

Abstract
The developing CNS is exposed to physiological hypoxia, under which hypoxia-inducible factor α (HIFα) is stabilized and plays a crucial role in regulating neural development. The cellular and molecular mechanisms of HIFα in developmental myelination remain incompletely understood. A previous concept proposes that HIFα regulates CNS developmental myelination by activating the autocrine Wnt/β-catenin signaling in oligodendrocyte progenitor cells (OPCs). Here, by analyzing a battery of genetic mice of both sexes, we presented in vivo evidence supporting an alternative understanding of oligodendroglial HIFα-regulated developmental myelination. At the cellular level, we found that HIFα was required for developmental myelination by transiently controlling upstream OPC differentiation but not downstream oligodendrocyte maturation and that HIFα dysregulation in OPCs but not oligodendrocytes disturbed normal developmental myelination. We demonstrated that HIFα played a minor, if any, role in regulating canonical Wnt signaling in the oligodendroglial lineage or in the CNS. At the molecular level, blocking autocrine Wnt signaling did not affect HIFα-regulated OPC differentiation and myelination. We further identified HIFα–Sox9 regulatory axis as an underlying molecular mechanism in HIFα-regulated OPC differentiation. Our findings support a concept shift in our mechanistic understanding of HIFα-regulated CNS myelination from the previous Wnt-dependent view to a Wnt-independent one and unveil a previously unappreciated HIFα–Sox9 pathway in regulating OPC differentiation.
SIGNIFICANCE STATEMENT Promoting disturbed developmental myelination is a promising option in treating diffuse white matter injury, previously called periventricular leukomalacia, a major form of brain injury affecting premature infants. In the developing CNS, hypoxia-inducible factor α (HIFα) is a key regulator that adapts neural cells to physiological and pathologic hypoxic cues. The role and mechanism of HIFα in oligodendroglial myelination, which is severely disturbed in preterm infants affected with diffuse white matter injury, is incompletely understood. Our findings presented here represent a concept shift in our mechanistic understanding of HIFα-regulated developmental myelination and suggest the potential of intervening with an oligodendroglial HIFα-mediated signaling pathway to mitigate disturbed myelination in premature white matter injury.
Keywords: hypoxia inducible factor, myelination, oligodendrocyte progenitor cells, oligodendrocytes, oligodendroglial differentiation, oligodendroglial maturation
Introduction
Hypoxia-inducible factor α (HIFα) is a master transcriptional regulator of the adaptive response to hypoxia (Semenza, 2012). The oxygen concentration in the developing CNS was reported ranging from 0.5% to 7% (Ivanovic, 2009; Zhang et al., 2011). HIFα protein (HIF1α and HIF2α) is constitutively translated but subjected to rapid turnover (half-life, <5 min) by the proteasome-mediated degradation in which the von Hippel-Lindau (VHL) protein plays an essential role (Semenza, 2012). Under physiological or pathologic hypoxia or VHL mutation, HIFα accumulates in the nucleus where it complexes with the stable HIFβ subunit and other coactivators to activate target gene expression.
The role of HIFα, particularly the representative HIF1α, in neural precursor cells under physiological conditions has been extensively investigated (Tomita et al., 2003; Milosevic et al., 2007; Cunningham et al., 2012; Li et al., 2014). Aggravated hypoxia and/or ischemia causes severe disturbance of normal oligodendroglial myelination in the preterm infants born between the 28th and 37th weeks of gestational age (Volpe, 2009). Animal studies demonstrate that pathologic hypoxia insult delays developmental myelination in preterm equivalent early postnatal murine CNS (Liu et al., 2011, 2013; Jablonska et al., 2012). The role of HIFα in developmental myelination had not been defined until an important study reported that HIFα plays a major role in hypoxia-elicited myelination disturbance in cell/brain slice culture systems (Yuen et al., 2014). However, the cellular and molecular mechanisms underlying oligodendroglial HIFα-regulated myelination remain incompletely defined. The current popular hypothesis states that HIFα dysregulation disturbs developmental myelination by activating autocrine Wnt/β-catenin signaling (Yuen et al., 2014). This “Wnt-dependent” mechanistic hypothesis is in line with the inhibitory effect of intracellular Wnt/β-catenin activation on CNS myelination (Guo et al., 2015), but it was only tested in the cell/slice culture systems in combination with pharmacological manipulations. Given the intrinsic caveats of noncellular specificity and/or off-target effects of pharmacological application, it is imperative to determine, by using in vivo genetic approaches, the involvement of oligodendroglial HIFα-derived Wnt signaling in developmental myelination.
CNS developmental myelination consists of at least the following two major sequential steps: oligodendrocyte progenitor cell (OPC) differentiation into oligodendrocytes [OLs (referred to as OPC differentiation)] and oligodendrocyte maturation and myelination (Huang et al., 2013; Guo et al., 2015). In humans, developmental myelination starts in the third trimester of gestational ages, which is equivalent to the perinatal and early postnatal ages in rodents (Semple et al., 2013). Therefore, we used the murine CNS of perinatal and early postnatal ages as a third trimester-equivalent in vivo model to dissect the cellular and molecular mechanisms underlying development myelination. By analyzing a series of cell-specific HIFα and Wnt genetic mutant mice, we provided convincing evidence supporting an alternative HIFα model of CNS myelination: HIFα transiently regulates developmental myelination by controlling OPC differentiation, but subsequent OL maturation and its hyperactivation disturbs OPC differentiation in a manner independent of autocrine Wnt/β-catenin signaling. Our results further demonstrate that sustained Sox9 activation is a downstream mechanism underlying disturbed OPC differentiation elicited by HIFα hyperactivation.
Materials and Methods
Transgenic animals
The following transgenic mice were used in our study: Cnp-Cre mice (RRID:MGI_3051754) provided by Nave (Lappe-Siefke et al., 2003); Sox10-Cre (RRID:IMSR_JAX:025807); Sox10-CreERT2 (RRID:IMSR_JAX:027651); Pdgfrα-CreERT2 (RRID:IMSR_JAX:018280); Hif1α-floxed (RRID:IMSR_JAX:007561); Hif2α-floxed (RRID:IMSR_JAX:008407); Vhl-floxed (RRID:IMSR_JAX:012933); and Wls-floxed (RRID:IMSR_JAX:012888). In our study, we found no difference in oligodendroglial phenotypes between non-Cre mice carrying floxed alleles and Cre transgenic mice carrying no floxed alleles. Therefore, we used non-Cre and/or Cre transgenic mice from same litters of cKO (conditional knock-out) mice as littermate control (Ctrl) mice. All mice were maintained on the C57BL/6 background and housed on a 12 h light/dark cycle with free access to water and food. Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of California, Davis.
Tamoxifen treatment
Tamoxifen (TM; catalog #T5648, Sigma-Aldrich) was prepared at a concentration of 30 mg/ml in a mixture of ethanol and sunflower seed oil (1:9, v/v). All study mice including littermate control and conditional knock-out mice were received TM intraperitoneally at a dose of 200 µg/g body weight.
Hypoxia/ischemia-induced brain white matter injury
We used our established protocols (Shen et al., 2010; Chung et al., 2013) to induce hypoxia/ischemia (H/I) injury in C57BL/6J background mice at postnatal day 6 (P6), a time point when OPC differentiation barely occurs in the brain. In brief, P6 mice were anesthetized with indirect cooling by wet ice, which were immediately subjected to permanent occlusion (by cauterizers) of the right side common carotid artery (ischemia). One hour after the artery occlusion, mice were exposed to 6% O2 for 45 min (hypoxia). After hypoxia exposure, the mice were returned to the dam. Sham operation was performed in the same way as H/I injury, including anesthesia, neck skin incision, and exposure of the right common carotid artery, but without artery cauterization or hypoxia. Previous study demonstrates that H/I injury results in widespread periventricular white matter injury (WMI) including arrested OPC differentiation and microglial/astroglial activation (Liu et al., 2011).
Tissue processing and immunohistochemistry
After anesthetization by ketamine/xylazine mixture, mice were perfused with cold PBS. Tissues were collected, fixed with fresh 4% PFA for 2 h at room temperature, cryopreserved in 30% sucrose overnight at 4°C, and embedded in OCT (optimal cutting temperature) medium (VWR International). Serial coronal sections (14 µm) were cut by a Cryostat (model CM 1900–3-1, Leica). Tissue sections were incubated with primary antibody overnight at 4°C after blocking with 10% donkey serum at room temperature for at least 1 h. After washing three times, the fluorescence-conjugated secondary antibody (Jackson ImmunoResearch) was applied and incubated for 1.5 h. Then the slice was protected by mounting medium after DAPI nucleus staining. The following primary antibodies were used in immunohistochemical study: HIF-1α (1:200; catalog #100642, Cayman Chemical; RRID:AB_409037); Sox10 (RRID:AB_2195374; 1:100; catalog #sc-17 342, Santa Cruz Biotechnology; 1:500; ab27655, Abcam; RRID:AB_778021); Olig2 (1:200; catalog #AB9610, Millipore; RRID:AB_570666); myelin basic protein (MBP; 1:500; catalog #SMI-99, BioLegend; RRID:AB_2564741); SMI312 (1:1000; catalog #SMI-312R, Covance; RRID:AB_2566782); CC1 (1:200; catalog #OP80, Calbiochem; RRID:AB_213434); PDGFRα (1:200; catalog #AF1062, R&D Systems; RRID:AB_2236897); SMI32 (1:1000; SMI-32P, BioLegend; RRID:AB_2314912); and Sox9 (1:200; catalog #AF3075, R&D Systems; RRID:AB_2194160).
In situ hybridization and transmission electron microscopy
Briefly, the slices were treated with Proteinase K (catalog #AM2548, Thermo Fisher Scientific) and acetylated by triethanolamine (catalog #90279, Sigma-Aldrich) and acetic anhydride (catalog #320102, Sigma-Aldrich). The tissue was incubated with 100 ng/μl DIG-labeled cRNA at 65°C overnight. Then the slices were incubated with alkaline phosphatase anti-DIG secondary antibody (1:100; catalog #11093274910, Sigma-Aldrich) overnight after blocking with 10% donkey serum. The signal was determined by nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (toluidine salt; catalog #72091, Sigma-Aldrich). Semithin sections for toluidine blue myelin staining and ultrathin section for transmission electron microscopy (TEM) imaging were prepared according to our previous protocols (Zhang et al., 2018a,b). TEM imaging was conducted by the professionals at UC Davis Core Facility, who were blinded to the mouse genotypes.
ELISA
The Wnt3a protein level was measured by the mouse Wnt3a ELISA kit (M13146365, CSB-EL026136MO, Cusabio) according to the manufacturer instruction.
Primary OPC culture and gene manipulation
The mouse cortex between P0 and P2 was collected and dissociated with papain kit and DNase I (catalog #LK003176, Worthington; 250 U/ml; catalog #D5025, Sigma-Aldrich). OPCs were purified by immunopanning approach according to our previous protocol (Lang et al., 2013). Purified OPCs were used for experiments of Wls knockdown, Wnt3a overexpression, and Sox9 overexpression. Wls-shRNA (TRCN 0000 234932; Mission ShRNA Bacterial Glycerol; stock #NM_026582) and scramble control (Mission TRC2 PlkO.5-PURO Non-Mammalian shRNA Control Plasmid) were purchased from Sigma-Aldrich. Wnt3a plasmid pLCN-Wnt3a-HA (catalog #18030, Addgene) and empty pLCN-exp (catalog #64865, Addgene) were used for Wnt3a overexpression experiment. The transfection of Wls-shRNA and pLCN-Wnt3a-HA and controls was performed by using HiPerFect transfection reagent (catalog #301704, QIAGEN) according to the manual. p-WPXL-Sox9 (catalog #36979, Addgene) and control plasmid p-WPXL (catalog #12257, Addgene) were used for Sox9 overexpression, and the transfection was performed by using FuGENE6 Transfection reagent (catalog #E2691, lot #000371257, Promega).
Primary rat OPCs were used in Sox9 knockdown and dimethyloxalylglycine (DMOG) treatment. DMOG (0.5 mm dissolved in DMSO; catalog #D3695, Sigma-Aldrich), and Sox9 siRNA (SASI_Rn02_00372739, Sigma-Aldrich) or negative control siRNA (SIC001-5X1NMOL, Sigma-Aldrich) were used according to the experimental design (see Fig. 12H). Transfection of Sox9 siRNA was conducted by using HiPerFect transfection reagent (catalog #301704, QIAGEN).
Figure 12.
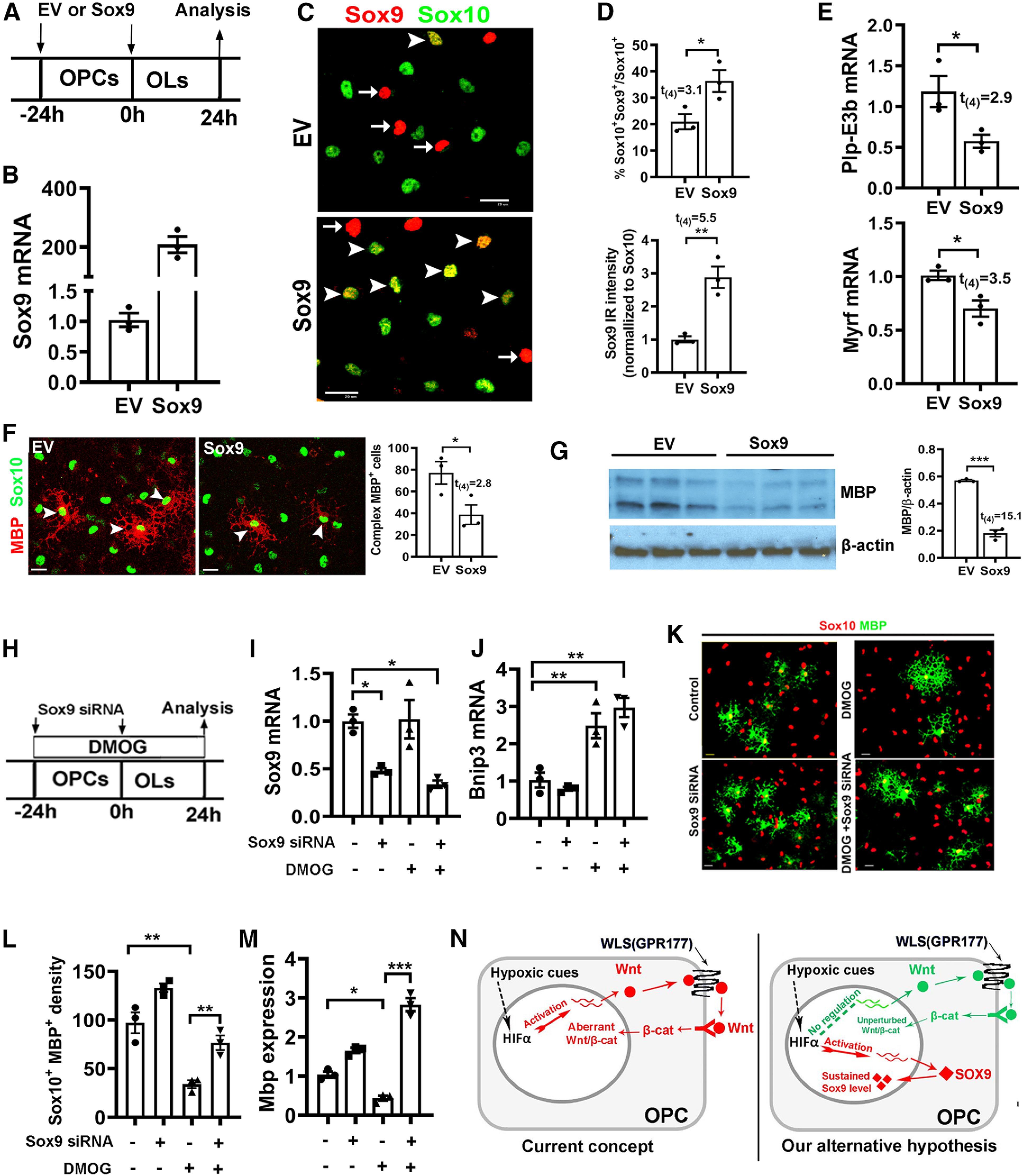
Sustained Sox9 expression inhibits OPC differentiation. A, Experimental design for B–G. Cortical primary OPCs in growth medium were transfected with Sox9-expressing plasmid (Sox9, n = 3) or EV (n = 3), then differentiated for 24 h in serum-free differentiating medium. B, qRT-PCR quantification of Sox9 mRNA expression. C, Representative immunocytochemical images of Sox9/Sox10. Arrowheads point to Sox9+/Sox10+ oligodendrocytes and arrows point to Sox9+/Sox10– cells, which are presumably astrocytes. Scale bar, 20 μm. D, Percentage of Sox10+ oligodendroglial lineage cells that express Sox9 (top) and Sox9-immunoreactive intensity normalized to Sox10 (bottom). E, qRT-PCR assay of exon3b-containing Plp (Plp-E3b), which specifically expresses in differentiated OLs, and myelin regulatory factor (Myrf), a potent prodifferentiation gene. F, Representative immunocytochemical images of MBP/Sox10 and density (#/mm2) of Sox10+ oligodendroglial cells of complex ramified morphology indicated by MBP staining (F, arrowheads), Scale bar, 10 μm. G, Western blot images and quantification of MBP and internal loading control β-actin. H, Experimental designs for I–M, primary OPCs were treated with HIFα stabilizer DMOG and transfected with Sox9 siRNA twice with 24 h apart. I, J, Expression of Sox9 (I) and HIFα target gene Bnip3 (J) quantified by qRT-PCR. One-way ANOVA followed by Tukey's test: Sox9, F(3,8) = 10.33, p = 0.004; Bnip3: F(3,8) = 20.83, p = 0.0004. K, Representative immunostaining images of differentiated OLs labeled by MBP and Sox10. Scale bar, 10 µm. L, M, Density (#/mm2) of MBP+/Sox10+ differentiated OLs (L) and qRT-PCR assay of Mbp mRNA expression (M). One-way ANOVA followed by Tukey's test: MBP+/Sox10+, F(3,8) = 33.29, p < 0.0001; Mbp, F(3,8) = 97.96, p < 0.0001. N, Previously proposed and our alternative working model explaining the role of HIFα in OPC differentiation. Endogenous HIFα regulates CNS myelination by controlling upstream OPC differentiation but not downstream OL maturation in perinatal and early postnatal CNS, whereas aberrant HIFα activation (via VHL mutation of hypoxia-ischemia insult) inhibits OPC differentiation through the Wnt-independent and Sox9-dependent pathway.
Purification of brain OPCs by magnetic-activated cell sorting
Magnetic-activated cell sorting (MACS) purification of OPCs was performed according to the protocols in the kit instruction from Miltenyi Biotec. The mouse forebrain was dissociated using the papain dissociation system (catalog #LK003176, Worthington) and gentleMACS Dissociator (catalog #130–092-235, Miltenyi Biotec). Astrocytes and microglia in the cell suspension were removed by using anti-ACSA-2 microbeads (catalog #130–097-679, Miltenyi Biotec) and anti-CD11b microbeads (catalog #130–049-601, Miltenyi Biotec), respectively. After astroglial and microglial removal, the cell suspension was then incubated with anti-O4 micro beads (catalog #130–094-543, Miltenyi Biotec) for OPC purification. O4+ cells were collected for RNA preparation.
RNA preparation, RT-PCR, and quantitative real-time PCR
Total RNA was obtained using QIAGEN RNeasy for lipid tissue kit (catalog #74804, QIAGEN) according to the manual. We used on-column DNase I digestion to eliminate DNA contamination. cDNA was prepared using QIAGEN Omniscript RT Kit (catalog #205111, QIAGEN). Mouse and rat gene expression was normalized to the internal control Hsp90 and β-actin, respectively, and calculated by the equation 2^(Ct(cycle threshold) of Hsp90-Ct of indicated genes). The value of control groups was normalized to 1 throughout the study. Quantitative real-time PCR (qRT-PCR) was performed by using QuantiTect SYBR Green PCR Kit (catalog #204145, QIAGEN) and thermocycler (catalog #MP3005P, Stratagene). The quantitative PCR (qPCR) primers used in the study were as follows: Mouse Wls: AGGGCAAGGAAGAAGGAGAG/ATCCCTCCAACAATGCAGAG; Mouse Glut1: CAGTTCGGCTATAACACTGGTG/GCCCCCGACAGAGAAGATG; Mouse Ldha: CATTGTCAAGTACAGTCCACACT/TTCCAATTACTCGGTTTTTGGGA; Mouse Hk2: TGATCGCCTGCTTATTCACGG/AACCGCCTAGAAATCTCCAGA; Mouse Axin2: AACCTATGCCCGTTTCCTCTA/GAGTGTAAAGACTTGGTCCACC; Mouse Naked1: CAGCTTGCTGCATACCATCTAT/GTTGAAAAGGACGCTCCTCTTA; Mouse Wnt7a: CGACTGTGGCTGCGACAAG/CTTCATGTTCTCCTCCAGGATCTTC; Mouse Wnt7b: CTTCACCTATGCCATCACGG/TGGTTGTAGTAGCCTTGCTTCT; Mouse Vegfa: GCACATAGAGAGAATGAGCTTCC/CTCCGCTCTGAACAAGGCT; Mouse Mbp: ACACGAGAACTACCCATTATGGC/CCAGCTAAATCTGCTGAGGGA; Mouse Plp (exon3b-specific): CCAGAATGTATGGTGTTCTCCC/GGCCCATGAGTTTAAGGACG; Mouse Cnp: TTTACCCGCAAAAGCCACACA/CACCGTGTCCTCATCTTGAAG; Mouse Mobp: AACTCCAAGCGTGAGATCGT/CAGAGGCTGTCCATTCACAA; Mouse Opalin: CTGCCTCTCACTCAACATCA/GCTGGATCAAAGTAAACAGC; Mouse Mog: AGCTGCTTCCTCTCCCTTCTC/ACTAAAGCCCGGATGGGATAC; Mosue Qk: CTGGACGAAGAAATTAGCAGAGT/ACTGCCATTTAACGTGTCATTGT; Mouse Myrf: CAGACCCAGGTGCTACAC/TCCTGCTTGATCATTCCGTTC; Mouse Sox9: AGTACCCGCATCTGCACAAC/ACGAAGGGTCTCTTCTCGCT; Mouse Hsp90: AAACAAGGAGATTTTCCTCCGC/CCGTCAGGCTCTCATATCGAAT; Rat Bnip3: GCTCCCAGACACCACAAGAT/TGAGAGTAGCTGTGCGCTTC; Rat Mbp: TTGACTCCATCGGGCGCTTCTTTA/TTCATCTTGGGTCCTCTGCGACTT; Rat Sox9: CTGAAGGGCTACGACTGGAC/TACTGGTCTGCCAGCTTCCT; and Rat β-actin: CGTCTTCCCCTCCATCGT/GGAGTCCTTCTGACCCATACC.
Chromatin immunoprecipitation by HIFα antibody and qPCR verification
Chromatin immunoprecipitation (ChIP) was performed using the SimpleChIP Plus Enzymatic Chromatin IP kit (catalog #9005, Cell Signaling Technology) following the manufacturer protocol with some modifications. Briefly, primary cultured rat OPCs (4 × 106 cells) treated with 1 mm DMOG for 7 h were cross-linked in culture medium containing 1% formaldehyde at room temperature for 10 min. After the addition of glycine solution to stop the reaction, the cells were then collected, centrifuged, and lysed. Nuclei were collected and treated with 0.5 μl micrococcal nuclease per IP for 20 min at 37°C. The reaction was stopped using 0.05 m EDTA, and nuclear membranes were destroyed by sonication. Then the supernatant was collected by centrifugation. Chromatin solutions were incubated with 10 μg of anti-HIF1α (rabbit, catalog #10006421, Cayman Chemical) antibody or anti-rabbit IgG (catalog #2729, Cell Signaling Technology) overnight at 4°C, and immunoprecipitated with protein G magnetic beads for 2 h at 4°C with rotation. Bound DNA–protein complexes were washed and eluted, and cross-links were reversed according to kit instructions. DNA was purified using spin columns and used for ChIP-qPCR analysis. The fold enrichment of specific genomic regions was assessed relative to the qPCR data from the IPs normalized to the control IgG values. Fold enrichment = 2^ – [(Ct IP) – (Ct IgG)]. qPCR primer sets for ChIP binding site verification are as follows: negative site: forward, TTCCTCTTGGGATGGTTGTC/reverse, CCACCTCTGGGGAGTATGAA; –75 bp site: forward, TTTCAAAATCCGGTCCAATC/reverse, CCCCCTTCACCTTAGAGACC; –828 bp site: forward, CGGGGAAGGACTTGTCAGT/reverse, ATGAAAACCAAAGCCAAGCA; –1211 bp site: forward, CGGCTCCAAGCACTCTTAAA/reverse, GCGTCCTTTTAGACCTGCAC; –1928 bp site: forward, GTCGTTCTCGCTGCCTTTAG/reverse, TTGAAGAGACCAGGGACCAC; and –2098 bp site: forward, CTCTGGATGTTGCCGAAAAT/reverse, CTCCACGCAAGCGTTTTTAT.
Western blot and antibodies
Twenty microgram protein was loaded into AnykD Mini-PROTEAN gel (catalog #4569035, BIO-RAD). After blocking, the membrane was incubated with primary antibodies overnight. The membrane was washed three times by TBST (TBS with 1% Tween 20). Then the HRP-conjugated secondary antibody was reacted with membrane for 1.5 h. Protein signals were developed by using an enhanced chemiluminescence kit (NEL103001EA, PerkinElmer). Quantification of interested protein bands were conducted by using NIH ImageJ software. Antibodies used in Western blot are MBP (1:2000; catalog #SMI-99, BioLegend; RRID:AB_2564741), active β-catenin (1:1000; catalog #05–665, Millipore; RRID:AB_309887), Axin-2 (1:1000; catalog #6163, Prosci; RRID:AB_10904353), and β-actin (1:2000; catalog #3700, Cell Signaling Technology; RRID:AB_2242334).
Experimental design and statistical analyses
Data collection was conducted by laboratory members who were blinded to mouse genotypes. We used Shapiro–Wilk approach to test data normality. F test and Browne–Forsythe test were used for variance equality of two groups and three or more groups, respectively. We used scatter graphs to present the quantification data throughout the current article. Both male and female mice were included in our data analysis. All data were presented as the mean ± SEM where each dot represents one mouse. Unpaired two-tailed Student's t test was used for statistically analyzing two groups of data. The t values and the degree of freedom (df) were shown as t(df) in each graph. One-way ANOVA followed by Tukey's post hoc test was used for statistically analyzing three or more groups of data. The F ratio, and DFn and DFd was presented as F(DFn, DFd) in the figure legends, where DFn stands for degree of freedom numerator, and DFd for degree of freedom of denominator. The exact p value of the one-way ANOVA was also described in the figure legends. All data graphing and statistical analyses were performed using GraphPad Prism version 8.0. The p values were designated as follows: *p < 0.05; **p < 0.01; ***p < 0.001 ns, not significant p > 0.05.
Results
Transient HIFα stabilization during early CNS development
HIF1α and HIF2α share high sequence homology and function in a similar manner through partnering with HIF1β and subsequently activating HIFα target genes (Semenza, 2012). Therefore, we used antibodies against the representative HIF1α to determine whether HIFα protein is stabilized in the perinatal and early postnatal CNS, where the vasculature is still in immature state (Harb et al., 2013; Yuen et al., 2014). We found that HIF1α protein was detected by immunohistochemistry (IHC) in Sox10+ oligodendroglial lineage cells in the white matter areas of the spinal cord (Fig. 1A) and the forebrain (Fig. 1C) at P5. As positive controls, HIF1α-immunoreactive signals were markedly increased in age-matched transgenic Sox10-Cre:Vhlfl/fl mice (Fig. 1B,D), in which Sox10-Cre-mediated VHL disruption prevents HIFα from degradation and causes HIFα accumulation specifically in oligodendroglial lineage cells. HIF1α signals became barely detectable in the forebrain white matter by P10 (Fig. 1E), which temporally approximates to the peak of CNS angiogenesis (Harb et al., 2013; Yuen et al., 2014), and remarkably upregulated in mice that had been challenged by hypoxia/ischemia injury at P6 (Fig. 1F; Shen et al., 2010; Liu et al., 2011; Chung et al., 2013). Our quantification data showed that the density of HIF1α+Sox10+ cells were significantly increased in P10 H/I-injured forebrain white matter compared with sham controls (mean ± SEM; sham, 4.2 ± 4.2; H/I, 312 ± 9.7; n = 3; t(4) = 29.3, p < 0.0001). Consistent with HIF1α histologic upregulation, Western blot assays of microdissected subcortical white matter demonstrated a substantial elevation of HIF1α and its canonical target gene PKM2 in H/I-injured brain (Fig. 1G), which was also confirmed by PKM2 IHC (Fig. 1H). Concomitant with HIFα activation, myelin staining by MBP showed severe disturbance of developmental myelination in H/I-injured white matter compared with sham controls (Fig. 1I1–I3). These results demonstrate that HIFα is transiently stabilized in the early postnatal CNS but rapidly downregulated in the second postnatal week and that hypoxia/ischemia injury results in sustained HIFα stabilization in the brain white matter.
Figure 1.
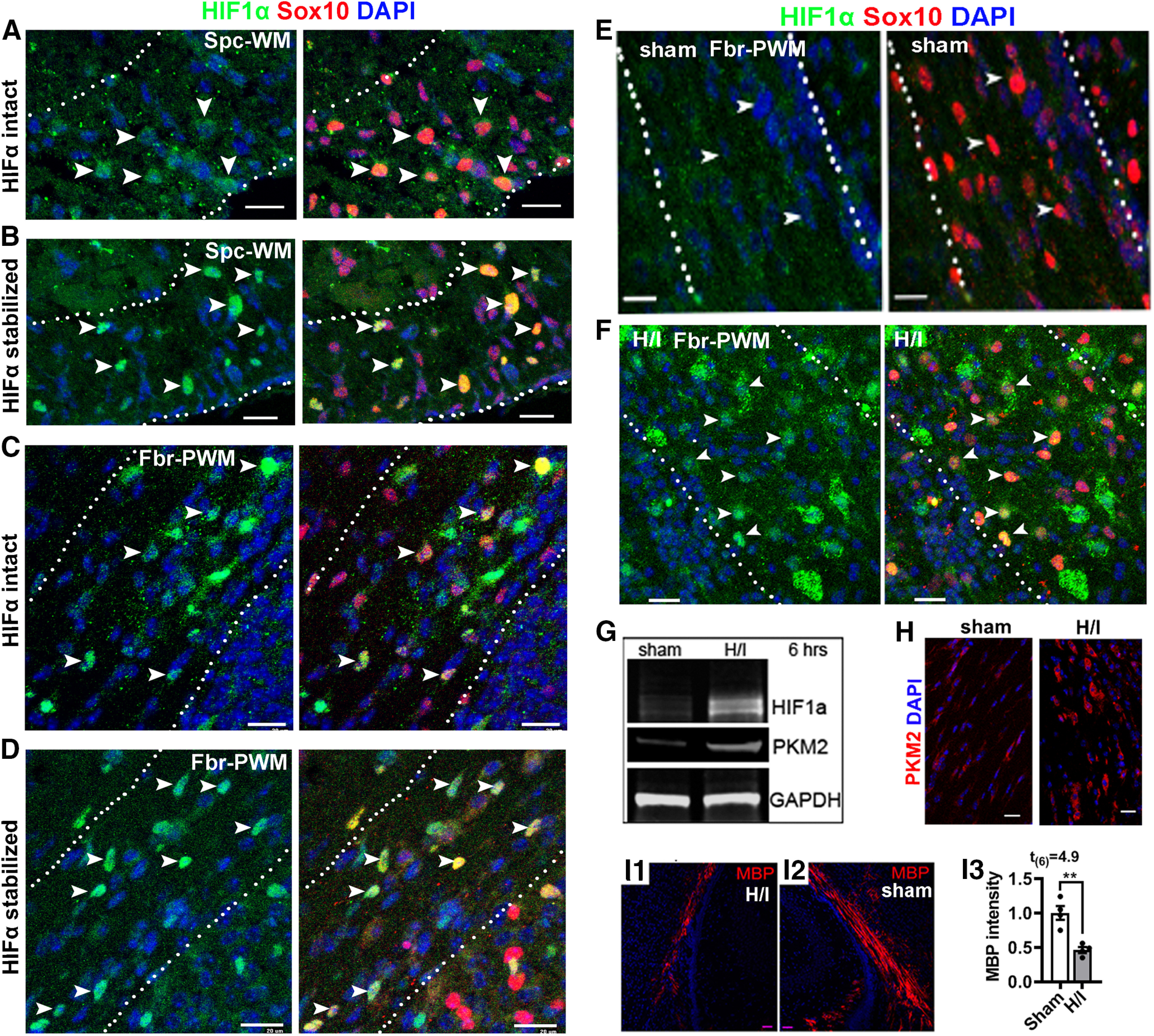
HIFα stabilization in the early postnatal murine CNS. A, B, IHC of HIF1α and oligodendroglial lineage marker Sox10 in the spinal cord ventral white matter (Spc-WM, marked by dotted lines) of HIFα intact mice (A) and HIFα stabilized (Sox10-Cre:Vhlfl/fl) mice (B) at P5. C, D, IHC of HIF1α and Sox10 in the forebrain periventricular white matter (Fbr-PWM; marked by dotted lines) of HIFα intact mice (C) and HIFα stabilized (Sox10-Cre:Vhlfl/fl) mice (D) at P5. E, F, IHC of HIF1α and Sox10 in the Fbr-PWM of P10 mice that had been subjected to sham operation (E) and H/I injury on P6 (i.e., 4 d post-H/I or sham; F; for details, see Materials and Methods). Arrowheads in A–F point to HIF1α+Sox10+ cells. G, Western blot of microdissected subcortical white matter for HIF1α and canonical HIFα target PKM2 at 6 h post-H/I or sham. GAPDH, Internal loading control. H, PKM2 IHC in the Fbr-PWM at P10, 4 d post-H/I or sham. I1–I3, IHC and quantification of MBP staining in the Fbr-PWM at P10, 4 d post-H/I or sham. MBP intensity was normalized to that of the contralateral brain hemispheres to the occluded artery of the same mouse. Scale bars, 20 µm.
HIFα hyperactivity impairs OPC differentiation but not oligodendrocyte maturation
Pathologic hypoxia–ischemia insults caused aberrant HIFα activation in the white matter (Fig. 1F–H) and hypomyelination (Fig. 1I1–I3). To determine whether aberrant HIFα stabilization disturbs normal developmental myelination, we induced HIFα hyperactivity by conditionally disrupting the negative regulator VHL (Fig. 1B,D). Constitutive Sox10-Cre:Vhlfl/fl (Sox10:VHL cKO) pups were much smaller than littermate controls (Vhlfl/+, or Vhlfl/fl) at birth and rarely survived beyond P7. Our prior data show that Sox10-Cre-mediated gene disruption initiates in early OPCs during embryonic CNS development (Zhang et al., 2018a). Sox10-Cre-elicited HIFα hyperactivity (see Fig. 6A) caused severe hypomyelination in the developing postnatal spinal cord (Fig. 2A). The number of CC1+ mature OLs (Fig. 2B,C) and the mRNA level of major myelin protein (Fig. 2D) were reduced by ∼40–60% in Sox10:VHL cKO mutants compared with littermate controls. However, the number of PDGFRα+ OPCs was unaffected in Sox10:VHL cKO mutants (Fig. 2C). These data suggest that constitutive HIFα activation in OPCs inhibits OPC differentiation and disturbs developmental myelination.
Figure 6.

Stabilizing HIFα activates HIFα-mediated signaling but does not perturb Wnt/β-catenin signaling in the oligodendroglial lineage and in the CNS. A, B, Relative expression of HIFα and Wnt target genes from unbiased RNAseq analysis. FPKM, Fragments per kilobase of transcript per million mapped reads, a normalized measurement of gene expression. The RNA was prepared from the spinal cord of P5 Sox10-Cre:Vhlfl/fl mice (n = 3, white bars) and littermate control (Vhlfl/+ or Vhlfl/fl; n = 3, gray bars) mice. C–E, Top three significant enriched KEGG Pathway and GO terms among 255 upregulated and 351 downregulated genes derived from unbiased RNAseq. Note that HIFα signaling pathway (C) and response to hypoxia (D) were significantly upregulated, validating HIFα hyperactivation in Sox10-Cre:Vhlfl/fl mice and that oligodendroglial cell differentiation was significantly downregulated (E), which is in agreement with the data of Figure 2A–D. F, Experimental design for G and H. PDGFRα:VHL cKO (n = 3, white bars) and littermate control (Vhlfl/fl, n = 3, gray bars) mice were treated with TM at P5, and forebrain OPCs at P6 were acutely purified by MACS. G, H, qRT-PCR quantification of HIFα target gene (G) and Wnt target gene (H) expression in purified OPCs. I, Expression of Wnt target gene (Axin2 and Naked1) and Wnt7a/7b in P8 spinal cord (n = 3, Ctrl, gray bars; n = 5, PDGFRα cKO, white bars) quantified by qRT-PCR. J, Western blot images and relative expression (normalized to the internal loading control β-actin) of the active form of β-catenin (ABC) and Axin2 in P14 forebrain (n = 3 each group). K, Expression of Wnt target genes (Axin2 and Naked1) and Wnt7a/7b in P8 spinal cord measured by qRT-PCR (n = 4 each group).
Figure 2.
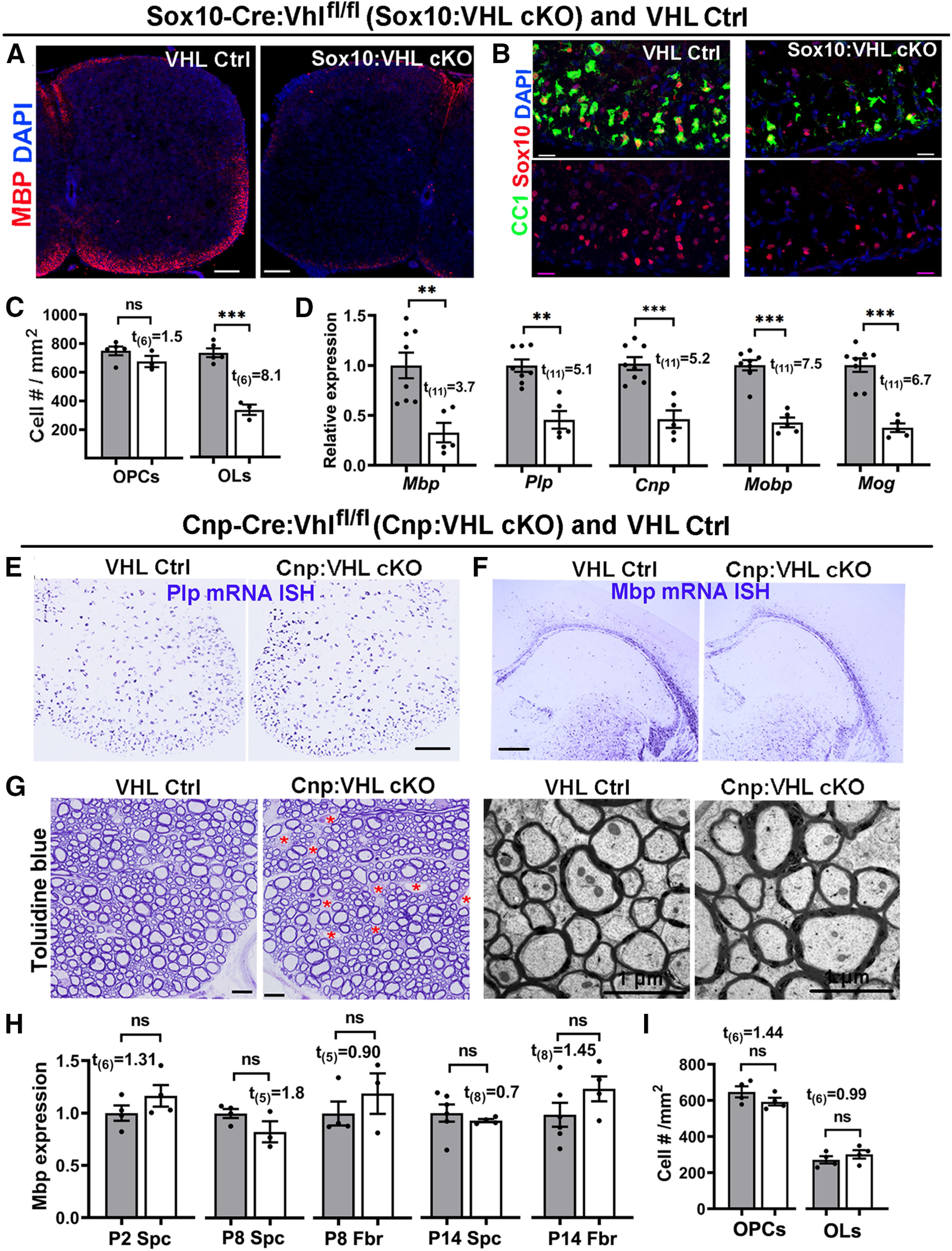
HIFα activation impairs developmental myelination by inhibiting OPC differentiation but not subsequent OL maturation. A, Myelin staining of MBP in the spinal cord of Sox10-Cre:Vhlfl/fl (Sox10:VHL cKO) and littermate Ctrl (Vhlfl/+ or Vhlfl/fl) pups at P5. Most Sox10:VHL cKO pups died by the first postnatal week. B, Representative confocal images of Sox10 (red) and differentiated OL marker CC1 (green) in P5 spinal cord (Ctrl, n = 5; cKO, n = 3). Blue, DAPI nuclear counterstaining. C, Quantification of Sox10+CC1+ OLs and Sox10+PDGFRa+ OPCs (right) in P5 spinal cord (n = 5 Ctrl, n = 3 cKO). D, qRT-PCR assay for myelin-specific genes in the spinal cord at P5. Plp, Proteolipid protein; Cnp, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase; Mobp, myelin oligodendrocyte basic protein; Mog, myelin oligodendrocyte glycoprotein. E, F, mRNA in situ hybridization (ISH) of Plp and Mbp in the spinal cord (E) and forebrain (F) of Cnp-Cre:Vhlfl/fl (Cnp:VHL cKO) and littermate control (Cnp-Cre:Vhlfl/+, Vhlfl/+, or Vhlfl/fl) mice at P14. G, Myelination in the adult spinal cord indicated by toluidine blue staining of semithin sections (left) and TEM of ultrathin section (right). Red asterisks in G denotes blood vessels, the density of which is elevated in Cnp:VHL cKO mice (Guo et al., 2015). H, Mbp mRNA levels quantified by qRT-PCR in Cnp:VHL cKO and Ctrl mice at different time points. Spc, Spinal cord; Fbr, forebrain. I, densities of Sox10+CC1+ OLs and Sox10+PDGFRα+ OPCs in P8 spinal cord of Cnp:VHL cKO and Ctrl mice. Scale bars: A, 100 µm; B, 20 µm; E, F, 200 µm; G, left, 10 µm; G, right, 1 µm. Data are presented as the mean ± SEM; gray bars are from Ctrl, white bars from cKO; two-tailed Student's t test, *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. The above data presentation and statistics are applied to all figures unless otherwise indicated).
To determine the effect of HIFα hyperactivity on subsequent oligodendrocyte maturation, we analyzed Cnp-Cre:Vhlfl/fl (Cnp:VHL cKO) mutants. Previous studies have demonstrated that, compared with Sox10-Cre, Cnp-Cre drives gene disruption in later stages of oligodendrocyte development (Moyon et al., 2016; Zhang et al., 2018a,b). Cnp:VHL cKO mice were viable and indistinguishable from their littermate controls (Vhlfl/fl, Vhlfl/+, or Cnp-Cre:Vhlfl/+) throughout the early postnatal and adult ages. In sharp contrast to Sox10:VHL cKO mice, Cnp:VHL cKO mice had normal levels of myelin gene expression (Fig. 2E,F,H); unperturbed myelination, evidenced by toluidine blue myelin staining (Fig. 2G, left) and TEM myelination assay (Fig. 2G, right); and normal numbers of OLs and OPCs (Fig. 2I) despite the elevated density of blood vessels in the CNS we recently reported (Guo et al., 2015; Zhang et al., 2020). These data indicate that HIFα hyperactivity in OLs does not affect their maturation. Together, our genetic evidence convincingly demonstrates that elevated HIFα activity perturbs developmental myelination by inhibiting OPC differentiation but not OL maturation, suggesting a lineage stage-dependent role of HIFα in developmental myelination.
We next used a time-conditional and OPC-specific genetic model to circumvent the early death of Sox10:VHL cKO mice and investigate whether time-conditional HIFα activation in postnatal OPCs impacts their differentiation. Toward this end, we generated Pdgfrα-CreERT2:Vhlfl/fl (PDGFRα:VHL cKO) mutants. Our data showed that OPC-specific HIFα activation during neonatal ages (Fig. 3A,B) significantly reduced the expression of genes encoding major myelin proteins (Fig. 3C) and decreased the densities of mature OLs, but not OPCs, in the spinal cord (Fig. 3D). IHC of myelin protein PLP demonstrated a marked hypomyelination (Fig. 3E). Further analysis of myelination by transmission electron microscopy (Fig. 3F) demonstrated that the density of myelinated axons was reduced by ∼42% in the ventral spinal white matter of PDGFRα:VHL cKO mice compared with littermate controls (Fig. 3G), which is consistent with histologic myelin protein assays (Fig. 3F). To assess the long-term effect of HIFα activation, we induced HIFα activation by tamoxifen injections to neonatal mice (Fig. 3H,I) and analyzed oligodendroglial phenotypes in the adult CNS (P60–P70). We found that the number of OLs, but not OPCs, was persistently reduced in the corpus callosum of PDGFRα:VHL cKO mice compared with control mice (Fig. 3J), suggesting that neonatal HIFα activation inhibits OPC differentiation in the adult CNS. Given that virtually all axons in the optic nerve are myelinated in the adult rodents (Bercury and Macklin, 2015), we then analyzed myelinated axons visualized by toluidine blue myelin staining of semithin resin sections (Fig. 3K1–K4). Our quantification demonstrated an ∼44% reduction in the density of myelinated axons in the optic nerve of adult PDGFRα:VHL cKO mutants compared with age-matched fully myelinated control optic nerves (Fig. 3K5). Together, our data suggest that HIFα activation inhibits long-term OPC differentiation and results in hypomyelination throughout postnatal CNS development.
Figure 3.
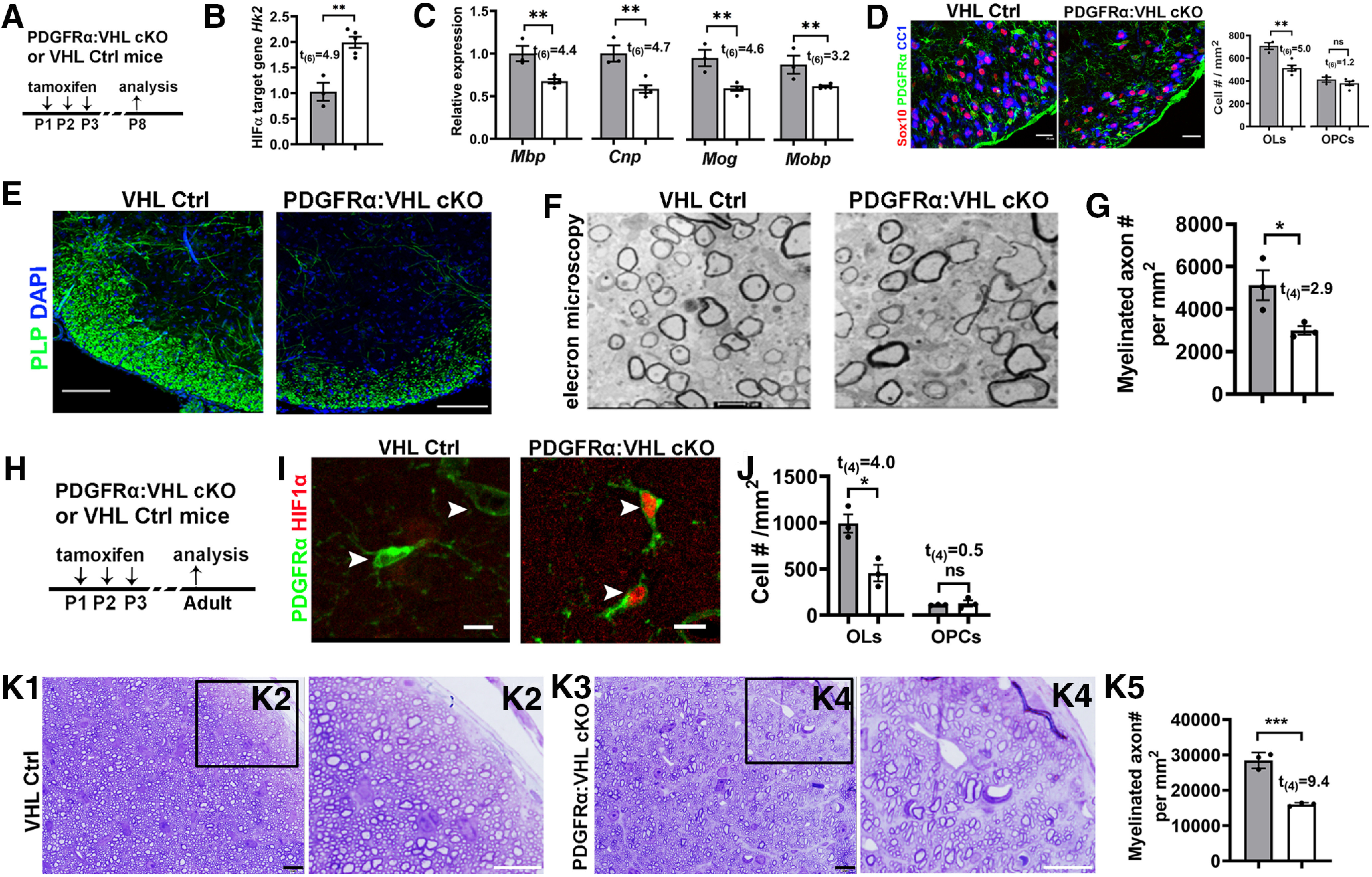
Inducible HIFα stabilization in postnatal OPCs inhibits OPC differentiation. A, experimental design for B–F. PDGFRα:VHL cKO (Pdgfrα-CreERT2:Vhlfl/fl) and littermate control (Vhlfl/fl) mice were treated with tamoxifen intraperitoneally at P1, P2, and P3, and analyzed at P8. B, qRT-PCR quantification of HIFα target gene hexokinase 2 Hk2 in the spinal cord. C, Expression of major myelin proteins in the spinal cord quantified by qRT-PCR. D, Representative confocal images and the density of Sox10+/CC1+ OLs and Sox10+/PDGFRα+ OPCs. E, IHC of PLP showing myelination in the spinal cord ventral white matter. F, G, Representative TEM images (F) and the density of myelinated axons (G) in the spinal cord ventral white matter. H, Experimental design for I–K. PDGFRα:VHL cKO and littermate control mice were treated with tamoxifen intraperitoneally at P1, P2, and P3, and analyzed at P60–P70. I, Representative confocal images showing HIF1α stabilization in PDGFRα+ OPCs (arrowheads) of PDGFRα:VHL cKO mice. J, Densities of CC1+ mature OLs and PDGFRα+ OPCs in the corpus callosum. K1–K4, Toluidine blue staining for myelin on semithin (500 nm) resin sections showing hypomyelination in the optic nerve of adult PDGFRα:VHL cKO mice. Boxed areas in K1 and K3 were shown at higher-magnification images in K2 and K4, respectively. K5, Density of myelinated axons in the adult optic nerves. Scale bars: D, 25 µm; E, 100 µm; F, 0.5 µm; I–K5, 10 µm.
Together, our in vivo data derived from three different genetic mouse models suggest that HIFα hyperactivity disturbs developmental myelination by inhibiting OPC differentiation.
Disrupting HIFα in OPCs but not OLs delays developmental myelination
Transient HIFα stabilization (Fig. 1A–E) suggests that HIFα may regulate OPC differentiation and/or myelination. To define the role of physiological HIFα stabilization in developmental myelination, we analyzed oligodendroglial phenotypes in HIFα cKO genetic mice. Our extensive analysis of HIF1α or HIF2α conditional knock-out mice driven by Pdgfrα-CreERT2 and Cnp-Cre revealed no phenotypic abnormalities of the oligodendroglial lineage, suggesting functional redundancy between HIF1α and HIF2α. We therefore generated HIF1α and HIF2α double conditional knockout (referred to as HIFα cKO).
To determine the role of HIFα in OPC differentiation, we used Pdgfrα-CreERT2:Hif1αfl/fl:Hif2αfl/fl (PDGFRα:HIFα cKO) transgenic mice. Tamoxifen was administered to PDGFRα:HIFα cKO and littermate control (Hif1αfl/fl:Hif2αfl/fl) pups at P1, P2, and P3, and the CNS tissue was analyzed at P8. Our previous data show that this tamoxifen paradigm induces >85% of gene cKO efficiency in OPCs (Zhang et al., 2018a,b). The density of differentiated OLs (CC1+Olig2+), but not OPCs (PDGFRα+Olig2+), was significantly decreased in the spinal cord white matter of PDGFRα:HIFα cKO mice compared with HIFα controls (Fig. 4A,B), suggesting that HIFα regulates OPC differentiation and is dispensable for OPC population expansion. In agreement with impaired OPC differentiation, the mRNA levels of major myelin protein (Mbp and Plp) and mature OL-enriched protein [Opalin and QK (called CC1)] were significantly reduced in PDGFRα:HIFα cKO mice compared with littermate controls (Fig. 4C). In the forebrain, developmental myelination in the periventricular white matter was delayed in PDGFRα:HIFα cKO mice at P8 (Fig. 4D) and became indistinguishable from that in littermate control mice at P14 (Fig. 4E). These data suggest that HIFα regulates the timing of developmental myelination.
Figure 4.
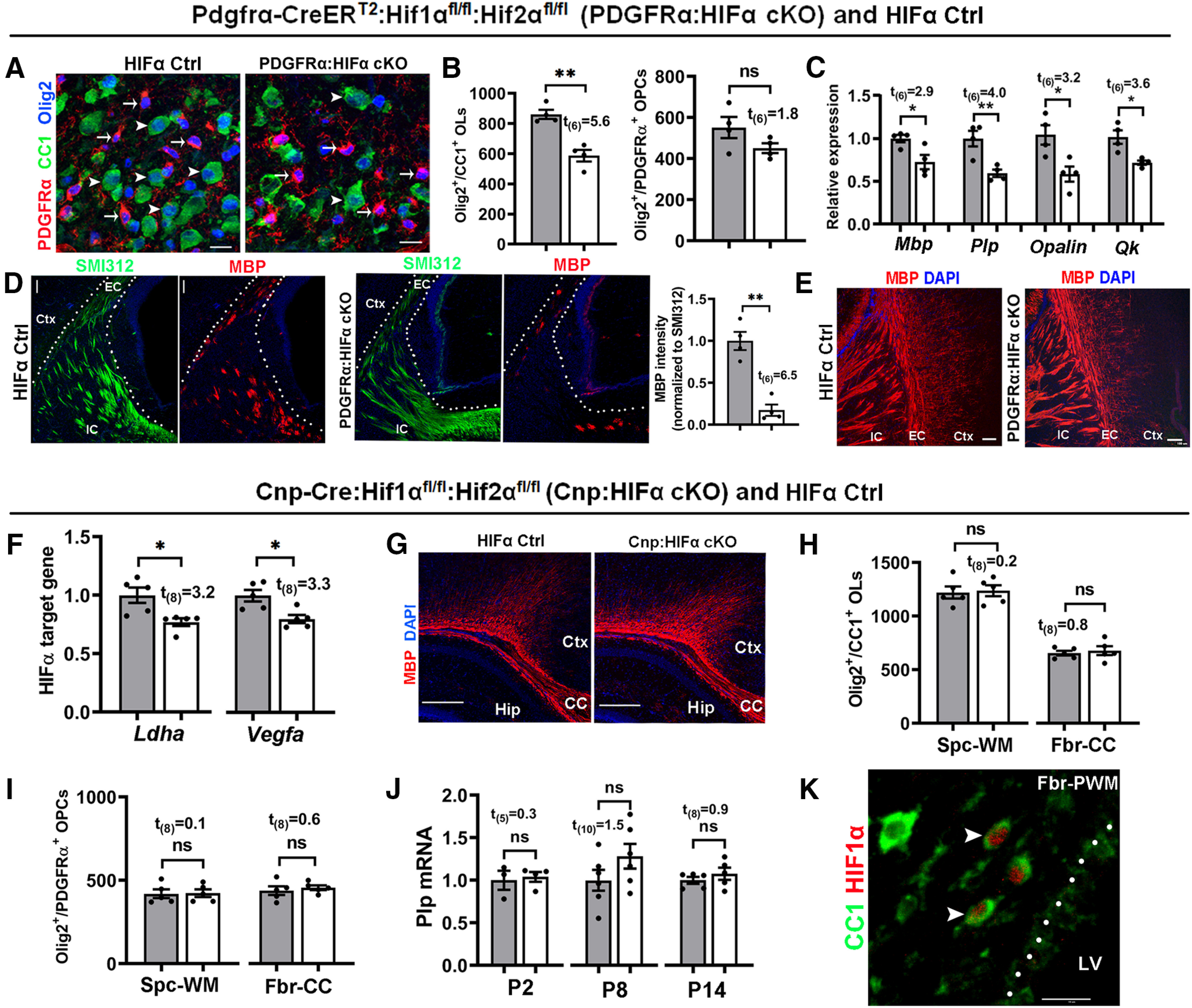
HIFα inactivation transiently delays developmental myelination by controlling OPC differentiation but not subsequent OL maturation. A–E, PDGFRα:HIFα cKO and littermate control (Hif1αfl/fl:Hif2αfl/fl) mice were treated with tamoxifen at P1, P2, and P3, and analyzed at P8 (n = 4 each group) and P14 (n = 3 each group). A, B, IHC (A) and quantification (B) of Olig2+CC1+OLs (arrowheads) and Olig2+PDGFRα+ OPCs (arrows) in the spinal cord. C, Relative expression of myelin protein genes of Mbp and Plp and mature OL-specific gene of Opalin and quake [Qk (called CC1)] in the forebrain measured by qRT-PCR. D, IHC of myelin marker MBP and pan-axonal marker SMI312 in forebrain periventricular white matter (left, marked by dotted area) and quantification (right) at P8. EC, External capsule; IC, internal capsule; Ctx, cortex. E, IHC of MBP in forebrain periventricular white matter at P14 (tamoxifen at P1, P2, and P3). F–J, Cnp:HIFα cKO and littermate control (Hif1αfl/fl:Hif2αfl/fl) mice were analyzed at P2, P8, and P14. F, qRT-PCR quantification of HIFα target genes Ldha and Vegfa in P8 spinal cord. G, Myelin staining by MBP in the forebrain. CC, Corpus callosum; Hip, hippocampus. H, I, Densities (#/mm2) of Olig2+CC1+ OLs (H) and Olig2+PDGFRα+ OPCs (I) at P14. Spc-WM, Spinal cord white matter; Fbr-CC, forebrain corpus callosum. J, qRT-PCR assay of exon3b-containing Plp mRNA, which is specific to mature OLs, in the spinal cord at different time points. K, IHC of CC1 and HIF1α in P8 forebrain periventricular white matter (Fbr-PWM). Arrowheads point to double-positive cells. LV, Lateral ventricle. Scale bars: A, K, 10 μm; D, 20 μm; E, 100 μm; G, 250 μm.
To determine the role of HIFα in oligodendroglial maturation, we used Cnp-Cre:Hif1αfl/fl:Hif2αfl/fl (Cnp:HIFα cKO) and littermate control (Hif1αfl/fl:Hif2αfl/fl) mice. Cnp:HIFα cKO mice were born at expected Mendelian ratios and displayed no behavioral abnormalities during postnatal development and throughout adult ages. The expression of canonical HIFα target genes Ldha and Vegfa (Sharp and Bernaudin, 2004) was significantly reduced in Cnp:HIFα cKO mice (Fig. 4F), thus validating the disruption of HIFα function. Unexpectedly, we found no significant difference in developmental myelination (Fig. 4G), the number of CC1+Olig2+ mature OLs (Fig. 4H) and PDGFRα+Olig2+ OPCs (Fig. 4I), and myelin gene expression (Fig. 4J) between Cnp:HIFα cKO and littermate control mice at different ages (P2, P8, and P14). Double IHC showed that some CC1+ OLs were positive for HIF1α in the periventricular white matter of the brain at P8 (Fig. 4K, arrowheads) but not at P14 (data not shown).
These data indicate that HIFα plays a minor role in oligodendrocyte maturation. Collectively, the contrast phenotypes derived from PDGFRα:HIFα cKO versus Cnp:HIFα cKO mice support a working model that HIFα controls the timing of developmental myelination by regulating the differentiation of OPCs into OLs but not subsequent oligodendrocyte maturation.
Disrupting oligodendroglial HIFα does not affect axonal integrity or cell survival
HIFα is required for neuronal survival, as HIFα deficiency in Nestin-expressing neural cells results in massive neuronal death and neurodegeneration (Tomita et al., 2003). Recent data reported that HIFα deficiency in Olig1-expressing neural cells leads to neuronal death and widespread axonal injury in the corpus callosum at the perinatal ages (Yuen et al., 2014). To determine whether OPC- or OL-specific HIFα deficiency causes axonal damage and cell death, we used IHC of SMI32 monoclonal antibody and cleaved Caspase 3 (CC3) antibody. SMI32 labels functionally compromised axons (Soulika et al., 2009; Yuen et al., 2014) and a subset of pyramidal neuronal cell bodies and dendrites during normal development (Campbell and Morrison, 1989; Voelker et al., 2004; Haynes et al., 2005; Fig. 5A, Ctx). We did not observe SMI32-immunoreactive signals in the corpus callosum of Cnp:HIFα cKO (Fig. 5A) and PDGFRα:HIFα cKO (Fig. 5C) mice compared with respective littermate controls. In contrast, many tightly packed axons running through the corpus callosum were clearly labeled by the pan-axonal marker SMI312 (Fig. 5B,D). Furthermore, the densities of CC3+ apoptotic cells and CC3+/Sox10+ apoptotic oligodendroglial lineage cells were statistically indistinguishable between PDGFRα (or Cnp):HIFα cKO mutants and their respective littermate controls (Fig. 5E–G). We used TEM to assess the structural integrity of axons in the corpus callosum of PDGFRα:HIFα cKO mice at P8, a time point at which few axons are myelinated. The density of axons with diameter ≥400 nm, which will be myelinated by oligodendrocyte during postnatal development (Lee et al., 2012; Hines et al., 2015), was similar between PDGFRα:HIFα cKO mice (458,401 ± 59,957 axons/mm2) and littermate controls (485,558 ± 38,780 axons/mm2; t(4) = 0.4, p = 0.72), suggesting that axons in HIFα cKO mice are not subjected to degeneration. We did not find visible morphologic differences in axonal structure (Fig. 5H,I, Ax) and axonal mitochondria (Fig. 5H,I, Mt) between PDGFRα:HIFα cKO and littermate control mice. Thus, disrupting oligodendroglial HIFα does not affect neuronal cell survival and axonal integrity in the white matter in the early postnatal CNS.
Figure 5.
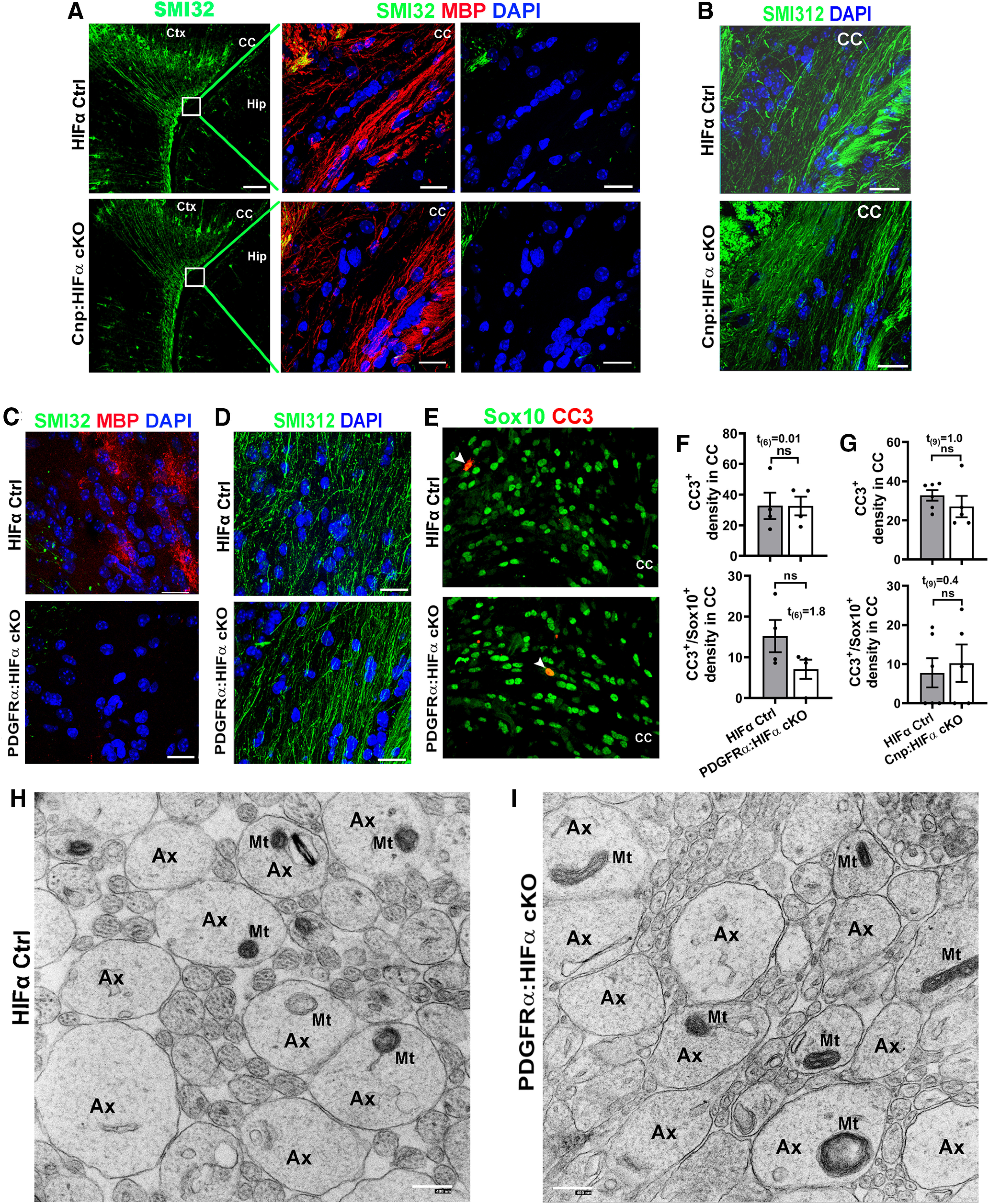
Oligodendroglial HIFα is dispensable for axonal integrity or cell death. A, Immunostaining images of P14 forebrain using SMI32, a monoclonal antibody recognizing the nonphosphorylated neurofilament proteins of heavy chain (NFH), which has been reported labeling injured axons and a subset of pyramidal neuron bodies and dendrites. Boxed areas show the corpus callosum (CC) in higher-magnification images of SMI32 and MBP in the right panels. Ctx, Cortex; Hip, hippocampus. Note the absence of SMI32-positive signals in the CC of both Cnp:HIFα cKO and Ctrl mice. B, Immunostaining images of P14 CC using SMI312, a monoclonal antibody recognizing both phosphorylated and nonphosphorylated NFH (a pan-axonal marker). C, D, IHC of SMI32/MBP (C) and SMI312 (D) in the CC of P8 non-Cre Ctrl and PDGFRα:HIFα cKO and littermate control (Hif1αfl/fl:Hif2αfl/fl) mice (tamoxifen injection at P1, P2, and P3). E, F, Representative confocal images and quantification of cells positive for cleaved CC3 and/or pan-oligodendroglial lineage marker Sox10 in P8 CC of PDGFRα:HIFα cKO and control mice. Arrowheads point to CC3+/Sox10+ cells. G, Quantification of CC3+ and CC3+/Sox10+ cells in P14 CC of Cnp:HIFα cKO and control mice. H, I, Representative TEM images showing the cross-sections of axons in the corpus callosum of HIFα Ctrl (I) and cKO (H) mice at P8 (tamoxifen injections at P1, P2, and P3) when myelination is barely detectable at the TEM level. Note that the morphology of axons (Ax) and axonal mitochondria (Mt) were indistinguishable between HIFα Ctrl and cKO mice. Scale bars: A, low magnification, 100 µm; A, high magnification, 20 µm; B–E, 20 µm; H, I, 400 nm.
Oligodendroglial HIFα plays a minor role, if any, in regulating Wnt/β-catenin activity
A previous study reported that aberrant HIFα stabilization in OPCs activates Wnt7a/Wnt7b expression and canonical Wnt signaling (i.e., Wnt/β-catenin; Yuen et al., 2014). The unbiased RNA sequencing (RNAseq) analysis of Sox10-Cre:Vhlfl/fl mice prompted us to revisit HIFα-Wnt regulation in oligodendroglial lineage cells (Fig. 6A–E). Sox10-Cre-mediated VHL cKO markedly elevated HIFα activity, as evidenced by the increased expression of canonical HIFα target genes Hk2, Ldha, Glut1, Bnip3, and Vegfa (Fig. 6A). In stark contrast to HIFα activation, there were no significant changes in the expression of Wnt genes Wnt7a and Wnt7b, and canonical Wnt signaling target genes Axin2 and Naked1 (Fig. 6B). The KEGG Pathway of HIF-1 signaling and the gene ontology (GO) term of response to hypoxia were significantly overrepresented among 255 upregulated genes (Fig. 6C,D), whereas the GO terms of oligodendrocyte differentiation and sterol biosynthesis were significantly overrepresented among 351 downregulated genes (Fig. 6E), which is in agreement with elevated HIFα signaling and diminished OPC differentiation in the CNS of Sox10:VHL cKO mice we documented in Figure 2A–D. However, we did not find overrepresentation of Wnt-related KEGG Pathways or GO terms among the differentially regulated genes. The unbiased analysis of gene regulation at the transcriptomic level strongly suggests that oligodendroglial HIFα stabilization plays a minor role in regulating canonical Wnt signaling.
To strengthen our conclusion derived from RNAseq, we analyzed OPCs isolated from PDGFRα:VHL cKO mice. Tamoxifen was injected to PDGFRα:VHL cKO and littermate control mice at P5, and brain OPCs were acutely purified at P6 (Fig. 6F) by MACS. As shown in Figure 6G, HIFα was indeed functionally stabilized in purified VHL-disrupted OPCs, as evidenced by the elevated expression of HIFα target genes Hk2, Ldha, and Glut1. However, there were no significant changes in the expression of canonical Wnt/β-catenin signaling target genes Axin2 and Naked1 and Wnt ligands Wnt7a and Wnt7b in VHL-disrupted OPCs compared with VHL-intact OPCs (Fig. 6H), suggesting that genetically stabilizing HIFα does not perturb the activity of autocrine Wnt/β-catenin or the expression of Wnt7a and Wnt7b in OPCs.
To corroborate our findings, stabilizing HIFα in PDGFRα+ OPCs did not affect the activity of Wnt/β-catenin signaling in the spinal cord at P8 (tamoxifen at P1, P2, and P3) and in the forebrain at P14 (tamoxifen at P6 and P7) of PDGFRα:VHL cKO mice compared with littermate controls, which was supported by the unaltered expression of canonical Wnt target genes at the mRNA and protein levels (Fig. 6I,J). We also found that the expression of Axin2, Naked1, Wnt7a, and Wnt7b was not perturbed in PDGFRα:HIFα cKO mice compared with littermate controls (Fig. 6K). Thus, HIFα does not regulate canonical Wnt signaling activity or Wnt7a/Wnt7b expression in the oligodendroglial lineage and in the CNS.
Blocking oligodendroglial-derived Wnt signaling does not affect HIFα hyperactivation-elicited inhibition of OPC differentiation and myelination
That nonperturbation of Wnt signaling in HIFα-stabilized OPCs led us to hypothesize that HIFα hyperactivation inhibits myelination in a manner independent of autocrine Wnt activation in OPCs. To test our hypothesis, we stabilized HIFα function and simultaneously blocked oligodendroglial-derived autocrine Wnt signaling.
Previous studies demonstrated that disrupting WLS blocks the secretion of Wnt ligands from Wnt-producing cells and inhibits intracellular Wnt signaling activity in Wnt-receiving cells (Bänziger et al., 2006; Bartscherer et al., 2006). To determine whether WLS deficiency blocks autocrine Wnt/β-catenin activity in OPCs, we knocked down WLS in Wnt3a-expressing primary OPCs (Fig. 7A–C). We chose Wnt3a because it is a typical Wnt ligand that activates the canonical Wnt signaling in Wnt-receiving cells. Wnt3a expression in OPCs significantly increased Wnt3a concentration and simultaneous WLS knockdown abrogated Wnt3a elevation in the culture medium of primary OPCs (Fig. 7D), suggesting that WLS is required for Wnt3a secretion from OPCs. We found that the activity of the canonical Wnt signaling, as assessed by Wnt target genes Axin2 and Sp5, was upregulated in Wnt3a-expressing OPCs but blocked in Wnt3a-expressing/WLS-deficient OPCs (Fig. 7E,F). These results demonstrate that disrupting WLS blocks OPC-derived autocrine Wnt/β-catenin signaling activity.
Figure 7.
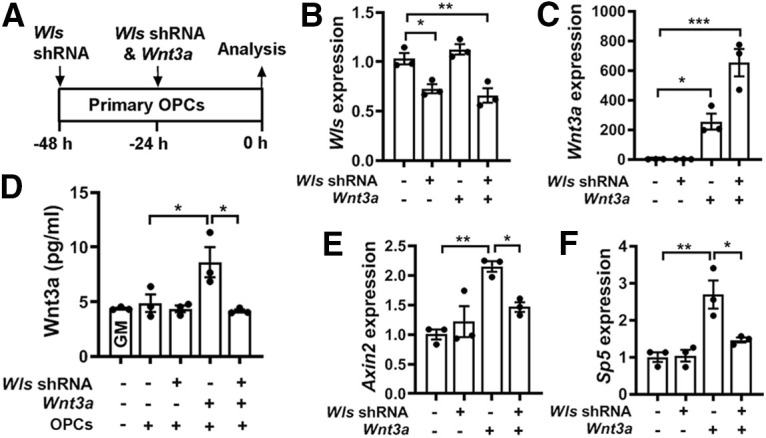
WLS deficiency inhibits Wnt3a secretion and blocks autocrine Wnt/β-catenin signaling activity in OPCs. A, Experimental design of cell transfection of Wls shRNA and Wnt3a-expressing plasmids to primary brain OPC maintained in the growth medium. B, C, qRT-PCR assay of Wls (B) and Wnt3a (C) expression in transfected primary OPCs. One-way ANOVA followed by Tukey's multiple-comparisons test: Wls, F(3,8) = 15.39, p = 0.001; Wnt3a, F(3,8) = 32.81, p < 0.0001. D, ELISA quantification of Wnt3a in the growth medium in the absence or presence of OPCs transfected by Wls shRNA and/or Wnt3a. One-way ANOVA followed by Tukey's multiple-comparisons test, ***p < 0.001; ns, not significant. F(4,10) = 6.70, p < 0.0069. Note that Wnt3a concentration in the GM in the presence of primary OPCs is statistically indistinguishable from that in the GM alone, suggesting that intact primary OPCs do not secrete Wnt3a. E, F, qRT-PCR quantification of Wnt target genes Axin2 and Sp5 in OPCs transfected with Wls shRNA and/or Wnt3a. One-way ANOVA followed by Tukey's multiple-comparisons test: Axin2, F(3,8) = 11.06, p = 0.0032; Sp5, F(3,8) = 13.25, p = 0.0018.
Genetically disrupting WLS in PDGFRα+ OPCs had no rescuing effects on HIFα hyperactivation-elicited hypomyelination (Fig. 8A–C), impaired OPC differentiation (Fig. 8D,E), and diminished myelin gene expression (Fig. 8F) in PDGFRα:VHL/WLS cKO mice, suggesting that autocrine Wnt signaling is unlikely a downstream pathway in mediating HIFα stabilization-induced inhibition of OPC differentiation. To corroborate this conclusion, we used a different inducible genetic model, Sox10-CreERT2:Vhlfl/fl:Wlsfl/fl (iSox10:VHL/WLS cKO; Fig. 9A). Conditionally stabilizing HIFα in Sox10-expressing oligodendroglial lineage cells resulted in impaired OPC differentiation and hypomyelination in iSox10:VHL cKO mice; however, simultaneous WLS cKO did not affect the degree of inhibited OPC differentiation and hypomyelination in iSox10:VHL/WLS cKO mice (Fig. 9B–E). We further demonstrated that disrupting WLS alone in Sox10-expressing oligodendroglial lineage cells had no detectable effect on normal OPC differentiation and developmental myelination in Sox10-CreERT2:Wlsfl/fl (iSox10:WLS cKO) mice compared with non-Cre littermate control mice (Fig. 10). Collectively, our results derived from cell-specific genetic approaches suggest that autocrine Wnt/β-catenin signaling plays a dispensable role in HIFα-regulated OPC differentiation and myelination.
Figure 8.
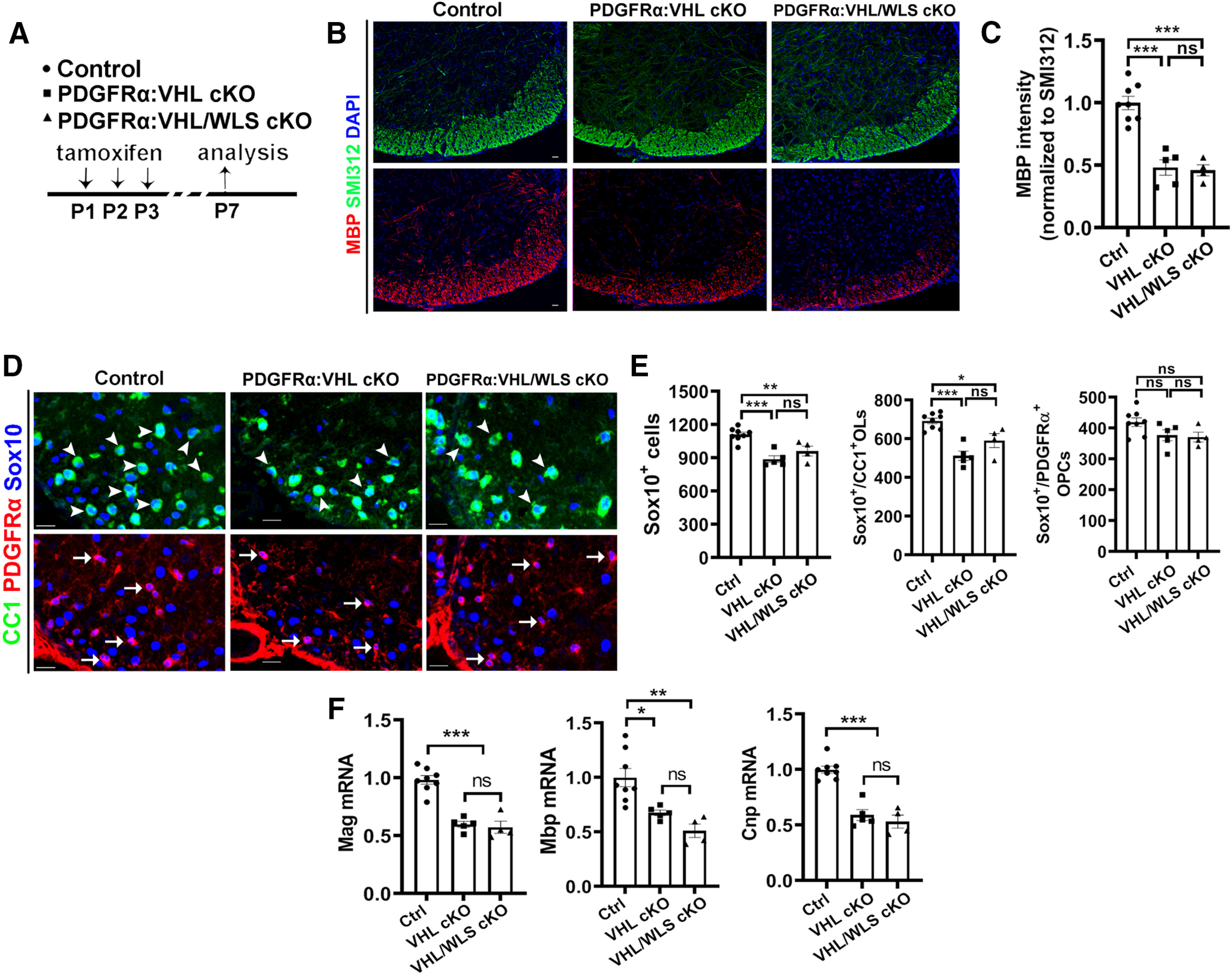
Disrupting WLS in PDGFRα-expressing OPCs does not affect the degree of inhibited OPC differentiation and hypomyelination elicited by HIFα hyperactivation. A, Experimental design for B–F. PDGFRα:VHL cKO, Pdgfrα-CreERT2:Vhlfl/fl (n = 5); PDGFRα:VHL/WLS cKO, Pdgfrα-CreERT2:Vhlfl/fl:Wlsfl/fl (n = 4); non-Cre Ctrl carrying Vhlfl/fl and/or Wlsfl/fl (n = 8). B, C, Representative confocal images (B) and quantification of myelination (C) by MBP staining. The signal of SMI312, a pan-axonal marker is indistinguishable among each group and used as an internal control of MBP quantification. One-way ANOVA followed by Tukey's multiple-comparisons test: F(2, 14)=31.93 p < 0.0001. D, E, Representative confocal images (D) and quantification (E) of Sox10+/CC1+ differentiated OLs (D, arrowheads), Sox10+/PDGFRα+ OPCs (D, arrows), and Sox10+ oligodendroglial lineage cells in the spinal cord. One-way ANOVA followed by Tukey's multiple-comparisons test: Sox10+, F(2,14) = 17.34, p = 0.0002; Sox10+/CC1+, F(2,14) = 18.44, p = 0.0001; Sox10+/PDGFRα+, F(2,14) = 2.967, p = 0.0843. F, qRT-PCR quantification of mature OL-enriched genes in the spinal cord. One-way ANOVA followed by Tukey's multiple-comparisons test: Mbp, F(2,14) = 11.53, p = 0.0011; Mag, F(2,14) = 39.88, p < 0.0001; Cnp, F(2,14) = 38.78, p < 0.0001. Scale bars: B, D, 20 µm.
Figure 9.
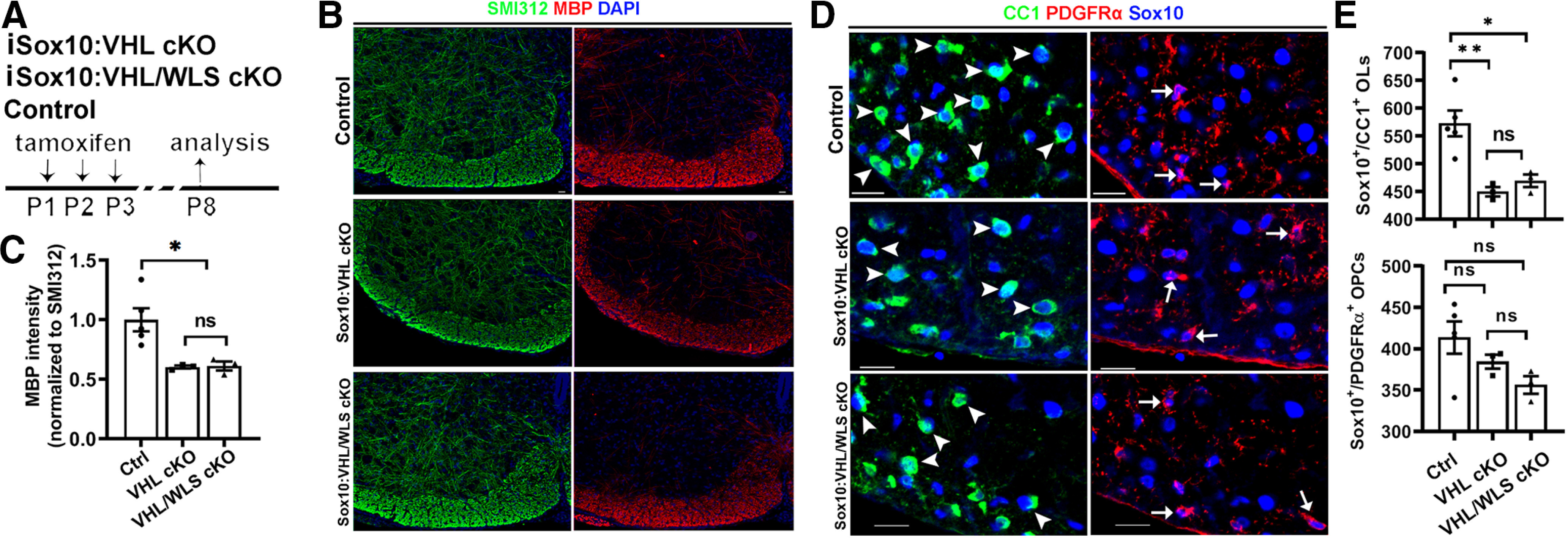
Disrupting WLS in Sox10-expressing oligodendroglial lineage cells does not affect HIFα hyperactivation-elicited inhibition of OPC differentiation and hypomyelination. A, Experimental design for B–E. Tamoxifen-inducible Sox10-CreERT2:Vhlfl/fl (iSox10:VHL cKO, n = 3); Sox10-CreERT2:Vhlfl/fl:Wlsfl/fl (iSox10:VHL/WLS cKO, n = 3); non-Cre Ctrl carrying Vhlfl/fl and/or Wlsfl/fl (n = 5).& B, C, Representative confocal images (B) and quantification (C) of myelination by MBP staining of the spinal cord. SMI312 signal was used as an internal control of MBP quantification. One-way ANOVA followed by Tukey's multiple-comparisons test: F(2,8) = 8.484, p = 0.0105. D, E, Representative confocal images (D) and quantification (E) of Sox10+/CC1+ differentiated OLs (D, arrowheads) and Sox10+/PDGFRα+ OPCs (D, arrows) in the spinal cord. One-way ANOVA followed by Tukey's multiple-comparisons test: Sox10+/CC1+, F(2,8) = 11.90, p = 0.004; Sox10+/PDGFRα+, F(2, 14) = 2.880, p = 0.1143. Scale bars: B, D, 20 µm.
Figure 10.
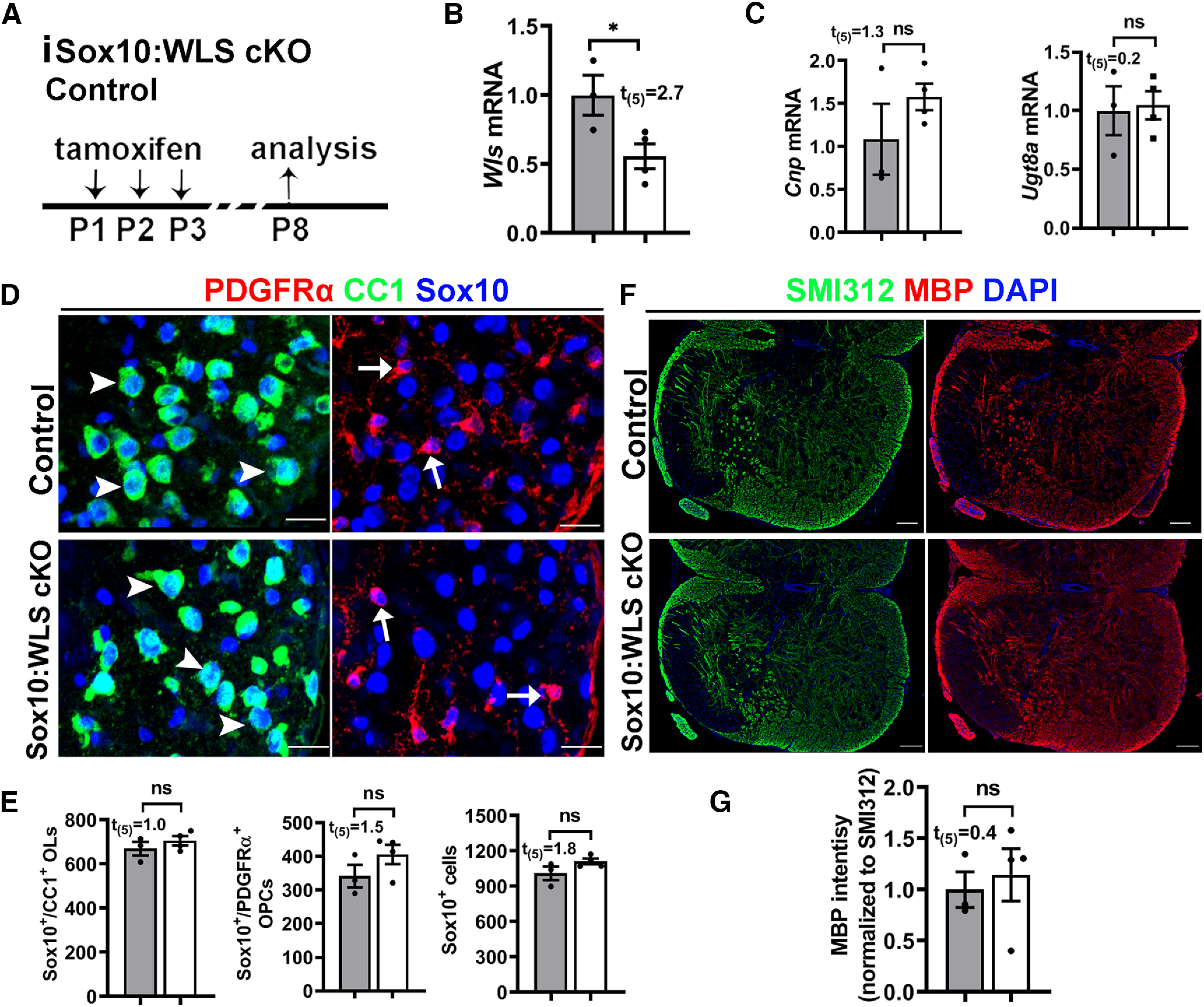
Disrupting WLS in Sox10-expressing oligodendroglial lineage cells does not perturb normal OPC differentiation and myelination. A, Experimental design for B–G. Tamoxifen-inducible Sox10-CreERT2:Wlsfl/fl (iSox10:WLS cKO, n = 4); littermate non-Cre control Wlsfl/fl mice (n = 3). B, C, qRT-PCR quantification of Wls (B) and differentiated OL-enriched gene Cnp and Ugt8a (C) in the spinal cord. D, E, Representative confocal images (D) and quantification (E) of Sox10+/CC1+ differentiated OLs (D, arrowheads), Sox10+/PDGFRα+ OPCs (D, arrows), and Sox10+ oligodendroglial lineage cells in the spinal cord. F, G, Representative confocal images (B) and quantification (C) of myelination by MBP staining of the spinal cord. SMI312 signal was used as an internal control of MBP quantification. Scale bars: D, 20 µm; E, 50 µm.
Sustained Sox9 activation mediates HIFα stabilization-elicited inhibition of OPC differentiation
The mRNA level of Sox9 was significantly upregulated in primary OPCs maintained in growth medium that had been treated with DMOG, a potent HIFα stabilizer (Yuen et al., 2014), and this upregulation was abrogated on simultaneous treatment of chetomin (Fig. 11A), an inhibitor of HIFα transcriptional activity (Kung et al., 2004; Viziteu et al., 2016). To support the functional regulation, our assay of ChIP followed by qPCR assay (ChIP-qPCR) showed that HIFα physically bound to the promoter region (−828 bp) of the rat Sox9 gene (Fig. 11B). We found that Sox9 mRNA expression was significantly upregulated in the spinal cord of PDGFRα:VHL cKO mice compared with littermate controls at P8 (Fig. 11C) and P14 (Fig. 11D). At the cellular level, sustained Sox9 expression was observed in HIFα-stabilized Sox10-expessing oligodendroglial lineage cells in Sox10:VHL cKO mice (Fig. 11E, right, arrowheads), in sharp contrast to control mice in which Sox9 was expressed at a much lower level or was barely detectable in Sox10-expressing cells (Fig. 11E, left, arrowheads). Our quantification showed that the percentage of Sox10+ cells expressing Sox9 (Fig. 11F) and the level of Sox9 mRNA (Fig. 11G) were significantly increased in Sox10:VHL cKO mice. Furthermore, Sox9 expression was significantly elevated in primary OPCs, which were purified from Sox10:VHL cKO neonatal brain (Fig. 11H). Sox9 is expressed in virtually all astrocytes in the postnatal CNS (Sun et al., 2017; Zhang et al., 2018b). We found that the density of Sox9+GFAP+ astrocytes was not significantly different in the spinal cords of Sox10:VHL cKO mutants from that in control mice (Fig. 11I). We also found that the percentage of Sox10+ oligodendroglial cells expressing Sox9 (Fig. 11J1,J2) was unchanged in PDGFRα:HIFα cKO mice (Fig. 11J3).
Figure 11.
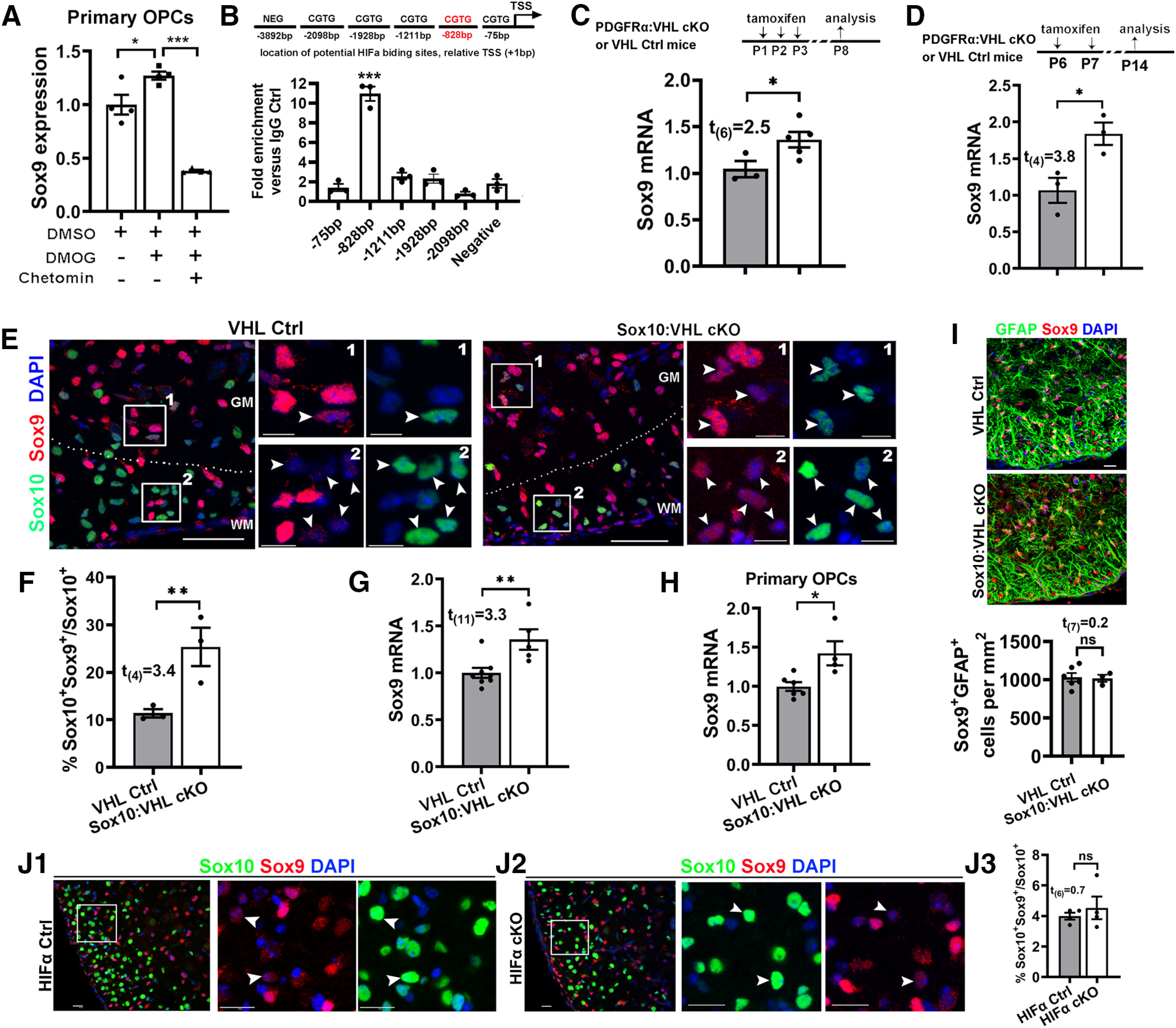
HIFα stabilization activates Sox9 expression. A, Sox9 expression in the purified primary OPCs assessed by qRT-PCR. Purified cortical OPCs from neonatal rat brains were treated with HIFα stabilizer DMOG (1 mm) in the absence and presence of HIFα signaling blocker chetomin (100 nm) for 7 h. n = 4 each group. One-way ANOVA followed by Tukey's multiple-comparisons test: F(2,9) = 62.53, p < 0.0001. B, Potential HIFα binding sites (CGTG) in the upstream sequence of rat Sox9 genes [+1 bp is defined as the transcription start site (TSS)] and ChIP-qPCR verification of physical binding of HIFα to the promoter region of −828 bp in primary OPCs treated with DMOG. One-way ANOVA with Dunnett post hoc test comparing each group with negative control: F(2,12) = 69.21 p < 0.0001. C, qRT-PCR quantification of Sox9 expression in P8 spinal cord of PDGFRα:VHL cKO (n = 5) and littermate control (Vhlfl/fl, n = 3) mice that had been treated with tamoxifen at P1, P2, and P3. D, qRT-PCR quantification of Sox9 expression in P14 spinal cords of PDGFRα:VHL cKO (n = 3) and littermate control (Vhlfl/fl, n = 3) mice that had received tamoxifen at P6 and P7. E, Confocal images showing that Sox9 is barely expressed in Sox10+ oligodendroglial lineage cells in the control spinal cord but upregulated in Sox10:VHL cKO mice at P5. WM, White matter; GM, gray matter. The boxed areas of #1 and #2 were shown at higher magnification at the right. F, Percentage of Sox10+ oligodendroglial lineage cells expressing Sox9 in the spinal cord (n = 3 each group). G, qRT-PCR quantification of Sox9 mRNA in the spinal cord of Sox10:VHL cKO (n = 5) and littermate control (Vhlfl/+ and/or Vhlfl/fl, n = 8) mice at P5. H, qRT-PCR assay of Sox9 expression in primary OPCs isolated from the neonatal brain of Sox10-Cre:Vhlfl/fl (n = 4) and littermate control (n = 6) pups. I, Representative confocal images and quantification of Sox9+GFAP+ astrocytes in the ventral spinal cord of P5 Sox10-Cre:Vhlfl/fl (n = 3) and littermate control (n = 6) mice. J1–J3, Representative confocal images and quantification of Sox10 and Sox9 in the ventral spinal cord of P8 PDGFRα:HIFα cKO (n = 4) and littermate control (n = 4) mice (tamoxifen at P1, P2, and P3). Arrowheads point to Sox10+Sox9+ cells. Scale bars: E, 50 μm; I, 10 μm; E, boxed areas, 10 μm; J1, J2, 20 μm.
Recent data demonstrate that Sox9 is rapidly downregulated on OPC differentiation (Reiprich et al., 2017), prompting us to hypothesize that sustained Sox9 expression may inhibits OPC differentiation. To test this hypothesis, we overexpressed Sox9 in primary OPCs in growth medium and assessed OPC differentiation in differentiating medium (Fig. 12A). Sox9 mRNA and protein were significantly increased in primary OPCs transfected with Sox9-expressing plasmid compared with those transfected with empty vector (EV; Fig. 12B–D). Sox9 overexpression reduced the mRNA expression of the most abundant myelin protein Plp and the potent prodifferentiation factor Myrf (Emery et al., 2009) assessed by qRT-PCR (Fig. 12E), the number of ramified MBP+/Sox10+ oligodendrocytes assessed by immunocytochemistry (Fig. 12F), and the protein level of MBP assessed by Western blot (Fig. 12G). To define the role of Sox9 activation in HIFα-regulated OPC differentiation, we stabilized HIFα in OPCs by DMOG treatment and simultaneously knocked down Sox9 expression by siRNA (Fig. 12H–J). Our data showed that Sox9 downregulation rescued the degree of myelin gene inhibition elicited by DMOG treatment, as evidenced by the density of MBP+/Sox10+ differentiated OLs with ramified morphology (Fig. 12K,L) and the expression of myelin gene Mbp (Fig. 12M). Collectively, our results suggest that sustained Sox9 expression is one of the downstream molecular pathways in mediating HIFα-regulated OPC differentiation.
Discussion
Summary of key findings and conclusions
Through the analysis of a series of HIFα genetic models (stage-dependent HIFα loss-of-function or gain-of-function in OPCs and OLs), our study provides compelling evidence supporting an alternative understanding of the cellular and molecular mechanisms underlying HIFα-regulated CNS myelination (Fig. 12N). At the cellular level, our in vivo genetic data support a new working model in which physiological HIFα transiently regulates OPC differentiation, whereas persistent HIFα activation directly causes arrested OPC differentiation, leading to developmental hypomyelination. At the molecular level, HIFα regulates OPC differentiation in a manner independent of the autocrine Wnt/β-catenin signaling. Instead, dysregulated Sox9 expression is one of the downstream pathways in mediating HIFα activation-elicited inhibition of OPC differentiation (Fig. 12N).
HIFα regulates developmental myelination by controlling OPC differentiation but not subsequent oligodendrocyte maturation
One of the unexpected observations is the differential responses of OPCs and OLs to HIFα disruption and hyperactivation. Disrupting or stabilizing HIFα function results in severe disturbance of myelin gene expression and developmental myelination in the CNS of OPC-specific PDGFRα:HIFα (VHL) cKO mice. In sharp contrast, HIFα disruption or stabilization in Cnp:HIFα (VHL) cKO mice does not perturb normal myelin gene expression and myelination. Previous genetic studies suggest that Cnp-Cre elicits gene disruption primarily in the later stages of oligodendrocyte development. For example, disrupting DNA methyltransferase 1 (DNMT1) by Cnp-Cre line shows no phenotypes in OPC proliferation and expansion, in sharp contrast to DMNT1 disruption by Olig1-Cre, which targets the progenitor stages of the oligodendroglial lineage (Moyon et al., 2016). Our recent studies also demonstrated that disrupting Sox2 by Cnp-Cre shows no phenotypes in OPC proliferation and expansion, in sharp contrast to Sox2 disruption by Sox10-Cre or Pdgfrα-CreERT2, which targets OPCs (Zhang et al., 2018a,b). Therefore, the discrepant phenotypes in HIFα (or VHL) deletion driven by Cnp-Cre versus Sox10-Cre or Pdgfrα-CreERT2 (Figs. 2, 4, 5) support the interpretation that HIFα play a major role in OPC differentiation into OLs and is dispensable for oligodendrocyte maturation.
We observed impaired OPC differentiation in HIFα activation (Figs. 2A–C, 3) and inactivation (Fig. 4A–D) genetic models. These observations suggest delicate downstream pathways regulated by HIFα. We recently reported that HIFα depletion reduces the canonical HIFα target genes involving in glucose metabolism and CNS angiogenesis (Zhang et al., 2020), both of which entail energy supply for OPC differentiation(Yuen et al., 2014). It is possible that HIFα inactivation may transiently affect OPC differentiation through limiting the energy supply regulated by physiological HIFα, whereas sustained HIFα activation might stimulate additional noncanonical HIFα target genes, which are otherwise nonresponsive to physiological HIFα during normal development. Our experimental data identified noncanonical targets such as Sox9 whose expression is upregulated in HIFα activation mice and unaltered in HIFα inactivation mice. Furthermore, the unaltered developmental myelination in OL-specific HIFα activation and inactivation models indicates that OLs are resistant to HIFα dysregulation, which is in line with the previous concept that OLs are remarkably resistant to hypoxia/ischemia injury (Back et al., 2002, 2007).
Molecular mechanisms underlying HIFα-regulated OPC differentiation
Previous study used cerebellar slice/cell culture as a studying model and proposed that the autocrine activation of Wnt/β-catenin signaling in OPCs by Wnt7a/Wnt7b plays a crucial role in HIFα activation-induced hypomyelination (Yuen et al., 2014). Our data derived from in vivo genetic models do not support this concept. First, unbiased RNAseq analysis did not reveal dysregulation of the Wnt/β-catenin signaling activity and Wnt7a/Wnt7b expression. Second, neither the activity of Wnt/β-catenin signaling nor the expression of Wnt7a/Wnt7b is perturbed in HIFα-stabilized OPCs. Third, disrupting the Wnt secretion mediator WLS blocks OPC-derived autocrine Wnt/β-catenin signaling, but it does not affect the degree of inhibited OPC differentiation elicited by HIFα hyperactivation. Our findings indicate that Wnt7a/Wnt7b or Wnt/β-catenin signaling is unlikely a direct target of HIFα in the oligodendroglial lineage cells (Fig. 12N), which is supported by a recently submitted study from an independent group (Allan et al., 2020). The reasons for the different observations between our current study and prior study (Yuen et al., 2014) are unclear. It is possible that noncellular specificity and/or being off-target of pharmacological compounds may underlie the discrepancy. HIFα stabilization induced by hypoxia treatment or by pharmacological DMOG application occurs not only in oligodendroglial lineage cells but also in other lineages of neural cells or vascular cells. Similarly, Wnt/β-catenin inhibition in cultured slices by pharmacological XAV939 or IWP2 treatment may also happen in other lineage cells (Yuen et al., 2014). We used cell-specific Cre-loxP genetic approaches to circumvent the potential caveats of pharmacological application and convincingly demonstrated that OPC-derived autocrine Wnt/β-catenin plays a minor role, if any, in mediating HIFα-regulated OPC differentiation and myelination.
Another novel finding of this study is the identification of Sox9 as a potential target that mediates HIFα stabilization-elicited inhibition of OPC differentiation (Fig. 12N, right). Sox9 is highly expressed in neural stem cells (Scott et al., 2010), downregulated in OPCs (Stolt et al., 2003), and completely absent from differentiated OLs (Sun et al., 2017). Sox9 disruption affects OPC specification but not OPC terminal differentiation (Stolt et al., 2003; Finzsch et al., 2008). HIFα stabilization activated Sox9 expression in OPCs both in vitro and in vivo, which is in agreement with previous data showing that Sox9 is transcriptionally activated by HIFα in chondrocytes (Amarilio et al., 2007) and pancreatic β-cell precursor cells (Puri et al., 2013). We found that elevated Sox9 expression perturbed OPC differentiation and that Sox9 knockdown rescued the extent of inhibited OPC differentiation elicited by pharmacological HIFα stabilization in primary OPC culture system.
It should be pointed out that the nonregulation of the HIFα–Wnt axis does not negate the crucial role of Wnt/β-catenin in oligodendroglial lineage cells. Previous studies have revealed an inhibitory effect of Wnt/β-catenin activation on OPC differentiation (Guo et al., 2015). However, these studies modulated the signaling activity through manipulating the intracellular components or relevant regulatory molecules within OPCs, for instance, by disrupting intracellular β-catenin (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009), Axin2 (Fancy et al., 2011), APC (Lang et al., 2013; Fancy et al., 2014), APCDD1 (Lee et al., 2015b), and Daam2 (Lee et al., 2015a). The Wnt-producing cells, which play a crucial role in activating the intracellular Wnt/β-catenin signaling axis in OPCs, remain unknown (Guo et al., 2015). Our WLS cKO data demonstrate that oligodendroglial lineage-derived autocrine Wnt plays a minor role in OPC differentiation and myelination in the early postnatal CNS under physiological (Fig. 10) or HIFα-stabilized (Figs. 8, 9) conditions, suggesting that Wnt ligands derived from other lineage cells may be responsible for control OPC differentiation through paracrine Wnt signaling. Previous RNAseq data (Zhang et al., 2014) indicate that astrocytes may be the major cellular source of Wnt proteins, for example Wnt7a and Wnt5a. Future studies are needed to test the tempting hypothesis that astrocyte-derived paracrine Wnt signaling may play major a role in controlling OPC differentiation under normal or HIFα-stabilized conditions.
Implications of our findings in hypoxia/ischemia-induced oligodendroglial pathology in preterm white matter injury
Disturbance of normal developmental myelination is one of the established pathologic hallmarks in preterm (or premature) infants affected by diffuse WMI (Deng, 2010; Back, 2017). Hypoxia/ischemia-induced diffuse WMI frequently occurs in the brains of preterm infants because of the anatomic and physiological immaturity of the respiratory system and the brain white matter vasculature. We demonstrated that, compared with healthy controls, HIF1α was markedly elevated in the brain white matter of mice subjected to hypoxia-ischemia injury at the time equivalent to developmental myelination in human brains (Fig. 1F–I). Our findings—that sustained HIFα activation disturbs OPC differentiation and results in hypomyelination—suggest that oligodendroglial HIFα upregulation may play a crucial role in hypoxia/ischemia-induced myelination disturbance. The function and molecular mechanisms of oligodendroglial HIFα in hypoxia/ischemia-induced hypomyelination (Liu et al., 2011) have yet to be determined in genetic animal models. Our in vivo data indicate that sustained HIFα activation in response to hypoxia-ischemia injury may inhibit OPC differentiation and cause hypomyelination, and that the underlying mechanisms may be independent of OPC autocrine canonical Wnt signaling as previously proposed. Future studies using cell-specific genetic HIFα models (HIFα loss-of-function and gain-of-function) and preterm equivalent mouse models of diffuse white matter injury are needed to address these important questions.
Footnotes
The authors declare no competing financial interests.
This study was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke (Grants R21-NS-109790 and R01-NS-094559 to F.G.) and Shriners Hospitals for Children (Grants 86100, 85200-NCA16, 85107-NCA-19 to F.G.; Grant 84307-NCAL to S.Z.).
References
- Allan KC, Hu LR, Morton AR, Scavuzzo MA, Gevorgyan AS, Clayton BLL, Bederman IR, Hung S, Bartels CF, Madhavan M, Tesar PJ (2020) Non-canonical targets of HIF1a drive cell-type-specific dysfunction. BioRxiv. Advance online publication. Retrieved November 16, 2020 Doi: 10.1101/2020.04.03.003632 [DOI] [Google Scholar]
- Amarilio R, Viukov SV, Sharir A, Eshkar-Oren I, Johnson RS, Zelzer E (2007) HIF1alpha regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 134:3917–3928. 10.1242/dev.008441 [DOI] [PubMed] [Google Scholar]
- Back SA. (2017) White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol 134:331–349. 10.1007/s00401-017-1718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM (2002) Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci 22:455–463. 10.1523/JNEUROSCI.22-02-00455.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Riddle A, McClure MM (2007) Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 38:724–730. 10.1161/01.STR.0000254729.27386.05 [DOI] [PubMed] [Google Scholar]
- Bänziger C, Soldini D, Schütt C, Zipperlen P, Hausmann G, Basler K (2006) Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 125:509–522. 10.1016/j.cell.2006.02.049 [DOI] [PubMed] [Google Scholar]
- Bartscherer K, Pelte N, Ingelfinger D, Boutros M (2006) Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 125:523–533. 10.1016/j.cell.2006.04.009 [DOI] [PubMed] [Google Scholar]
- Bercury KK, Macklin WB (2015) Dynamics and mechanisms of CNS myelination. Dev Cell 32:447–458. 10.1016/j.devcel.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH (1989) Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol 282:191–205. 10.1002/cne.902820204 [DOI] [PubMed] [Google Scholar]
- Chung SH, Guo F, Jiang P, Pleasure DE, Deng W (2013) Olig2/Plp-positive progenitor cells give rise to Bergmann glia in the cerebellum. Cell Death Dis 4:e546. 10.1038/cddis.2013.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham LA, Candelario K, Li L (2012) Roles for HIF-1α in neural stem cell function and the regenerative response to stroke. Behav Brain Res 227:410–417. 10.1016/j.bbr.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W. (2010) Neurobiology of injury to the developing brain. Nat Rev Neurol 6:328–336. 10.1038/nrneurol.2010.53 [DOI] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA (2009) Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138:172–185. 10.1016/j.cell.2009.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH (2009) Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 23:1571–1585. 10.1101/gad.1806309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S, Rowitch DH (2014) Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci 17:1841 10.1038/nn1214-1841a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ, Rowitch DH (2011) Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci 14:1009–1016. 10.1038/nn.2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson K, Reid M, See J, Crenshaw EB 3rd, Grinspan JB (2009) Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci 42:255–265. 10.1016/j.mcn.2009.07.010 [DOI] [PubMed] [Google Scholar]
- Finzsch M, Stolt CC, Lommes P, Wegner M (2008) Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development 135:637–646. 10.1242/dev.010454 [DOI] [PubMed] [Google Scholar]
- Guo F, Lang J, Sohn J, Hammond E, Chang M, Pleasure D (2015) Canonical Wnt signaling in the oligodendroglial lineage-puzzles remain. Glia 63:1671–1693. 10.1002/glia.22813 [DOI] [PubMed] [Google Scholar]
- Harb R, Whiteus C, Freitas C, Grutzendler J (2013) In vivo imaging of cerebral microvascular plasticity from birth to death. J Cereb Blood Flow Metab 33:146–156. 10.1038/jcbfm.2012.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL, Borenstein NS, Desilva TM, Folkerth RD, Liu LG, Volpe JJ, Kinney HC (2005) Axonal development in the cerebral white matter of the human fetus and infant. J Comp Neurol 484:156–167. 10.1002/cne.20453 [DOI] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, Appel B (2015) Neuronal activity biases axon selection for myelination in vivo. Nat Neurosci 18:683–689. 10.1038/nn.3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhao XF, Zheng K, Qiu M (2013) Regulation of the timing of oligodendrocyte differentiation: mechanisms and perspectives. Neurosci Bull 29:155–164. 10.1007/s12264-013-1314-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z. (2009) Hypoxia or in situ normoxia: the stem cell paradigm. J Cell Physiol 219:271–275. 10.1002/jcp.21690 [DOI] [PubMed] [Google Scholar]
- Jablonska B, Scafidi J, Aguirre A, Vaccarino F, Nguyen V, Borok E, Horvath TL, Rowitch DH, Gallo V (2012) Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27Kip1 expression. J Neurosci 32:14775–14793. 10.1523/JNEUROSCI.2060-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM (2004) Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 6:33–43. 10.1016/j.ccr.2004.06.009 [DOI] [PubMed] [Google Scholar]
- Lang J, Maeda Y, Bannerman P, Xu J, Horiuchi M, Pleasure D, Guo F (2013) Adenomatous polyposis coli regulates oligodendroglial development. J Neurosci 33:3113–3130. 10.1523/JNEUROSCI.3467-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lee HK, Chaboub LS, Zhu W, Zollinger D, Rasband MN, Fancy SP, Deneen B (2015a) Daam2-PIP5K Is a Regulatory Pathway for Wnt Signaling and Therapeutic Target for Remyelination in the CNS. Neuron 85:1227–1243. 10.1016/j.neuron.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Laug D, Zhu W, Patel JM, Ung K, Arenkiel BR, Fancy SP, Mohila C, Deneen B (2015b) Apcdd1 stimulates oligodendrocyte differentiation after white matter injury. Glia 63:1840–1849. 10.1002/glia.22848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Leach MK, Redmond SA, Chong SY, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, Chan JR (2012) A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods 9:917–922. 10.1038/nmeth.2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Candelario KM, Thomas K, Wang R, Wright K, Messier A, Cunningham LA (2014) Hypoxia inducible factor-1α (HIF-1α) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J Neurosci 34:16713–16719. 10.1523/JNEUROSCI.4590-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Shen Y, Plane JM, Pleasure DE, Deng W (2011) Neuroprotective potential of erythropoietin and its derivative carbamylated erythropoietin in periventricular leukomalacia. Exp Neurol 230:227–239. 10.1016/j.expneurol.2011.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Shen Y, Plane JM, Deng W (2013) Vulnerability of premyelinating oligodendrocytes to white-matter damage in neonatal brain injury. Neurosci Bull 29:229–238. 10.1007/s12264-013-1311-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic J, Maisel M, Wegner F, Leuchtenberger J, Wenger RH, Gerlach M, Storch A, Schwarz J (2007) Lack of hypoxia-inducible factor-1α impairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J Neurosci 27:412–421. 10.1523/JNEUROSCI.2482-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyon S, Huynh JL, Dutta D, Zhang F, Ma D, Yoo S, Lawrence R, Wegner M, John GR, Emery B, Lubetzki C, Franklin RJ, Fan G, Zhu J, Dupree JL, Casaccia P (2016) Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep 15:748–760. 10.1016/j.celrep.2016.03.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri S, Akiyama H, Hebrok M (2013) VHL-mediated disruption of Sox9 activity compromises β-cell identity and results in diabetes mellitus. Genes Dev 27:2563–2575. 10.1101/gad.227785.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiprich S, Cantone M, Weider M, Baroti T, Wittstatt J, Schmitt C, Küspert M, Vera J, Wegner M (2017) Transcription factor Sox10 regulates oligodendroglial Sox9 levels via microRNAs. Glia 65:1089–1102. 10.1002/glia.23146 [DOI] [PubMed] [Google Scholar]
- Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV, Booth S, Gao B, Cheah KS, Lovell-Badge R, Briscoe J (2010) SOX9 induces and maintains neural stem cells. Nat Neurosci 13:1181–1189. 10.1038/nn.2646 [DOI] [PubMed] [Google Scholar]
- Semenza GL. (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148:399–408. 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ (2013) Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106-107:1–16. 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M (2004) HIF1 and oxygen sensing in the brain. Nat Rev Neurosci 5:437–448. 10.1038/nrn1408 [DOI] [PubMed] [Google Scholar]
- Shen Y, Plane JM, Deng W (2010) Mouse models of periventricular leukomalacia. J Vis Exp 18:1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D (2009) Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci 29:14965–14979. 10.1523/JNEUROSCI.3794-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M (2003) The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev 17:1677–1689. 10.1101/gad.259003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Cornwell A, Li J, Peng S, Osorio MJ, Aalling N, Wang S, Benraiss A, Lou N, Goldman SA, Nedergaard M (2017) SOX9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J Neurosci 37:4493–4507. 10.1523/JNEUROSCI.3199-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N, Sakamoto H, Gassmann M, Kageyama R, Ueda N, Gonzalez FJ, Takahama Y (2003) Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol Cell Biol 23:6739–6749. 10.1128/mcb.23.19.6739-6749.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viziteu E, Grandmougin C, Goldschmidt H, Seckinger A, Hose D, Klein B, Moreaux J (2016) Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br J Cancer 114:519–523. 10.1038/bjc.2016.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker CC, Garin N, Taylor JS, Gähwiler BH, Hornung JP, Molnár Z (2004) Selective neurofilament (SMI-32, FNP-7 and N200) expression in subpopulations of layer V pyramidal neurons in vivo and in vitro. Cereb Cortex 14:1276–1286. 10.1093/cercor/bhh089 [DOI] [PubMed] [Google Scholar]
- Volpe JJ. (2009) Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 8:110–124. 10.1016/S1474-4422(08)70294-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR (2009) HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci 12:829–838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH (2014) Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158:383–396. 10.1016/j.cell.2014.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhu L, Fan M (2011) Oxygen, a key factor regulating cell behavior during neurogenesis and cerebral diseases. Front Mol Neurosci 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kim B, Zhu X, Gui X, Wang Y, Lan Z, Prabhu P, Fond K, Wang A, Guo F (2020) Glial type specific regulation of CNS angiogenesis by HIFα-activated different signaling pathways. Nat Commun 11:2027. 10.1038/s41467-020-15656-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Rasai A, Wang Y, Xu J, Bannerman P, Erol D, Tsegaye D, Wang A, Soulika A, Zhan X, Guo F (2018a) The stem cell factor Sox2 is a positive timer of oligodendrocyte development in the postnatal murine spinal cord. Mol Neurobiol 55:9001–9015. 10.1007/s12035-018-1035-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Zhu X, Gui C, Croteau L, Song J, Xu A, Wang P, Bannerman F Guo (2018b) Sox2 is essential for oligodendroglial proliferation and differentiation during postnatal brain myelination and CNS remyelination. J Neurosci 38:1802–1820. 10.1523/JNEUROSCI.1291-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]