

Abstract
Dysfunction of neuronal circuits is an important determinant of neurodegenerative diseases. Synaptic dysfunction, death, and intrinsic activity of neurons are thought to contribute to the demise of normal behavior in the disease state. However, the interplay between these major pathogenic events during disease progression is poorly understood. Spinal muscular atrophy (SMA) is a neurodegenerative disease caused by a deficiency in the ubiquitously expressed protein SMN and is characterized by motor neuron death, skeletal muscle atrophy, as well as dysfunction and loss of both central and peripheral excitatory synapses. These disease hallmarks result in an overall reduction of neuronal activity in the spinal sensory-motor circuit. Here, we show that increasing neuronal activity by chronic treatment with the FDA-approved potassium channel blocker 4-aminopyridine (4-AP) improves motor behavior in both sexes of a severe mouse model of SMA. 4-AP restores neurotransmission and number of proprioceptive synapses and neuromuscular junctions (NMJs), while having no effects on motor neuron death. In addition, 4-AP treatment with pharmacological inhibition of p53-dependent motor neuron death results in additive effects, leading to full correction of sensory-motor circuit pathology and enhanced phenotypic benefit in SMA mice. Our in vivo study reveals that 4-AP-induced increase of neuronal activity restores synaptic connectivity and function in the sensory-motor circuit to improve the SMA motor phenotype.
SIGNIFICANCE STATEMENT Spinal muscular atrophy (SMA) is a neurodegenerative disease, characterized by synaptic loss, motor neuron death, and reduced neuronal activity in spinal sensory-motor circuits. However, whether these are parallel or dependent events is unclear. We show here that long-term increase of neuronal activity by the FDA-approved drug 4-aminopyridine (4-AP) rescues the number and function of central and peripheral synapses in a SMA mouse model, resulting in an improvement of the sensory-motor circuit and motor behavior. Combinatorial treatment of pharmacological inhibition of p53, which is responsible for motor neuron death and 4-AP, results in additive beneficial effects on the sensory-motor circuit in SMA. Thus, neuronal activity restores synaptic connections and improves significantly the severe SMA phenotype.
Keywords: motor neuron death, neurodegeneration, neuronal activity, sensory-motor circuit, spinal muscular atrophy, synaptic dysfunction
Introduction
Neuronal activity plays key roles in the formation, refinement and maintenance of neuronal circuits by shaping synaptic function, axonal plasticity, and survival of neurons in health and disease. Because of its broad effects on gene regulation, calcium homeostasis and neurotrophic support within the nervous system (Tam and Gordon, 2003; West and Greenberg, 2011; Dekkers et al., 2013), imbalance of neuronal activity contributes significantly to neurodegenerative diseases (Chopra and Shakkottai, 2014; Leroy and Zytnicki, 2015; Nelson and Valakh, 2015; Shorrock et al., 2019). Aside from altered neuronal activity, the hallmarks of neurodegenerative diseases are selective neuronal death, dysfunction, and synaptic elimination (Gillingwater and Wishart, 2013; Chi et al., 2018).
The two most common neurodegenerative diseases affecting motor neurons, spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS), are characterized by impairment of sensory-motor circuits, including motor neuron death and degeneration of excitatory synapses, resulting in muscle paralysis and patient's death (Mentis et al., 2011; Torres-Benito et al., 2012; Vinsant et al., 2013; Tisdale and Pellizzoni, 2015; Vaughan et al., 2015; Fletcher et al., 2017; Baczyk et al., 2020). While the onset of ALS typically occurs in adulthood (Mejzini et al., 2019), SMA is the most frequent genetic cause of infant mortality resulting from a homozygous deletion or mutation of the survival motor neuron 1 (SMN1) gene with the retention of the hypomorphic SMN2 gene leading to the ubiquitous deficiency of the SMN protein (Lefebvre et al., 1995). The SMNΔ7 mouse model of SMA recapitulates key features of the most severe form of the human disease (Le et al., 2005; Mentis et al., 2011; Martinez et al., 2012; Tisdale and Pellizzoni, 2015). SMA mice exhibit impaired motor neuron firing (Fletcher et al., 2017), reduced function and number of the excitatory proprioceptive synapses and neuromuscular junctions (NMJs) before motor neuron death (Ling et al., 2010, 2012; Ruiz et al., 2010; Mentis et al., 2011). Thus, these findings suggest an early systemic decrease of neuronal activity as one of the earliest pathologic alterations in the sensory-motor circuit in motor neuron diseases. However, whether the decrease of activity causes neuronal death and synaptic loss has not been resolved and is of importance for many neurodegenerative diseases.
The above findings raise the possibility that an increase of neuronal activity might benefit the sensory-motor circuit pathology. Accordingly, exercise or pharmacological increase of neuronal activity improves proprioceptive synaptic function and cellular disease-related markers of motor neurons in ALS (Saxena et al., 2013; Baczyk et al., 2020) and the phenotype of SMA mouse models (Biondi et al., 2008; Fletcher et al., 2017). Interestingly, the FDA-approved voltage-activated potassium channel blocker 4-aminopyridine (4-AP) corrected motor circuit defects in a Drosophila model of SMA by increasing neuronal activity (Hayes, 2004; Imlach et al., 2012), making it a promising candidate to address whether activity-dependent mechanisms contribute to motor neuron death and synaptic loss in vivo.
Here, we investigate the chronic effects of neuronal activity on the SMA sensory-motor circuit by administering 4-AP into SMA mice. We found that 4-AP-induced neuronal activity ameliorates the SMA motor phenotype, which coincided with improved function and number of proprioceptive synapses and NMJs, while it had no effect on motor neuron survival. Combinatorial treatment of 4-AP with pharmacological inhibition of p53-dependent motor neuron death resulted in additive beneficial effects on the sensory-motor circuit and phenotype of SMA mice. Taken together, our results establish that increase of neuronal activity restores function and connectivity of the sensory-motor circuit in SMA and could be beneficial in other neurodegenerative diseases.
Materials and Methods
Animal procedure and behavioral analysis
Breeding and experiments were performed in the animal facilities of the Faculty of Medicine, University of Leipzig according to NHI, European (Council Directive 86/609/EEC) and German (Tierschutzgesetz) guidelines for the welfare of experimental animals and the regional directorate (Landesdirektion) Leipzig, as well as Columbia University and approved by the Institutional Laboratory Animal Care and Use Committee (IACUC) of Columbia University. Mice were housed in a 12/12 h light/dark cycle with access to food and water ad libitum. The original breeding pairs for SMNΔ7 (Smn+/−; SMN2+/+; SMNΔ7+/+) mice (stock #005025) were obtained from The Jackson Laboratory. Tail DNA PCR genotyping was performed as described previously (Fletcher et al., 2017). 4-AP was dissolved in 0.9% NaCl and administered daily twice at a concentration of 1 mg/kg by intraperitoneal injection starting at P0. For c-Fos in vivo immunohistochemistry, P11 mice were injected with 4-AP once at a concentration of 1 mg/kg and after 2 h processed as described in the immunohistochemistry section. Pifithrin-α (PFT) was dissolved in DMSO and delivered daily at a concentration of 2.2 mg/kg by intraperitoneal injection starting at postnatal day (P)0 as described previously (Simon et al., 2017). Approximately equal proportions of mice of both sexes were used and aggregated data are presented since gender-specific differences were not found nor have they been previously reported.
For behavioral analysis, mice from all experimental groups were monitored daily, body weight measurements and the righting reflex were timed and averaged as described previously (Mentis et al., 2011). Mice with a 25% reduction of body weight and an inability to right were euthanized to comply with IACUC and German guidelines. Righting time was defined as the time for the pup to turn over after being placed on its back. The cutoff test time for the righting reflex was 60s to comply with IACUC and German guidelines.
Electrophysiology
To record the monosynaptic ventral root response, we conducted the experiment as previously described (Mentis et al., 2011; Simon et al., 2017). The animals were decapitated, the spinal cords dissected and removed under cold (∼12°C) artificial CSF (aCSF) containing 128.35 mm NaCl, 4 mm KCl, 0.58 mm NaH2PO4.H2O, 21 mm NaHCO3, 30 mm D-glucose, 1.5 mm CaCl2.H2O, and 1 mm MgSO4.7H2O. The spinal cord was then transferred to a customized recording chamber. The intact ex vivo spinal cord preparation was perfused continuously with oxygenated (95%O2/5%CO2) aCSF (∼13 ml/min). The dorsal root and ventral root of the L1 segment were placed into suction electrodes for stimulation or recording respectively. The extracellular recorded potentials were recorded (DC – 3 kHz, Cyberamp, Molecular Devices) in response to a brief (0.2 ms) orthodromic stimulation (A365, current stimulus isolator, WPI) of the L1 dorsal root. The stimulus threshold was defined as the current at which the minimal evoked response was recorded in three out of five trials. Recordings were fed to an A/D interface (Digidata 1440A, Molecular Devices) and acquired with Clampex (v10.2, Molecular Devices) at a sampling rate of 10 kHz. Data were analyzed off-line using Clampfit (v10.2, Molecular Devices). The temperature of the physiological solution ranged between 21°C and 23°C. Synaptic depression experiments were performed and analyzed for all experimental groups at P11. The dorsal root was stimulated at 10 Hz for five stimuli and the resulting monosynaptic component of the amplitude recorded and analyzed off-line. The amplitude of the second to fifth stimuli was expressed as a percentage of the amplitude of the first stimulus.
To assess functionally NMJs of the quadratus lumborum (QL) muscle at P11, we applied the same technique as described previously (Fletcher et al., 2017). Motor neurons axons in the ventral root L1 supplying the QL muscle were stimulated by drawing the ventral root into a suction electrode, having removed the spinal cord, and recorded the compound muscle action potential (CMAP) from the muscle using a concentric bipolar electrode. L1 motor neuron axons were stimulated with five stimuli at 0.1 Hz. The maximum CMAP amplitude (baseline-to-peak) was measured from five averages.
Whole-cell recordings were performed as previously described (Mentis et al., 2011; Fletcher et al., 2017) at room temperature (∼21°C) and obtained with patch electrodes advanced through the lateral aspect of the spinal cord. Patch electrodes were pulled from thin-walled borosilicate glass capillary with filament (Sutter Instruments) using a P-1000 puller (Sutter Instruments) to resistances between 5–8 MΩ. The electrodes were filled with intracellular solution containing the following: 10 mm NaCl, 130 mm K-gluconate, 10 mm HEPES, 11 mm EGTA, 1 mm MgCl2, 0.1 mm CaCl2, and 1 mm Na2ATP, pH was adjusted to 7.2–7.3 with KOH (the final osmolarity of the intracellular solution was 295–305 mOsm). Motor neurons were targeted blindly after removal of the dura and pia mater from the lateral aspect of the cord over the L1 spinal segments. The identity of recorded neurons as motor neurons was confirmed by evoking an antidromic action potential by stimulation of the cut ventral root. Motor neurons were accepted for further analysis only if the following three criteria were met: (1) stable resting membrane potential of −50 mV or more negative (2) an overshooting antidromically-evoked action potential and (3) at least 30 min of recording. γ motor neurons were not included in our analysis. γ motor neurons were identified by the presence of an antidromic action potential, but lack of direct monosynaptic activation from proprioceptive sensory fibers. For the measurements of action potential shape, motor neurons were injected with sequential steps positive currents for 100 ms in 10-pA steps at −60 mV membrane potential. After electrophysiological recording of motor neuron properties under “normal condition,” a 100 μm 4-AP extracellular solution was administered via the gravity perfusion system to record the effect under 4-AP exposure. For the ex vivo investigation of c-Fos expression, the native P4 spinal cord was in the bath chamber with aCSF under 4-AP exposure for 2 h and then processed as indicated below in the immunohistochemistry section.
Immunohistochemistry
For immunostainings of the spinal cord, mice were killed, the spinal cord was removed natively and postfixed in 4% paraformaldehyde (PFA) overnight at 4°C. On the following day, the spinal cords were briefly washed with PBS, subsequently embedded in warm 5% Agar and serial transverse sections (75 μm) were cut at the Vibratome. The sections were blocked with 10% normal donkey serum in 0.01 M PBS with 0.3% Triton X-100 (PBS-T; pH 7.4) for 90 min and incubated overnight at room temperature in different combinations of the primary antibodies (for antibody details, see Table 1). The following day, after 5 × 10-min PBS washes, secondary antibody incubations were performed for 3 h with the appropriate species-specific antiserum coupled to FITC, Cy3, or Cy5 (The Jackson Laboratory) diluted at 1:250 in PBS-T. After secondary antibody incubations, the sections were washed five times for 10 min in PBS and mounted on slides and cover-slipped with an anti-fading solution made of glycerol:PBS (3:7; Simon et al., 2017).
Table 1.
List of antibodies
| Name | Company | Catalog # | Host | Application | Dilution |
|---|---|---|---|---|---|
| SMN clone 8 | BD Transd. Lab | 610646 | Mouse | WB | 1:10,000 |
| Tubulin DM1A | Sigma | T9026 | Mouse | WB | 1:50,000 |
| p53 | Leica Novocastra | NCL-p53-CM5p | Rabbit | IF | 1:1000 |
| p-p53S15 | Cell Signaling | 9284 (lots: 12, 15) | Rabbit | IF | 1:200 |
| VGluT1 | Synaptic Systems | 135 304 | Guinea pig | IF | 1:5000 |
| VGluT1 | Covance (custom made) | N/A | Guinea pig | IF | 1:5000 |
| Synaptophysin | Synaptic Systems | 101-004 | Guinea pig | IF | 1:500 |
| Neurofilament | Millipore | AB1987 | Rabbit | IF | 1:500 |
| c-Fos | Abcam | AB190289 | Rabbit | IF | 1:20,000 |
| ChAT | Millipore | AB144P | Goat | IF | 1:100 |
| Bungarotoxin | Invitrogen | B35451 | N/A | IF | 1:500 |
| Kv2.1 | Neuromab | K89/34 concent. | Mouse | IF | 1:500 |
| Parvalbumin | Synaptic Systems | 195004 | Guinea pig | IF | 1:5000 |
For immunostaining of NMJs, mice were killed and the QL muscle was dissected and immediately fixed with 4% PFA for 20 min. After fixation, single muscle fibers were teased and washed 5 times in PBS for 10 min each followed by staining of the postsynaptic part of the NMJ with α-bungarotoxin Alexa Fluor 555 in PBS for 20 min. Subsequently, the muscle fibers were washed 5 times in PBS for 10 min and blocked with 10% donkey serum in 0.01 m PBS with 0.3% Triton X-100 for 1 h. Rabbit anti-Neurofilament-M and anti-Synaptophysin-1 antibodies to immunolabel the presynaptic part of the NMJ were applied in blocking solution overnight at 4°C (for antibody details, see Table 1). The muscle fibers were then washed three times for 10 min in PBS. Secondary antibodies were applied for 1 h in blocking solution at room temperature. Finally, the muscle fibers were washed three times in PBS for 10 min and mounted with Vectashield (Vector Laboratories; Simon et al., 2010, 2017).
Confocal microscopy and analysis
Spinal cord sections were imaged using SP8 Leica confocal microscopes. Sections were scanned using a 20×, 40×, or 63× objective. Motor neurons were counted off-line from z-stack images (collected at 3-μm intervals in the z-axis) from 11 sections of the selected spinal segment at P11 from L1, as we previously described (Van Alstyne et al., 2018; Simon et al., 2019). Only Choline acetyltransferase (ChAT)+ motor neurons that contained the nucleus were counted to avoid double counting from adjoining sections. Quantitative analysis of Vesicular glutamate transporter 1 (VGluT1) immunoreactive synaptic densities on motor neurons at P11 was performed on image stacks of optical sections scanned using a 40× oil or 63× glycerol objective throughout the whole section thickness at 0.4-μm z-steps to include the whole cell body and dendrites of ChAT+ motor neurons. The number of VGluT1+ synapses were counted over the entire surface of the motor neuron soma as well as on primary dendrites up to a distance of 50 µm from the soma using Leica LASAF software as previously described (Mentis et al., 2011). For the analysis of muscle innervation, at least 200 randomly selected NMJs per muscle sample were quantified for each biological replicate. Only BTX+ endplates that lack any presynaptic coverage by both synaptophysin and NF-M were scored as fully denervated. For axonal sprouting and polysynaptic elimination analysis, at least n = 40 NMJs per animal were scanned with the 63× objective at 1-μm z-steps. Axonal sprouting events were determined by counting the number of NMJs innervated from the terminal part (up to 50 µm away from NMJ) of each axon. For polysynaptic innervation analysis, number of axons innervating a single NMJ were quantified. Kv2.1 coverage was quantified as previously described (Fletcher et al., 2017).
Protein analysis
Whole spinal cords were prepared in 1× LDS buffer (Invitrogen) and resolved using the NuPAGE precast gel system (Invitrogen) by SDS-PAGE. Extracts (20 μg) were ran on Novex Bis-Tris 12% gels and transferred onto a iBlot2 transfer stack nitrocellulose membrane (Invitrogen) using the iBlot2 Dry Blotting system unit (Invitrogen). After protein transfer, the membranes were blocked for 1 h in 5% non-fat dry milk prepared in 1 × PBS with 0.1% Tween 20. The membranes were then incubated with the corresponding antibodies overnight. Thereafter, the membranes were incubated with IRDye 680RD or 800CW secondary antibodies (LI-COR) followed by visualization using a near-infrared imager (Odyssey; LI-COR; Blanco-Redondo et al., 2019).
Statistics
Results are expressed as mean ± SEM from at least three independent experiments using three or more animals per group. The number of observations and animals used are shown in each figure legend. Gaussian distribution was analyzed by Shapiro–Wilk normality test for each dataset. Differences between two groups were analyzed by a two-tailed Student's t test, whereas differences among three or more groups were analyzed by one-way ANOVA followed by Tukey's correction for multiple comparisons as applicable. Body weight and righting time was analyzed by the multiple t test with Holm–Sidak method; Kaplan–Meyer survival curve was analyzed by Mantel–Cox test. GraphPad Prism 7 was used for all statistical analyses and p values are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
4-AP treatment increases neuronal activity and improves motor behavior in SMA mice
To investigate the role of neuronal activity in motor neuron death and synaptic loss, we treated SMA mice chronically with 4-AP, a potassium channel blocker, which is known to increase neuronal activity (Mathie et al., 1998; Imlach et al., 2012; Avoli and Jefferys, 2016) and improves motor function in patients with myasthenia gravis, Lambert–Eaton syndrome, spinal cord injury, and multiple sclerosis (Hayes, 2004; Verschuuren et al., 2006). To assess whether 4-AP reliably increases neuronal activity in the spinal sensory-motor circuit, we used whole-cell patch clamp to record intracellularly from lumbar motor neurons in P4 wild-type (WT) mice during the first postnatal week. To do so, we used the intact ex vivo spinal cord preparation before and after acute exposure to 100 μm 4-AP, a concentration that has been reported to block Kv3.1 and Kv1 potassium channels (Kirsch and Drewe, 1993; Grissmer et al., 1994).
As expected (Greensmith et al., 1996; Hayes, 2004), 4-AP increased significantly the half-width and repolarization time of action potentials by blocking potassium channels (Fig. 1A,B), while the rise time and amplitude of the action potential were largely unaffected (Fig. 1A). In addition, 4-AP depolarized the resting membrane potential to levels above the firing threshold, resulting in an increase of neuronal activity, reflected by an increase in sustained spontaneous repetitive firing (Fig. 1C,D). Furthermore, 4-AP exposure for 2 h resulted in the broad upregulation of c-Fos, a known marker of neuronal activity (Chung, 2015), in motor neurons and sensory neurons in dorsal root ganglia in ex vivo P4 spinal cord preparations (Fig. 1E,F). To test whether 4-AP would also result in c-Fos upregulation in vivo, we tested a single intraperitoneal injection of 4-AP (1 mg/kg). We found that there was a significant increase in the percentage of c-Fos+ L1 motor neurons (Fig. 1G,H) as well as other spinal cord neurons and dorsal root ganglia sensory neurons, including proprioceptive neurons (Fig. 1I) of both P11 WT and SMA mice 2 h after injection. Thus, 4-AP robustly increases the activity of the sensory-motor circuit following intraperitoneal injection in vivo.
Figure 1.
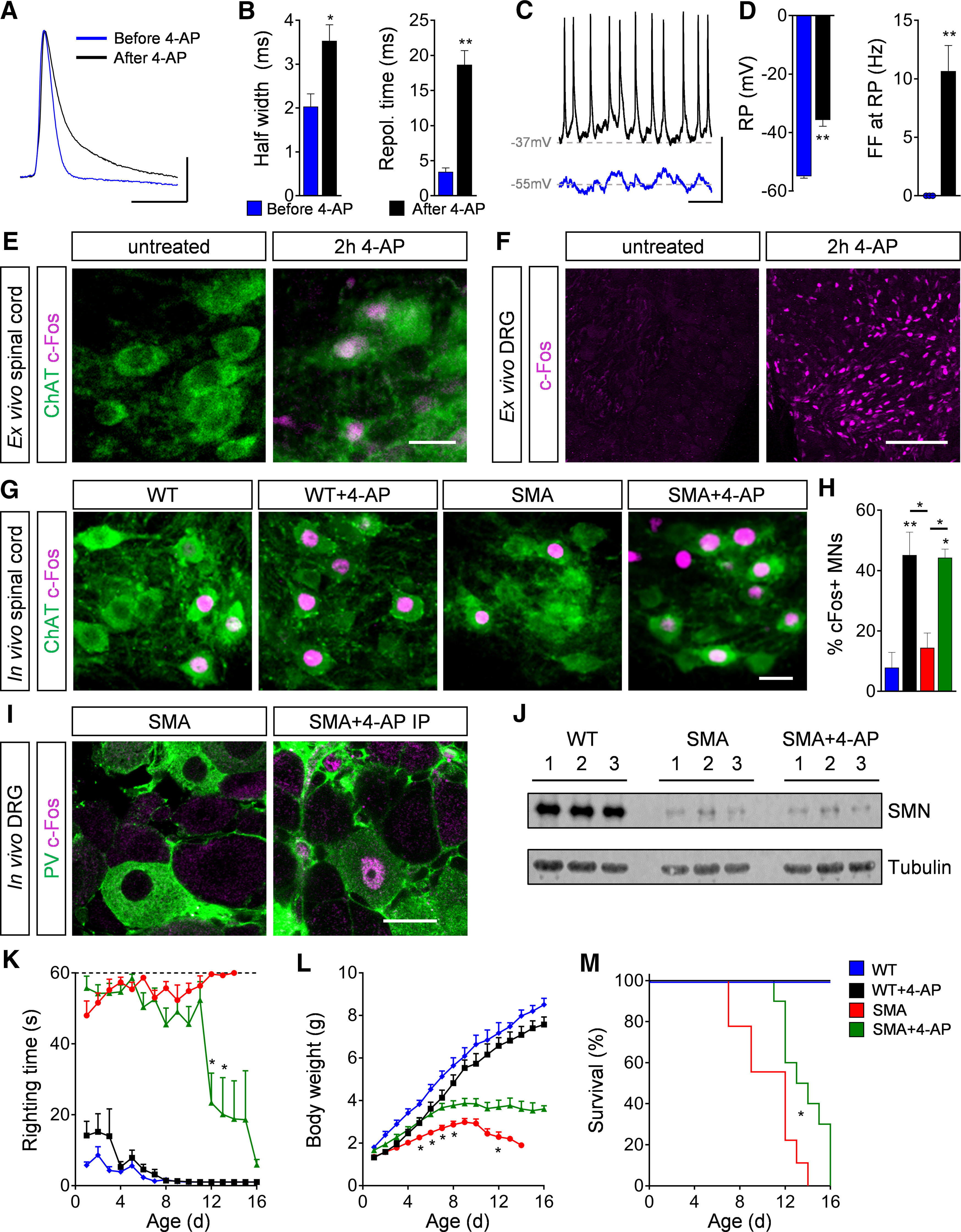
4-AP increases motor neuron (MN) activity and SMA motor function in vivo. A, Action potential of a P4 WT MN before and after 4-AP (100 μm) exposure to an ex vivo spinal cord preparation. Scale bar: 20 mV, 10 ms. B, Quantification of half width and repolarization time of P4 WT MNs before and after 4-AP exposure ex vivo. Half width in milliseconds: before 4-AP = 2.03 ± 0.29, after 4-AP = 3.53 ± 0.37, n = 3 MNs from 3 spinal cords, p = 0.033, two-tailed t test; Repol. Time in milliseconds: before 4-AP = 3.40 ± 0.57, after 4-AP = 18.67 ± 2.04, n = 3 MNs from 3 spinal cords, p = 0.002, two-tailed t test. C, Spontaneous firing of P4 WT MNs at resting potential (RP) before and after 4-AP treatment. Scale bar: 20 mV, 200 ms. D, Quantification of RP and spontaneous firing frequency (FF) at RP of P4 WT MNs before and after 4-AP treatment. RP in millivolts: before 4-AP = −55 ± 0.58, after 4-AP = −35.67 ± 2.19, n = 3 MNs from 3 spinal cords, p = 0.001, two-tailed t test; FF in Hertz: before 4-AP = 0 ± 0, after 4-AP = 10.67 ± 2.19, n = 3 MNs from 3 spinal cords, p = 0.008, two-tailed t test. E, F, Immunostaining of ChAT+ MNs (green) and c-Fos as neuronal activity marker (magenta) in L1 spinal cord (E) and DRG (F) sections of P4 WT ex vivo spinal cord preparation without and with 2-h 4-AP (100 μm) exposure. Scale bar: 30 µm (for spinal cord) and 100 µm (for DRG). G, Immunostaining of ChAT+ MNs (green) and c-Fos as neuronal activity marker (magenta) in L1 spinal cord sections from WT and SMA mice without and with acute 4-AP intraperitoneal injections (WT, WT + 4-AP, SMA, and SMA + 4-AP) at P11. Scale bar: 25 µm. H, Percentage of c-Fos+ MNs from the same groups as in G. WT (3) = 7.88 ± 5.03 WT + 4-AP (4) = 45.12 ± 7.63, SMA (3) = 14.44 ± 4.87, SMA + 4-AP (3) = 44.25 ± 2.88, p for WT versus WT + 4-AP = 0.006, p for WT versus SMA + 4-AP = 0.011, p for WT + 4-AP versus SMA p = 0.020, p for SMA versus SMA + 4-AP = 0.033, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. I, Immunostaining of PV+ proprioceptive neurons (green) and c-Fos (magenta) in L1 DRG sections from P11 SMA and SMA + 4-AP mice. Scale bar: 20 µm. J, Western blot analysis of SMN and tubulin (loading control) protein levels in the spinal cord of WT, SMA, and SMA + 4-AP at P11. K–M, Righting time (K), body weight (L), and Kaplan–Meyer analysis of survival (M) from the same groups as in (G) with chronic 4-AP treatment. Number of mice for WT = 7, WT + 4-AP = 11, SMA = 12, SMA + 4-AP = 18; righting time: SMA n = 12 (P7), 11 (P8), 9 (P11), 6 (P12), 3 (P13), 1 (P14); SMA + 4-AP n = 18 (P10), 12 (P11), 7 (P12), 6, (P13), 5 (P14), 4 (P15), 3 (P16); p for SMA versus SMA + 4-AP indicate with asterisks at P12 = 0.002 and P13 = 0.036; body weight: p for SMA versus SMA + 4-AP indicate with asterisk at P5 = 0.034, P6 = 0.024, P7 = 0.019, P8 = 0.049, and P12 = 0.049, multiple t test with Holm–Sidak method; Kaplan–Meyer analysis: p for SMA versus SMA + 4-AP = 0.014 indicated with asterisk, Mantel–Cox test. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; *p < 0.05, **p < 0.01, ***p < 0.001.
To investigate the long-term effect of elevated neuronal activity on the SMA phenotype, we performed chronic treatment with 4-AP by intraperitoneal injections at a dose of 1 mg/kg (Jankowska et al., 1982; Pinter et al., 1997; Verschuuren et al., 2006) twice a day in WT and SMA mice starting at birth until death. Chronic 4-AP treatment did not result in any significant differences in the righting time, body weight and survival of WT mice (Fig. 1K–M). In striking contrast, 4-AP-treated SMA mice exhibited significantly improved late onset motor behavior comprising of the ability to right (Fig. 1K) and walking as well as a moderate increase in body weight and survival (Fig. 1L,M) compared with untreated SMA littermates. To rule out any potential effects of chronic 4-AP treatment on SMN expression, we confirmed by Western blot analysis that SMN protein levels were not increased in spinal cords of 4-AP-treated SMA mice (Fig. 1J). To conclude, increased neuronal activity elicited by chronic 4-AP treatment improves the motor phenotype of SMA mice through SMN-independent mechanisms.
Enhanced neuronal activity induces sprouting of peripheral and central axons
To identify the neuronal alterations underlying the improved motor phenotype in 4-AP-treated SMA mice, we first sought to investigate whether 4-AP rescued motor neurons from death. We focused on the vulnerable motor neurons in the first and second lumbar spinal segments (L1 and L2). Confocal analysis revealed that 4-AP treatment in SMA mice did neither rescue nor delay the death of lumbar L1 and L2 motor neurons which innervate disease-relevant axial muscles (Fig. 2A–D; Fletcher et al., 2017). In accordance, nuclear accumulation and phosphorylation of p53 at serine 18 (p-p53S18), two converging mechanisms mediating motor neuron death in SMA (Simon et al., 2017, 2019; Van Alstyne et al., 2018), were not altered following 4-AP treatment (Fig. 2E–G), suggesting that motor neuron death occurs independently of neuronal activity.
Figure 2.
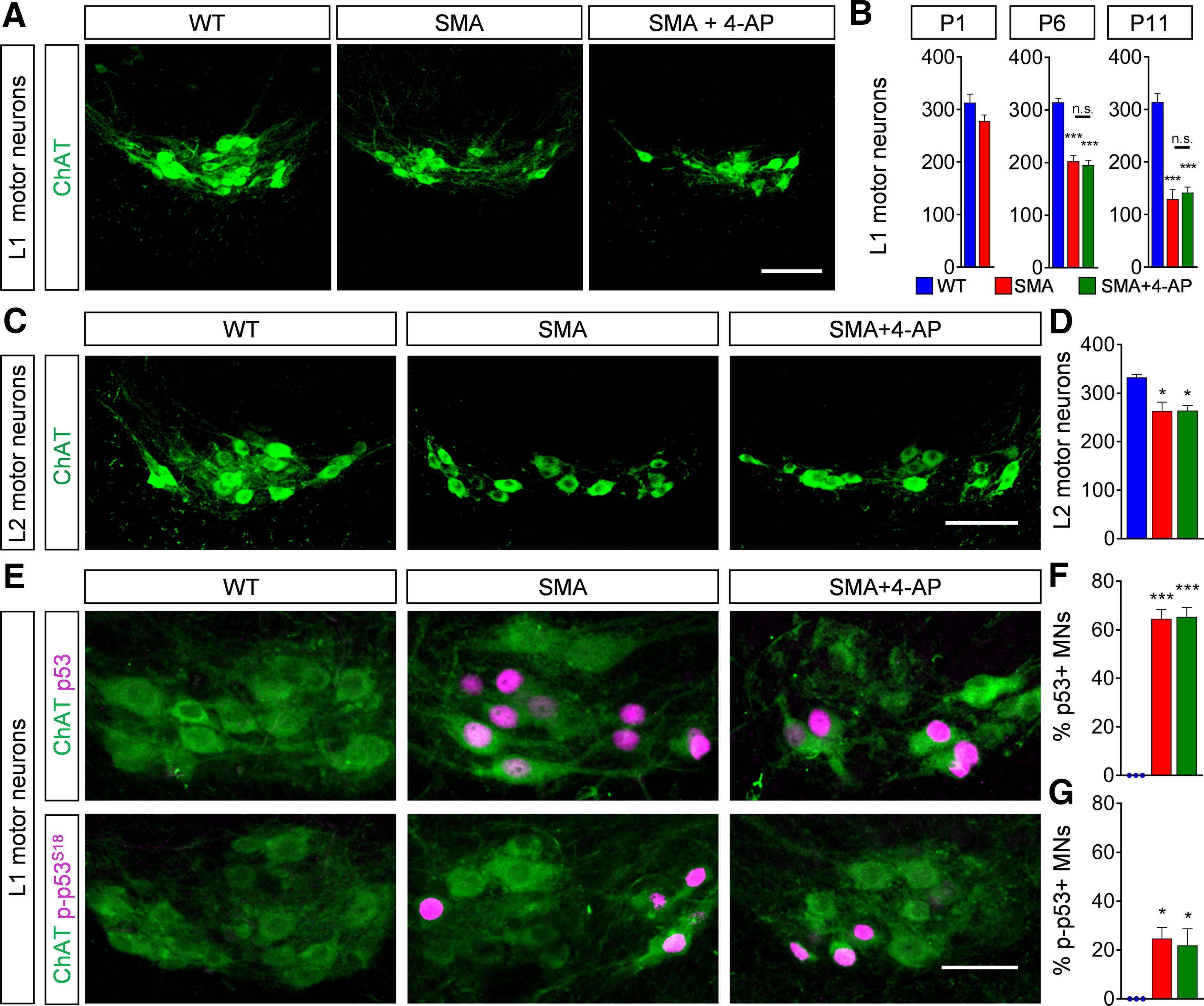
Increased neuronal activity does not prevent motor neuron death. A, Immunostaining of ChAT+ motor neurons (green) in L1 spinal cord sections from WT, SMA, and SMA + 4-AP mice with chronic intraperitoneal injections at P11. Scale bar: 100 µm. B, Quantification of L1 motor neuron counts of the same groups as in A at P1, P6, and P11. P1: WT (3) = 313 ± 16.09, SMA (3) = 278 ± 11.27, p = 0.149, two-tailed t test; P6: WT (3) = 314.7 ± 7.31, SMA (5) = 202.6 ± 10.84, SMA + 4-AP (3) = 195.7 ± 8.99, p for WT versus SMA = 0.0001, p for WT versus SMA + 4-AP = 0.0002, one-way ANOVA with Tukey's correction; P11: WT (4) = 314 ± 16.2, SMA (4) = 129.5 ± 17.81, SMA + 4-AP (6) = 141.7 ± 10.78, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP < 0.0001, one-way ANOVA with Tukey's correction. n.s. = not significant. Number of mice is reported in parentheses. C, Immunostaining of ChAT+ motor neurons (green) in L2 spinal cord sections from the same groups as in A at P11. Scale bar: 100 µm. D, Quantification of L2 motor neuron counts of the same groups as in A at P11. WT (4) = 332 ± 6.22, SMA (4) = 263.9 ± 17.69, SMA + 4-AP (3) = 264.2 ± 10.31, p for WT versus SMA = 0.0107, p for WT versus SMA + 4-AP = 0.0164, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. E, Immunostaining of ChAT+ motor neurons (green) and p53 or p-p53S18 (magenta) of L1 spinal cords from the same groups as in A at P11. Scale bar: 50 µm. F, G, Percentage of p53+ L1 motor neurons (F) and p-p53S18+ L1 motor neurons (G) in the same groups as in A. p53: WT (3) = 0 ± 0, SMA (3) = 64.60 ± 3.86, SMA + 4-AP (3) = 65.4 ± 3.85, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP < 0.0001; p-p53: WT (3) = 0 ± 0, SMA (3) = 24.76 ± 4.46, SMA + 4-AP (3) = 21.93 ± 6.78, p for WT versus SMA = 0.0167, p for WT versus SMA + 4-AP = 0.0388, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we investigated the number of VGluT1+ proprioceptive synapses onto L1 motor neurons, since their reduction contributes significantly to motor dysfunction of SMA mice (Fletcher et al., 2017). While untreated SMA mice exhibited a progressive loss of synaptic connections, 4-AP-treated SMA mice showed a significant increase of proprioceptive synapses on the somata and proximal dendrites of motor neurons at end-stage (investigated at P11, treated from P0 to P11), but not at earlier stages (investigated at P6, treated from P0 to P6; Fig. 3A–C). To study the effect of 4-AP on peripheral synapses, we examined the NMJ innervation of the axial QL muscle, which is innervated by L1–L3 motor neurons and severely affected in SMA (Fletcher et al., 2017; Simon et al., 2017, 2019), using presynaptic (synaptophysin, neurofilament) and postsynaptic (α-bungarotoxin) markers. The QL of untreated SMA mice showed ∼50% NMJ denervation at P11, in contrast to a fully innervated QL muscle in WT mice. Strikingly, chronic 4-AP treatment resulted in significantly improved QL innervation (∼20% denervation) in SMA mice at end-stage (P11, treated from P0 to P11), but not before (∼40% denervation at P6, treated from P0 to P6; Fig. 3D,E).
Figure 3.
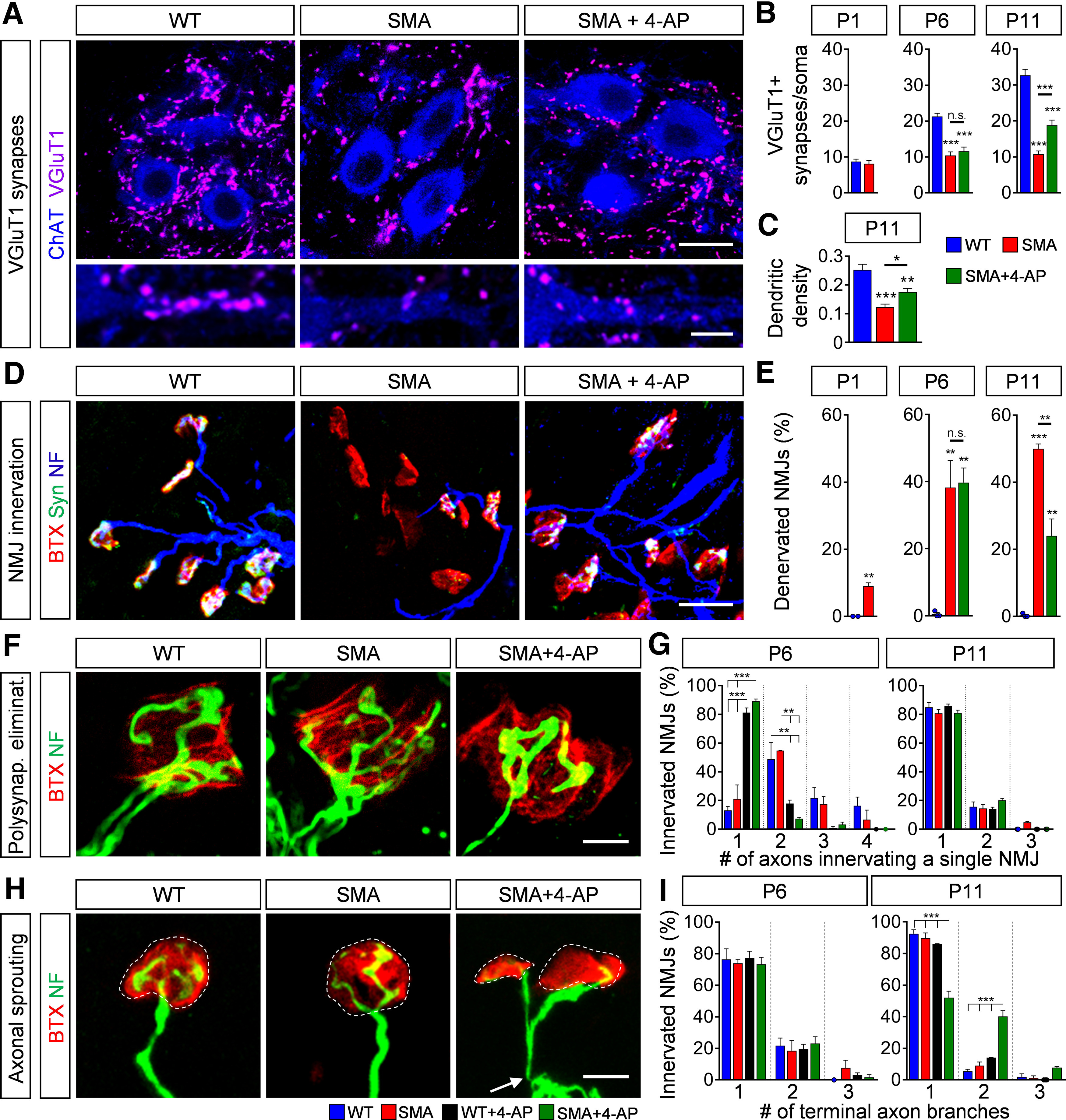
4-AP restores synaptic connections via sprouting in the SMA sensory-motor circuit. A, Immunostaining of VGluT1+ synapses (magenta) on ChAT+ motor neuron (blue) somata (upper panel) and proximal dendrites (lower panel) in L1 spinal cord sections from WT, SMA, and SMA + 4-AP mice at P11. Scale bar: 20µm (upper panel) and 10 µm (lower panel). B, Number of VGluT1+ synapses on the somata of L1 motor neurons from the same groups as A at P1, P6, and P11. P1: WT (10) = 8.7 ± 0.68, SMA (10) = 8.1 ± 0.92, p = 0.6082, two-tailed t test; P6: WT (12) = 21.25 ± 0.90, SMA (10) = 10.4 ± 1.03, SMA + 4-AP (16) = 11.56 ± 1.19, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP < 0.0001, one-way ANOVA with Tukey's correction; P11: WT (14) = 32.71 ± 1.66, SMA (14) = 10.71 ± 0.96, SMA + 4-AP (15) = 18.8 ± 1.45, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP < 0.0001, p for SMA versus SMA + 4-AP = 0.0005, one-way ANOVA with Tukey's correction. Number of motor neurons from three mice is reported in parentheses. C, Number of VGluT1+ synapses on the proximal dendrites (0–50 µm from soma) of L1 ChAT+ motor neurons from the same groups as A at P11. WT (12) = 0.253 ± 0.019, SMA (16) = 0.123 ± 0.011, SMA + 4-AP (10) = 0.176 ± 0.012, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP = 0.003, p for SMA versus SMA + 4-AP = 0.035, one-way ANOVA with Tukey's correction. Number of motor neurons from three mice is reported in parentheses. D, NMJ staining with bungarotoxin (BTX; red), synaptophysin (Syn; green), and neurofilament (NF; blue) of QL muscles from the same groups as in A at P11. Scale bar: 30 µm. E, Quantification of denervated NMJs in the QL muscle of the same groups as in A at P1, P6, and P11. P1: WT (3) = 0 ± 0, SMA (4) = 9.06 ± 0.93, p = 0.0004, two-tailed t test; P6: WT (3) = 0.49 ± 0.49, SMA (3) = 38.21 ± 8.05, SMA + 4-AP (3) = 39.69 ± 8.05, p for WT versus SMA = 0.0056, p for WT versus SMA + 4-AP = 0.0047, one-way ANOVA with Tukey's correction; P11: WT (3) = 0.32 ± 0.32, SMA (4) = 49.96 ± 1.47, SMA + 4-AP (6) = 24.03 ± 5.02, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP = 0.0090, p for SMA versus SMA + 4-AP = 0.0028, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses, >200 NMJs were analyzed per animal. F, High magnification of axons innervating a single NMJ of the QL muscle visualized with NF (green) and BTX (red) from the same groups as in A at P11. Scale bar: 5 µm. G, Proportion of axons innervating a single NMJ in the QL muscle from WT, WT + 4-AP, SMA, and SMA + 4-AP mice at P6 and P11. P6 one axon innervating a single NMJ: WT = 13.1 ± 2.96, SMA = 21.15 ± 9.68, WT + 4-AP = 81.12 ± 3.28, SMA + 4-AP = 89.28 ± 1.47, p for WT versus WT + 4-AP < 0.0001, p for WT versus SMA + 4-AP < 0.0001; p for SMA versus WT + 4AP = 0.0002, p for SMA versus SMA + 4-AP < 0.0001, one-way ANOVA with Tukey's correction; P6 two axons innervating a single NMJ: WT = 48.76 ± 11.73, SMA = 54.62 ± 0.38, WT + 4-AP = 17.78 ± 2.50, SMA + 4-AP = 7.42 ± 0.95, p for WT versus WT + 4-AP = 0.0273, p for WT versus SMA + 4-AP = 0.0055, p for SMA versus SMA + 4-AP = 0.0108, p for SMA versus SMA + 4-AP = 0.0024, one-way ANOVA with Tukey's correction. More than 40 NMJs per animal were analyzed and the mean was formed. Three mice of each group were analyzed. H, High magnification of sprouting events at the NMJs of the QL muscle visualized with NF (green) and BTX (red) from the same groups as in A at P11. White dashed circles mark single NMJs. Arrow indicates a single motor axon. Scale bar: 10 µm. I, Proportion of NMJ innervating terminal branches of a single axon in the QL from the same groups as in G at P6 and P11. P11 one terminal axon branch: WT = 92.53 ± 2.47, SMA = 89.78 ± 3.27, WT + 4-AP = 85.95 ± 0.33, SMA + 4-AP = 52.05 ± 4.13, p for WT versus SMA + 4-AP < 0.0001, p for SMA versus SMA + 4-AP < 0.0001; p for SMA + 4-AP versus WT + 4AP = 0.0002, one-way ANOVA with Tukey's correction; P11 two terminal axon branches: WT = 5.51 ± 1.22, SMA = 8.94 ± 2.39, WT + 4-AP = 14.05 ± 0.33, SMA + 4-AP = 40.13 ± 3.72, p for WT versus SMA-4-AP <0.0001, p for SMA versus SMA + 4-AP <0.0001, p for WT + 4-AP versus SMA + 4-AP = 0.0002, one-way ANOVA with Tukey's correction. More than 40 NMJs per animal were analyzed and the mean was formed. Three mice of each group were analyzed. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; n.s. = not significant; *p < 0.05, **p < 0.01, ***p < 0.001.
Taking into account that the number of proprioceptive neurons located in the DRG are unaltered in SMA (Mentis et al., 2011) and 4-AP treatment does not prevent the death of QL-innervating motor neurons (Fig. 2), these results suggest that the observed elevation of central and peripheral synapses at end-stage is induced by activity-dependent compensatory mechanisms such as axonal sprouting in SMA mice. To test whether chronic increase in neuronal activity modifies axonal plasticity, we first investigated the level of normal occurring elimination of polysynaptic innervation at the NMJ. While elimination of polysynaptic innervation was not altered in untreated WT and SMA mice, it was accelerated by activity-induced 4-AP treatment in both genotypes (Fig. 3F,G), as reported previously (Sanes and Lichtman, 1999). In contrast to its broad effect on the acceleration of the elimination of polysynaptic innervation, 4-AP treatment selectively increased terminal axonal branches in P11 SMA but not in WT innervated NMJs (Fig. 3H,I), suggesting activity-dependent compensatory axonal sprouting during the second postnatal week in SMA, while 4-AP had no effect on sensory-motor circuit of WT animals (Fig. 4).
Figure 4.
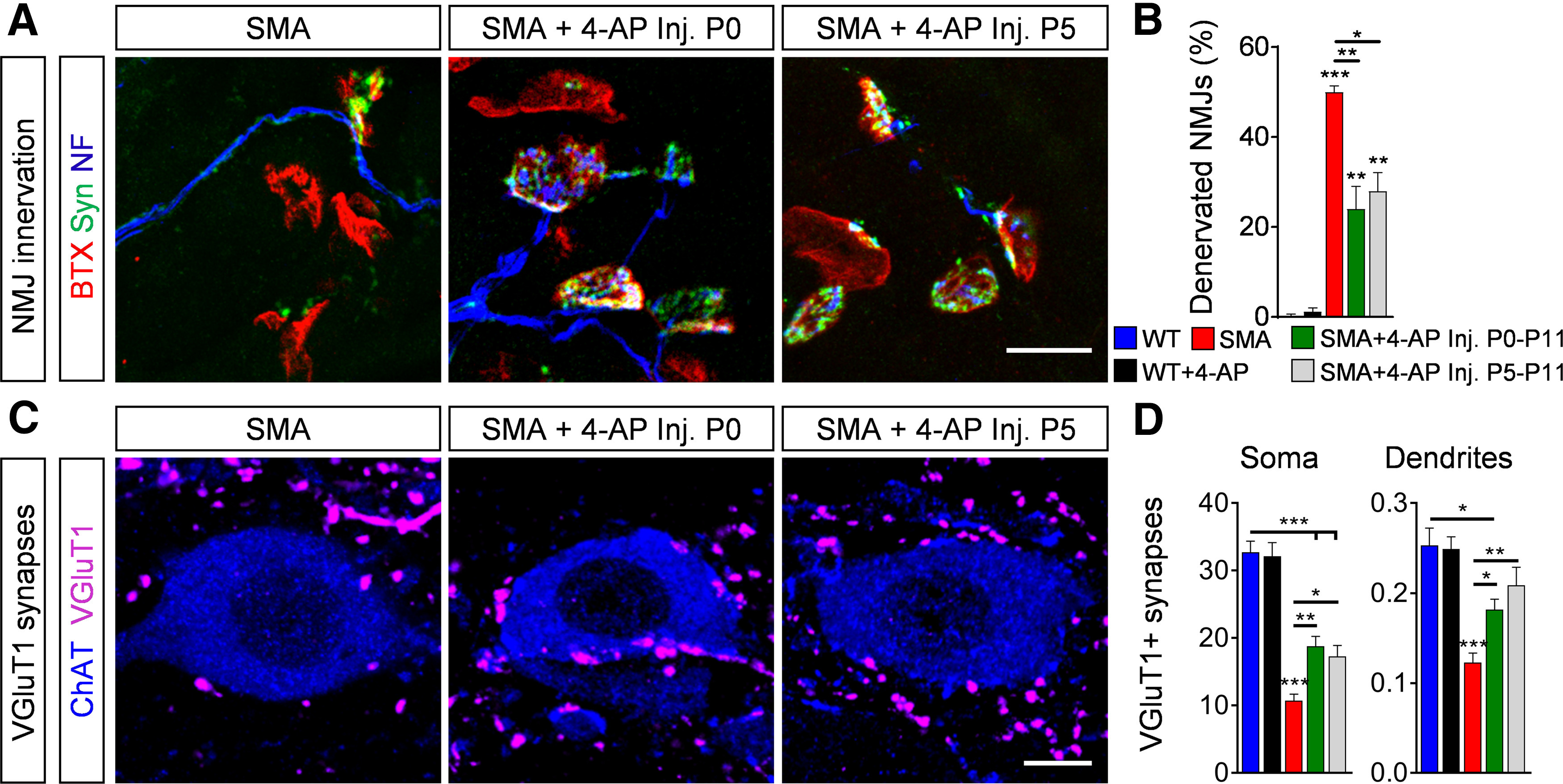
4-AP-induced synaptic connectivity requires a more mature neuronal network. A, NMJ staining with bungarotoxin (BTX; red), synaptophysin (Syn; green), and neurofilament (NF; blue) in QL muscles from P11 SMA mice and SMA mice with 4-AP injections started at birth until P11 (Inj. P0–P11) or at P5 until P11 (Inj. P5–P11). Scale bar: 20 µm. B, Quantification of denervated NMJs in percent of the QL muscle of P11 WT, WT + 4-AP, SMA, SMA + 4-AP inj. P0–P11 and SMA + 4-AP inj. P5–P11. WT (3) = 0.32 ± 0.32, WT + 4-AP (3) = 1.15 ± 0.83, SMA (4) = 49.96 ± 1.47, SMA + 4-AP P0–P11 (6) = 24.03 ± 5.02, SMA + 4-AP (3) P5–P11 = 27.91 ± 4.19, p for WT versus SMA <0.0001, p for WT versus SMA + 4-AP P0–P11 = 0.0067, p for WT versus SMA + 4-AP P5–P11 = 0.0063, p for SMA versus SMA + 4-AP P0–P11 = 0.0014, p for SMA versus SMA + 4-AP P5–P11 = 0.0198, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses, >200 NMJs were analyzed per animal. C, Immunostaining of VGluT1+ synapses (magenta) and ChAT+ motor neurons (blue) in L1 spinal cord sections from the same groups as in A. Scale bar: 10 µm. D, Number of VGluT1+ synapses on the somata and proximal dendrites (0–50 µm from the soma) of L1 motor neurons from the same groups as B at P11. Soma: WT (14) = 32.71 ± 1.66, WT + 4-AP (10) = 32.1 ± 2.05, SMA (14) = 10.71 ± 0.96, SMA + 4-AP P0–P11 (15) = 18.8 ± 1.45, SMA + 4-AP (10) P5–P11 = 17.3 ± 1.61, p for WT versus SMA + 4-AP P0–P11 and P5–P11 < 0.0001, p for WT versus SMA <0.0001, p for SMA versus SMA + 4-AP P0–P11 = 0.0018, p for SMA versus SMA + 4-AP P5–P11 = 0.0325, one-way ANOVA with Tukey's correction; dendrites: WT (12) = 0.253 ± 0.019, WT + 4-AP (6) = 0.2 ± 0.013, SMA (16) = 0.1229 ± 0.010, SMA + 4-AP P0–P11 (9) = 0.181 ± 0.012, SMA + 4-AP (6) P5–P11 = 0.208 ± 0.0201, p for WT versus SMA + 4-AP P0–P11 = 0.0144, p for WT versus SMA < 0.0001, p for SMA versus SMA + 4-AP P0–P11 = 0.0444, p for SMA versus SMA + 4-AP P5–P11 = 0.0055, one-way ANOVA with Tukey's correction. Number of motor neurons from three mice is reported in parentheses. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; *p < 0.05, **p < 0.01, ***p < 0.001.
To examine whether axonal sprouting events in the second postnatal week depend on the duration of 4-AP treatment or on the developmental stage of SMA mice, we treated SMA mice with 4-AP for a shorter period of time, starting at P5 instead of at birth and quantified synaptic inputs at end-stage (investigated at P11, treated from P5 to P11). Interestingly, 4-AP treatment for 6 d starting at P5 was sufficient to improve number of innervated NMJs (Fig. 4A,B) and proprioceptive synapses (Fig. 4C,D) in SMA mice, whereas the same duration of treatment starting at birth until P6 had no effect (investigated at P6; Fig. 3A–E), indicating that axonal sprouting requires a more mature nervous system to take effect instead of treatment duration.
Taken together, these results reveal that elevated neuronal activity induces axonal sprouting to partially compensate the loss of proprioceptive synapses and NMJs in SMA.
Increased neuronal activity improves function of central and peripheral SMA synapses
To investigate whether activity-induced restoration of synaptic connectivity led to functional improvements of the severely affected sensory-motor neurotransmission in SMA (Mentis et al., 2011; Fletcher et al., 2017; Simon et al., 2019), we used extracellular recordings in the ex vivo spinal cord and muscle preparations from WT, SMA, and SMA mice treated with 4-AP. To assess NMJ function in vulnerable SMA muscles, we stimulated the L1 ventral root in absence of the spinal cord and recorded the CMAP of the QL muscle as previously described (Fletcher et al., 2017; Simon et al., 2019). At end-stage, the CMAP of the QL in SMA mice was reduced by ∼90% compared with WT mice (Fig. 5A,B). Importantly, 4-AP-injected SMA mice showed a significant increase of CMAP amplitude compared with uninjected SMA littermates (Fig. 5A,B).
Figure 5.
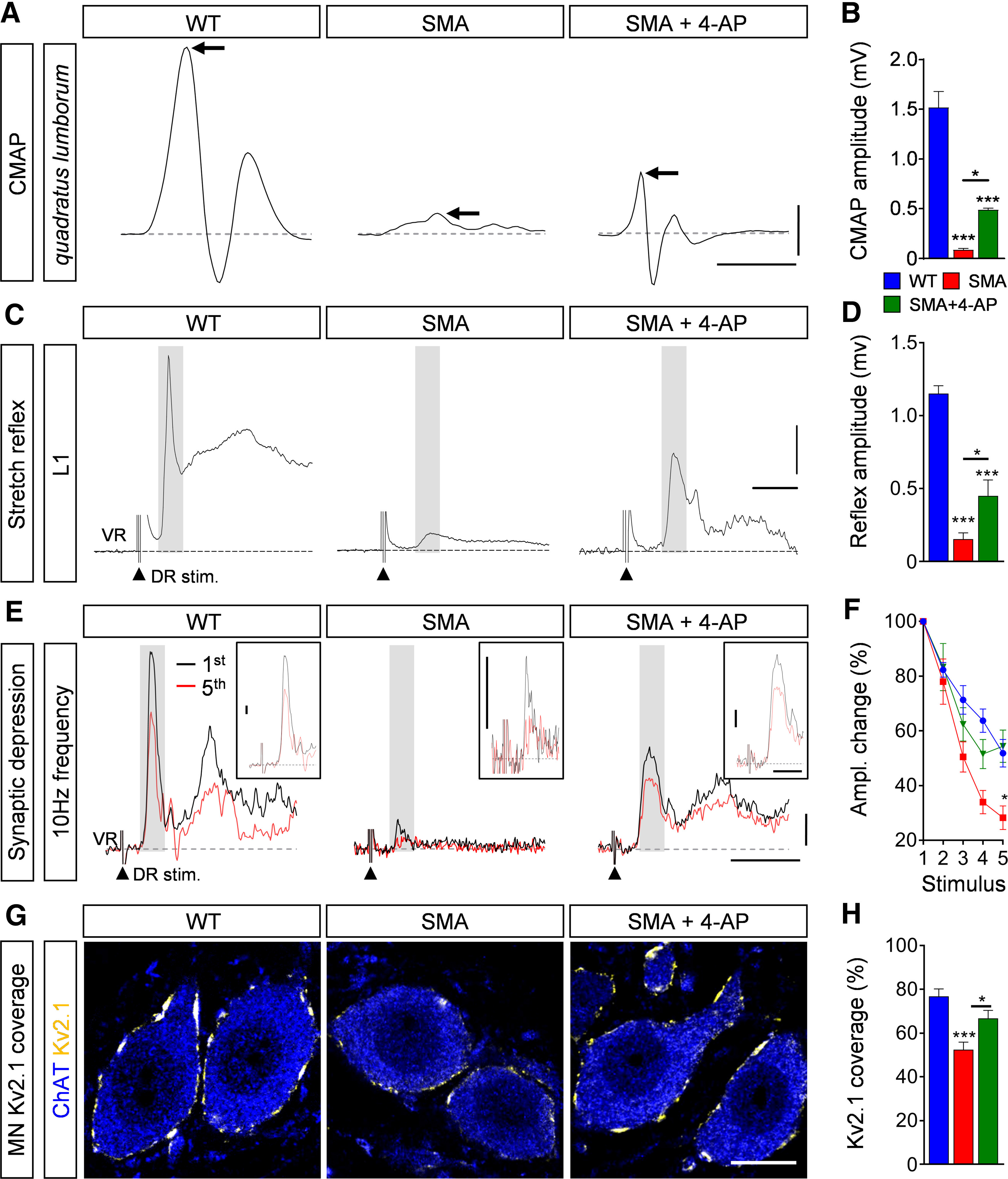
4-AP improves synaptic function of sensory-motor circuit in SMA mice. A, CMAP recordings from the QL muscle following L1 ventral root stimulation from WT, SMA, and SMA + 4-AP mice at P11. Arrows indicate maximum amplitude. Scale bars: 0.5 mV, 3 ms. B, Quantification of CMAP amplitudes recorded from the QL muscle in the same groups as in A. CMAP in millivolts: WT (6) = 1.52 ± 0.16, SMA (6) = 0.05 ± 0.01, SMA + 4-AP (6) = 0.45 ± 0.02, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP <0.0001, p for SMA versus SMA + 4-AP = 0.0228, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. C, Representative traces of extracellular recordings from L1 ventral root (VR) following L1 dorsal root (DR) stimulation from the same groups as in A. Shaded area marks the monosynaptic response. Arrowheads indicate the stimulus artifact. Scale bars: 0.2 mV, 5 ms. D, Quantification of spinal ventral root amplitudes recorded from the same groups as in A at P11. Amplitude in millivolts: WT (5) = 1.15 ± 0.05, SMA (6) = 0.15 ± 0.04, SMA + 4-AP (7) = 0.45 ± 0.10, p for WT versus SMA < 0.0001, p for WT versus SMA + 4-AP <0.0001, p for SMA versus SMA + 4-AP = 0.0458, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. E, Representative traces of the first (black) and fifth (red) VR responses recorded at P11 following stimulation of the homonymous L1 DR at 10 Hz from the same groups as in A. Scale bars: 0.2 mV and 10 ms. Inset scale bars: 0.1 mV, 5 ms. F, Quantification of amplitude changes in percent of the monosynaptic ventral root response following 10-Hz stimulation from the same groups as in A. Number of mice for WT = 7, SMA = 6, SMA + 4-AP = 6, p for SMA versus SMA + 4-AP at stimulus 5 = 0.0204, multiple t test with Holm–Sidak method. G, Single optical plane confocal images of L1 ChAT+ motor neurons (blue) expressing Kv2.1 channels (yellow) from the same groups as in A at P11. H, Percentage of somatic coverage of Kv2.1 expression in motor neurons for the same experimental groups shown in A. WT (16) = 76.83 ± 3.38, SMA (23) = 52.4 ± 3.49, SMA + 4-AP (21) = 66.77 ± 3.64, p for WT versus SMA < 0.0001, p for SMA versus SMA + 4-AP = 0.0113, one-way ANOVA with Tukey's correction. Number of motor neurons from three mice is reported in parentheses. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; *p < 0.05, **p < 0.01, ***p < 0.001.
To examine whether increased neuronal activity also improves proprioceptive synapses, we measured the monosynaptic ventral root response at end-stage by recording the ventral root following stimulation of the homonymous L1 dorsal root. 4-AP treatment of SMA mice resulted in a significant increase in the amplitude of the monosynaptic ventral root response, which is regulated by the sum of the number and function of proprioceptive synapses and motor neurons (Fig. 5C,D). To confirm that this increase does not only result from the increased number of proprioceptive synapses, but also from their synaptic neurotransmission onto motor neurons, we performed repetitive stimulation of proprioceptive synapses at high frequency (10 Hz). While the percentage change in the ventral root amplitude following five consecutive stimuli of 10 Hz decreased in WT proprioceptive synapses by 40%, untreated SMA proprioceptive synapses showed a depression of >70%, as reported previously (Simon et al., 2017, 2019). Notably, 4-AP chronic treatment improves synaptic depression in SMA mice to a level comparable to WT (Fig. 5E,F), leading to an overall increased excitatory synaptic drive. We have previously shown that excitatory synaptic drive controls expression of the potassium channel Kv2.1 in motor neurons, thereby shaping motor neuron function (Fletcher et al., 2017). Therefore, we investigated whether the 4-AP-induced improvement of excitatory drive in SMA influences the expression of Kv2.1. Strikingly, 4-AP normalizes Kv2.1 expression in SMA to WT levels (Fig. 5G,H).
Taken together, these results indicate that an increase of neuronal activity improves neurotransmission of proprioceptive synapses and NMJs as well as motor neuron function in SMA mice.
4-AP treatment and p53 inhibition have additive effects on SMA phenotype
We demonstrated here that the increase of neuronal activity by 4-AP improves the number and function of proprioceptive and NMJ synapses but does not prevent motor neuron loss in SMA mice. The opposite effects were observed in one of our recent studies in which inhibition of the p53 pathway by the small molecule PFT rescued SMA motor neurons from death, while having minimal effects on the number and function of synapses (Simon et al., 2017). The complementary effects of 4-AP and PFT in the sensory-motor circuit suggest potential added benefit from combinatorial treatment. To test this hypothesis, we co-injected daily 4-AP (bis in die (BID), 1 mg/kg) and PFT (2.2 mg/kg) in SMA mice starting at birth until death for phenotype analysis or P11 for functional and morphologic experiments. In agreement with our previous report (Simon et al., 2017), chronic PFT treatment prevented death of L1 motor neurons in P11 SMA mice (Fig. 6A,B). Combinatorial PFT and 4-AP treatment improved motor neuron survival (Fig. 6A,B), central (Fig. 6C–E) and peripheral synaptic connectivity (Fig. 6F,G), as well as central synaptic transmission (Fig. 6H,I) and peripheral NMJ transmission (Fig. 6J,K), without adding any effects to the specific improvements achieved by treatment with each drug individually in SMA mice.
Figure 6.
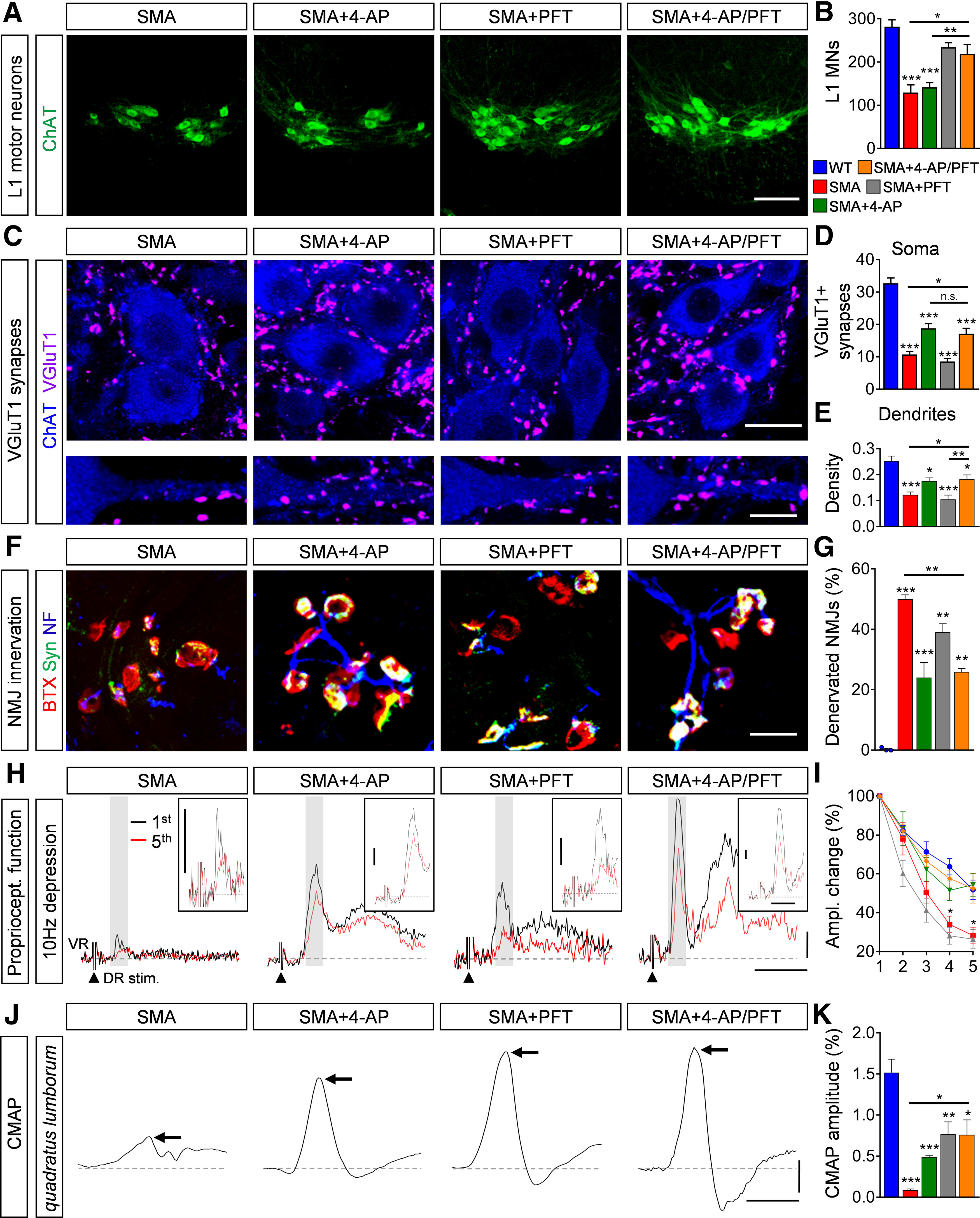
Pharmacological p53 inhibition and 4-AP treatment show complementary effects on the sensory-motor circuit in SMA. A, Immunostaining of ChAT+ motor neurons (green) in L1 spinal cord sections from SMA, SMA + 4-AP, SMA + PFT and SMA + 4-AP/PFT mice at P11. Scale bar: 100 µm. B, Quantification of L1 motor neuron counts from WT, SMA, SMA + 4-AP, SMA + PFT, and SMA + 4-AP/PFT mice at P11. WT (5) = 282.2 ± 15.6, SMA (4) = 129.5 ± 17.81, SMA + 4-AP (6) = 141.7 ± 10.78, SMA + PFT (5) = 234 ± 10.71, SMA + 4-AP/PFT (4) = 219 ± 21.6, p for WT versus SMA and SMA + 4-AP < 0.0001, p for SMA versus SMA + PFT = 0.0010, p for SMA versus SMA + 4-AP/PFT = 0.0076, p for SMA + PFT versus SMA + 4-AP = 0.0013, p for SMA + 4-AP versus SMA + 4-AP/PFT = 0.0119, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. C, Immunostaining of VGluT1+ synapses (magenta) and ChAT+ motor neuron (blue) somata (upper panel) and proximal dendrites (lower panel) in L1 spinal cord sections of the same groups as in A at P11. Scale bar: 20 µm (upper panel) and 10 µm (lower panel). D, E, Number of VGluT1+ synapses on the somata (D) and proximal dendrites (E) of L1 motor neurons from the same groups as B at P11. Soma: WT (14) = 32.71 ± 1.66, SMA (14) = 10.71 ± 0.96, SMA + 4-AP (15) = 18.8 ± 1.45, SMA + PFT (18) = 8.55 ± 0.95, SMA + 4-AP/PFT (13) = 17.08 ± 1.72, p for WT versus all other groups <0.0001, p for SMA versus SMA + 4-AP = 0.0007, p for SMA versus SMA + 4-AP/PFT = 0.0176, p for SMA + PFT versus SMA + 4-AP < 0.0001, p for SMA + PFT versus SMA + 4-AP/PFT = 0.0002, one-way ANOVA with Tukey's correction; dendrites: WT (12) = 0.253 ± 0.019, SMA (16) = 0.1229 ± 0.010, SMA + 4-AP (10) = 0.1758 ± 0.012, SMA + PFT (13) = 0.1047 ± 0.0167, SMA + 4-AP/PFT = 0.1828 ± 0.0166, p for WT versus SMA and SMA + PFT < 0.0001, p for WT versus SMA + 4-AP = 0.0127, p for WT versus SMA + 4-AP/PFT = 0.0195, p for SMA versus SMA + 4-AP/PFT = 0.0400, p for SMA + PFT versus SMA + 4-AP = 0.0221, p for SMA + PFT versus SMA + 4-AP/PFT = 0.0056, one-way ANOVA with Tukey's correction. Number of motor neurons from three mice is reported in parentheses. F, NMJ staining with bungarotoxin (BTX; red), synaptophysin (Syn; green), and neurofilament (NF; blue) of QL muscles from the same groups as in A at P11. Scale bar: 20 µm. G, Proportion of denervated NMJs in percent in the QL muscle of the same groups as in B at P11. WT (3) = 0.32 ± 0.32, SMA (4) = 49.96 ± 1.47, SMA + 4-AP (6) = 24.03 ± 5.02, SMA + PFT (4) = 39.07 ± 2.79, SMA + 4-AP/PFT (3) = 25.89 ± 1.17, p for WT versus SMA and SMA + PFT < 0.0001, p for WT versus SMA + 4-AP = 0.0043, p for WT versus SMA + 4-AP/PFT = 0.0075, p for SMA versus SMA + 4-AP = 0.0008, p for SMA versus SMA + 4AP/PFT = 0.0071, versus one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses, >200 NMJs were analyzed per animal. H, Representative traces of the first (black) and fifth (red) VR responses recorded at P11 following stimulation of the homonymous L1 DR at 10 Hz from the same groups as in A. Scale bars: 0.2 mV and 10 ms. Inset scale bars: 0.1 mV, 5 ms. I, Quantification of amplitude changes in percent of the monosynaptic ventral root response following 10-Hz stimulation from the same groups as in B. Number of mice for WT = 5, SMA = 5, SMA + 4-AP = 5, SMA + PFT = 5, SMA + 4-AP/PFT = 5, p for SMA versus SMA + 4-AP/PFT at stimulus 4 = 0.0237 and 5 = 0.0487, multiple t test with Holm–Sidak method. J, CMAP recordings from the QL muscle following L1 ventral root stimulation from the same groups as in A at P11. Arrows indicate maximum amplitude. Scale bars: 0.2 mV, 3 ms. K, Quantification of CMAP amplitudes in millivolts recorded from the QL muscle in the same groups as in B at P11. CMAP in millivolts: WT (6) = 1.52 ± 0.16, SMA (6) = 0.08 ± 0.02, SMA + 4-AP (6) = 0.45 ± 0.02, SMA + PFT (9) = 0.76 ± 0.15, SMA + 4-AP/PFT (3) = 0.76 ± 0.18, p for WT versus SMA and SMA + 4-AP < 0.0001, p for WT versus SMA + PFT = 0.0016, p for WT versus SMA + 4-AP/PFT = 0.0225, p for SMA versus SMA + PFT = 0.0043, p for SMA versus SMA + 4-AP/PFT = 0.0499, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; n.s. = not significant; *p < 0.05, **p < 0.01, ***p < 0.001.
Since the combinatorial 4-AP/PFT treatment rescued motor neurons (through PFT) and synaptic connections (through 4-AP), we wondered whether there might be an additive effect on functional output of the sensory-motor circuit and SMA phenotype. In striking contrast to treatment with either drug alone, 4-AP and PFT co-administration resulted in a full recovery of the monosynaptic ventral root response (Fig. 7A,B) at end-stage and enhanced improvement of motor function (Fig. 7C), without any significant changes in weight gain or the lifespan of SMA mice (Fig. 7D,E).
Figure 7.
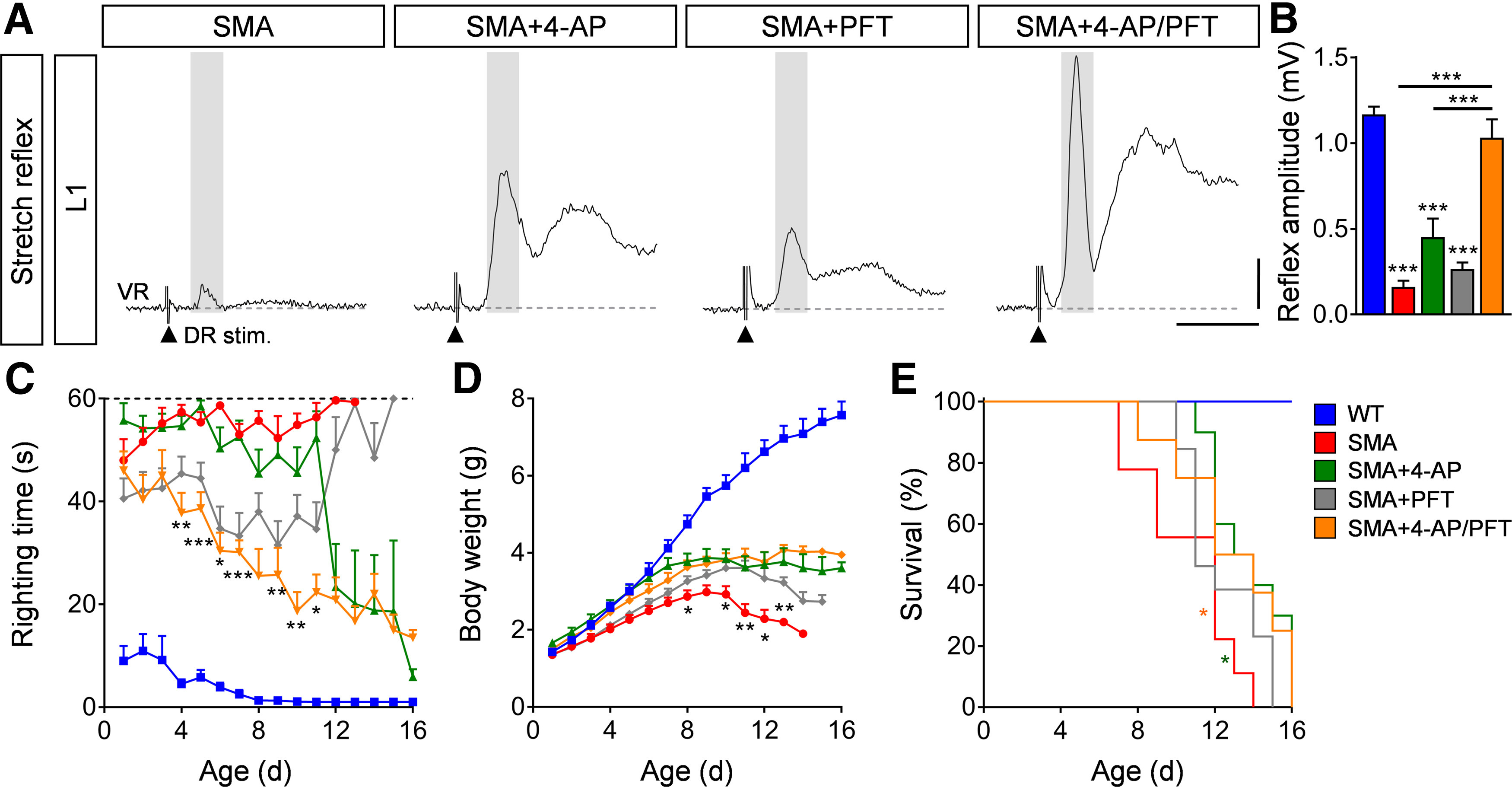
Combinatorial inhibition of motor neuron death and 4-AP treatment fully restores the sensory-motor circuit in SMA. A, Representative traces of extracellular recordings from L1 ventral root (VR) following L1 dorsal root (DR) stimulation from SMA, SMA + 4-AP, SMA + PFT, and SMA + 4-AP/PFT mice. Shaded area marks the monosynaptic response. Arrowheads indicate the stimulus artifact. Scale bars: 0.2 mV, 10 ms. B, Quantification of spinal ventral root amplitudes in millivolts recorded from WT, SMA, SMA + 4-AP, SMA + PFT, and SMA + 4-AP/PFT mice at P11. Amplitudes in millivolts: WT (6) = 1.17 ± 0.05, SMA (7) = 0.16 ± 0.04, SMA + 4-AP (7) = 0.45 ± 0.10, SMA + PFT (9) = 0.26 ± 0.04, SMA + 4-AP/PFT (5) = 1.03 ± 0.11; p for WT versus SMA, p for SMA + 4-AP and SMA + PFT < 0.0001, p for SMA versus SMA + 4-AP = 0.0426, p for SMA + 4-AP/PFT versus SMA, p for SMA + 4-AP and SMA + PFT < 0.0001, one-way ANOVA with Tukey's correction. Number of mice is reported in parentheses. C–E, Righting time (C), body weight (D), and Kaplan–Meyer analysis of survival (E) from the same groups as in B. Number of mice for SMA + PFT = 18, SMA + 4AP/PFT = 14, for other groups please see legend of Figure 1K–M. Righting time: SMA + PFT n = 18 (P10), 16 (P11), 5 (P12), 4 (P13), 3 (P13), 1 (P15); SMA + 4-AP/PFT n = 14 (P7), 13 (P8), 12 (P10), 6 (P11), 4 (P13), 3 (P15), 2 (P16); p for SMA + 4-AP versus SMA + 4-AP/PFT indicated with asterisks at P4 = 0.0038, P5 < 0.0001, P6 = 0.0121, P7 < 0.0001, P9 = 0.0072, P10 = 0.0058, P11 = 0.0144; Body weight: p for SMA versus SMA + 4-AP/PFT indicated with asterisks at P8 = 0.0494, P10 = 0.0814, P11 = 0.0065, P12 = 0.0143, and P13 = 0.0097, multiple t test with Holm–Sidak method; Kaplan–Meyer analysis: p for SMA versus SMA + 4-AP indicated with asterisks = 0.014, p for SMA versus SMA + 4-AP/PFT = 0.044, Mantel–Cox test. Data are presented as mean ± SEM. Asterisks on top of bars without a horizontal line indicate the significance compared with the WT group; *p < 0.05, **p < 0.01, ***p < 0.001.
These results demonstrate that simultaneous pharmacological induction of neuronal activity and neuroprotection have additive beneficial effects on the sensory-motor circuit and motor behavior in SMA.
Discussion
The hallmarks of neurologic disorders are neuronal death, synaptic loss and altered neuronal activity of select groups of neurons. However, the interaction of these neurodegenerative events and their contribution to the disease progression are not fully understood. The molecular causes of neuronal death and synaptic loss are beginning to emerge and vary among neurologic disorders (Gillingwater and Wishart, 2013; Kaplan et al., 2014; Tisdale and Pellizzoni, 2015; Chi et al., 2018; Baczyk et al., 2020; Shorrock et al., 2019; Simon et al., 2019). Currently, FDA-approved neuronal activity-modifying drugs are available to treat a subset of neurodegenerative diseases and to study activity-dependent mechanisms of neuronal death and synaptic loss (Hayes, 2004; Blackburn-Munro et al., 2005). Therefore, here we used 4-AP to investigate the effects of increased neuronal activity in a SMA mouse model which exhibits an overall reduction of excitatory drive (Ruiz et al., 2010; Mentis et al., 2011; Fletcher et al., 2017). 4-AP treatment improved motor behavior by increasing synaptic function and connectivity in the sensory-motor circuit in SMA mice. Notably, the combinatorial treatment of 4-AP together with the pharmacological inhibition of p53 resulted in an additive benefit for the recovery of sensory-motor neurotransmission and motor function in SMA mice. Taken together, our study reveals that the sufficient increase of neuronal activity improves synaptic connections and may have beneficial effects for patients with SMA and other neurodegenerative diseases.
Does network activity regulate neuronal death? The glutamatergic excitotoxicity model suggests that increased neuronal activity drives neuronal death by calcium influx following excessive excitatory synaptic input (Bading, 2017). However, there is mounting evidence in motor neuron diseases that selective degeneration of excitatory synapses leads to a reduction of excitation onto motor neurons (Fletcher et al., 2017; Baczyk et al., 2020; Seki et al., 2019). In addition, 4-AP-induced neuronal activity prevents death of human-derived ALS motor neurons (Naujock et al., 2016), contradicting the excitotoxicity model. Here, we demonstrate that 4-AP neither worsens nor improves motor neuron death, pointing to a complete uncoupling of neuronal activity and death in SMA. This is in agreement with our previous findings in which elimination or restoration of excitatory synapses onto WT and SMA motor neurons, respectively, had no effect on their survival both in vitro and in vivo (Simon et al., 2016; Fletcher et al., 2017). This view is also consistent with the cell autonomous mechanisms of p53-mediated death of SMA motor neurons, which is driven by the selective convergence in vulnerable neurons of specific splicing changes induced by SMN deficiency (Simon et al., 2017, 2019; Van Alstyne et al., 2018). These observations together with our results, show a clear uncoupling of motor neuron death and neuronal activity in SMA.
Although 4-AP has no effect on motor neuron survival, it significantly improves both the ability of remaining motor neurons to reinnervate skeletal muscles and the number of proprioceptive sensory synapses. Together with the previously reported resistance of proprioceptive neurons to cell death and reduced proprioceptive axonal arborization within the motor neuron pool of SMA mice (Mentis et al., 2011), our findings suggest that axonal sprouting of proprioceptive and motor axons likely act as a compensatory mechanism stimulated by neural activity. Intramuscular axonal sprouting is a well-established phenomenon that compensates functional loss in patients and mouse models with motor neuron diseases (Crawford and Pardo, 1996; Suzuki et al., 2010; Joyce and Carter, 2013; Querin et al., 2018).
Patients and mouse models with a mild form of SMA exhibit sprouting synapses (Monani et al., 2003; Simon et al., 2010; Murray et al., 2013; Querin et al., 2018), while severe SMA patients and mouse models with early onset exhibit no evidence of axonal sprouting (Crawford and Pardo, 1996; Cifuentes-Diaz et al., 2002), which is consistent with our present findings in untreated SMA mice. In agreement with a previous study (Kariya et al., 2008), we confirm that SMA mice do not display alterations of axonal pruning during natural occurring polysynaptic elimination compared with WT mice, suggesting a lack of disease-induced axonal plasticity in severe forms of SMA. In contrast, 4-AP treatment in WT and SMA mice is sufficient to accelerate activity-dependent polysynaptic elimination as reported previously (Sanes and Lichtman, 1999). Also, we find that 4-AP-induced neuronal activity leads to sprouting in motor and proprioceptive axons selectively in SMA mice. Accordingly, neuronal activity induces outgrowth and reinnervation of motor axons and proprioceptive afferents after nerve and spinal cord injuries (Ward et al., 2016; Takeoka and Arber, 2019). Taken together, our results indicate that enhancing neuronal activity is sufficient to induce axonal sprouting within the sensory-motor circuit in severe forms of SMA.
The molecular mechanisms of activity-induced sprouting are largely unknown and may differ for proprioceptive synapses and NMJs. Recent studies identified that the UBA1/GARS pathway (Shorrock et al., 2018), the classical complement system (Vukojicic et al., 2019) and the aberrant transmembrane protein Stasimon (Simon et al., 2019) cause proprioceptive synaptic loss, whereas the NMJ organizer Agrin is involved in denervation in SMA (Kim et al., 2017). However, the observation that 4-AP treatment neither prevents proprioceptive synapse removal nor NMJ denervation during the first postnatal week, but leads to an active increase of synapses at later stages, may indicate an independent mechanism for activity-induced sprouting. Presynaptic blockade of neuromuscular activity reduces the capability of axonal sprouting in axotomized muscles and mild SMA mice (Tam and Gordon, 2003; Murray et al., 2013), suggesting that synaptic neurotransmission plays a crucial role in sprouting mechanisms. 4-AP improves presynaptic transmission by activity-dependent calcium influx (Hayes, 2004; Kasatkina, 2016), which are both impaired in SMA (Jablonka et al., 2007; Ruiz et al., 2010; Mentis et al., 2011; Fletcher et al., 2017). Accordingly, 4-AP improves calcium-dependent transmission of NMJs and proprioceptive synapses in our study, and fully corrects defects in motor output in two other animal models of SMA (Pinter et al., 1997; Imlach et al., 2012). Axonal sprouting requires spatiotemporal rearrangements of actin and microtubule cytoskeletons. Similar to the 4-AP effects in our study, overexpression of the calcium-dependent actin-bundling protein plastin 3, a protective modifier of SMA families, increases the amount of filamentous actin in presynaptic terminals, the number of proprioceptive synapses and terminal arborization at NMJs in SMA mice (Oprea et al., 2008; Ackermann et al., 2013). Furthermore, the microtubule stabilizing protein signal transducer and activator of transcription-3 (STAT3) is calcium-dependently translated in lesioned motor axons and increases collateral sprouting of sensory neurons and axon elongation in ALS mice following activation of ciliary neurotrophic factor (CNTF; Bareyre et al., 2011; Ben-Yaakov et al., 2012; Selvaraj et al., 2012), which mediates sprouting in a mild SMA mouse model (Simon et al., 2010). Collectively, these findings suggest that neuronal activity acts on calcium-dependent mechanisms to remodel the cytoskeleton for axonal sprouting in neurodegeneration.
In this study, 4-AP ameliorates the severe motor phenotype of SMA mice during the second postnatal week, which coincides with the increased number and function of synaptic contacts, suggesting that the enhanced sensory-motor circuit is responsible for the observed activity-improved motor behavior. This is in agreement with previous findings in which exercise-induced or pharmacologically-induced enhancement of neuronal activity improves the phenotype of SMA and ALS mouse models (Biondi et al., 2008; Saxena et al., 2013; Fletcher et al., 2017), holding promise that 4-AP may be beneficial for patients with motor neuron diseases. 4-AP treatment (10 mg BID) of 11 SMA Type III adult patients over six weeks resulted in an improved H-reflex as a measurement of proprioceptive synaptic function and a trend toward increased functional motor abilities (Chiriboga et al., 2020). However, it did not reach significance likely because of ∼five times lower dosage compared with our study and the short treatment duration. In agreement, SMN targeting therapy requires at least three months of treatment to improve motor function in SMA patients (De Vivo et al., 2019). Additionally, one-year mean duration of 4-AP treatment in a case series resulted in improved quality of life of two ALS patients because of regain of facial motor function and decreased disease progression rate (Peikert et al., 2019), suggesting a beneficial long-term effect of 4-AP on muscle function in motor neuron diseases. We further demonstrate that a co-treatment of 4-AP with a p53 inhibitor that recues motor neurons has an additive effect on motor behavior and fully restores the spinal ventral root response in an SMN-independent manner. In contrast, rescue of motor neurons has little effect on the life span of SMA mice, which is in agreement with previous reports (Park et al., 2010; Martinez et al., 2012; Fletcher et al., 2017; Simon et al., 2019). Possible reasons for contributing to the lifespan of SMA mice except motor neuron survival, have been reported to be because of the systemic pathology resulting from ubiquitous SMN deficiency in other vital organs (Shababi et al., 2014). Because of its role as tumor suppression, direct inhibition of p53 is not suitable in patients. However, we recently showed that p53-dependent motor neuron death in SMA mice is inhibited by MW150 (Simon et al., 2019), a highly selective p38 MAPK inhibitor (Roy et al., 2015). Pursuit of 4-AP treatment combined with MW150 to enhance axonal sprouting and neuroprotection, respectively, might assist recently approved SMN-targeting therapies strategies as a new combinational approach for SMA patients.
In conclusion, our study emphasizes the important contribution of decreased neuronal activity to SMA disease progression. Elevation of neuronal activity improves muscle function and motor behavior by increasing synaptic transmission and rewiring synaptic connections in the sensory-motor circuit and possibly in other dysfunctional networks during neurodegeneration.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Grant SI-1969/2-1 (to C.M.S.). G.Z.M. was supported by the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH) Grant R01-NS078375, The NIH Blueprint for Neuroscience Research, the National Institute on Alcohol Abuse and Alcoholism and NINDS Grant R01-AA027079, The SMA Foundation, and Project-ALS. We thank Dr. Livio Pellizzoni for helpful comments. We also thank Dr. Stefan Hallermann, Dr. Johannes Hirrlinger, and Dr. Tobias Langenhan for providing reagents and facility access.
The authors declare no competing financial interests.
References
- Ackermann B, Kröber S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, Wunderlich TF, Riessland M, Tabares L, Wirth B (2013) Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 22:1328–1347. 10.1093/hmg/dds540 [DOI] [PubMed] [Google Scholar]
- Avoli M, Jefferys JG (2016) Models of drug-induced epileptiform synchronization in vitro. J Neurosci Methods 260:26–32. 10.1016/j.jneumeth.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baczyk M, Alami NO, Delestree N, Martinot C, Tang L, Commisso B, Bayer D, Doisne N, Frankel W, Manuel M, Roselli F, Zytnicki D (2020) Synaptic restoration by cAMP/PKA drives activity-dependent neuroprotection to motoneurons in ALS. J Exp Med 217:e20191734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2017) Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med 214:569–578. 10.1084/jem.20161673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Büning H, Kerschensteiner M (2011) In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci USA 108:6282–6287. 10.1073/pnas.1015239108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, Blesch A, Pilpel Y, Twiss JL, Fainzilber M (2012) Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J 31:1350–1363. 10.1038/emboj.2011.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O, Grondard C, Lécolle S, Deforges S, Pariset C, Lopes P, Cifuentes-Diaz C, Li H, della Gaspera B, Chanoine C, Charbonnier F (2008) Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 28:953–962. 10.1523/JNEUROSCI.3237-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn-Munro G, Dalby-Brown W, Mirza NR, Mikkelsen JD, Blackburn-Munro RE (2005) Retigabine: chemical synthesis to clinical application. CNS Drug Rev 11:1–20. 10.1111/j.1527-3458.2005.tb00033.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Redondo B, Nuwal N, Kneitz S, Nuwal T, Halder P, Liu Y, Ehmann N, Scholz N, Mayer A, Kleber J, Kähne T, Schmitt D, Sadanandappa MK, Funk N, Albertova V, Helfrich-Förster C, Ramaswami M, Hasan G, Kittel RJ, Langenhan T, et al. (2019) Implications of the Sap47 null mutation for synapsin phosphorylation, longevity, climbing proficiency and behavioural plasticity in adult Drosophila. J Exp Biol 222:jeb203505. 10.1242/jeb.203505 [DOI] [PubMed] [Google Scholar]
- Chi H, Chang HY, Sang TK (2018) Neuronal cell death mechanisms in major neurodegenerative diseases. Int J Mol Sci 19:3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriboga CA, Marra J, LaMarca NM, Dunaway Young S, Weimer LH, Levin B, McCabe B (2020) Lack of effect on ambulation of dalfampridine-ER (4-AP) treatment in adult SMA patients. Neuromuscul Disord 30:693–700. [DOI] [PubMed] [Google Scholar]
- Chopra R, Shakkottai VG (2014) The role for alterations in neuronal activity in the pathogenesis of polyglutamine repeat disorders. Neurotherapeutics 11:751–763. 10.1007/s13311-014-0289-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L (2015) A brief introduction to the transduction of neural activity into Fos signal. Dev Reprod 19:61–67. 10.12717/DR.2015.19.2.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J (2002) Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet 11:1439–1447. 10.1093/hmg/11.12.1439 [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3:97–110. 10.1006/nbdi.1996.0010 [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, Kirschner J, Kuntz NL, Parsons JA, Ryan MM, Butterfield RJ, Topaloglu H, Ben-Omran T, Sansone VA, Jong YJ, Shu F, Staropoli JF, Kerr D, Sandrock AW, Stebbins C, et al. (2019) Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord 29:842–856. 10.1016/j.nmd.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers MP, Nikoletopoulou V, Barde YA (2013) Cell biology in neuroscience: death of developing neurons: new insights and implications for connectivity. J Cell Biol 203:385–393. 10.1083/jcb.201306136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EV, Simon CM, Pagiazitis JG, Chalif JI, Vukojicic A, Drobac E, Wang X, Mentis GZ (2017) Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat Neurosci 20:905–916. 10.1038/nn.4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Wishart TM (2013) Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol Appl Neurobiol 39:320–334. 10.1111/nan.12014 [DOI] [PubMed] [Google Scholar]
- Greensmith L, Dick J, Emanuel AO, Vrbová G (1996) Induction of transmitter release at the neuromuscular junction prevents motoneuron death after axotomy in neonatal rats. Neuroscience 71:213–220. 10.1016/0306-4522(95)00446-7 [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG (1994) Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 45:1227–1234. [PubMed] [Google Scholar]
- Hayes KC (2004) The use of 4-aminopyridine (fampridine) in demyelinating disorders. CNS Drug Rev 10:295–316. 10.1111/j.1527-3458.2004.tb00029.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD (2012) SMN is required for sensory-motor circuit function in Drosophila. Cell 151:427–439. 10.1016/j.cell.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M (2007) Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 179:139–149. 10.1083/jcb.200703187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E, Lundberg A, Rudomin P, Sykova E (1982) Effects of 4-aminopyridine on synaptic transmission in the cat spinal cord. Brain Res 240:117–129. 10.1016/0006-8993(82)90649-7 [DOI] [PubMed] [Google Scholar]
- Joyce NC, Carter GT (2013) Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R 5:S89–S95. 10.1016/j.pmrj.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE (2014) Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 81:333–348. 10.1016/j.neuron.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR (2008) Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17:2552–2569. 10.1093/hmg/ddn156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasatkina LA (2016) 4-Аminopyridine sequesters sequesters intracellular Ca2+ which triggers exocytosis in excitable and non-excitable cells. Sci Rep 6:34749. 10.1038/srep34749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Caine C, Awano T, Herbst R, Monani UR (2017) Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice. Hum Mol Genet 26:2377–2385. 10.1093/hmg/ddx124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch GE, Drewe JA (1993) Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol 102:797–816. 10.1085/jgp.102.5.797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet 14:845–857. 10.1093/hmg/ddi078 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–165. 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- Leroy F, Zytnicki D (2015) Is hyperexcitability really guilty in amyotrophic lateral sclerosis? Neural Regen Res 10:1413–1415. 10.4103/1673-5374.165308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Lin MY, Zingg B, Feng Z, Ko CP (2010) Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One 5:e15457. 10.1371/journal.pone.0015457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Gibbs RM, Feng Z, Ko CP (2012) Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet 21:185–195. 10.1093/hmg/ddr453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez TL, Kong L, Wang X, Osborne MA, Crowder ME, Van Meerbeke JP, Xu X, Davis C, Wooley J, Goldhamer DJ, Lutz CM, Rich MM, Sumner CJ (2012) Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci 32:8703–8715. 10.1523/JNEUROSCI.0204-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Wooltorton JR, Watkins CS (1998) Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen Pharmacol 30:13–24. 10.1016/S0306-3623(97)00034-7 [DOI] [PubMed] [Google Scholar]
- Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA (2019) ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci 13:1310. 10.3389/fnins.2019.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69:453–467. 10.1016/j.neuron.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M, Burghes AHM (2003) A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol 160:41–52. 10.1083/jcb.200208079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LM, Beauvais A, Bhanot K, Kothary R (2013) Defects in neuromuscular junction remodelling in the Smn(2B/-) mouse model of spinal muscular atrophy. Neurobiol Dis 49:57–67. 10.1016/j.nbd.2012.08.019 [DOI] [PubMed] [Google Scholar]
- Naujock M, Stanslowsky N, Bufler S, Naumann M, Reinhardt P, Sterneckert J, Kefalakes E, Kassebaum C, Bursch F, Lojewski X, Storch A, Frickenhaus M, Boeckers TM, Putz S, Demestre M, Liebau S, Klingenstein M, Ludolph AC, Dengler R, Kim KS, et al. (2016) 4-Aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells 34:1563–1575. 10.1002/stem.2354 [DOI] [PubMed] [Google Scholar]
- Nelson SB, Valakh V (2015) Excitatory/Inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87:684–698. 10.1016/j.neuron.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320:524–527. 10.1126/science.1155085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, Monani UR (2010) Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci 30:12005–12019. 10.1523/JNEUROSCI.2208-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peikert K, Naumann M, Günther R, Wegner F, Hermann A (2019) Off-label treatment of 4 amyotrophic lateral sclerosis patients with 4-aminopyridine. J Clin Pharmacol 59:1400–1404. 10.1002/jcph.1437 [DOI] [PubMed] [Google Scholar]
- Pinter MJ, Waldeck RF, Cope TC, Cork LC (1997) Effects of 4-aminopyridine on muscle and motor unit force in canine motor neuron disease. J Neurosci 17:4500–4507. 10.1523/JNEUROSCI.17-11-04500.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querin G, Lenglet T, Debs R, Stojkovic T, Behin A, Salachas F, Le Forestier N, Amador MDM, Lacomblez L, Meininger V, Bruneteau G, Laforêt P, Blancho S, Marchand-Pauvert V, Bede P, Hogrel JY, Pradat PF (2018) The motor unit number index (MUNIX) profile of patients with adult spinal muscular atrophy. Clin Neurophysiol 129:2333–2340. 10.1016/j.clinph.2018.08.025 [DOI] [PubMed] [Google Scholar]
- Roy SM, Grum-Tokars VL, Schavocky JP, Saeed F, Staniszewski A, Teich AF, Arancio O, Bachstetter AD, Webster SJ, Van Eldik LJ, Minasov G, Anderson WF, Pelletier JC, Watterson DM (2015) Targeting human central nervous system protein kinases: an isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer's disease mouse models. ACS Chem Neurosci 6:666–680. 10.1021/acschemneuro.5b00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R, Casañas JJ, Torres-Benito L, Cano R, Tabares L (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30:849–857. 10.1523/JNEUROSCI.4496-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22:389–442. 10.1146/annurev.neuro.22.1.389 [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. 10.1016/j.neuron.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Seki S, Yamamoto T, Quinn K, Spigelman I, Pantazis A, Olcese R, Wiedau-Pazos M, Chandler SH, Venugopal S (2019) Circuit-specific early impairment of proprioceptive sensory neurons in the SOD1(G93A) mouse model for ALS. J Neurosci 39:8798–8815. 10.1523/JNEUROSCI.1214-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M (2012) Local axonal function of STAT3 rescues axon degeneration in the PMN model of motoneuron disease. J Cell Biol 199:437–451. 10.1083/jcb.201203109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M, Lorson CL, Rudnik-Schöneborn SS (2014) Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J Anat 224:15–28. 10.1111/joa.12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock HK, van der Hoorn D, Boyd PJ, Llavero Hurtado M, Lamont DJ, Wirth B, Sleigh JN, Schiavo G, Wishart TM, Groen EJN, Gillingwater TH (2018) UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain 141:2878–2894. 10.1093/brain/awy237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock HK, Gillingwater TH, Groen EJN (2019) Molecular mechanisms underlying sensory-motor circuit dysfunction in SMA. Front Mol Neurosci 12:59. 10.3389/fnmol.2019.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CM, Jablonka S, Ruiz R, Tabares L, Sendtner M (2010) Ciliary neurotrophic factor-induced sprouting preserves motor function in a mouse model of mild spinal muscular atrophy. Hum Mol Genet 19:973–986. 10.1093/hmg/ddp562 [DOI] [PubMed] [Google Scholar]
- Simon CM, Janas AM, Lotti F, Tapia JC, Pellizzoni L, Mentis GZ (2016) A stem cell model of the motor circuit uncouples motor neuron death from hyperexcitability induced by SMN deficiency. Cell Rep 16:1416–1430. 10.1016/j.celrep.2016.06.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CM, Dai Y, Van Alstyne M, Koutsioumpa C, Pagiazitis JG, Chalif JI, Wang X, Rabinowitz JE, Henderson CE, Pellizzoni L, Mentis GZ (2017) Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep 21:3767–3780. 10.1016/j.celrep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CM, Van Alstyne M, Lotti F, Bianchetti E, Tisdale S, Watterson DM, Mentis GZ, Pellizzoni L (2019) Stasimon contributes to the loss of sensory synapses and motor neuron death in a mouse model of spinal muscular atrophy. Cell Rep 29:3885–3901.e5. 10.1016/j.celrep.2019.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Katsuno M, Banno H, Takeuchi Y, Kawashima M, Suga N, Hashizume A, Hama T, Uchida K, Yamashita F, Nakamura T, Hirayama M, Tanaka F, Sobue G (2010) The profile of motor unit number estimation (MUNE) in spinal and bulbar muscular atrophy. J Neurol Neurosurg Psychiatry 81:567–571. 10.1136/jnnp.2009.190462 [DOI] [PubMed] [Google Scholar]
- Takeoka A, Arber S (2019) Functional local proprioceptive feedback circuits initiate and maintain locomotor recovery after spinal cord injury. Cell Rep 27:71–85.e3. 10.1016/j.celrep.2019.03.010 [DOI] [PubMed] [Google Scholar]
- Tam SL, Gordon T (2003) Mechanisms controlling axonal sprouting at the neuromuscular junction. J Neurocytol 32:961–974. 10.1023/B:NEUR.0000020635.41233.0f [DOI] [PubMed] [Google Scholar]
- Tisdale S, Pellizzoni L (2015) Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci 35:8691–8700. 10.1523/JNEUROSCI.0417-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Benito L, Ruiz R, Tabares L (2012) Synaptic defects in spinal muscular atrophy animal models. Dev Neurobiol 72:126–133. 10.1002/dneu.20912 [DOI] [PubMed] [Google Scholar]
- Van Alstyne M, Simon CM, Sardi SP, Shihabuddin LS, Mentis GZ, Pellizzoni L (2018) Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev 32:1045–1059. 10.1101/gad.316059.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan SK, Kemp Z, Hatzipetros T, Vieira F, Valdez G (2015) Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J Comp Neurol 523:2477–2494. 10.1002/cne.23848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschuuren JJ, Wirtz PW, Titulaer MJ, Willems LN, van Gerven J (2006) Available treatment options for the management of Lambert-Eaton myasthenic syndrome. Expert Opin Pharmacother 7:1323–1336. 10.1517/14656566.7.10.1323 [DOI] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C (2013) Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav 3:431–457. 10.1002/brb3.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukojicic A, Delestrée N, Fletcher EV, Pagiazitis JG, Sankaranarayanan S, Yednock TA, Barres BA, Mentis GZ (2019) The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell Rep 29:3087–3100.e7. 10.1016/j.celrep.2019.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PJ, Jones LN, Mulligan A, Goolsby W, Wilhelm JC, English AW (2016) Optically-induced neuronal activity is sufficient to promote functional motor axon regeneration in vivo. PLoS One 11:e0154243. 10.1371/journal.pone.0154243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Greenberg ME (2011) Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol 3:a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]