

Abstract
Gestational exposure to environmental stress induces fetal growth restriction (FGR), and thereby increasing the risk of infant death and chronic noncommunicable diseases in adults. However, the mechanism by which environmental stress induces FGR remains unclear. Based on case-control study, we found that the reduced level of melatonin (MT), a major secretory product from the pineal gland, was observed in placentae of FGR. This work was to investigate the protective effect of MT on environmental stress-caused FGR and its mechanisms. We used cadmium (Cd) as an environmental stressor to stimulate pregnant mice and thereby establishing a FGR model. The data showed that maternal Cd exposure lowered the P4 concentration in maternal sera, placentae and amniotic fluid, and caused FGR. Correspondingly, the expression of CYP11A1, a critical P4 synthase, was markedly downregulated in Cd-treated placentae. Simultaneously, Cd triggered BNIP3-dependent mitophagy in placental trophoblasts, as determined by the degradation of mitochondrial proteins, including HSP60 and COX IV, and the accumulation of puncta representing co-localization of TOM20 with LC3B or BNIP3 with LC3B. Based on our case-control study, we also found that activated BNIP3-dependent mitophagy and P4 synthesis inhibition occurred in SGA placentae. Most importantly, BNIP3 siRNA reversed Cd-induced P4 synthesis suppression in human placental trophoblasts. It is noteworthy that MT alleviated Cd-caused P4 synthesis suppression and FGR via antagonizing BNIP3-dependent mitophagy in placental trophoblasts. Further results confirmed that MT attenuated Cd-triggered BNIP3-dependent mitophagy via blocking GCN2/ATF4 signaling. Amusingly, Cd triggered oxidative stress and then activating GCN2/ATF4 signaling in placental trophoblasts. As expected, MT obviously suppressed Cd-caused reactive oxygen species (ROS) release. In the present study, we propose a neoteric mechanism by which MT protects against environmental stress-impaired P4 synthesis and fetal growth via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. As above, MT is a potential therapeutic agent antagonizing environmental stress-induced developmental toxicity.
Keywords: Melatonin, Mitophagy, Environmental stress, ROS, Fetal growth restriction, Placenta
Graphical abstract

Highlights
-
•
Melatonin protects against Cd-induced fetal growth restriction.
-
•
Melatonin attenuates Cd-induced placental P4 synthesis inhibition by mitophagy.
-
•
Melatonin suppresses Cd-triggered placental mitophagy via blocking GCN2/ATF4.
-
•
Melatonin blocks Cd-activated placental GCN2/ATF4 signaling via repressing ROS.
-
•
Activated mitophagy and reduced P4 synthesis occur in SGA placentae.
1. Introduction
Fetal growth restriction (FGR), containing low-birth-weight (LBW) and small-for-gestational-age (SGA) infants, refers to that the fetus fails to reach genetically determined growth range on account of gestational exposure to various harmful factors. Two population studies indicated that the incidence of global LBW was 14.6% and the incidence of SGA was 27% in low- and middle-income countries [1,2]. FGR is not only an important dangerous factor of infant death, but also increases the risk of chronic noncommunicable diseases in adults, such as cardiovascular diseases, cancer, metabolic disorders, chronic immune diseases and neurological impairments [[3], [4], [5], [6], [7], [8], [9]]. Numerous studies demonstrated that prenatal exposure to various stressors, such as noise, smoking, microorganisms, endocrine disruptors and heavy metals, could cause FGR [[10], [11], [12], [13], [14]]. Increasing evidences showed that Cd was a main environmental stressor inducing FGR. Epidemiological studies found that maternal exposure to Cd during gestation markedly elevated the risk of intrauterine growth retardation [[15], [16], [17], [18]]. Animal experiments further verified that gestational exposure to environmental Cd caused FGR [[19], [20], [21]]. Nevertheless, the mechanism by which environmental stress induces FGR remains unclear.
Mitophagy is the process in which the cell selectively eliminates mitochondria via lysosomes [22]. Temperately activated mitophagy contributes to remove damaged or superfluous mitochondria, and thereby maintaining mitochondrial function and adapting to stress [23,24]. However, overactivated mitophagy causes excessive mitochondrial loss, to result in cellular bioenergy deficit and cell death [[25], [26], [27]]. Recently, an animal study confirmed that placental mitochondria was highly adaptable over the course of normal gestation, which is benefit to improve fetal growth [28]. So far, there was no studies to clarify the effect of placental mitophagy on fetal growth in response to environmental stress. Accumulating evidences demonstrated that mitochondrial progesterone (P4) synthases, containing StAR, CYP11A1 and 3β-HSD, was crucial in the process whereby mitochondria synthesizes cholesterol into placental P4, a sterol hormone for supporting fetal growth [10,[29], [30], [31], [32], [33], [34], [35], [36], [37]]. Our recent study found that gestational exposure to the environmental stressor Cd reduced the level of placental mitochondrial proteins, including StAR and 3β-HSD [13]. Emerging studies found that exposure to environmental stress resulted in excessive mitophagy and the following mitochondrial proteins loss in hepatic cells [38,39]. Hereby, we speculate that environmental stress affects fetal growth maybe via triggering placental mitophagy. In the past, numerous studies focused on parkin-dependent mitophagy less abundantly in parkin-independent mitophagy. BNIP3, a novel mitophagy receptor, was detected to be expressed in animal and human placentae [[40], [41], [42]]. As above, we make a hypothesis that placental BNIP3-dependent mitophagy contributes to environmental stress-induced FGR.
Melatonin (N-acetyl-5-methoxytryptamine, MT), a major secretory product from the pineal gland in mammals and human, plays a pivotal role in the neuro-immuno-endocrine system [43,44]. MT has numerous physiological functions including anti-inflammation, circadian, anti-apoptotic properties and endocrine rhythm regulation, and its potent antioxidant activity [[45], [46], [47], [48]]. A previous study had demonstrated that MT alleviated infection-caused multifarious cellular stress response, such as oxidative stress, hypoxia and heat stress, in mouse placentae [49]. Furthermore, emerging evidences indicated that MT could antagonize stress-triggered mitophagy in nonpregnant tissues, such as platelet and cardiac microvasculature [50,51]. Until now, there is no studies to explore the effect of MT on placental BNIP3-dependent mitophagy in response to environmental stress.
In present study, we first found activated BNIP3-dependent mitophagy and P4 synthesis suppression occurred in SGA placentae based on our case-control study. Subsequently, we used Cd as an environment stressor to establish in vivo and in vitro models to promote our understanding for the role of placental BNIP3-dependent mitophagy in environmental stress-impaired P4 synthesis and fetal growth. Then, we further investigated the protective effect of MT on FGR and its mechanism upon environmental stress.
2. Materials and methods
2.1. Reagents
Cadmium chloride (202908), Melatonin (M5250), Chloroquine diphosphate (C6628) were from Sigma Chemical Co (St. Louis, MO). Antibodies against HSP60 (12165S), COX IV (4850S), LC3B-I/II (3868S), ATF4 (11815S), p-eIF2α (9721S), eIF2α (2103S), GCN2 (3302S) and β-Actin (4970S) were from Cell Signaling Technology (Beverley, MA). Antibodies against CYP11A1 (SC18043) and TOM20 (SC17764) were from Santa Cruz (California, US). Antibodies against BNIP3 (ab10433), PGC1α (ab54481) and LC3B-I/II (ab48394) were from Abcam (Cambridge, MA). The secondary antibody conjugated with Alexa Fluor 488 (711-545-152) and the secondary antibody conjugated with Cy3TM (715-165-150) were from Jackson ImmunoResearch (PA, USA). Progesterone (P4) ELISA kit (E-EL-0090C) was from Elabscience (Wuhan, China). Human short interfering RNAs targeting BNIP3 and GCN2 were from GenePharma (Shanghai, China).
2.2. Case-control study
All human placentae (n = 70) were from Department of Obstetrics and Gynecology of the First Affiliated Hospital of Anhui Medical University. The placentae were collected after term delivery. Meanwhile, the birth weight was also recorded. The placentae of large-for-gestational-age and premature infants were excluded in this study. According to a previous epidemiological study, we divided the placentae into appropriate-for-gestational-age (AGA) and small-for-gestational-age (SGA) placentae based on the birth weight [52]. We performed the experimental procedures in reference to the standard operating procedure (Ethical approval number: 20190297).
2.3. Animal experiments
8-week-old CD-1 mice including male and female were from Beijing Vital River (Beijing, China). All mice freely acquired water and food at any time and were fed under standard conditions (light/dark alternate for 12 h, 20–25 °C, 50–60% air humidity) to acclimatize for 2 weeks. Four females and two males spend the night in one cage for mating. In the following morning, the female with a vaginal plug was considered as gestational day 0 (GD0). Our in vivo experimental procedures were approved by the Association of Laboratory Animal Sciences at Anhui Medical University and implemented according to its guidelines for humane treatment. Melatonin (MT) was dissolved using dimethyl sulfoxide (DMSO) to gain the stock solution (500 mg/ml) and then diluted it with saline to working solution. In working solution, the final concentration of DMSO was no more than 0.1%. Cadmium (Cd) was directly dissolved in saline to produce working solution (0.45 mg/ml). Saline containing the same amount of DMSO was used as vehicle control.
To explore whether gestational exposure to environmental stress inhibits placental progesterone (P4) synthesis and BNIP3-dependent mitophagy and its mechanism, all pregnant mice except that in control and MT groups were received intraperitoneal injection with 4.5 mg/kg CdCl2 on GD8. The dose of Cd was designated based on our previous study [21]. Each pregnant mouse in control group was treated with saline. The pregnant mice were performed euthanasia on GD12 or GD16. Fetal sizes were recorded on GD16. Sera of maternal mice, placentae and amniotic fluid were collected on GD12 or GD16.
To investigate whether MT alleviates environmental stress-caused placental P4 synthesis inhibition and BNIP3-dependent mitophagy and its mechanism, we randomly divided forty-eight pregnant mice into control, Cd , MT and MT + Cd groups. Each pregnant mouse except which in the control and MT groups was received intraperitoneal injection with 4.5 mg/kg CdCl2 on GD8. In MT and MT + Cd groups, each pregnant mouse was received intraperitoneal injection with 5 mg/kg/day MT from GD7 to GD15. The pregnant mice treated with saline were regarded as controls. All pregnant mice were performed euthanasia on GD12 or GD16. Fetal sizes were recorded. Sera of maternal mice, placentae and amniotic fluid were collected.
2.4. Cell culture and treatment
Human JEG-3 cells were from Chinese Academy Sciences (TCHu195). The cells were grown in MEM supplemented with fetal bovine serum (10%), non-essential amino acids (1%), sodium pyruvate (1%) and penicillin-streptomycin (100 units/ml). We selected Cd concentration in medium in reference to our previous study [21]. MT was dissolved using DMSO as a stock solution (1 M) and diluted it in cell medium before use. Other cell medium contained 1% DMSO to exclude its effect in each in vitro assay.
We divided the in vitro study into six separate experiments. In order to investigate whether environmental stress inhibits progesterone (P4) synthesis and activates BNIP3-dependent mitophagy, human JEG-3 cells were stimulated using CdCl2 (20 μM) for 0, 2, 6 or 12 h. To explore whether BNIP3-dependent mitophagy mediates environmental stress-caused P4 synthesis suppression, the cells were pretreated using BNIP3 siRNA (siR) before Cd stimulation. To investigate the role of GCN2/ATF4 signaling in environmental stress-triggered BNIP3-dependent mitophagy, the cells were pretreated using GCN2 siR before Cd stimulation. To obtain evidence to support the hypothesis that MT confers powerful protective effects on environmental stress-induced P4 synthesis suppression and FGR via blocking GCN2/BNIP3/mitophagy pathways, the cells was pretreated with MT (1 mM) for 1 h before CdCl2 (20 μM) stimulation. To further confirm that Cd activated GCN2/ATF4 signaling via ROS release, human JEG-3 cells were pretreated with PBN (4 mM) for 1 h before CdCl2 (20 μM) stimulation. To obtain more evidence that MT blocked placental GCN2/ATF4 signaling via its the strong antioxidant effect, human JEG-3 cells were pretreated with MT (1 mM) for 1 h before H2O2 (1.5 mM) stimulation.
2.5. Immunoblotting
The total protein of placentae and cells was extracted using RIPA buffer containing protease inhibitor cocktail. Based on the manufacturer's protocol, the protein concentration was detected recurring to a Pierce BCA Protein Assay kit. After total protein being boiled, protein (10–50 μg per well) was separated in 8%–15% SDS-PAGE and then transferred onto a PVDF membrane. To block the proteins, the 5% milk without fat was used to incubate the PVDF membrane. Subsequently, the membrane was incubated for 1–3 h using primary antibodies, such as CYP11A1, HSP60, COX IV, LC3BI/II, GCN2, p-eIF2α, eIF2α and ATF4. Following wash, the corresponding secondary antibody was used to incubate the membrane. The signal was detected using the digital imaging equipment. The quantification for protein bands was performed by feat of Image-Pro Plus software.
2.6. Isolation of total RNA and real-time RT-PCR
The total RNA was extracted using the TRI reagent in human JEG-3 cells. Subsequently, the genomic DNA was removed via using DNase without RNase. Real-time RT-PCR was implemented after reverse transcription using AMV. The normalization was performed based on the level of 18S mRNA. The sequences of human BNIP3 gene-specific primer were 5′-CTTTAAACACCCGAAGCGCA-3′ (forward) and 5′-GTGCTGGTGGAGGTTGTCA’ (reverse). The sequences of human 18S gene-specific primer were 5′- CGGCTACCACATCCAAGGAA-3′ (forward) and 5′-GCTGGAATTACCGCGGCT-3′ (reverse).
2.7. Immunofluorescence
The frozen placental tissues were cut into 5 μm sections after dehydration using sucrose solution (30%). Human JEG-3 cells were grown on the slides, and then fixed using paraformaldehyde (4%) for 30 min after treatment. To block the nonspecific binding sites, the frozen sections and the cellular slides were incubated with PBS supplemented with 10% normal donkey serum for 1.5 h. Subsequently, the frozen sections were incubated with the mixture containing primary antibodies against LC3BI/II (1:200) and TOM20 (1:200) at 37 °C for 2 h. The cellular slides were incubated with the mixture containing primary antibodies against LC3BI/II (1:200) and BNIP3 (1:200) at 37 °C for 2 h. After being washed with PBS, the frozen sections and the cellular slides were incubated using the mixture of luciferin conjugated secondary antibodies. The nucleus was tagged using hoechst 33258 for 5 min. Each section was observed by a confocal microscope (LSM880, Zeiss) or fluorescence microscope (BX53F, Olympus).
2.8. RNA interference
Human JEG-3 cells were grown to 70–80% in 60-mm culture dishes. Homo sapiens BNIP3-, GCN2- or ATF4-specific small interfering RNA (siRNA) were mixed with lipofectamine 3000 for 20 min in Opti-MEM without serum. The scrambled RNA was used as vehicle control. The mixture was added into cell medium without serum and penicillin/streptomycin to transfect human JEG-3 cells for 6 h. Subsequently, the cells were grown in complete medium for 42 h before Cd stimulation. The cells treated with PBS were regarded as control. Finally, the cells were washed and then collected. The sequences of BNIP3 siRNA were 5′-GCAUCAAGUUACAGGUCUUTT-3′ (forward) and 5′-AAGACCUGUAACUUGAUGCTT-3′ (reverse). The sequences of GCN2 siRNA were 5′-GCCUCGGUUUCUAUUUAUATT-3′ (forward) and 5′-UAUAAAUAGAAACCGAGGCTT-3′ (reverse). The sequences of ATF4 siRNA were 5′-GUGAGAAACUGGAUAAGAATT -3′ (forward) and 5′-UUCUUAUCCAGUUUCUCACTT-3′ (reverse). The scrambled siRNA control sequences were 5′-UUCUCCGAACGUGUCACGUTT-3′ (forward) and 5′-ACGUGACACGUUCGGAGAATT-3′ (reverse).
2.9. ELISA assay
The level of progesterone and melatonin (MT) was detected in reference to the manufacturer's instructions. P4 level in serum of maternal mice, amniotic fluid and cells were shown as ng/ml. The frozen placentae were thawed and then 100 mg of tissue was homogenized in 900 μl PBS. After being centrifuged at 5000 g for 10 min at 4 °C, the supernatant was collected for subsequent detection. P4 and MT content on placentae are expressed as ng/g.
2.10. Measurement of GSH content
The concentration of total GSH (T-GSH) and GSSG in placentae was quantified by a GSH and GSSG Assay Kit following the manufacturer's instruction (Bioengineering Institute of Jincheng, Nanjing, China). The concentration of T-GSH and GSSG in placentae were shown as μmol/mg. The concentration of GSH was calculated as follows: GSH = T-GSH – 2GSSG.
2.11. Reactive oxygen species assay
Human JEG-3 cells were grown to 70–80% in MEM medium plus with fetal bovine serum (10%), non-essential amino acids (1%), sodium pyruvate (1%) and penicillin-streptomycin (100 units/ml). After Cd administration, the cells were washed three times with PBS. Subsequently, the level of intracellular ROS was examined using DCFH-DA (10 μM). The cells were incubated with DCFH-DA at 37 °C for 15 min, then washed three times with PBS. The fluorescence signal was detected by a fluorescence microscope (BX53F, Olympus)) or Multimode Reader (SYNERGY4, BioTek).
2.12. Statistical analysis
In the present study, all data were shown as mean ± SEM. All statistical analysis is performed recurring to SPSS 23.0. The differences between two groups were analyzed using Student's t-test. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. Difference was regarded obvious when P<0.05.
3. Results
3.1. Association of placental mitophagy, reduced melatonin (MT) level, inhibition of progesterone synthesis and all-cause fetal growth restriction
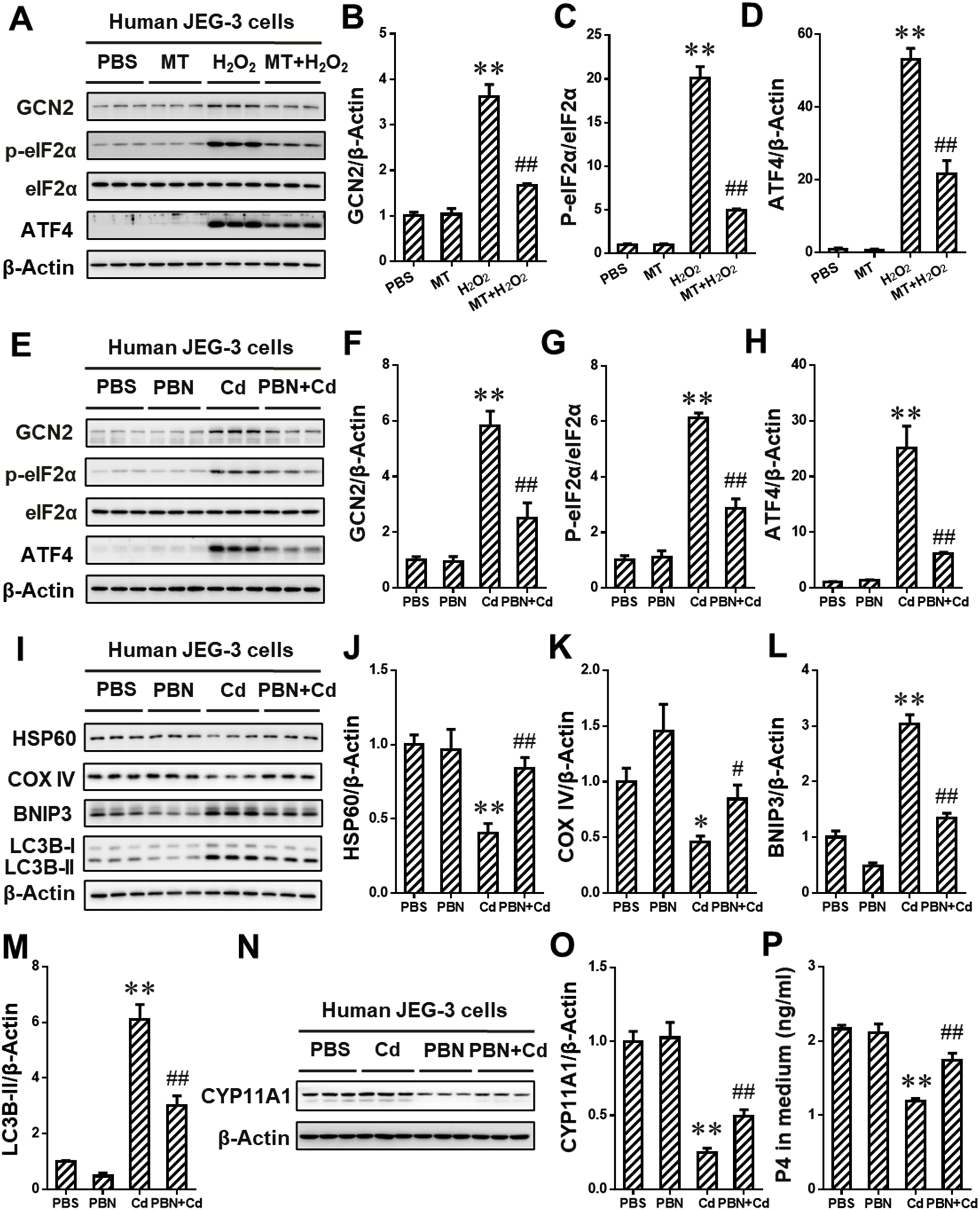
The human placentae (n = 70) were collected and the birth weight was recorded after delivery. The concentration of MT in human placentae was assessed. Notably, there was a positive correlation between the placental MT concentration and the birth weight (r = 0.869, P < 0.01; Fig. 1A). According to the birth weight, the placentae were divided into appropriate-for-gestational-age (AGA) and small-for-gestational-age (SGA) placentae. We further detected the level of CYP11A1, a key progesterone (P4) synthase, in placentae. As shown in Fig. 1B and C, the level of CYP11A1 protein was markedly reduced in SGA placentae. Moreover, the level of placental mitophagy was evaluated. The results suggested that mitochondrial proteins, such as HSP60 and COX IV, were reduced in SGA placentae when compared to AGA placentae (Fig. 1D–F). Meanwhile, the elevation of PGC1α,a regulator promoting mitochondrial biosynthesis, was observed in SGA placentae (Fig. 1D and G). In addition, the level of BNIP3 and LC3B-II proteins in SGA placentae was higher than that in AGA placentae (Fig. 1H–J). To provide more mitophagy-related evidences, we performed immunofluorescent staining of LC3B and TOM20. As expected, the accumulation of puncta (Fig. 1K and L), representing co-localization of LC3B with TOM20, occurred in SGA placentae. As above, there is a positive association of the placental mitophagy, reduced MT level, inhibition of progesterone synthesis and all-cause fetal growth restriction.
Fig. 1.

Association between placental mitophagy, reduced melatonin level, inhibition of progesterone synthesis and all-cause fetal growth restriction. All placentae (n = 70) were collected after term delivery. (A) The association between Birth weight and human placental MT level (ng/g). (B–L) According to the birth weight, the placentae were divided into appropriate-for-gestational-age (AGA) and small-for-gestational-age (SGA) placentae (n = 12 per group). (B) Representative immunoblots of CYP11A1 protein in placentae. (C) Quantification for CYP11A1. (D) Representative immunoblots of HSP60, COX IV and PGC1α proteins in placentae. (E–G) Quantification for HSP60, COX IV and PGC1α. (H) Representative immunoblots of LC3B-I/II and BNIP3 proteins in placentae. (I and J) Quantification for LC3B-I/II and BNIP3. (K) Quantification for yellow dots per cell. (L) Representative immunofluorescent images of AGA or SGA placentae. Data are expressed as the mean ± SEM. The differences between AGA and SGA were analyzed using Student's t-test. *P < 0.05, **P < 0.01 versus AGA group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Melatonin attenuates environmental stress-induced fetal growth (FGR) restriction in mice
Cd, a proverbial environmental stressor, was used to establish in vivo FGR model. The effects of MT on environmental stress-caused FGR were assessed on GD16. Notably, MT reversed Cd-lowered fetal weight and crown-rump length in mice (Fig. 2A and B). We further compared the rates of FGR among different groups. Results showed that MT significantly reversed Cd-elevated rate of FGR in mice (Fig. 2C). The number of fetuses per litter was also recorded on GD16. As shown in Fig. 2D, there was no significant statistical difference of fetuses per litter in each group. To sum up, MT could provide a powerful protective effect on environmental stress-caused FGR.
Fig. 2.

Melatonin (MT) attenuates environmental stress-induced fetal growth restriction in mice. Fig. 2 Melatonin (MT) attenuates environmental stress-induced fetal growth restriction in mice. Pregnant mice in Cd and MT + Cd groups received intraperitoneal injection of Cd on GD8. In MT and MT + Cd groups, pregnant mice were treated with MT from GD7 to GD15. Pregnant mice in Ctrl group were treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) Fetal weight. (B) Crown-rump length. (C) The rate of FGR. (D) Fetuses per litter. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. **P < 0.01 versus NS group. #P < 0.05 versus Cd group.
3.3. Melatonin alleviates environmental stress-induced progesterone synthesis suppression in placental trophoblasts
To explore the effects of MT on environmental stress-caused reduction of progesterone (P4) level in sera of maternal mice, placentae and amniotic fluid, the mice were exposed to environmental stressor Cd on GD8. As presented in Fig. 3A–C, MT markedly reversed Cd-induced reduction of P4 concentration in sera of maternal mice, placentae and amniotic fluid. We further assessed the effects of MT on Cd-induced inhibition of P4 synthesis. As presented in Fig. 3D and E, MT obviously alleviated Cd-caused downregulation of CYP11A1 protein expression in placentae. We also investigated whether MT alleviated Cd-induced P4 synthesis inhibition in human placental trophoblasts. As expected, MT attenuated Cd-reduced level of P4 and CYP11A1 protein in human JEG-3 cells (Fig. 3F–H). Collectively, our data confirmed that MT could reverse environmental stress-caused P4 synthesis inhibition in placental trophoblasts.
Fig. 3.

Melatonin (MT) alleviates environmental stress-induced progesterone synthesis suppression in placental trophoblasts. (A–F) Pregnant mice in Cd and MT + Cd groups received intraperitoneal injection of Cd on GD8. In MT and MT + Cd groups, pregnant mice were treated with MT from GD7 to GD15. Pregnant mice in Ctrl group were treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) Progesterone (P4) in maternal sera. (B) P4 in placentae. (C) P4 in amniotic fluid. (D) Representative immunoblots of CYP11A1 protein in placentae. (E) Quantification for CYP11A1. (F-H) Human JEG-3 cells were pretreated with MT before Cd stimulation (n = 3 per group). (F) P4 in cell medium. (G) Representative immunoblots of CYP11A1 protein in the cells. (H) Quantification for CYP11A1. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS/NS group; #P < 0.05, ##P < 0.01 versus Cd group.
3.4. Gestational exposure to environmental stress activates BNIP3-dependent mitophagy in placental trophoblasts
To investigate the effects of maternal exposure to environmental stress during pregnancy on BNIP3-dependent mitophagy, the level of mitophagy in environmental stressor Cd-treated placentae was detected on GD12 and GD16. As shown in Fig. 4A–C, maternal exposure to Cd during gestation obviously reduced the level of mitochondrial proteins, including HSP60 and COX IV, in placentae. Moreover, the level of PGC1α protein,a regulator promoting mitochondrial biosynthesis, was elevated in Cd-treated placentae when compared to NS group (Fig. 4A and D). The results about immunofluorescence staining showed that maternal exposure to Cd caused the increment of co-localization between TOM 20 and LC3B in placentae, as determined by the accumulation of puncta (Fig. 4E and F). The level of BNIP3 and LC3B-I/II proteins in placentae were also detected on GD12 and GD16. As presented in Fig. 5A–C, gestational Cd exposure significantly induced the elevation of BNIP3 and LC3B-I/II proteins in placentae. We further assessed the effect of Cd on BNIP3-mediated mitophagy in human JEG-3 cells. As shown in Fig. 4G–J, Cd treatment markedly caused the reduction of mitochondrial proteins, including HSP60 and COX IV, and the elevation of PGC1α. The level of BNIP3 mRNA level was also detected in the cells. As shown in Fig. 5D, Cd obviously elevated the level of BNIP3 mRNA in the cells. In addition, Cd also enhanced BNIP3 and LC3B-II protein level in a time-dependent manner (Fig. 5E–G). To explore whether Cd activated BNIP3-dependent mitophagy, human JEG-3 cells were pretreated with CQ to inhibit lysosomal acidification. As expected, CQ pretreatment obviously antagonized Cd-caused degradation of HSP60 and COX IV proteins (Fig. 4K-M). Furthermore, the results showed that there was a further upregulation of BNIP3 and LC3B-II protein expression in Cd-stimulate cells after CQ pretreatment (Fig. 5H–J). To obtain more evidences that BNIP3 mediated Cd-triggered mitophagy, we further performed the immunofluorescent staining for BNIP3 and LC3B in human JEG-3 cells. We found that there was a significant increment of puncta, representing co-localization of BNIP3 with LC3B, in Cd-simulated cells (Fig. 5K and L). As above, maternal exposure to environmental stress during gestation triggers BNIP3-dependent mitophagy in placental trophoblasts.
Fig. 4.

Gestational exposure to environmental stress activates mitophagy in placental trophoblasts. (A–F) Each pregnant mouse in Cd group received intraperitoneal injection of Cd on GD8. Each pregnant mouse in Ctrl group was treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) Representative immunoblots of HSP60, COX IV and PGC1α proteins in placentae. (B–D) Quantification for HSP60, COX IV and PGC1α. (E) Representative immunofluorescent images of mouse placentas from NS and Cd groups. Arrows: co-localizations of TOM20 with LC3B. The Hoechst was used to tagged the nucleus. Scale bar: 20 μm. (F) Quantification for yellow dots per cell. (G–J) Human JEG-3 cells were incubated with Cd for 0, 2, 6 or 12 h (n = 3 per group). (G) Representative immunoblots of HSP60, COX IV and PGC1α proteins in the cells. (H–J) Quantification for HSP60, COX IV and PGC1α. (K–M) Human JEG-3 cells were pretreated using CQ before Cd stimulation (n = 3 per group). (K) Representative immunoblots of HSP60 and COX IV proteins in the cells. (L and M) Quantification for HSP60 and COX IV. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS/NS group, ##P < 0.01 versus Cd group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 5.

BNIP3 mediates environmental stress-triggered mitophagy in placental trophoblasts. (A–D) Each pregnant mouse in Cd group received intraperitoneal injection of Cd on GD8. Each pregnant mouse in Ctrl group was treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) Representative immunoblots of BNIP3 and LC3B-I/II proteins in placentae. (B and C) Quantification for BNIP3 and LC3B-II. (D–G) Human JEG-3 cells were treated with Cd for 0 h, 2 h, 6 h or 12 h (n = 3 per group). (D) The level of relative BNIP3 mRNA. (E) Representative immunoblots of BNIP3 and LC3B-I/II proteins in the cells. (F and G) Quantification for BNIP3 and LC3B-II. (H–J) Human JEG-3 cells were pretreated using CQ before Cd stimulation (n = 3 per group). (H) Representative immunoblots of BNIP3 and LC3B-I/II proteins in the cells. (I and J) Quantification for BNIP3 and LC3B-II. (K and L) Human JEG-3 cells were treated with Cd for 6 h (n = 6 per group). (K) Quantification for yellow dots per cell. (L) Representative immunofluorescent images of PBS- or Cd-treated cells. The Hoechst was used to tagged the nucleus. Scale bar: 20 μm. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS/NS group, #P < 0.05, versus Cd group.
3.5. Gestational exposure to environmental stress inhibits progesterone synthesis via BNIP3-dependent mitophagy in placental trophoblasts
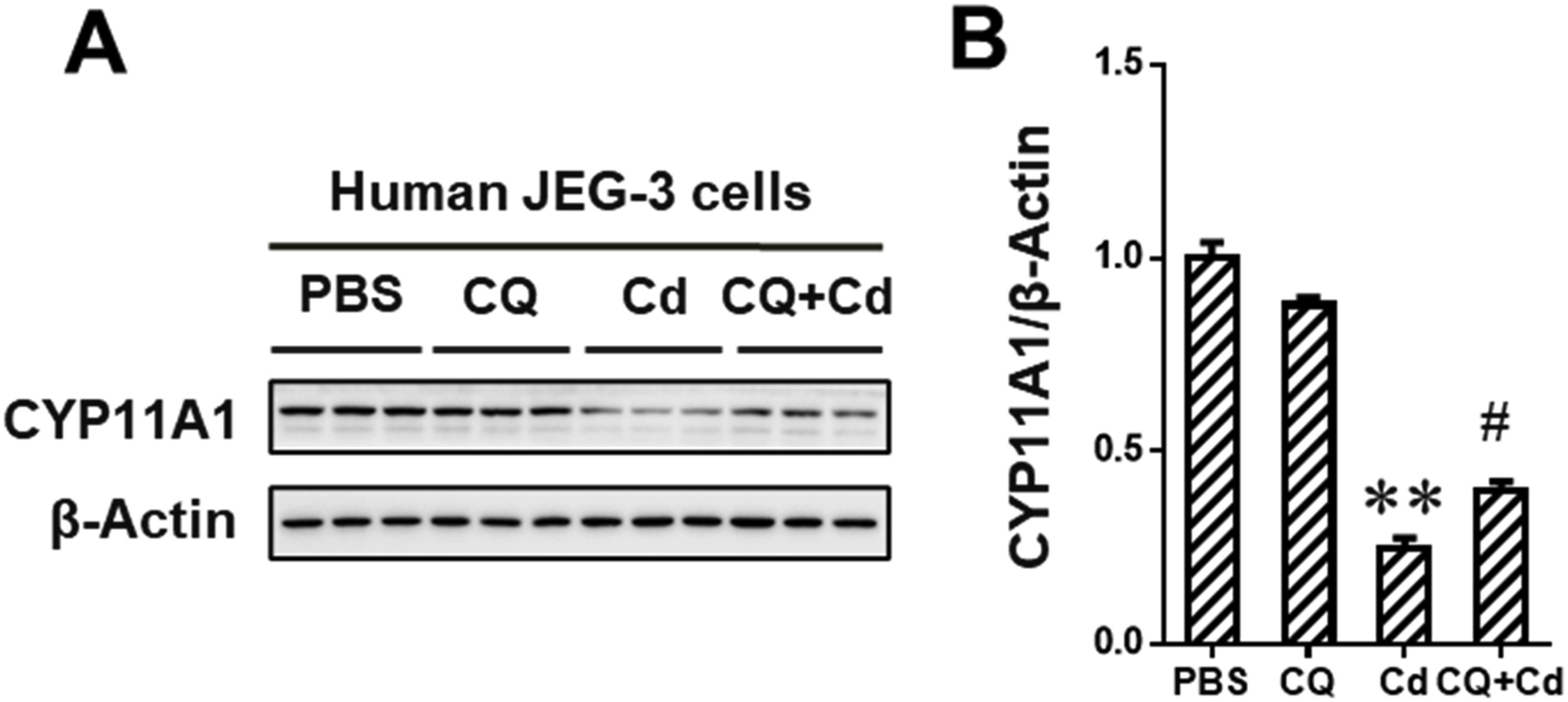
To explore whether BNIP3-dependent mitophagy mediated environmental stress-induced progesterone (P4) synthesis inhibition, human JEG-3 cells were pretreated with BNIP3 siRNA (siR) before environmental stressor Cd stimulation. Results on ELISA assay showed that BNIP3 siR markedly reversed Cd-caused reduction of P4 level in cell medium (Fig. 6A). Correspondingly, the expression of CYP11A1 protein, a critical P4 synthase, was markedly upregulated in Cd-stimulated cells after BNIP3 siR pretreatment (Fig. 6B and C). Furthermore, BNIP3 siR obviously reversed Cd-reduced the level of mitochondrial proteins, including HSP60 and COX IV, in the cells (Fig. 6D–F). Meanwhile, a significant reduction of BNIP3 and LC3B-II protein level was observed in Cd-treated cells after BNIP3 siR pretreatment (Fig. 6G and I). Furthermore, the expression of CYP11A1 protein was upregulated after CQ pretreatment in Cd-stimulated human placental trophoblasts (Figs. S2 A and B). Notably, environmental stress suppressed P4 synthesis via BNIP3-dependent mitophagy in placental trophoblasts.
Fig. 6.

Gestational exposure to environmental stress inhibits progesterone synthesis via BNIP3-dependent mitophagy in placental trophoblasts. (A–I) Human JEG-3 cells were pretreated with BNIP3 siRNA before Cd stimulation. (A) Progesterone in cell medium. (B) Representative immunoblots of CYP11A1 protein in the cells. (C) Quantification for CYP11A1. (D) Representative immunoblots of HSP60 and COX IV proteins in the cells. (E and F) Quantification for HSP60 and COX IV. (G) Representative immunoblots of LC3B-I/II and BNIP3 proteins in the cells. (H and I) Quantification for LC3B-II and BNIP3. Data are expressed as the mean ± SEM. n = 3 per group. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS group, #P < 0.05, ##P < 0.01 versus Cd group.
3.6. Melatonin inhibits environmental stress-triggered BNIP3-dependent mitophagy in placental trophoblasts
Based on the model by which environmental stressor causes FGR, the effects of MT on BNIP3-dependent mitophagy were evaluated in environmental stress-exposed placentae. As compared with the Cd group, the puncta representing co-localization of TOM 20 with LC3B were reduced in placenta from MT + Cd group (Fig. 7A and B). Furthermore, MT significantly antagonized Cd-elevated level of BNIP3 and LC3B-II proteins in placentae (Fig. 7C and D). Correspondingly, MT also alleviated Cd-reduced level of mitochondrial proteins, HSP60 and COX IV, in placentae (Fig. 7C and D). To further confirm the effects of MT on Cd-triggered BNIP3-dependent mitophagy, human JEG-3 cells were pretreated with MT. The results showed that MT obviously reversed Cd-induced elevation of BNIP3 and LC3B-II protein level in the cells (Fig. 7E and F). As we expected, the level of HSP60 and COX IV protein was obvious elevated in Cd-stimulated cells after MT pretreatment (Fig. 7E and F). Next, we performed the immunofluorescent staining for BNIP3 and LC3B in the cells. As shown in Fig. 7G and H, MT markedly reversed Cd-caused increment of puncta representing co-localization of BNIP3 with LC3B. Taken as a whole, these data showed that MT could inhibit environmental stress-triggered BNIP3-dependent mitophagy in placental trophoblasts.
Fig. 7.

Melatonin (MT) inhibits environmental stress-triggered BNIP3-dependent mitophagy in placental trophoblasts. (A–D) Pregnant mice in Cd and MT + Cd groups received intraperitoneal injection of Cd on GD8. In MT and MT + Cd groups, pregnant mice were treated with MT from GD7 to GD15. Pregnant mice in Ctrl group were treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) Representative immunofluorescent images of mouse placenta from Cd and MT + Cd groups. Arrows: co-localizations of TOM20 with LC3B. The Hoechst was used to tagged the nucleus. Scale bar: 20 μm. (B) Quantification for yellow dots per cell. (C) Representative immunoblots of BNIP3, LC3B-I/II, HSP60 and COX IV proteins in mouse placenta. (D) Quantification for BNIP3, LC3B-II, HSP60 and COX IV. (E–H) Human JEG-3 cells were pretreated using MT before Cd administration (n = 3 per group). (E) Representative immunoblots of BNIP3, LC3BI/II, HSP60 and COX IV proteins in mouse placenta. (F) Quantification for BNIP3, LC3BI/II, HSP60 and COX IV. (G) Quantification for yellow dots per cell. (H) Representative immunofluorescent images from Cd-treated cells with or without MT pretreatment. The Hoechst was used to tagged the nucleus. Scale bar: 20 μm. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS/NS group, #P < 0.05, ##P < 0.01 versus Cd group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.7. Gestational exposure to environmental stress triggers BNIP3-dependent mitophagy via activating GCN2/ATF4 signaling in human placental trophoblasts
To investigate the mechanism by which environmental stress activates BNIP3-dependent mitophagy in placental trophoblasts, we performed the detection for the GCN2/ATF4 signaling in environmental stressor Cd-stimulated human JEG-3 cells. As presented in Fig. 8A–D, Cd significantly upregulated the expression of GCN2, p-eIF2α and ATF4 proteins in the cells. To determine whether GCN2/ATF4 signaling mediates Cd-triggered BNIP3-dependent mitophagy, human JEG-3 cells were pretreated with GCN2 siRNA (siR). Our experimental data showed that GCN2 siR obviously alleviated Cd-elevated level of BNIP3 and LC3B-II proteins (Fig. 8F and G). Meanwhile, GCN2 siR markedly alleviated Cd-caused reduction of mitochondrial proteins, HSP60 and COX IV, in the cells (Fig. 8F and H). We further assessed the level of P4 synthase, including CYP11A1 protein, and P4 in the cells. As expected, GCN2 siR significantly elevated the level of CYP11A1 protein and P4 in Cd-stimulated cells (Fig. 8E, F and I). Meanwhile, GCN2 siR significantly blocked Cd-activated GCN2/ATF4 signaling in the cells (Fig. 8J-L). Mechanistically, the above data indicated that GCN2/ATF4 signaling regulated placental BNIP3-dependent mitophagy in response to environmental stress.
Fig. 8.

Gestational exposure to environmental stress triggers BNIP3-dependent mitophagy via activating GCN2/ATF4 signaling in human placental trophoblasts. (A–D) Human JEG-3 cells were treated with Cd for 0, 2, 6 or 12 h (n = 3 per group). (A) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (B–D) Quantification for GCN2, p-eIF2α and ATF4. (E–L) Human JEG-3 cells were pretreated with GCN2 siR before Cd stimulation (n = 3 per group). (E) P4 in the medium. (F) Representative immunoblots of BNIP3, LC3BI/II, HSP60, COX IV and CYP11A1 proteins in the cells. (G–I) Quantification for BNIP3, LC3BI/II, HSP60, COX IV and CYP11A1. (J) Representative immunoblots of ATF4 and GCN2 proteins in the cells. (K and L) Quantification ATF4 for and GCN2. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS group, #P < 0.05, ##P < 0.01 versus Cd group.
3.8. Melatonin blocks environmental stress-activated GCN2/ATF4 signaling via suppressing ROS release in placental trophoblasts
The placental GSH/GSGG ratio has been measured on GD12. As shown in Fig. 9A, MT abolished Cd-induced elevation of the GSH/GSGG ratio in mouse placentae. To determine whether MT blocking GCN2/ATF4 signaling in environmental stress-exposed mouse placentae, the pregnant mice were pretreated MT before environmental stressor Cd stimulation. As presented in Fig. 9B–E, MT significantly reversed Cd-elevated level of GCN2, p-eIF2α and ATF4 proteins in mouse placentae. We proposed a hypothesis that MT blocked maternal stress-induced GCN2/ATF4 signaling activation via suppressing ROS release based on the strong antioxidant effect of MT. The data revealed that the level of NOX2, NOX4 and HO-1 proteins was significantly reduced in placenta from MT + Cd group when compared to Cd group (Fig. 9F and G). We further explored the effects of MT on Cd-activated GCN2/ATF4 signaling in human placental trophoblasts. Expectedly, MT also attenuated Cd-upregulated protein expression of GCN2, p-eIF2α and ATF4 in JEG-3 cells (Fig. 9H–K). Furthermore, the result about fluorescent staining showed that MT suppressed Cd-induced ROS release (Figs. 9L and M). Meanwhile, we evaluate the effect of MT on Cd-triggered oxidative stress. As shown in Fig. 9N–Q, MT alleviated Cd-elevated the level of NOX2, NOX4 and HO-1 proteins in placental trophoblasts. To obtain more evidences that MT blocked placental GCN2/ATF4 signaling via its the strong antioxidant effect, human JEG-3 cells were pretreated with MT before H2O2 stimulation. The result showed that MT blocked H2O2-induced GCN2/ATF4 signaling activation (Figs. S1A-D). To further confirm that Cd activates GCN2/ATF4 signaling via ROS release, human JEG-3 cells were pretreated with PBN before Cd stimulation. As shown in Figs. S1E-H, the level of GCN2, p-eIF2α and ATF4 proteins was obviously reduced in Cd-treated cells after PBN pretreatment. Notably, PBN also inhibited Cd-induced mitophagy, as evidenced by the elevation of mitochondrial proteins, such as HSP60 and COX IV, in Cd-stimulated cells (Figs. S1 I-K). Meanwhile, PBN mitigated Cd-induced elevation of BNIP3 and LC3B-II in human placental trophoblasts. As expected, PBN also abolished Cd-induced reduction of CYP11A1 protein and P4 level in human placental trophoblasts (Figs. S1 M − O). To further explore whether ATF4 regulates the expression of NOXs, ATF4 siRNA (siR) was used to pretreat human placental trophoblasts before Cd-stimulation. The data showed that there was no significant change of NOX4 protein level after ATF4 siR pretreatment in Cd-stimulated cells (Figs. S3 A-C). All in all, our results further verify that MT blocks environmental stress-induced GCN2/ATF4 signaling activation via suppressing ROS release in placental trophoblasts.
Fig. 9.

Melatonin (MT) blocks environmental stress-activated GCN2/ATF4 singaling via suppressing ROS release in placental trophoblasts. (A–H) Pregnant mice in Cd and MT + Cd groups received intraperitoneal injection of Cd on GD8. In MT and MT + Cd groups, pregnant mice were treated with MT from GD7 to GD15. Pregnant mice in Ctrl group were treated with saline. n = 10–12 from 10 to 12 different pregnant mice. (A) GSH/GSSG ratio in mouse placentae. (B) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in placentae. (C–E) Quantification for GCN2, p-eIF2α and ATF4. (F) Representative immunoblots of NOX2, NOX4 and HO-1 proteins in placentae. (G) Quantification for NOX2, NOX4 and HO-1. (H–Q) Human JEG-3 cells were pretreated with MT before Cd stimulation (n = 3 per group). (H) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (I-K) Quantification for GCN2, p-eIF2α and ATF4. (L) Representative fluorescent images of ROS detection. (M) Quantification for fluorescence recur to a Multimode Reader. (N) Representative immunoblots of NOX2, NOX4 and HO-1 proteins in placenta. (O–Q) Quantification for NOX2, NOX4 and HO-1. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P < 0.05, **P < 0.01 versus PBS/NS group, #P < 0.05, ##P < 0.01 versus Cd group.
4. Discussion
Fetal growth restriction (FGR) is an important inducement of neonatal death and high incidence on chronic disease in adulthood [9,[53], [54], [55], [56]]. Cadmium (Cd), as a widely distributed heavy metal, is a main environmental stressor inducing developmental toxicity. In the present study, we used Cd as an environmental stress to stimulated pregnant mice and then found that maternal exposure to Cd stress on gestational day 8 (GD8) resulted in the reduction of fetal weight and crown-rump length. Furthermore, the rate of fetal growth restriction (FGR) was obviously elevated in Cd-stimulated mice when compaired to controls. In concert with our results, several previous studies also demonstrated that maternal exposure to Cd during pregnancy caused FGR in mice [13,50]. Melatonin (N-acetyl-5-methoxytryptamine, MT) is a major secretory product from the pineal gland, and has numerous physiological functions. An earlier study found that MT could confer powerful protective effects on gestational infection-induced FGR in mice [57]. Subsequent studies indicated that MT protected against Cd-induced injury in nonpregnant tissues, such as liver, brain and testes [48,58,59]. In this study, we firstly found that MT alleviated Cd-caused reduction of fetal weight and crown-rump length in mice. MT also protected against Cd-induced elevation of FGR rate in mice. Meanwhile, our case-control study further showed that the reduced level of MT was occurred in placentae of all-cause FGR. As above, MT could confer protection against environmental stress-caused FGR in mice.
Progesterone (P4) is essential for pregnancy success and contributing to fetal growth [35,60]. A previous study revealed that prenatal exposure to noise stress impaired fetal growth via lowering maternal P4 level in mice [10]. Similarly, our results showed that the stressor Cd induced the reduction of P4 level in maternal sera and then caused FGR in mice. Human studies and murine experiments found that the placenta, the main organ synthetizing progesterone in middle and late gestation, is essential for keeping maternal P4 level and promoting fetal growth [29,30,61]. In the present study, there was a significant reduction of placental P4 concentration in Cd-treated mice when compared to controls. These results are in agreement with the previous study [13]. An early human study found that MT contributed to progesterone synthesis in nonpregnant woman [62]. Our experiment further found that MT provided powerful protective effects on Cd-induced reduction of P4 level in sera of maternal mice, placentae and amniotic fluid. Moreover, MT also elevated the P4 concentration in medium of Cd-treated cells. To sum up, MT protected against environmental stress-induced FGR via elevating the level of P4 in placenta.
The P4 synthases, including StAR, CYP11A1 and 3β-HSD, are located in mitochondria and play a pivotal role in P4 synthesis [[31], [32], [33],36,63,64]. Our experiments found that gestational exposure to Cd lowered the level of placental CYP11A1, a crucial P4 synthase, and then reduced P4 level in sera of maternal mice, placentae and amniotic fluid. Analogously, our in vitro experiments further verified that the stressor Cd downregulated the protein expression of CYP11A1, thereby lowering the P4 level in the medium from human placental trophoblasts. In line with our data, another animal research also found that maternal exposure to the stressor lipopolysaccharide (LPS) during gestation significantly lowered the level of P4 via reducing the mRNA and the protein level of P4 synthases, such as CYP11A1 and 3β-HSD, in murine placentae [30]. Moreover, our population case-control study further showed that P4 synthesis was inhibited in SGA placentae when compared to AGA placentae. Most importantly, our results further showed that MT could protect against Cd-induced reduction of CYP11A1 protein level in mouse placentae and human placental trophoblasts. Collectively, MT alleviated environmental stress-induced reduction of P4 level via reversing P4 synthesis inhibition in placental trophoblasts.
Mitophagy is a process in which cells target mitochondria for degradation via lysosomes [24]. In the present study, our data showed that gestational exposure to Cd significantly reduced mitochondrial proteins, such as HSP60 and COX IV. We suspected that the placental trophoblasts compensated for this effect via up-regulating the expression of PGC-1a, a transcriptional co-activator promoting mitochondrial biogenesis [65]. As above, we concluded that gestational exposure to Cd decreased placental mitochondrial proteins via promoting mitochondrial degradation rather than inhibiting mitochondrial biogenesis. Thus, we further explored the role of placental mitophagy, a novel mitochondrial protein degradation pathway, in Cd-caused FGR. There are Parkin-dependent mitophagy and Parkin-independent mitophagy involving other mediators, such as BNIP3, NIX, FUNDC1 and cardiolipin [66]. Several studies found that the stressors, including cadmium and ketoconazole, triggered hyperactive mitophagy and led to mitochondrial proteins loss in hepatic cells [38,39]. In our present study, we firstly found gestational exposure to Cd stress significantly triggered BNIP3-dependent mitophagy in mouse placentae and human placental trophoblasts, as determined by the degradation of mitochondrial proteins, including HSP60 and COX IV, and the accumulation of puncta representing co-localization of TOM20 with LC3B and BNIP3 with LC3B. We further pretreated human JEG-3 cells with BINP3 siRNA (siR) to investigate the role of BNIP3-dependent mitophagy in Cd-caused placental P4 synthesis suppression. Results showed that the protein expression of CYP11A1 was elevated in Cd-treated human JEG-3 cells after BINP3 siR pretreatment. Furthermore, the expression of CYP11A1 protein was upregulated after CQ pretreatment in Cd-stimulated human placental trophoblasts. Our population case-control study also found BNIP3-dependent mitophagy was activated in SGA placentae. Furthermore, we also found MT could confer powerful inhibitory effects on Cd-caused elevation of BNIP3 and LC3B-II proteins and reduction of HSP60 and COX IV proteins in mouse placenta. Similarly, MT also reversed Cd-induced degradation of mitochondrial proteins, such as HSP60 and COX IV, and accumulation of puncta, representing co-localization of TOM20 with LC3B and BNIP3 with LC3B in human JEG-3 cells. As above, MT attenuated environmental stress-induced P4 synthesis inhibition via suppressing BNIP3-dependent mitophagy in placental trophoblasts.
The GCN2/ATF4 signaling, one response pathway of the mitochondrial stress, is activated upon mitochondrial stress and thereby regulating protein synthesis [67]. A previous study found maternal exposure to stressors including nicotine induced GCN2 signaling activation in murine placenta [68]. Until now, the role of GCN2/ATF4 signaling in mitophagy activation remains unclear in response to environmental stress. In the present study, we found Cd stress obviously elevated the level of GCN2 and ATF4 proteins in placental trophoblasts. ATF4, a well-known nuclear transcription factor, regulated multifarious gene transcription and protein synthesis [69]. Our results showed that Cd stress elevated the level of BNIP3 mRNA and then upregulated the expression of BNIP3 protein in human JEG-3 cells. Most importantly, GCN2 siR pretreatment significantly lowered the level of BNIP3 protein. Meanwhile, the level of HSP60 and COX IV proteins was elevated in Cd-stimulated cells after GCN2 siR pretreatment. Further study showed that MT conferred powerful inhibitive effects on GCN2/ATF4 signaling in Cd-treated placentae and cells. Mechanistically, MT antagonized environmental stress-triggered BNIP3-dependent mitophagy via blocking GCN2/ATF4 signaling activation in placental trophoblasts.
Oxidative stress is the result from imbalance between reactive oxygen species (ROS) overproduction and endogenous antioxidants. Excessive ROS triggered multifarious cellular stress response, such as endoplasmic reticulum (ER) stress, inflammatory reaction, genotoxic stress and hypoxia [[70], [71], [72], [73]]. A recent plant study found ROS induced the activation of GCN2 kinase in Arabidopsis [74]. Our previous studies confirmed that gestational exposure to environmental stress induced oxidative stress [49,75]. In present study, we firstly explored the effect of ROS on placental GCN2/ATF4 signaling activation. The data showed that the level of NOX2/4, two NADPH oxidases inducing ROS production, was elevated in placental trophoblasts after Cd treatment. Further results showed that PBN, a free radical scavenger, obviously blocked Cd-activated GCN2/ATF4 signaling. Numerous studies have confirmed that MT is a powerful antioxidant [[76], [77], [78]]. In this study, we also found MT inhibited ROS release, and thereby reducing the level of GCN2 and ATF4 proteins in Cd-treated placental trophoblasts. Our results further confirmed that MT inhibited H2O2-activated GCN2/ATF4 signaling activation in placental trophoblasts. In conclusion, MT blocked environmental stress-induced GCN2/ATF4 signaling activation via suppressing ROS production in placental trophoblasts.
In the present study, we find activated BNIP3-dependent mitophagy and P4 synthesis suppression is observed in SGA placentae based on our case-control study. Subsequently, we propose a neoteric mechanism by which gestational exposure to environmental stress causes P4 synthesis inhibition and FGR via activating BNIP3-dependent mitophagy in placental trophoblasts. More importantly, our data suggest that MT confers protection against environmental stress-impaired P4 synthesis and fetal growth via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. In conclusion, our findings provide new pharmacological evidence for the application of MT as a potential therapeutic agent antagonizing environmental stress-induced developmental toxicity.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgments
The authors thank the Center for Scientific Research of Anhui Medical University for valuable help in our experiment.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101854.
Contributor Information
De-Xiang Xu, Email: xudex@126.com.
Hua Wang, Email: wanghuadev@126.com.
Funding
This work was supported by National Natural Science Foundation of China (81973079, 81473016 and 81930093), Anhui Provincial Natural Science Foundation (2008085J38), Academic Funding Project for Top Talents in Colleges and Universities (gxbjZD2020059) and Young Scholars of Wan Jiang in Anhui Province, Anhui Provincial Academic and Technical Leader Reserve Candidate Research Funding (2020H208) and Scientific Research Promotion Plan of Anhui Medical University (2020xkjT005).
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Melatonin (MT) blocks environmental stress-activated GCN2/ATF4 singaling via suppressing ROS release in placental trophoblasts. (A-D) Human JEG-3 cells were pretreated with MT before H2O2 stimulation (n=3 per group). (A) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (B-D) Quantification for GCN2, p-eIF2α and ATF4. (E-H) Human JEG-3 cells were pretreated with PBN before Cd stimulation (n=3 per group). (E) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (F-H) Quantification for GCN2, p-eIF2α and ATF4. (I) Representative immunoblots of HSP60, COX IV, BNIP3 and LC3B in the cells. (J-M) Quantification for HSP60, COX IV, BNIP3 and LC3B-II. (N) Representative immunoblots of CYP11A1 in the cells. (O) Quantification for CYP11A1. (P) P4 in the medium. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P<0.05, **P<0.01 versus PBS group, #P<0.05, ##P<0.01 versus H2O2/Cd group.
{kind=link}
CQ inhibited Cd-downregulated expression of CYP11A protein in human placental trophoblasts. Human JEG-3 cells were pretreated with CQ (1h) before Cd stimulation (n=3 per group). (A) Representative immunoblots of CYP11A1 protein in the cells. (B) Quantification for CYP11A1. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. **P<0.01 versus PBS group, #P<0.05 versus Cd group.
{kind=link}
ATF4 has no effect on the expression of NOX4. (A-C) Human JEG-3 cells were pretreated with ATF4 siRNA before Cd stimulation (n=3 per group). (A) Representative immunoblots of NOX4 and ATF4 proteins in the cells. (B and C) Quantification for NOX4 and ATF4. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. **P<0.01 versus PBS group, ##P<0.01 versus Cd group.
{kind=link}
References
- 1.Lee A.C., Katz J., Blencowe H., Cousens S., Kozuki N., Vogel J.P. National and regional estimates of term and preterm babies born small for gestational age in 138 low-income and middle-income countries in 2010. Lancet Glob Health. 2013;1:e26–36. doi: 10.1016/S2214-109X(13)70006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blencowe H., Krasevec J., de Onis M., Black R.E., An X., Stevens G.A. National, regional, and worldwide estimates of low birthweight in 2015, with trends from 2000: a systematic analysis. Lancet Glob Health. 2019;7:e849–e860. doi: 10.1016/S2214-109X(18)30565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaiserman A., Lushchak O. Developmental origins of type 2 diabetes: focus on epigenetics. Ageing Res. Rev. 2019;55 doi: 10.1016/j.arr.2019.100957. [DOI] [PubMed] [Google Scholar]
- 4.Flenady V., Koopmans L., Middleton P., Froen J.F., Smith G.C., Gibbons K. Major risk factors for stillbirth in high-income countries: a systematic review and meta-analysis. Lancet. 2011;377:1331–1340. doi: 10.1016/S0140-6736(10)62233-7. [DOI] [PubMed] [Google Scholar]
- 5.Christian P., Lee S.E., Angel M.D., Adair L.S., Arifeen S.E., Ashorn P. Risk of childhood undernutrition related to small-for-gestational age and preterm birth in low- and middle-income countries. Int. J. Epidemiol. 2013;42:1340–1355. doi: 10.1093/ije/dyt109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Donnell K.J., Meaney M.J. Fetal origins of mental health: the developmental origins of health and disease hypothesis. Am. J. Psychiatr. 2017;174:319–328. doi: 10.1176/appi.ajp.2016.16020138. [DOI] [PubMed] [Google Scholar]
- 7.Abitbol C.L., Rodriguez M.M. The long-term renal and cardiovascular consequences of prematurity. Nat. Rev. Nephrol. 2012;8:265–274. doi: 10.1038/nrneph.2012.38. [DOI] [PubMed] [Google Scholar]
- 8.Crispi F., Bijnens B., Figueras F., Bartrons J., Eixarch E., Le Noble F. Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation. 2010;121:2427–2436. doi: 10.1161/CIRCULATIONAHA.110.937995. [DOI] [PubMed] [Google Scholar]
- 9.Oestreich A.K., Moley K.H. Developmental and transmittable origins of obesity-associated health disorders. Trends Genet. 2017;33:399–407. doi: 10.1016/j.tig.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solano M.E., Kowal M.K., O'Rourke G.E., Horst A.K., Modest K., Plosch T. Progesterone and HMOX-1 promote fetal growth by CD8+ T cell modulation. J. Clin. Invest. 2015;125:1726–1738. doi: 10.1172/JCI68140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang A., Zsengeller Z.K., Hecht J.L., Buccafusca R., Burke S.D., Rajakumar A. Excess placental secreted frizzled-related protein 1 in maternal smokers impairs fetal growth. J. Clin. Invest. 2015;125:4021–4025. doi: 10.1172/JCI80457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotechini T., Komisarenko M., Sperou A., Macdonald-Goodfellow S., Adams M.A., Graham C.H. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J. Exp. Med. 2014;211:165–179. doi: 10.1084/jem.20130295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong Y.W., Zhu H.L., Nan Y., Cao X.L., Shi X.T., Yi S.J. Maternal cadmium exposure during late pregnancy causes fetal growth restriction via inhibiting placental progesterone synthesis. Ecotoxicol. Environ. Saf. 2020;187:109879. doi: 10.1016/j.ecoenv.2019.109879. [DOI] [PubMed] [Google Scholar]
- 14.Lee S., Hong Y.C., Park H., Kim Y., Ha M., Ha E. Combined effects of multiple prenatal exposure to pollutants on birth weight: the Mothers and Children's Environmental Health (MOCEH) study. Environ. Res. 2019 doi: 10.1016/j.envres.2019.108832. [DOI] [PubMed] [Google Scholar]
- 15.Wang H., Liu L., Hu Y.F., Hao J.H., Chen Y.H., Su P.Y. Association of maternal serum cadmium level during pregnancy with risk of preterm birth in a Chinese population. Environ. Pollut. 2016;216:851–857. doi: 10.1016/j.envpol.2016.06.058. [DOI] [PubMed] [Google Scholar]
- 16.Wang H., Liu L., Hu Y.F., Hao J.H., Chen Y.H., Su P.Y. Maternal serum cadmium level during pregnancy and its association with small for gestational age infants: a population-based birth cohort study. Sci. Rep. 2016;6:22631. doi: 10.1038/srep22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filippini T., Torres D., Lopes C., Carvalho C., Moreira P., Naska A. Cadmium exposure and risk of breast cancer: a dose-response meta-analysis of cohort studies. Environ. Int. 2020;142 doi: 10.1016/j.envint.2020.105879. [DOI] [PubMed] [Google Scholar]
- 18.Kim S.S., Xu X., Zhang Y., Zheng X., Liu R., Dietrich K.N. Birth outcomes associated with maternal exposure to metals from informal electronic waste recycling in Guiyu, China. Environ. Int. 2020;137 doi: 10.1016/j.envint.2020.105580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G.B., Wang H., Hu J., Guo M.Y., Wang Y., Zhou Y. Cadmium-induced neural tube defects and fetal growth restriction: association with disturbance of placental folate transport. Toxicol. Appl. Pharmacol. 2016;306:79–85. doi: 10.1016/j.taap.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Wang H., Wang Y., Bo Q.L., Ji Y.L., Liu L., Hu Y.F. Maternal cadmium exposure reduces placental zinc transport and induces fetal growth restriction in mice. Reprod. Toxicol. 2016;63:174–182. doi: 10.1016/j.reprotox.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H.L., Xu X.F., Shi X.T., Feng Y.J., Xiong Y.W., Nan Y. Activation of autophagy inhibits cadmium-triggered apoptosis in human placental trophoblasts and mouse placenta. Environ. Pollut. 2019;254 doi: 10.1016/j.envpol.2019.112991. [DOI] [PubMed] [Google Scholar]
- 22.Strack R. A clearer view of mitophagy. Nat. Methods. 2020;17:656. doi: 10.1038/s41592-020-0903-z. [DOI] [PubMed] [Google Scholar]
- 23.Kessel D., Reiners J.J. Photodynamic therapy: autophagy and mitophagy, apoptosis and paraptosis. Autophagy. 2020:1–4. doi: 10.1080/15548627.2020.1783823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafsson A.B., Dorn G.W., 2nd Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 2019;99:853–892. doi: 10.1152/physrev.00005.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choubey V., Safiulina D., Vaarmann A., Cagalinec M., Wareski P., Kuum M. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011;286:10814–10824. doi: 10.1074/jbc.M110.132514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampaio-Marques B., Felgueiras C., Silva A., Rodrigues M., Tenreiro S., Franssens V. SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir 2)-mediated mitophagy. Autophagy. 2012;8:1494–1509. doi: 10.4161/auto.21275. [DOI] [PubMed] [Google Scholar]
- 27.Sansanwal P., Yen B., Gahl W.A., Ma Y., Ying L., Wong L.J. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J. Am. Soc. Nephrol. 2010;21:272–283. doi: 10.1681/ASN.2009040383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sferruzzi-Perri A.N., Higgins J.S., Vaughan O.R., Murray A.J., Fowden A.L. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc. Natl. Acad. Sci. U. S. A. 2019;116:1621–1626. doi: 10.1073/pnas.1816056116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naruse M., Ono R., Irie M., Nakamura K., Furuse T., Hino T. Sirh7/Ldoc1 knockout mice exhibit placental P4 overproduction and delayed parturition. Development. 2014;141:4763–4771. doi: 10.1242/dev.114520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu L., Chen Y.H., Xu S., Yu Z., Zhang Z.H., Zhang C. Oral cholecalciferol supplementation alleviates lipopolysaccharide-induced preterm delivery partially through regulating placental steroid hormones and prostaglandins in mice. Int. Immunopharm. 2019;69:235–244. doi: 10.1016/j.intimp.2019.01.052. [DOI] [PubMed] [Google Scholar]
- 31.Albrecht E.D., Pepe G.J. Placental steroid hormone biosynthesis in primate pregnancy. Endocr. Rev. 1990;11:124–150. doi: 10.1210/edrv-11-1-124. [DOI] [PubMed] [Google Scholar]
- 32.Strushkevich N., MacKenzie F., Cherkesova T., Grabovec I., Usanov S., Park H.W. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10139–10143. doi: 10.1073/pnas.1019441108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas J.L., Bose H.S. Regulation of human 3-beta-hydroxysteroid dehydrogenase type-2 (3betaHSD2) by molecular chaperones and the mitochondrial environment affects steroidogenesis. J. Steroid Biochem. Mol. Biol. 2015;151:74–84. doi: 10.1016/j.jsbmb.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Schatz F., Guzeloglu-Kayisli O., Arlier S., Kayisli U.A., Lockwood C.J. The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding. Hum. Reprod. Update. 2016;22:497–515. doi: 10.1093/humupd/dmw004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartwig I.R., Pincus M.K., Diemert A., Hecher K., Arck P.C. Sex-specific effect of first-trimester maternal progesterone on birthweight. Hum. Reprod. 2013;28:77–86. doi: 10.1093/humrep/des367. [DOI] [PubMed] [Google Scholar]
- 36.Barrera D., Avila E., Hernandez G., Halhali A., Biruete B., Larrea F. Estradiol and progesterone synthesis in human placenta is stimulated by calcitriol. J. Steroid Biochem. Mol. Biol. 2007;103:529–532. doi: 10.1016/j.jsbmb.2006.12.097. [DOI] [PubMed] [Google Scholar]
- 37.Mizutani T., Ishikane S., Kawabe S., Umezawa A., Miyamoto K. Transcriptional regulation of genes related to progesterone production. Endocr. J. 2015;62:757–763. doi: 10.1507/endocrj.EJ15-0260. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y., Chen H.N., Wang K., Zhang L., Huang Z., Liu J. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J. Hepatol. 2019;70:66–77. doi: 10.1016/j.jhep.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 39.Pi H., Xu S., Zhang L., Guo P., Li Y., Xie J. Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy. 2013;9:1780–1800. doi: 10.4161/auto.25665. [DOI] [PubMed] [Google Scholar]
- 40.Trollmann R., Rehrauer H., Schneider C., Krischke G., Huemmler N., Keller S. Late-gestational systemic hypoxia leads to a similar early gene response in mouse placenta and developing brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R1489–R1499. doi: 10.1152/ajpregu.00697.2009. [DOI] [PubMed] [Google Scholar]
- 41.Yasuda M., Han J.W., Dionne C.A., Boyd J.M., Chinnadurai G. BNIP3alpha: a human homolog of mitochondrial proapoptotic protein BNIP3. Canc. Res. 1999;59:533–537. [PubMed] [Google Scholar]
- 42.Marinkovic M., Sprung M., Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. 2020:1–12. doi: 10.1080/15548627.2020.1755120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stehle J.H., Saade A., Rawashdeh O., Ackermann K., Jilg A., Sebesteny T. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 2011;51:17–43. doi: 10.1111/j.1600-079X.2011.00856.x. [DOI] [PubMed] [Google Scholar]
- 44.Tan D.X., Hardeland R., Back K., Manchester L.C., Alatorre-Jimenez M.A., Reiter R.J. On the significance of an alternate pathway of melatonin synthesis via 5-methoxytryptamine: comparisons across species. J. Pineal Res. 2016;61:27–40. doi: 10.1111/jpi.12336. [DOI] [PubMed] [Google Scholar]
- 45.Xia M.Z., Liang Y.L., Wang H., Chen X., Huang Y.Y., Zhang Z.H. Melatonin modulates TLR4-mediated inflammatory genes through MyD88- and TRIF-dependent signaling pathways in lipopolysaccharide-stimulated RAW264.7 cells. J. Pineal Res. 2012;53:325–334. doi: 10.1111/j.1600-079X.2012.01002.x. [DOI] [PubMed] [Google Scholar]
- 46.Hardeland R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018;65 doi: 10.1111/jpi.12525. [DOI] [PubMed] [Google Scholar]
- 47.Majidinia M., Reiter R.J., Shakouri S.K., Mohebbi I., Rastegar M., Kaviani M. The multiple functions of melatonin in regenerative medicine. Ageing Res. Rev. 2018;45:33–52. doi: 10.1016/j.arr.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 48.Ji Y.L., Wang H., Meng C., Zhao X.F., Zhang C., Zhang Y. Melatonin alleviates cadmium-induced cellular stress and germ cell apoptosis in testes. J. Pineal Res. 2012;52:71–79. doi: 10.1111/j.1600-079X.2011.00921.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang H., Li L., Zhao M., Chen Y.H., Zhang Z.H., Zhang C. Melatonin alleviates lipopolysaccharide-induced placental cellular stress response in mice. J. Pineal Res. 2011;50:418–426. doi: 10.1111/j.1600-079X.2011.00860.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhou H., Li D., Zhu P., Hu S., Hu N., Ma S. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J. Pineal Res. 2017;63 doi: 10.1111/jpi.12438. [DOI] [PubMed] [Google Scholar]
- 51.Zhou H., Zhang Y., Hu S., Shi C., Zhu P., Ma Q. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017;63 doi: 10.1111/jpi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thamotharan S., Chu A., Kempf K., Janzen C., Grogan T., Elashoff D.A. Differential microRNA expression in human placentas of term intra-uterine growth restriction that regulates target genes mediating angiogenesis and amino acid transport. PloS One. 2017;12 doi: 10.1371/journal.pone.0176493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Y., Liew Z., Wang A., Arah O.A., Li J., Olsen J. Mediating roles of preterm birth and restricted fetal growth in the relationship between maternal education and infant mortality: a Danish population-based cohort study. PLoS Med. 2019;16 doi: 10.1371/journal.pmed.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Donnell K.J., Meaney M.J. Fetal origins of mental health: the developmental origins of health and disease hypothesis. Am. J. Psychiatr. 2017;174:319–328. doi: 10.1176/appi.ajp.2016.16020138. [DOI] [PubMed] [Google Scholar]
- 55.Barker D.J. Fetal programming of coronary heart disease. Trends Endocrinol. Metabol. 2002;13:364–368. doi: 10.1016/s1043-2760(02)00689-6. [DOI] [PubMed] [Google Scholar]
- 56.Law C.M., Shiell A.W., Newsome C.A., Syddall H.E., Shinebourne E.A., Fayers P.M. Fetal, infant, and childhood growth and adult blood pressure: a longitudinal study from birth to 22 years of age. Circulation. 2002;105:1088–1092. doi: 10.1161/hc0902.104677. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y.H., Xu D.X., Wang J.P., Wang H., Wei L.Z., Sun M.F. Melatonin protects against lipopolysaccharide-induced intra-uterine fetal death and growth retardation in mice. J. Pineal Res. 2006;40:40–47. doi: 10.1111/j.1600-079X.2005.00274.x. [DOI] [PubMed] [Google Scholar]
- 58.Cao Z., Fang Y., Lu Y., Tan D., Du C., Li Y. Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J. Pineal Res. 2017;62 doi: 10.1111/jpi.12389. [DOI] [PubMed] [Google Scholar]
- 59.Li M., Pi H., Yang Z., Reiter R.J., Xu S., Chen X. Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J. Pineal Res. 2016;61:353–369. doi: 10.1111/jpi.12353. [DOI] [PubMed] [Google Scholar]
- 60.Mucci L.A., Lagiou P., Tamimi R.M., Hsieh C.C., Adami H.O., Trichopoulos D. Pregnancy estriol, estradiol, progesterone and prolactin in relation to birth weight and other birth size variables (United States) Cancer Causes Control. 2003;14:311–318. doi: 10.1023/a:1023966813330. [DOI] [PubMed] [Google Scholar]
- 61.Malassine A., Frendo J.L., Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum. Reprod. Update. 2003;9:531–539. doi: 10.1093/humupd/dmg043. [DOI] [PubMed] [Google Scholar]
- 62.Taketani T., Tamura H., Takasaki A., Lee L., Kizuka F., Tamura I. Protective role of melatonin in progesterone production by human luteal cells. J. Pineal Res. 2011;51:207–213. doi: 10.1111/j.1600-079X.2011.00878.x. [DOI] [PubMed] [Google Scholar]
- 63.Walsh L.P., McCormick C., Martin C., Stocco D.M. Roundup inhibits steroidogenesis by disrupting steroidogenic acute regulatory (StAR) protein expression. Environ. Health Perspect. 2000;108:769–776. doi: 10.1289/ehp.00108769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tuckey R.C. Progesterone synthesis by the human placenta. Placenta. 2005;26:273–281. doi: 10.1016/j.placenta.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Bhargava P., Schnellmann R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017;13:629–646. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ni H.M., Williams J.A., Ding W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Melber A., Haynes C.M. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018;28:281–295. doi: 10.1038/cr.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong M.K., Nicholson C.J., Holloway A.C., Hardy D.B. Maternal nicotine exposure leads to impaired disulfide bond formation and augmented endoplasmic reticulum stress in the rat placenta. PloS One. 2015;10 doi: 10.1371/journal.pone.0122295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kilberg M.S., Shan J., Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metabol. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fuhrmann D.C., Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215. doi: 10.1016/j.redox.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Z., Zhang L., Zhou L., Lei Y., Zhang Y., Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019;25 doi: 10.1016/j.redox.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Masi A., Fortini P., Krokidis M.G., Romeo E.F., Bascietto C., De Angelis P. Increased levels of 5',8-Cyclopurine DNA lesions in inflammatory bowel diseases. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ziegler K., Kunert A.T., Reinmuth-Selzle K., Leifke A.L., Widera D., Weller M.G. Chemical modification of pro-inflammatory proteins by peroxynitrite increases activation of TLR4 and NF-kappaB: implications for the health effects of air pollution and oxidative stress. Redox Biol. 2020 doi: 10.1016/j.redox.2020.101581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lokdarshi A., Guan J., Urquidi Camacho R.A., Cho S.K., Morgan P.W., Leonard M. Light activates the translational regulatory kinase GCN2 via reactive oxygen species emanating from the chloroplast. Plant Cell. 2020;32:1161–1178. doi: 10.1105/tpc.19.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Z., Wang H., Xu Z.M., Ji Y.L., Chen Y.H., Zhang Z.H. Cadmium-induced teratogenicity: association with ROS-mediated endoplasmic reticulum stress in placenta. Toxicol. Appl. Pharmacol. 2012;259:236–247. doi: 10.1016/j.taap.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 76.Jiang J., Liang S., Zhang J., Du Z., Xu Q., Duan J. Melatonin ameliorates PM2.5 -induced cardiac perivascular fibrosis through regulating mitochondrial redox homeostasis. J. Pineal Res. 2020 doi: 10.1111/jpi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Siddiqui M.H., Alamri S., Nasir Khan M., Corpas F.J., Al-Amri A.A., Alsubaie Q.D. Melatonin and calcium function synergistically to promote the resilience through ROS metabolism under arsenic-induced stress. J. Hazard Mater. 2020;398 doi: 10.1016/j.jhazmat.2020.122882. [DOI] [PubMed] [Google Scholar]
- 78.Gonzalez-Candia A., Veliz M., Carrasco-Pozo C., Castillo R.L., Cardenas J.C., Ebensperger G. Antenatal melatonin modulates an enhanced antioxidant/pro-oxidant ratio in pulmonary hypertensive newborn sheep. Redox Biol. 2019;22 doi: 10.1016/j.redox.2019.101128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Melatonin (MT) blocks environmental stress-activated GCN2/ATF4 singaling via suppressing ROS release in placental trophoblasts. (A-D) Human JEG-3 cells were pretreated with MT before H2O2 stimulation (n=3 per group). (A) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (B-D) Quantification for GCN2, p-eIF2α and ATF4. (E-H) Human JEG-3 cells were pretreated with PBN before Cd stimulation (n=3 per group). (E) Representative immunoblots of GCN2, p-eIF2α/eIF2α and ATF4 proteins in the cells. (F-H) Quantification for GCN2, p-eIF2α and ATF4. (I) Representative immunoblots of HSP60, COX IV, BNIP3 and LC3B in the cells. (J-M) Quantification for HSP60, COX IV, BNIP3 and LC3B-II. (N) Representative immunoblots of CYP11A1 in the cells. (O) Quantification for CYP11A1. (P) P4 in the medium. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. *P<0.05, **P<0.01 versus PBS group, #P<0.05, ##P<0.01 versus H2O2/Cd group.
CQ inhibited Cd-downregulated expression of CYP11A protein in human placental trophoblasts. Human JEG-3 cells were pretreated with CQ (1h) before Cd stimulation (n=3 per group). (A) Representative immunoblots of CYP11A1 protein in the cells. (B) Quantification for CYP11A1. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. **P<0.01 versus PBS group, #P<0.05 versus Cd group.
ATF4 has no effect on the expression of NOX4. (A-C) Human JEG-3 cells were pretreated with ATF4 siRNA before Cd stimulation (n=3 per group). (A) Representative immunoblots of NOX4 and ATF4 proteins in the cells. (B and C) Quantification for NOX4 and ATF4. Data are expressed as the mean ± SEM. Multiple comparisons were performed using ANOVA. The post hoc test is executed adopting Bonferroni or Tamhane's T2 method following the result of homogeneity of variance test. **P<0.01 versus PBS group, ##P<0.01 versus Cd group.