers IL-1β and TNF-α and M2 markers CCL17, CCL22, and CD163 after transfecting miR-101-mimic in M1 macrophages (A) and transfecting miR-101-inhibitor in M2 macrophages (B). (C and D) The cell viabilities of MCF-7 and OV cells cocultured with miR-101-mimic/NC-mimic-transfected M1 macrophages (C) and cocultured with miR-101-inhibitor/NC-inhibitor-transfected M2 macrophages (D) by MTT assays. (E and F) The migration abilities of MCF-7 and OV cells cocultured with miR-101-mimic/NC-mimic-transfected M1 macrophages were validated by wound-healing (E) and Transwell (F) assays. (G and H) The migration abilities of MCF-7 and OV cells cocultured with miR-101-inhibitor/NC-inhibitor-transfected M2 macrophages were validated by wound-healing (G) and Transwell (H) assays. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3; mean ± SD.
In addition, we assessed the effects of miR-101-activated macrophage polarization to the M2 phenotype on the tumor proliferation and migration abilities. We cocultured MCF-7 and OV cells with M1 conditional medium after transfected with the miR-101-mimic and cocultured MCF-7 and OV cells with M2 conditional medium after transfected with the miR-101-inhibitor. We found that the cell viabilities of MCF-7 and OV cells cocultured with miR-101-mimic-transfected M1 macrophages were increased compared to the NC-mimic group (Figure 5C), whereas the proliferation abilities of MCF-7 and OV cells cocultured with miR-101-inhibitor-transfected M2 macrophages were decreased compared to the NC-inhibitor group (Figure 5D). The wound-healing and Transwell results showed that the migration abilities of MCF-7 and OV cells in miR-101-mimic groups were strengthened compared to the NC-mimic group (Figures 5E and 5F), whereas the migration abilities of MCF-7 and OV cells in miR-101-inhibitor groups were weakened compared to the NC-inhibitor group (Figures 5G and 5H). Overall, miR-101 could promote proliferation and migration abilities of MCF-7 and OV cells by inducing polarization of macrophages to the M2 phenotype.
The Xist/miR-101-3p/KLF6/C/EBPα axis mediates macrophage polarization to promote tumor cell proliferation and migration
We cotransfected WT/MT-Xist plasmids with miR-101-mimic or NC-mimic into 293T cells and performed a luciferase reporter assay to investigate the correlation between Xist and miR-101. The results showed that compared with NC groups, miR-101 could significantly reduce the luciferase activity of a reporter that contained a binding site. Conversely, luciferase activity had little change in cells transfected with miR-101 and MT-Xist plasmids (Figure 6A). Additionally, the expression of miR-101 was increased after transfecting sh-Xist plasmids in M1 macrophages (Figure 6B). These results suggested that the interaction of Xist and miR-101 was realized by the putative binding site. Next, we, respectively, transfected sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor in M1 macrophages and detected the mRNA expression of Xist, miR-101, C/EBPα, and KLF6 and the protein expression of C/EBPα, KLF6, PU.1, and PPARγ. Compared with the sh-NC + NC-inhibitor group, the expression of Xist, C/EBPα, and KLF6 was decreased, whereas miR-101 expression was increased in the sh-Xist + NC-inhibitor group. However, the expression of Xist, C/EBPα, and KLF6 was increased, whereas miR-101 expression was decreased in the sh-NC + 101-inhibitor group, and the expression of Xist, C/EBPα, KLF6, and miR-101 had little changes in the sh-Xist + 101-inhibitor group (Figure 6C). The data suggested that the suppression of miR-101 expression could restore the inhibition of C/EBPα and KLF6 expression caused by silencing Xist. These consistent changes were also observed from the protein expression of C/EBPα, KLF6, PU.1, and PPARγ (Figure 6D). In addition, we also detected the protein levels of C/EBPα and PPARγ in MCF-7 and OV cells and found that the suppression of miR-101 expression in M1 macrophages could restore the inhibition of C/EBPα and KLF6 expression in MCF-7 and OV cells caused by silencing Xist in M1 macrophages (Figure S1).
Figure 6.

The Xist/miR-101/C/EBPα/KLF6 axis mediates macrophage polarization to regulate tumor cell proliferation and migration
(A) The sequence of Xist with a highly conserved putative miR-101-binding site from the starBase 2.0 prediction and the luciferase activity of the 293T cells cotransfected with the miR-101-mimic or NC-mimic and containing WT/ MT-Xist by dual luciferase reporter assays. (B) The relative expression of miR-101 in the M1 macrophages after silencing Xist by quantitative real-time PCR analysis. (C−F) sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor were, respectively, transfected in M1 macrophages. (C) Quantitative real-time PCR results showing the relative mRNA expression of Xist, miR-101, C/EBPα, and KLF6. (D) Western blot results showing the protein expression of C/EBPα, KLF6, PU.1, and PPARγ. (E) Quantitative real-time PCR results showing the relative mRNA expression of IL-1β, TNF-α, CCL17, CCL22, and CD163. (F) Flow cytometry results showing the surface expression of CD86 and CD206. (G and H) MCF-7 and OV cells were cocultured with M1 conditional medium transfected with sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor. (G) MTT results showing the proliferation abilities of MCF-7 and OV cells. Transwell results showing the migration abilities of MCF-7 and OV cells (H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3; mean ± SD.
Next, we detected the changes of M1 and M2 marker expression. As expected, silencing Xist decreased the expression of M1 markers IL-1β, TNF-α, and CD86, whereas increased the expression of M2 markers CCL17, CCL22, CD163, and CD206. However, 101-inhibitor blocked the effects of silencing Xist on these marker expressions (Figures 6E and 6F). Furthermore, we cocultured MCF-7 and OV cells with the M1 conditional medium transfected with sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, or sh-Xist + 101-inhibitor to explore the changes of tumor proliferation and migration abilities. The MTT assay results showed that silencing Xist resulted in the increased cell viabilities of MCF-7 and OV cells, and the suppression of miR-101 expression restored the promotion of proliferation of MCF-7 and OV cells caused by silencing Xist (Figure 6G). Consistently, the migration abilities of MCF-7 and OV cells were significantly strengthened by silencing Xist, and the increased migration abilities were restored by the 101-inhibitor (Figure 6H). Taken together, these results indicated that the Xist/miR-101/C/EBPα/KLF6 axis could mediate macrophage polarization to regulate cell proliferation and migration of breast and ovarian cancer.
The Xist/miR-101-3p/KLF6/C/EBPα axis mediates macrophage polarization to promote breast and ovarian cancer development
To verify the Xist/miR-101-3p/KLF6/C/EBPα axis in vivo, MCF-7 and M1 macrophages transfected with the sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor or OV and M1 macrophages transfected with the sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor were inoculated into mice to construct subcutaneous tumors, respectively. After tumor formation, M1 macrophages transfected with the sh-NC + NC-inhibitor or sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, and sh-Xist + 101-inhibitor were injected into tumor tissues once per 3 days until the mice were killed. Tumor volume was measured after the tumor volume of the control group reached 125 mm3. During the process, tumor volumes were measured every 3 days.33 The results showed that compared with the sh-NC + NC-inhibitor group, the tumor volumes and weights were increased in the sh-Xist + NC-inhibitor group, were decreased in the sh-NC + 101-inhibitor group, and were not obviously changed in the sh-Xist + 101-inhibitor group (Figure 7A). Furthermore, bone marrow-derived macrophages (BMDMs)34 were isolated from breast or ovarian cancer-bearing mice to detect the mRNA expression of Xist, miR-101, C/EBPα, and KLF6 and the protein expression of C/EBPα, KLF6, PU.1, and PPARγ. We found the consistent changes of these markers with the above in vitro test (Figures 7B and 7C).
Figure 7.

The Xist/miR-101-3p/KLF6/C/EBPα axis mediates macrophage polarization to promote breast and ovarian cancer development
MCF-7 and macrophage with sh-NC + NC-inhibitor, MCF-7 and macrophage with sh-Xist + NC-inhibitor, MCF-7 and macrophage with sh-NC + 101-inhibitor, MCF-7 and macrophage with sh-Xist + 101-inhibitor, OV and macrophage with sh-NC + NC-inhibitor, OV and macrophage with sh-Xist + NC-inhibitor, OV and macrophage with sh-NC + 101-inhibitor, and OV and macrophage with sh-Xist + 101-inhibitor were inoculated into mice to construct subcutaneous tumors, respectively (n = 6 mice in each group). After inoculation, the macrophage with sh-NC + NC-inhibitor, macrophage with sh-Xist + NC-inhibitor, macrophage with sh-NC + 101-inhibitor, and macrophage with sh-Xist + 101-inhibitor were injected into tissues one time/3 days until the mice were killed; the volumes of tumors were measured every 3 days. (A) Tumor size, tumor weight, and tumor volumes after treatment. (B) Quantitative real-time PCR results showing the relative mRNA expression of Xist, miR-101, C/EBPα, and KLF6. (C) Western blot results showing the protein expression of C/EBPα, KLF6, PU.1, and PPARγ. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3; mean ± SD.
Furthermore, we cocultured MCF-7 and OV cells with macrophages isolated from human medium transfected with the sh-NC + NC-inhibitor, sh-Xist + NC-inhibitor, sh-NC + 101-inhibitor, or sh-Xist + 101-inhibitor to explore the changes of tumor proliferation and migration abilities. The MTT assay results showed that silencing Xist resulted in the increased cell viabilities of MCF-7 and OV cells, and the suppression of miR-101 expression restored the promotion of proliferation of MCF-7 and OV cells caused by silencing Xist (Figure S2A). Consistently, the migration abilities of MCF-7 and OV cells were significantly strengthened by silencing Xist, and the increased migration abilities were restored by the 101-inhibitor (Figure S2B). Taken together, these results indicated that the Xist/miR-101/C/EBPα/KLF6 axis could mediate macrophage polarization to regulate cell proliferation and migration of breast and ovarian cancer.
Discussion
Increasing evidence has shown that TAMs play a crucial role in tumorigenesis and tumor treatment, especially M2 macrophages, which exert anti-inflammatory and tumor-promoting effects.35 Apart from TAM-derived cytokines and chemokines,36, 37, 38, 39 some common signaling pathways are also involved in the interaction between macrophages and cancer cells.40,41 However, it is still necessary to further explore specific mechanisms of how macrophages affect tumorigenesis and development. Therefore, targeting TAMs is an extremely promising immunotherapy strategy to inhibit cancers. Over the past few years, noncoding RNAs, including lncRNAs and miRNAs, have been shown to play important roles in tumorigenesis.42,43 Nevertheless, it is rarely reported that these noncoding RNAs can promote or suppress cancers by regulating macrophage polarization.
Xist is an essential lncRNA for maintaining the stable inactivation of the X chromosome in female mammals.15 In recent years, increasing studies demonstrated that Xist had a low expression and played a suppressed role in breast cancer, ovarian cancer, and cervical cancer.21, 22, 23 In this study, we found that Xist was significantly higher in M1 macrophages (TAM-M1) successfully induced by LPS and IFN-γ than in M2 macrophages (TAM-M2) successfully induced by IL-4. The results were verified in the GEO: GSE46903 dataset, which was a macrophage expression chip. When IL-4 was added to TAM-M1, Xist was decreased, accompanied by the enhanced expression of M2 markers CCL17, CCL22, and CD163 and the reduced expression of M1 markers IL-1β and TNF-α. In contrast, the addition of LPS and IFN-γ to TAM-M2 increased Xist expression, accompanied by enhanced expression of M1 markers and reduced expression of M2 markers. Furthermore, the knockdown of Xist expression in TAM-M1 could result in the decreased expression of M1 markers and the increased expression of M2 markers and also obviously promote the proliferation and migration abilities of conditioned cultured MCF-7 and OV cells. Our study indicated that Xist could inhibit the proliferation and migration of breast and ovarian cancer cells by regulating TAM polarization, which had some differences from the previous reports that Xist directly inhibited the development of breast and ovarian cancer proposed by Zheng et al.21 and Benoît et al.22 Recently, several studies have proven that lncRNA could regulate tumorigenesis and progression by altering TAM polarization. For example, lncRNA-GNAS-AS1 promoted migration and invasion of non-small cell lung cancer cells by altering macrophage polarization.44 Knockdown of lncRNA-p21 could alleviate breast cancer development by promoting macrophages to the M1 phenotype.33 These reports and our study have suggested that lncRNA plays an essential role in regulating tumor progression by mediating macrophage polarization.
KLF6 and C/EBPα are important regulators of macrophage polarization.28 KLF6 is a member of the KLFs family. In human and mouse macrophages, LPS and IFN-γ, which stimulate M1 macrophages, can increase the expression of KLF6, whereas IL-4 and IL-13, which stimulate M2 macrophages, can decrease the expression of KLF6. Additionally, KLF6 can inhibit M2 polarization by decreasing PPARγ expression.28 C/EBPα is highly expressed in M1 macrophages. C/EBPα and its target gene PU.1 take part in activating M1 macrophages induced by TLR ligands.29 In this study, we detected that C/EBPα, KLF6, and PU.1 had a higher expression, whereas PPARγ had a lower expression in M1 macrophages. Furthermore, the suppression of Xist expression in M1 macrophages reduced the protein expression of C/EBPα, KLF6, and PU.1, whereas promoted the protein expression of PPARγ. These results indicate that silencing Xist in M1 macrophages stimulates TAM to M2 polarization and promotes the proliferation and migration of breast and ovarian cancer cells by inhibiting C/EBPα and KLF6 expression. In addition, several studies have shown that C/EBPα had a low expression in many kinds of tumors, such as liver cancer,45 breast cancer,46 and lung cancer.47 Furthermore, Sarker et al.45 reported that MTL-CEBPA, a first-in-class small activating RNA (saRNA) oligonucleotide drug that upregulates C/EBPα, showed an acceptable safety profile and potential synergistic efficacy with tyrosine kinase inhibitors (TKIs) in Hepatocellular carcinoma (HCC). Thus, it will provide a new method for cancer treatment by targeting CEBPa. In our study, it might be another novel idea to suppress breast and ovarian cancer proliferation by targeting Xist to activate CEBPa in macrophages.
Some studies have illustrated that lncRNAs, as a new class of noncoding RNA, can be considered as miRNA sponges.48 For example, lncRNA-MT1JP inhibited the proliferation, invasion, and metastasis of gastric cancer cells by regulating FBXW7 through competitively binding to miR-92-3p.49 lncRNA-HOXD-AS1 facilitated liver cancer metastasis by regulating the HOXD-AS1/miR-130a-3p/SOX2 axis.50 According to bioinformatics analysis, we found that Xist had an MRE that competed with C/EBPα and KLF6 to bind miR-101. Moreover, dual luciferase reporter assays verified that miR-101 was able to bind to both Xist and C/EBPα and KLF6, which provide direct evidence for Xist as an miR-101 sponge to regulate the expression of C/EBPα and KLF6.
It has been reported that miR-101 played a positive or negative role in tumorigenesis and progression, including breast cancer, ovarian cancer, and other cancers. Med19/miR-101 promoted breast cancer progression by regulating the epidermal growth factor receptor (EGFR)/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway.51 miR-101 could inhibit the proliferation and invasion of ovarian cancer cells by reducing the expression of SOCS2.52 Additionally, Chen et al.53 and Li et al.54 revealed that miR-101 could increase the risk of breast cancer and was associated with poor prognosis of breast cancer. Nevertheless, it is imperative to explore the expression of miR-101 in macrophages and its effects on regulating macrophage polarization. In the present study, we found that miR-101 was highly expressed in M2 macrophages, and inhibition of miR-101 expression in M2 macrophages could polarize macrophages to M1 macrophages. In contrast, overexpression of miR-101 in M1 macrophages could promote M1 to M2 macrophages. Moreover, miR-101 could promote the proliferation and migration of MCF-7 and OV cells by inducing M2 macrophages. These results indicated that miR-101 and Xist had a reverse effect on macrophage polarization and promoting tumor proliferation and migration. Furthermore, suppression of miR-101 expression could restore a series of changes caused by silencing Xist, including stimulating M1 to M2 macrophages and the functions of promoting tumor cell proliferation and migration.
In conclusion, our study elucidates the mechanism by which lncRNA-Xist mediates macrophage polarization and affects cell proliferation and migration of breast and ovarian cancer by competing with miR-101 to regulate the expression of C/EBPα and KLF6 (Figure 8). Therefore, the promotion of Xist expression in M1 macrophages and inhibition of miR-101 expression in M2 macrophages might play important roles in inhibiting breast and ovarian tumor proliferation and migration abilities.
Figure 8.

The diagram that lncRNA-Xist mediates macrophage polarization and affects cell proliferation and migration of breast and ovarian cancers by competing with miR-101 to regulate the expression of C/EBPα and KLF6
Materials and methods
Cell culture
Cell lines were purchased from Cell Resource Center of the Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (China), including human monocyte cell lines (THP-1), human breast cancer cell lines (MCF-7), human OV cancer cell lines (OVCAR3), and human embryonic kidney cell lines (HEK293T). These cells were cultured at 37°C with 5% CO2, according to the standard protocols. THP-1 was grown in suspension culture flasks and cultured in RPMI-1640 medium (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA), antibiotics (penicillin 100 U/mL, streptomycin 0.1 mg/mL), and 1% N-2-hydroxyethylpiperazine-N9-2-ethanesufonic acid (HEPES). MCF-7 and OV cells were adherent cultured in RPMI-1640 medium with 10% FBS and antibiotics. The HEK293T cell was adherent cultured in DMEM with 10% FBS and antibiotics.
Generation of M1 and M2 macrophages
THP-1 cells were seeded into six-well plates with a density of 15 × 105 cells/well and differentiated to macrophages by treating with 200 ng/mL PMA (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. After the above-described treatment, the obtained macrophages were activated to an M1 phenotype by treating with 50 ng/mL IFN-γ (PeproTech, Princeton, NJ, USA) and 100 ng/mL LPS (Sigma-Aldrich, St. Louis, MO, USA) for 24 h and M2 phenotype by treating with 20 ng/mL IL-4 (PeproTech, Princeton, NJ, USA) for 24 h.
Preparation of conditional medium
M1 or M2 macrophages were further cultured in RPMI 1640 without FBS for 24 h. The supernatant was centrifuged at 2,000 × g for 15 min and collected for use as a conditional medium in subsequent experiments.
Cell transfection
The shRNA targeting lncRNA-Xist and NC plasmids was purchased from Shanghai GeneChem. Human miR-101-mimic, NC-mimic, miR-101-inhibitor, and NC-inhibitor were purchased from RIBOBIO (Guangzhou, China). M1 macrophages were transfected with 1.5 μg/well plasmids or 2 nM (final concentration) miR-101-mimic or NC-mimic in six-well plates. M2 macrophages were transfected with 2 nM (final concentration) miR-101-inhibitor or NC-inhibitor in six-well plates. Above-mentioned oligonucleotides and plasmids were transfected by using Lipofectamine 3000 (Invitrogen, Eugene, OR, USA), according to the manufacturer’s instruction.
Quantitative real-time PCR
Total RNA was extracted using TRIzol (Invitrogen, Eugene, OR, USA). The miRNA was extracted using TRIzol and the miRNA Purification Kit (CWBIO, Beijing, China). The mRNA was reverse transcribed into cDNA using the TaKaRa kit, and the reaction conditions were as follows: 37°C for 15 min and 85°C for 5 s. The miRNA was reverse transcribed into cDNA using Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (TaKaRa, Japan), and the reaction conditions were follows: 42°C for 1 h and 70°C for 10 min. The PCR primers of U6 and miR-101 were purchased from RIBOBIO (Guangzhou, China). The expression levels of mRNA and miRNA were determined using the SYBR Green PCR Master Mix Kit (TaKaRa, Japan) and quantitative real-time PCR instrument (Applied Biosystems, USA). β-actin and U6 were used as endogenous control for the quantification of the mRNA and miRNA levels. The reaction conditions were followed by 95°C for 30 s, 56°C for 45 s, and 72°C for 20 s for 40 cycles of amplification. Primer sequences were as follows: Xist forward 5′-CATTGCTAGGCATTGGGGATG-3′, reverse 5′-CCAGGAAGCATGTATCTTCTGG-3′; β-actin forward 5′-TCCTCCCTGGAGAAGAGCTA-3′, reverse 5′-TCCTGCTTGCTGATCCACAT-3′; IL-1β forward 5′-CTCGCCAGTGAAATGATGGCT-3′, reverse 5′-GTCGGAGATTCGTAGCTGGAT-3′; TNF-α forward 5′-CCAGCTGGAGAAGGGTGAC-3′, reverse 5′-AGGCGTTTGGGAAGGTTG-3′; CCL17 forward 5′-CTTCTCTGCAGCACATCCAC-3′, reverse 5′-AGTACTCCAGGCAGCACTCC-3′; CCL22 forward 5′-TGCCGTGATTACGTCCGTTA-3′, reverse 5′-AAGGTTAGCAACACCACGCC-3′; CD163 forward 5′-CAATGGGGTGGACTTACCTG-3′, reverse 5′-AACCAGTCTGGGTTCCCTGT-3′; C/EBPα forward 5′-TTGTGCCTTGGAAATGCAAC-3′, reverse 5′-TCGGGAAGGAGGCAGGAAAC-3′; KLF6 forward 5′-CAAGGGAAATGGCGATGCCT-3′, reverse 5′-CTTTTCTCCTGTGTGCGTCC-3′. The 2−ΔΔCt methods were used to analyze the relative fold changes.
MTT assays
Cells (2 × 103 cells/mL) were seeded into 96-well plates (200 μL/well) and placed into an incubator with 5% CO2 at 37°C for 24 h. Then, the cells were incubated with 50% RPMI 1640 containing 10% FBS and 50% conditioned medium without FBS for 12 h, 24 h, 48 h, and 72 h, respectively. After the cells were cultured for different time periods, 20 μL of MTT solution was added into cells of each well for 4 h. After removing the culture medium, the precipitate was dissolved in 200 μL of DMSO. The absorbance at 570 nm was measured to evaluate cell viability using the Anthos 2010 Microplate Reader (Anthos Labtec Instruments, Austria).
Dual luciferase reporter gene assays
For the construction of WT and MT of C/EBPα 3′ UTR, KLF6 3′ UTR, or XIST luciferase reporter vectors, the fragment of C/EBPα 3′ UTR, KLF6 3′ UTR, or XIST was cloned downstream of the GV272 vector, purchased from Shanghai Jikai Gene Chemical Technology (China). The C/EBPα 3′ UTR, KLF6 3′ UTR, or XIST MT reporter vectors contained a 7-bp or 9-bp MT in the putative miR-101-3p binding site. HEK293T cells were cotransfected with the indicated vectors and miR-101-mimic or NC-mimic. Firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter Gene Assay Kit (Promega, Madison, WI, USA). Relative luciferase activity normalized to the NC was used for comparison among groups.
Transwell assays
Transwell chambers (8 μm pore size; Corning Costar, USA) were used to measure cell migration ability according to the vendor’s instructions. MCF-7 or OV cells (104 cells/well) were seeded into the upper chambers with 100 μL of serum-free RPMI-1640 medium, which had been inserted into wells of 24-well plates containing 10% FBS-supplemented conditional medium. After 24 h, the cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for 30 min. The migrated cells in the upper chambers were photographed under a phase-contrast microscope and counted.
Wound-healing analysis
Wound-healing assay was performed to analyze the cell migration ability. MCF-7 or OV cells (5 × 105 cells/well) were seeded into six-well plates. After 24 h, the cells were incubated with conditioned medium without FBS. Images of a specific position on the scratched areas were taken using an inverted microscope (Nikon Eclipse TE2000-U, Japan) at 0 h, 12 h, 24 h, and 48 h. ImageJ software was used to measure the scratched area at different incubation times. The wound-closure percent was calculated as (the scratch area before incubation – the scratch area after incubation)/(the scratch area before incubation) × 100%.
Flow cytometry assay
Macrophages were trypsinized and suspended in phosphate-buffered saline (PBS), followed by incubation with 1 μg/mL fluorescein isothiocyanate-conjugated human anti-CD206 (BioLegend, San Diego, CA, USA) or phycoerythrin-conjugated human anti-CD86 (BioLegend, San Diego, CA, USA) monoclonal antibodies for 30 min at 4°C in the dark. The samples were subjected to flow cytometric analysis within 1 h.
Immunoblotting analysis
Immunoblotting analysis was conducted as previously described.55 Cells were lysed in radioimmunoprecipitation assay (RIPA) (Beyotime, Shanghai, China) buffer. Protein concentrations were determined using a bicinchoninic acid (BCA) protein quantification kit (Beyotime, Jiangsu, China). The primary antibodies included β-actin (1:1,000; Absin, China), KLF6 (1:1,000; Abcam, UK), C/EBPα (1:1,000; Abcam, UK), PU.1 (1:1,000; CST, USA), and PPARγ (1:1,000; CST, USA). The secondary antibodies were horseradish peroxidase-linked goat anti-rabbit or goat anti-mouse secondary antibodies (Santa Cruz, CA, USA). Protein expression was detected using a chemiluminescence detection system (ProteinSimple, USA). β-actin was used as an endogenous protein for normalization. Gray intensity analysis was performed using ImageJ software.
TCGA and GEO dataset collection and process
TCGA breast cancer expression profiles (HTSeq-Counts) were downloaded from TCGA database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). GEO: GSE26712, including ovarian cancer expression profiles, and GEO: GSE46903, including macrophages expression profiles, were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The differential expression and correlation analyses were performed using GraphPad Prism 7 software (GraphPad Software, USA).
Animal models of tumors
Specific pathogen-free (SPF) female BALB/c nu/nu mice (4−6 weeks old) were purchased from Beijing Huafukang. All of the mice were bred in the Animal Research Center of China Medical University (Liaoning, China) in compliance with the Guide for the Care and Use of Laboratory Animals. All of the protocols were approved by the Committee for Ethical Affairs of China Medical University (Liaoning, China), and the methods were carried out in “accordance” with the approved guidelines. To establish different tumor models, BALB/c nu/nu mice were subcutaneously injected in the flank with MCF-7 or OV cells (1 × 106/mouse) and macrophages with/without lncRNA-Xist knockdown and miR-101-inhibitor (1 × 105/mouse) in 100 μL of PBS. Tumor volume was measured after the tumor volume reached 125 mm3. Tumor growth was observed with tumor measurements using a caliper every 3 days, and tumor volume was calculated using the formula volume = length × width2 × 0.5 cm3. All of the tumor-bearing mice were killed, and every tumor was isolated and weighed.34
Isolation of macrophages from mice
Mice were sacrificed by cervical dislocation. Bone marrow cells were recovered from tibiae and femora, and erythrocytes were lysed. For differentiation, cells were cultured on nontissue-culture dishes in RPMI-1640 medium (HyClone, Logan, UT, USA), supplemented with 10% FBS (HyClone, Logan, UT, USA), antibiotics (penicillin 100 U/mL, streptomycin 0.1 mg/mL), and 20 ng/mL mouse recombinant macrophage colony-stimulating factor (M-CSF) (MedChemExpress, Shanghai, China). After 7 days in culture, cells were used to detected expression of mRNAs and proteins.34
Statistics
All experiments were repeated at least three times. All data of three independent experiments were presented as the mean ± SD. Statistical analysis was performed by using SPSS22.0 software (SPSS, Chicago, IL, USA) and GraphPad Prism 7 software (GraphPad Software, USA). Two-tailed Student’s t test was used between two groups. The p <0.05 was considered statistically significant.
Acknowledgments
The authors would like to acknowledge the Key Laboratory of Precision Diagnosis and Treatment of Gastrointestinal Tumors, Ministry of Education (China Medical University, Shenyang, China), for providing the space and equipment for conducting the experiments. This work was supported by the National Natural Science Foundation of China (NSFC; numbers 81601370, 81972794, 81573462, and 81902708); NSFC-Liaoning Joint Fund Key Program (number U1608281); Major Special S&T Projects in Liaoning Province (2019JH1/10300005); Key R&D Guidance Plan Projects in Liaoning Province (2019JH8/10300011); Liaoning Revitalization Talents Program (number XLYC1807201); Shenyang S&T Projects (19-109-4-09); Liaoning Provincial Department of Education Scientific Research Project (QN2019034); Liaoning Province Scientific Research Foundation (JC2019032); and Science Foundation for Young Scholars of China Medical University (QGZ2018083).
Author contributions
M.W., M.H., Z.Y., L. Zhao, and H.X. designed the research study. Y. Zhao, Z.Y., R.M., and Y. Zhang conducted experiments. Y. Zhao, Z.Y., R.M., Y.Y., X.L., L. Zhang, and P.S. acquired data. Y. Zhao, M.H., M.W., J.B., and L. Zhao analyzed data. M.W., M.H., Z.Y., and H.X. provided reagents. Y. Zhao, M.H., and M.W. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.005.
Contributor Information
Hong Xu, Email: xh4015@163.com.
Miao He, Email: hemiao_cmu@126.com.
Minjie Wei, Email: weiminjiecmu@163.com.
Supplemental information
References
- 1.Chanmee T., Ontong P., Konno K., Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alahari S.V., Dong S., Alahari S.K. Are macrophages in tumors good targets for novel therapeutic approaches? Mol. Cells. 2015;38:95–104. doi: 10.14348/molcells.2015.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinushi M., Komohara Y. Tumor-associated macrophages as an emerging target against tumors: Creating a new path from bench to bedside. Biochim. Biophys. Acta. 2015;1855:123–130. doi: 10.1016/j.bbcan.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J., Yao H., Song G., Liao X., Xian Y., Li W. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am. J. Transl. Res. 2015;7:1699–1711. [PMC free article] [PubMed] [Google Scholar]
- 5.Komohara Y., Fujiwara Y., Ohnishi K., Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016;99(Pt B):180–185. doi: 10.1016/j.addr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Friedl P., Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 8.Ehrt S., Schnappinger D., Bekiranov S., Drenkow J., Shi S., Gingeras T.R., Gaasterland T., Schoolnik G., Nathan C. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A., Sozzani S., Locati M., Allavena P., Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 10.Ramanathan S., Jagannathan N. Tumor associated macrophage: a review on the phenotypes, traits and functions. Iran. J. Cancer Prev. 2014;7:1–8. [PMC free article] [PubMed] [Google Scholar]
- 11.Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Z.Y., Luo R.Z., Peng R.J., Wang S.S., Xue C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. OncoTargets Ther. 2014;7:1475–1480. doi: 10.2147/OTT.S61838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren F., Fan M., Mei J., Wu Y., Liu C., Pu Q., You Z., Liu L. Interferon-γ and celecoxib inhibit lung-tumor growth through modulating M2/M1 macrophage ratio in the tumor microenvironment. Drug Des. Devel. Ther. 2014;8:1527–1538. doi: 10.2147/DDDT.S66302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt A.M., Chang H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown C.J., Ballabio A., Rupert J.L., Lafreniere R.G., Grompe M., Tonlorenzi R., Willard H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- 16.Sarkar M.K., Gayen S., Kumar S., Maclary E., Buttigieg E., Hinten M., Kumari A., Harris C., Sado T., Kalantry S. An Xist-activating antisense RNA required for X-chromosome inactivation. Nat. Commun. 2015;6:8564. doi: 10.1038/ncomms9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X., Zhang S., He C., Xue P., Zhang L., He Z., Zang L., Feng B., Sun J., Zheng M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer. 2020;19:46. doi: 10.1186/s12943-020-1146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen D.L., Ju H.Q., Lu Y.X., Chen L.Z., Zeng Z.L., Zhang D.S., Luo H.Y., Wang F., Qiu M.Z., Wang D.S. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res. 2016;35:142. doi: 10.1186/s13046-016-0420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li C., Wan L., Liu Z., Xu G., Wang S., Su Z., Zhang Y., Zhang C., Liu X., Lei Z., Zhang H.T. Long non-coding RNA XIST promotes TGF-β-induced epithelial-mesenchymal transition by regulating miR-367/141-ZEB2 axis in non-small-cell lung cancer. Cancer Lett. 2018;418:185–195. doi: 10.1016/j.canlet.2018.01.036. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Zhu Z., Huang S., Zhao Q., Huang C., Tang Y., Sun C., Zhang Z., Wang L., Chen H. lncRNA XIST regulates proliferation and migration of hepatocellular carcinoma cells by acting as miR-497-5p molecular sponge and targeting PDCD4. Cancer Cell Int. 2019;19:198. doi: 10.1186/s12935-019-0909-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng R., Lin S., Guan L., Yuan H., Liu K., Liu C., Ye W., Liao Y., Jia J., Zhang R. Long non-coding RNA XIST inhibited breast cancer cell growth, migration, and invasion via miR-155/CDX1 axis. Biochem. Biophys. Res. Commun. 2018;498:1002–1008. doi: 10.1016/j.bbrc.2018.03.104. [DOI] [PubMed] [Google Scholar]
- 22.Benoît M.H., Hudson T.J., Maire G., Squire J.A., Arcand S.L., Provencher D., Mes-Masson A.M., Tonin P.N. Global analysis of chromosome X gene expression in primary cultures of normal ovarian surface epithelial cells and epithelial ovarian cancer cell lines. Int. J. Oncol. 2007;30:5–17. [PubMed] [Google Scholar]
- 23.Kobayashi R., Miyagawa R., Yamashita H., Morikawa T., Okuma K., Fukayama M., Ohtomo K., Nakagawa K. Increased expression of long non-coding RNA XIST predicts favorable prognosis of cervical squamous cell carcinoma subsequent to definitive chemoradiation therapy. Oncol. Lett. 2016;12:3066–3074. doi: 10.3892/ol.2016.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L., Lv G., Wang B., Kuang L. XIST/miR-376c-5p/OPN axis modulates the influence of proinflammatory M1 macrophages on osteoarthritis chondrocyte apoptosis. J. Cell. Physiol. 2020;235:281–293. doi: 10.1002/jcp.28968. [DOI] [PubMed] [Google Scholar]
- 25.Chanput W., Mes J.J., Wichers H.J. THP-1 cell line: an in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014;23:37–45. doi: 10.1016/j.intimp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Liu W.S., Heckman C.A. The sevenfold way of PKC regulation. Cell. Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 27.Ansa-Addo E.A., Lange S., Stratton D., Antwi-Baffour S., Cestari I., Ramirez M.I., McCrossan M.V., Inal J.M. Human plasma membrane-derived vesicles halt proliferation and induce differentiation of THP-1 acute monocytic leukemia cells. J. Immunol. 2010;185:5236–5246. doi: 10.4049/jimmunol.1001656. [DOI] [PubMed] [Google Scholar]
- 28.Date D., Das R., Narla G., Simon D.I., Jain M.K., Mahabeleshwar G.H. Kruppel-like transcription factor 6 regulates inflammatory macrophage polarization. J. Biol. Chem. 2014;289:10318–10329. doi: 10.1074/jbc.M113.526749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee B., Qiao L., Lu M., Yoo H.S., Cheung W., Mak R., Schaack J., Feng G.S., Chi N.W., Olefsky J.M., Shao J. C/EBPα regulates macrophage activation and systemic metabolism. Am. J. Physiol. Endocrinol. Metab. 2014;306:E1144–E1154. doi: 10.1152/ajpendo.00002.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hua F., Tian Y., Gao Y., Li C., Liu X. Colony-stimulating factor 1 receptor inhibition blocks macrophage infiltration and endometrial cancer cell proliferation. Mol. Med. Rep. 2019;19:3139–3147. doi: 10.3892/mmr.2019.9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei C., Yang C., Wang S., Shi D., Zhang C., Lin X., Liu Q., Dou R., Xiong B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer. 2019;18:64. doi: 10.1186/s12943-019-0976-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X., Luo G., Zhang K., Cao J., Huang C., Jiang T., Liu B., Su L., Qiu Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018;78:4586–4598. doi: 10.1158/0008-5472.CAN-17-3841. [DOI] [PubMed] [Google Scholar]
- 33.Zhou L., Tian Y., Guo F., Yu B., Li J., Xu H., Su Z. LincRNA-p21 knockdown reversed tumor-associated macrophages function by promoting MDM2 to antagonize∗ p53 activation and alleviate breast cancer development. Cancer Immunol. Immunother. 2020;69:835–846. doi: 10.1007/s00262-020-02511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z., Xie Y., Xiong Y., Liu S., Qiu C., Zhu Z., Mao H., Yu M., Wang X. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett. 2020;469:173–185. doi: 10.1016/j.canlet.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Murray P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017;79:541–566. doi: 10.1146/annurev-physiol-022516-034339. [DOI] [PubMed] [Google Scholar]
- 36.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 37.Guo L., Cheng X., Chen H., Chen C., Xie S., Zhao M., Liu D., Deng Q., Liu Y., Wang X. Induction of breast cancer stem cells by M1 macrophages through Lin-28B-let-7-HMGA2 axis. Cancer Lett. 2019;452:213–225. doi: 10.1016/j.canlet.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 38.Vasiljeva O., Papazoglou A., Krüger A., Brodoefel H., Korovin M., Deussing J., Augustin N., Nielsen B.S., Almholt K., Bogyo M. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66:5242–5250. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 39.Arima K., Komohara Y., Bu L., Tsukamoto M., Itoyama R., Miyake K., Uchihara T., Ogata Y., Nakagawa S., Okabe H. Downregulation of 15-hydroxyprostaglandin dehydrogenase by interleukin-1β from activated macrophages leads to poor prognosis in pancreatic cancer. Cancer Sci. 2018;109:462–470. doi: 10.1111/cas.13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu G., Gao T., Zhang L., Chen X., Pang Q., Wang Y., Wang D., Li J., Liu Q. NKAP alters tumor immune microenvironment and promotes glioma growth via Notch1 signaling. J. Exp. Clin. Cancer Res. 2019;38:291. doi: 10.1186/s13046-019-1281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M., Liu F., Zhou P., Wang Q., Xu C., Li Y., Bian L., Liu Y., Zhou J., Wang F. The MTOR signaling pathway regulates macrophage differentiation from mouse myeloid progenitors by inhibiting autophagy. Autophagy. 2019;15:1150–1162. doi: 10.1080/15548627.2019.1578040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shuai Y., Ma Z., Liu W., Yu T., Yan C., Jiang H., Tian S., Xu T., Shu Y. TEAD4 modulated LncRNA MNX1-AS1 contributes to gastric cancer progression partly through suppressing BTG2 and activating BCL2. Mol. Cancer. 2020;19:6. doi: 10.1186/s12943-019-1104-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C., Ding D., Gao Y., Li Y. MicroRNA3651 promotes colorectal cancer cell proliferation through directly repressing Tbox transcription factor 1. Int. J. Mol. Med. 2020;45:956–966. doi: 10.3892/ijmm.2020.4458. [DOI] [PubMed] [Google Scholar]
- 44.Li Z., Feng C., Guo J., Hu X., Xie D. GNAS-AS1/miR-4319/NECAB3 axis promotes migration and invasion of non-small cell lung cancer cells by altering macrophage polarization. Funct. Integr. Genomics. 2020;20:17–28. doi: 10.1007/s10142-019-00696-x. [DOI] [PubMed] [Google Scholar]
- 45.Sarker D., Plummer R., Meyer T., Sodergren M.H., Basu B., Chee C.E., Huang K.W., Palmer D.H., Ma Y.T., Evans T.R.J. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020;26:3936–3946. doi: 10.1158/1078-0432.CCR-20-0414. [DOI] [PubMed] [Google Scholar]
- 46.Gery S., Tanosaki S., Bose S., Bose N., Vadgama J., Koeffler H.P. Down-regulation and growth inhibitory role of C/EBPalpha in breast cancer. Clin. Cancer Res. 2005;11:3184–3190. doi: 10.1158/1078-0432.CCR-04-2625. [DOI] [PubMed] [Google Scholar]
- 47.Halmos B., Huettner C.S., Kocher O., Ferenczi K., Karp D.D., Tenen D.G. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 2002;62:528–534. [PubMed] [Google Scholar]
- 48.Thomson D.W., Dinger M.E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 49.Zhang G., Li S., Lu J., Ge Y., Wang Q., Ma G., Zhao Q., Wu D., Gong W., Du M. LncRNA MT1JP functions as a ceRNA in regulating FBXW7 through competitively binding to miR-92a-3p in gastric cancer. Mol. Cancer. 2018;17:87. doi: 10.1186/s12943-018-0829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H., Huo X., Yang X.R., He J., Cheng L., Wang N., Deng X., Jin H., Wang N., Wang C. STAT3-mediated upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer metastasis by regulating SOX4. Mol. Cancer. 2017;16:136. doi: 10.1186/s12943-017-0680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X., Gao D., Fang K., Guo Z., Li L. Med19 is targeted by miR-101-3p/miR-422a and promotes breast cancer progression by regulating the EGFR/MEK/ERK signaling pathway. Cancer Lett. 2019;444:105–115. doi: 10.1016/j.canlet.2018.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Zheng H.B., Zheng X.G., Liu B.P. miRNA-101 inhibits ovarian cancer cells proliferation and invasion by down-regulating expression of SOCS-2. Int. J. Clin. Exp. Med. 2015;8:20263–20270. [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J., Qin Z., Jiang Y., Wang Y., He Y., Dai J., Jin G., Ma H., Hu Z., Yin Y., Shen H. Genetic variations in the flanking regions of miR-101-2 are associated with increased risk of breast cancer. PLoS ONE. 2014;9:e86319. doi: 10.1371/journal.pone.0086319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li C.Y., Xiong D.D., Huang C.Q., He R.Q., Liang H.W., Pan D.H., Wang H.L., Wang Y.W., Zhu H.W., Chen G. Clinical Value of miR-101-3p and Biological Analysis of its Prospective Targets in Breast Cancer: A Study Based on The Cancer Genome Atlas (TCGA) and Bioinformatics. Med. Sci. Monit. 2017;23:1857–1871. doi: 10.12659/MSM.900030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma M.-T., He M., Wang Y., Jiao X.-Y., Zhao L., Bai X.-F., Yu Z.-J., Wu H.-Z., Sun M.-L., Song Z.-G., Wei M.J. MiR-487a resensitizes mitoxantrone (MX)-resistant breast cancer cells (MCF-7/MX) to MX by targeting breast cancer resistance protein (BCRP/ABCG2) Cancer Lett. 2013;339:107–115. doi: 10.1016/j.canlet.2013.07.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.