

Summary
Histone lysine demethylases (KDMs) play critical roles in oncogenesis and therefore may be effective targets for anticancer therapy. Using a time-resolved fluorescence resonance energy transfer demethylation screen assay, in combination with multiple orthogonal validation approaches, we identified geldanamycin and its analog 17-DMAG as KDM inhibitors. In addition, we found that these Hsp90 inhibitors increase degradation of the alveolar rhabdomyosarcoma (aRMS) driver oncoprotein PAX3-FOXO1 and induce the repressive epigenetic mark H3K9me3 and H3K36me3 at genomic loci of PAX3-FOXO1 targets. We found that as monotherapy 17-DMAG significantly inhibits expression of PAX3-FOXO1 target genes and multiple oncogenic pathways, induces a muscle differentiation signature, delays tumor growth and extends survival in aRMS xenograft mouse models. The combination of 17-DMAG with conventional chemotherapy significantly enhances therapeutic efficacy, indicating that targeting KDM in combination with chemotherapy may serve as a therapeutic approach to PAX3-FOXO1-positive aRMS.
Subject areas: Molecular Biology, Cancer, Omics
Graphical Abstract
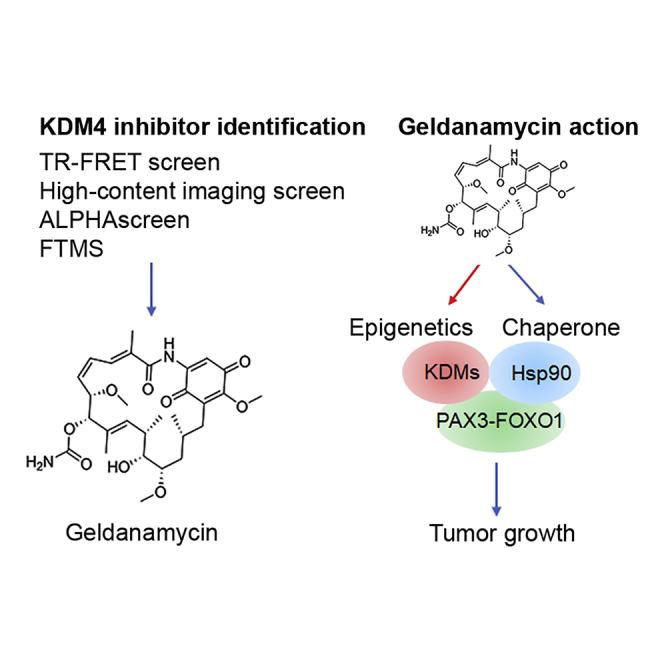
Highlights
-
•
Identification of geldanamycin/17-DMAG as histone lysine demethylase inhibitors
-
•
Geldanamycin/17-DMAG causes degradation of PAX3-FOXO1, an Hsp90 client
-
•
Geldanamycin/17-DMAG induces epigenetic changes and targets PAX3-FOXO1 pathway
-
•
17-DMAG alone or combined with chemotherapy show potency to PAX3-FOXO1 xenografts
Molecular Biology; Cancer; Omics
Introduction
Histone lysine methylation mediates multiple important biological processes including DNA replication, DNA repair, and gene expression (Greer and Shi, 2012; Martin and Zhang, 2005; Rodriguez-Paredes and Esteller, 2011). Histone methylation was initially felt to be irreversible until lysine-specific demethylase 1 (LSD1, KDM1A) was identified (Shi et al., 2004). Subsequent studies identified another family of histone demethylases, Jumonji C (JmjC) domain-containing demethylases (Hojfeldt et al., 2013), which requires iron and 2-oxoglutarate for their activities. The JmjC histone lysine demethylases (KDMs) are composed of 17 members and are responsible for reversing most of the histone methyl marks in the human epigenome. Aberrant histone lysine methylation is commonly seen in a variety of cancers (Esteller, 2008), due to genetic alteration or dysregulated expression of histone lysine methyltransferases and KDMs (Chi et al., 2010; Hojfeldt et al., 2013; Huether et al., 2014; Kandoth et al., 2013; Shi, 2007; Shi and Whetstine, 2007).
The KDM4 (KDM4A-D) subfamily are Jumonji-domain containing KDMs which are responsible for removing methyl groups from tri- and dimethylated H3K9 and H3K36 (H3K9me2/me3, H3K36me2/me3) (Kooistra and Helin, 2012). KDM4B is particularly important as it is involved in a variety of physiological functions (Wilson and Krieg, 2019). It regulates DNA repair (Young et al., 2013), self-renewal of embryonic stem cells (Das et al., 2014), conversion of induced pluripotent stem cells (Chen et al., 2013), osteogenic differentiation (Ye et al., 2012), obesity (Cheng et al., 2018), and mammary development (Kawazu et al., 2011). KDM4B is also involved in regulation of copy number gain at specific chromosomal loci (Mishra et al., 2018), topoisomerase-II accessibility to chromatin (Seoane et al., 2019) and demethylation of non-histone proteins such as AKT (Guo et al., 2019). We and others have shown that KDM4B is a direct target of estrogen receptor alpha (ERα) and hypoxia-inducible factor 1 (HIF1) in ERα+ breast cancer (Hahm et al., 2011; Kawazu et al., 2011; Yang et al., 2009, 2010), and that it epigenetically regulates G2/M phase cell cycle gene expression (Yang et al., 2010). In addition, KDM4B is a key molecule in androgen receptor signaling in prostate cancer (Coffey et al., 2013; Duan et al., 2019). Recently, we further showed that KDM4B is involved in neuroblastoma growth and tumor maintenance (Yang et al., 2015). Another study has shown that KDM4B is involved in the regulation of unfolded protein response (UPR) in PTEN-deficient triple-negative breast cancers, and genetic depletion or small molecule inhibition of KDM4B activates the UPR pathway, resulting in preferential apoptosis (Wang et al., 2018). These data indicate that KDM4B is a potential target for cancer therapy and so several groups, including us, have attempted to develop KDM4 inhibitors (Chen et al., 2017; Chu et al., 2014; Duan et al., 2015; King et al., 2010; Yang et al., 2017). We previously identified ciclopirox as a KDM4B inhibitor that has antitumor activity (Yang et al., 2017), targeting the Myc pathway and inducing neuroblastoma differentiation. Unfortunately, however, ciclopirox has poor in vivo pharmacokinetics. To develop more potent KDM4B inhibitors that are suitable for in vivo treatment, we employed multiple orthogonal validation approaches, and identified the Hsp90 inhibitors, geldanamycin and its analog 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17-DMAG) (Jez et al., 2003; Whitesell et al., 1994), as potent KDM inhibitors. Geldanamycin inhibited KDMs enzymatic activity, leading to an increase of the repressive transcription mark H3K9me3, at multiple target gene loci of PAX3-FOXO1, an oncogenic fusion oncoprotein that drives clinically unfavorable alveolar rhabdomyosarcoma (aRMS). We also found that PAX3-FOXO1 is an Hsp90 client, and is destabilized by geldanamycin and 17-DMAG. In contrast to other reported Hsp90 inhibitors (Kang et al., 2012; Lock et al., 2013), 17-DMAG significantly delayed tumor growth of PAX3-FOXO1 aRMS, affected multiple oncogenic pathways, and induced a muscle differentiation signature. The combination of 17-DMAG with conventional chemotherapy further enhanced antitumor efficacy in mouse xenograft models, indicating that targeting KDMs in combination with chemotherapy may serve as a therapeutic approach to PAX3-FOXO1-positive aRMS.
Results
TR-FRET demethylation assay identifies geldanamycin as a potent KDM4B inhibitor
We developed a primary screening, time-resolved fluorescence resonance energy transfer (TR-FRET) demethylation functional assay, to identify novel KDM4B inhibitors (Figure 1A). While KDM4A-4C are known to demethylate both H3K9me3 and H3K36me3, in vitro kinetic studies showed that KDM4A-4C are less efficient to demethylate H3K36me3(Hillringhaus et al., 2011). Thus, H3K36me3 may not be a good KDM4 substrate in vitro for a TR FRET assay. The assay uses a Terbium (Tb)-labeled anti-H3K9me2 antibody as a fluorescence donor, and an AF488 tagged streptavidin as a fluorescence acceptor that detects the biotinylated histone H3K9me2 peptide. When uninhibited, KDM4B converts the substrate H3K9me3 peptide to the product H3K9me2 peptide, which is recognized and bound by both the donor Tb-labeled antibody and acceptor AF488-labeled streptavidin. The resulting proximity of the Tb donor and the AF488 acceptor elicits a fluorescence emission at 520 nm when excited at 340 nm. When the KDM4B activity is inhibited, less biotin-H3K9me2 is generated and the 520 nm emission signal (and the 520 nm/490 nm ratio) is reduced. We optimized the conditions by (1) confirmation of the specificity of the Tb-labeled anti-H3K9me2 antibody to the Biotin-H3K9me2 peptide among the 4 relevant peptides (Biotin-H3K9me0, Biotin-H3K9me1, Biotin-H3K9me2 and Biotin-H3K9me3), (2) confirmation of the specificity of Tb-anti-H3K9me2 antibody to the product Biotin-H3K9me2 peptide over the substrate Biotin-H3K9me3 peptide over a wide concentration range (0.3 nM–312.5 nM), (3) optimization of the KDM4B concentrations, (4) optimization of incubation time, (5) optimization of KDM4B activity in three selected buffers, (6) optimization of the salt types and their concentrations in buffers, and (7) evaluation of DMSO tolerance of the assay (Figure S1). Our TR-FRET assay displayed predictability and reproducibility of responses to known KDM4B inhibitors and showed a clear threshold between positive and negative responses (Figures 1B and 1C). The high-throughput screening (HTS) statistical parameter Z′(Z-prime) had an average value of 0.73 (0.55–0.87) from our pilot screen (Figure 1D), indicating that the assay is robust and reproducible. On the basis of the optimized parameters, we performed a pilot screening of 3262 FDA-approved drugs and bioactive molecules and identified the Hsp90 inhibitor geldanamycin, a member of ansamycin family molecules, as a potent KDM4B inhibitor with an IC50 of 50.1 nM (Figures 1E and 1F).
Figure 1.

Identification of geldanamycin as a potent KDM4B inhibitor
(A) The KDM4B TR-FRET demethylation functional assay: As a KDM4B substrate, the Biotin-linked H3K9me3 peptide is converted to the product Biotin-H3K9me2 peptide, which is bound by both the Terbium-labeled anti-H3K9me2 antibody and AF488-streptavidin and brings the donor fluorophore Tb and acceptor fluorophore AF488 into close proximity. A high TR-FRET signal in the form of light emission at 520 nm from the acceptor fluorophore AF488 is generated when the donor fluorophore Tb was excited at 340 nm. (B = biotin, SA = streptavidin, Tb = Terbium).
(B) Fold ratio between positive and negative controls from the validation assay. The positive control is the group without KDM4B protein (mimic 100% inhibition) and the negative control is the group with 750 nM in-house KDM4B protein (mimic 0% inhibition).
(C) Activity scatterplot of all compounds tested in the TR-FRET assay (Green: positive control; Blue: primary hits; Black: inactive compounds; Red: negative control).
(D) Z-prime factor from the screen. The Z-prime factor value was calculated using the Equation 1 for each plate based on the method reported in the literature(Zhang et al., 1999). Each dot represents Z-prime factor value for one assay plate.
(E) The chemical structure of geldanamycin.
(F) Representative TR-FRET dose-response inhibitory activity curve of geldanamycin against KDM4B. Red dots are activity values of geldanamycin at its respective concentrations in the assay plate of NT00162940. Green dots are activity values of geldanamycin at its respective concentrations in the assay plate of NT00162941. Blue curve is the dose-response curve derived from the average inhibitory activities or pooled activities of geldanamycin at its corresponding concentrations.
(G) Representative HTRF dose-response inhibitory activity curve of geldanamycin against KDM4B. Red dots are activity values of geldanamycin at its respective concentrations in the assay plate of NT00162904. Green dots are activity values of geldanamycin at its respective concentrations in the assay plate of NT00162905. Blue curve is the dose-response curve derived from the average inhibitory activities or pooled activities of geldanamycin at its corresponding concentrations.
(H) Representative background TR-FRET dose-response curve of geldanamycin without KDM4B protein. Red dots are activity values of geldanamycin at its respective concentrations in the assay plate of NT00162907. Green curve is the dose-response curve derived from the inhibitory activities or pooled activities of geldanamycin at its corresponding concentrations.
See also Figures S1 and S2.
In the screening library, there were other non-ansamycin Hsp90 inhibitors (Ganetespib, KW2478, SNX-5422, AT13387, NVP-AUY922, STA-4783 and XL888). However, none of them showed an activity >15% of inhibition to KDM4B (Figure S2). Because the KDM4B inhibitory activities of these non-ansamycin Hsp90 inhibitors were relatively low (%Inhibition <15% at 15 μM), they were not further tested in the dose response confirmation.
We also designed a homogeneous TR-FRET assay (HTRF) that is similar to TR-FRET, but the fluorophore AF488 is replaced with AF647, which switches the light detection wavelength from 520 nm to 665 nm to avoid interference by intrinsically fluorescent compounds and reduce false positives. The HTRF assay obtained a comparable IC50 value (84.4 nM) for geldanamycin (Figure 1G). Omitting KDM4B abrogated the drug response relationship, confirming that inhibition of KDM4B-mediated conversion of H3K9me3 to H3K9me2 by geldanamycin appears to be KDM4B-mediated (Figure 1H).
Orthogonal identification of geldanamycin as a KDM4B inhibitor by high-content immunofluorescence imaging and a matrix-assisted laser desorption/ionization-Fourier transform ion cyclotron resonance (MALDI-FTICR) mass spectrometry-based approach
We also developed an orthogonal phenotypic assay in parallel with the TR-FRET assay as a validation approach. We made a retroviral vector to transfer the wild-type KDM4B gene and a catalytically dead mutant of KDM4B (H189A), in which histidine 189 was replaced by alanine, resulting in the loss of iron binding activity, leading to the loss of demethylation function. We then transduced U2OS cells with these retroviral vectors (Figure 2A), as this osteosarcoma cell line was able to tolerate overexpression of KDM4B and the differential H3K9me3 signals can be easily detected by microscope. We monitored the H3K9me3 levels with immunofluorescence imaging. We again found that geldanamycin inhibited KDM4B, with an IC50 of 21.2 nM in this cell-based assay (Figures 2B and 2C). We further assessed the effect of geldanamycin on KDM4B activity via Western blot analysis. 293T cells expressing KDM4B or the catalytically dead mutant were treated with geldanamycin and 17-DMAG, a geldanamycin analog which is better suited for in vivo use. Ciclopirox was included as a positive control (Figure S3). Geldanamycin, 17-DMAG, and ciclopirox greatly inhibited KDM4B activity in demethylating H3K9me3 in the cells expressing KDM4B, compared to those expressing the control or catalytically dead mutant (Figure S3). Similar results were obtained in U2OS cells in that geldanamycin and 17-DMAG blocked KDM4B catalytic activity on its H3K9me3 and H3K36me3 substrates but not on H3K4me3, which is not a substrate of KDM4B (Figure 2D). These data indicate that both geldanamycin and its analog 17-DMAG inhibit KDM4B activity in cells.
Figure 2.

Orthogonal identification of geldanamycin as a KDM4B inhibitor by high-content immunofluorescence imaging screen and a MALDI-FTICR mass spectrometry-based approach
(A) Western blot for KDM4B expression in U2OS cells using an anti-KDM4B antibody.
(B) High-content immunofluorescence assay shows geldanamycin inhibits KDM4B activity. The U2OS cells expressing KDM4B were treated with 149nM of geldanamycin for 24 hr. The cells were fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100, followed by primary anti-H3K9me3 antibody incubation overnight at 4°C. The secondary goat α-rabbit-Alexa-488 and Hoechst 34,580 were added for imaging. The images were captured of each well at 10X using a GE Healthcare InCell 6,000 at 405nm to detect nuclear staining and 488 nm to detect H3K9me3. Green = H3K9me3, Blue = Hoechst 34,580.
(C) Dose-response curve shows IC50 of KDM4B inhibition by immunofluorescence assay. Purple curve is the dose-response curve derived from the average inhibitory activities or pooled activities of geldanamycin at its corresponding concentrations. Y axis = geldanamycin inhibitory activity; x axis = geldanamycin concentration.
(D)Western blot shows 1uM of geldanamycin (GA) and its analog 17-DMAG block KDM4B activity in U2OS cells, as evidenced by changes in histone methyl marks assessed by specific antibodies, and inhibit the chaperone activity of HSP90 as evidenced by HSP70 compensatory upregulation.
(E) MALDI-FTICR mass spectrometry shows that KDM4B converts H3K9me3 to H3K9me2. Representative mass spectra of histone H3 peptide in the absence (upper panel) and presence (lower panel) of KDM4B. m/z = 1530.871 and 1516.854 peaks correspond to trimethylated H3K9 (substrate) and dimethylated H3K9 (product) peptides, respectively. Y axis indicates the signal intensity of H3K9me3 and H3K9me2 peaks. m/z represents mass divided by charge number and the x axis is expressed in units of m/z.
(F) Dose-response curve of geldanamycin shows the inhibition of KDM4B with IC50 of 1.24 μM, measured by MALDI-FTICR. KDM4B inhibition plot in the presence of geldanamycin. Value for Ki was calculated upon fitting the plot with Morrison equation. Data are shown as mean ± standard deviation (n = 3). Y axis indicates the turnover rate of H3K9me3 to H3K9me2, catalyzed by KDM4B. X axis indicates the concentration of geldanamycin.
(G) Microscale Thermophoresis assay of direct binding of KDM4B and geldanamycin. Y axis represents the fractions of KDM4B protein bound by geldanamycin. X axis represents geldanamycin concentration. Kd = dissociation constant.
To further validate geldanamycin as a KDM4B inhibitor, we developed a MALDI-FTICR mass spectrometry-based approach, which is detailed in the Transparent methods. We validated MALDI-FTICR by showing that it was able to detect the KDM4B substrate (H3K9me3) and the product H3K9me2 (Figure 2E). Again, we found that geldanamycin inhibited KDM4B activity with an IC50 of 1.24μM (Figure 2F). Although this method was less sensitive than our TR-FRET and immunofluorescence assays, probably due to the distinct platform and parameters used in this assay, it validated that KDM4B activity was inhibited by geldanamycin in vitro.
Microscale Thermophoresis (MST) was employed to measure the binding affinity of geldanamycin with the catalytic domain of KDM4B. MST measures the diffusion of molecules in the response to a microscale temperature change. The diffusion of molecules depends on size, solvation entropy, and surface charge. These properties differ from unbound to bound molecules. Introducing a microscale temperature change induces a difference in fluorescence between unbound and bound target molecules (Seidel et al., 2013). The normalized florescence intensities are used to fit the data to extract a binding affinity. A second generation labeling dye was conjugated to the cysteine residues of the KDM4B catalytic domain and geldanamycin was titrated into the labeled protein. Binding of geldanamycin with the catalytic domain of KDM4B revealed a Kd value of 540 nM (Figure 2G), indicating a direct binding of geldanamycin to the catalytic domain of KDM4B.
Profiling of geldanamycin and its analogs activity against KDMs using ALPHA screen
To determine the selectivity of geldanamycin against KDMs, a bead-based, non-radioactive Amplified Luminescent Proximity Homogeneous Assay (ALPHA) screen was used to assess the KDM inhibitory activity of geldanamycin at 5 μM. We also included geldanamycin analogs 17-DMAG, 17-AAG (17-N-allylamino-17-demethoxy-geldanamycin) and 17-AG (17-Amino Geldanamycin), which is only different at the 17-position of the benzoquinone ring of these compounds (Figure 3A). Geldanamycin showed greatest inhibition to KDM4 and KDM5 subfamilies (Figure 3B), which have the highest sequence homology in comparison to other KDMs with a phylogenetic tree showing that KDM4 and KDM5 members are the closest neighbors (Figure 3B). Interestingly, 17-DMAG had a broader inhibitory activity to KDMs including KDM3-6 (Figure 3B). In contrast, 17-AAG showed a high inhibition to KDM4A and KDM4C but not to KDM4B (Figure 3B). However, another analog, 17-AG, showed activity to KDM4B, KDM4C, KDM5A, and KDM6B but no activity to KDM2A and KDM1A (Figure 3B). We further determined the IC50 of the three ansamycin analogs (Geldanamycin, 17-DMAG and 17-AAG) to KDMs, and we chose 1–2 KDMs that were representatives of each subfamily. Geldanamycin had an IC50 below 1μM to KDM4B, KDM4C and KDM5A (Figure 3C), while 17-DMAG had an IC50 below 1μM to KDM3A, KDM4B, KDM4C, KDM5A, and KDM6B (Figure 3C). However, 17-AAG had an IC50 below 1μM to KDM4C and KDM5A (Figure 3C). All three compounds were much less effective at inhibiting KDM1A (IC50 > 10μM), a flavin adenine dinucleotide-dependent amine oxidase domain containing KDM, which is structurally distinct from JmjC KDMs. All three compounds showed a reasonable dose-response relationship (Figures 3D and 3E). These data indicate that the inhibitory activity of ansamycins to KDMs depends on the specific JmjC domains and is impacted by the modifications at 17-position of the benzoquinone ring of the compounds, which do not inhibit non-JmjC KDMs.
Figure 3.

Profiling of geldanamycin activity against KDMs using ALPHA screen
(A) 17-position of benzoquinone moieties of Geldanamycin, 17-DMAG, 17-AAG and 17-AG are circled (structures were obtained from htps://scifinder.cas.org).
(B) Phylogenetic tree of KDMs (left) and KDM selectivity profiling of geldanamycin, 17-DMAG, 17-AAG AND 17-AG at 5μM (right) assayed by ALPHA Screen.
(C) IC50 of geldanamycin (GA), 17-DMAG and 17-AAG for selected KDMs assayed by ALPHA Screen.
(D-E) Dose-response curve of geldanamycin, 17-DMAG and 17-AAG against KDM4B and KDM6B.
See also Figure S3.
Geldanamycin and its analog 17-DMAG promotes PAX3-FOXO1 degradation in aRMS cells
Previous studies have shown that Hsp90 also serves to stabilize fusion oncoproteins such as BCR-ABL (Peng et al., 2007), EML4-ALK (Richards et al., 2014) and EWS-FLI1 (Ambati et al., 2014), which have a chaperone dependency as they are Hsp90 clients and are destabilized by Hsp90 inhibitors. Since chimeric oncoproteins do not exist in normal cells, we hypothesized that fusion proteins such as PAX3-FOXO1 might also be more prone to degradation in the absence of cellular Hsp90 chaperone protein function. Therefore, we chose to test the antitumor efficacy of geldanamycin and its analogs in aRMS cell lines which have pathognomonic PAX3-FOXO1 translocation as a driver. We hypothesized that in addition to inhibiting KDM4B, that geldanamycin and 17-DMAG would downregulate PAX3-FOXO1 expression. Indeed, PAX3-FOXO1 expression was reduced by geldanamycin and 17-DMAG (Figure 4A). Immunoprecipitation confirmed that PAX3-FOXO1 and Hsp90 physically complexed (Figure 4B). Inhibition of Hsp90 often leads to proteasomal degradation of its clients (Butler et al., 2015). Consistent with this, the downregulation of PAX3-FOXO1 by geldanamycin or 17-DMAG was rescued by MG132 (Figure 4C), a reversible proteasome inhibitor. To test if this could be due to alterations in the PAX3-FOXO1 transcript levels, we performed quantitative PCR using fusion specific primers (spanning the fusion junction) and results showed that PAX3-FOXO1 gene expression remained unchanged (Figure 4D). We tested the effect of another Hsp90 inhibitor, AT13387, on PAX3-FOXO1 expression. While this compound showed minimal effect on KDM4B activity (Figure S2), it reduced the expression of PAX3-FOXO1 (Figure 4E). These data demonstrate that the PAX3-FOXO1 protein needs Hsp90 for its stabilization, providing the rational to target PAX3-FOXO1 protein stability by inhibiting Hsp90 activity. Thus, geldanamycin not only inhibits the enzymatic activity of KDM4B but also downregulates PAX3-FOXO1 oncoprotein levels by targeting Hsp90, thus making it a unique dual inhibitor.
Figure 4.

Geldanamycin promotes PAX3-FOXO1 degradation in aRMS cells
(A) After 24-hr treatment with geldanamycin or 17-DMAG, Rh30 PAX3-FOXO1 fusion-positive rhabdomyosarcoma cells were lysed for western blotting with the indicated antibodies.
(B) Immunoprecipitation of Rh30 cell lysates with mouse IgG or monoclonal Hsp90 antibody, followed by western blotting with indicated antibodies.
(C) Rh30 cells were treated with 200nM of geldanamycin or 17-DMAG with or without 5μM of MG132 for 24 hr. Western blot was performed with the indicated antibodies.
(D) Quantitative real-time PCR determines the mRNA of PAX3-FOXO1 after 48h treatment of Rh30 cells with 200nM of 17 DMAG (biological triplicates, Student t test). Data are represented as mean ± SEM.
(E) The whole cell lysates of Rh30 cells treated with AT13387 for 24 hr are assessed by Western blot with indicated antibodies.
Geldanamycin induces the KDM4 substrates H3K9me3 and H3K36me3 in aRMS cancer cells
PAX3-FOXO1 is a master transcription factor that mainly occupies enhancer regions(Gryder et al., 2017), driving the expression of its downstream target genes such as FGFR4, MYOD1, and MYCN. To test the biological relevance of geldanamycin as a KDM inhibitor in cancer, we treated PAX3-FOXO1 positive Rh30 cells with geldanamycin and 17-DMAG for 24 hr and assessed changes in the KDM4 substrates H3K9me3 and H3K36me3. Western blot showed that geldanamycin and 17-DMAG induced an increase of H3K9me3 and H3K36me3 (Figure 5A), suggesting that KDM4 activity is inhibited in aRMS cells. We then performed chromatin immunoprecipitation with sequencing (ChIP-seq) to assess the global epigenetic alterations induced by geldanamycin. The results showed that the H3K9me3 and H3K36me3 peaks were profoundly increased by geldanamycin (Figures 5B and 5C and Table S1). Four thousand four hundred six peaks of H3K9me3 were induced by geldanamycin but only one peak was found to be reduced (p < 0.05, fold change threshold logFC = 0.5). For H3K36me3, 1430 peaks were increased and only 213 peaks were decreased (Figure 5B). The impacted H3K9me3 peaks were mainly identified at promoters (35.6%), intergenic regions (20.7%), and introns (31.4%) (Figure 5D). The upregulated H3K36me3 peaks were mainly identified at intergenic regions (38%) and introns (54.3%), while the downregulated H3K36me3 peaks were predominantly at intron regions (77.4%) (Figure 5D). Gene set enrichment pathway analysis showed that the intergenic regions and introns with elevated H3K9me3 were significantly enriched with enhancers associated with PAX3-FOXO1 binding sites (Figure 5E and Table S2), which was similar to the H3K36me3 that was increased at the intergenic and intron regions (Table S2). However, the promoters with increased H3K9me3 were enriched with Myc targets (Figure 5E). The genes associated with the downregulated H3K36me3 at introns were associated with cell cycle related transcription factor binding (Table S2). H3K9me3 is a heterochromatin mark, representing repressive gene transcription(Kouzarides, 2007). These data indicate that KDM inhibition by geldanamycin impacts the transcriptional activity of two oncogenic transcription factors, PAX3-FOXO1 and Myc. H3K36me3 is mainly enriched at gene bodies with active transcription(Kouzarides, 2007). The elevated H3K36me3 at intergenic regions and introns may disrupt active gene transcription. Thus, geldanamycin not only inhibits the enzymatic activity of KDM and increases the heterochromatic state of the PAX3-FOXO1 targets but also downregulates PAX3-FOXO1 oncoprotein levels by targeting Hsp90, thus making it a unique dual inhibitor.
Figure 5.

Geldanamycin increases the KDM4B substrates H3K9me3 and H3K36me3 in aRMS cells.
(A) Western blot assessment of H3K9me3 and H3K36me3 levels after Rh30 cells are treated with different concentrations of geldanamycin or 17-DMAG for 24 hr.
(B) Rh30 cells are treated with 1μM of geldanamycin for 24 hr and the H3K9me3 and H3K36me3 peaks across the genome are analyzed by ChIP-seq. Significant change is defined for each peak when p < 0.05 and log fold change (LogFC) is no less than 0.5.
(C) The examples of H3K9me3 and H3K36me3 peaks at genomic loci of FGFR4 and DDIT4, respectively, snapshot using IGV program. (D) The global distribution of H3K9me3 and H3K36me3 peaks at different regions of annotated genes. 3′UTR = 3′ untranslated region, 5′UTR = 5′ untranslated region, TSS = transcription start site.
(E) Gene set enrichment analysis defines the gene sets with elevated H3K9me3 peaks that are most significantly impacted by geldanamycin treatment, at different regions of the annotated genes.
The geldanamycin analog 17-DMAG suppresses tumor growth and disrupts multiple oncogenic pathways and induces muscle differentiation signature
Previous studies reported that two specific and more potent Hsp90 inhibitors, AT13387 and Ganetespib, showed no effect on aRMS xenograft growth(Kang et al., 2012; Lock et al., 2013). Importantly, however, these two compounds did not inhibit KDM4B activity based on our assays (Figure S2). We hypothesized that geldanamycin and 17-DMAG might be more effective for PAX3-FOXO1-positive aRMS based on: (1) their ability to inhibit KDM activity and induction of heterochromatin marks of PAX3-FOXO1 and Myc targets; and (2) their ability to target PAX3-FOXO1 for proteasomal degradation. As geldanamycin has unfavorable pharmacokinetics in vivo and is associated with liver toxicity, we chose 17-DMAG and 17-AAG for in vivo assessment. After PAX3-FOXO1-positive Rh30 xenografts implanted in CB17 scid mice reached about 200 mm3 in size, 17-DMAG or 17-AAG was given intraperitoneally at a dose of 25mg/kg or 50mg/kg, respectively, twice daily, every 4 days. 17-DMAG treatment significantly delayed tumor growth (Figure 6A) and extended mouse survival (Figure 6B). While 17-AAG has a more potent activity than 17-DMAG to inhibit Hsp90 (Asgharzadeh et al., 2006; Kamal et al., 2003), 17-AAG had only a very modest effect on tumor growth and survival (Figures 6A and 6B), which is consistent with its weaker potency in inhibiting KDM4B (Figure 3). These in vivo data were consistent with a previous study (Smith et al., 2008), which showed that 17-DMAG significantly inhibited tumor growth of aRMS. To understand how 17-DMAG affected PAX3-FOXO1-positive tumor growth, we performed RNA-seq to define the signaling pathways impacted by 17-DMAG in xenografts. RNA-seq analysis of the treated xenografts showed that PAX3-FOXO1 targets such as FGFR4 and MYCN were significantly downregulated by 17-DMAG but not 17-AAG (Figure 6C and Table S3), in line with the combined effects of PAX3-FOXO1 degradation and the epigenetic impact on PAX3-FOXO1 target genes (Figures 4 and 5). Gene set enrichment analysis (GSEA) showed that 17-DMAG significantly inhibited MYC, E2F and NOTCH pathways (Ignatius et al., 2017; Slemmons et al., 2017; Takahashi et al., 2004; Tonelli et al., 2012) (Figure 6D), all of which are essential to cancer cell growth, proliferation and survival. Consistent with the in vitro results of differentiation marker induction by 17-DMAG (Figure S4), the muscle gene signature was significantly induced (Figure 6E), indicating that 17-DMAG induces aRMS cell differentiation in tumors. Immunohistochemical staining for the apoptosis marker Caspase 3 and a cell death marker TUNEL showed that 17-DMAG induced significant cancer cell death (Figures 6F and 6G). Taken together, 17-DMAG has potent antitumor effects and targets multiple oncogenic pathways in aRMS. Interestingly, a previous report showed that 17-DMAG was not effective against the PAX3-FOXO1-negative embryonal RMS (eRMS) (Smith et al., 2008), suggesting the specific activity of 17-DMAG against PAX3-FOXO1-positive tumors. Additionally, two specific and more potent Hsp90 inhibitors, AT13387 and Ganetespib, showed no effect on either aRMS or eRMS xenograft growth(Kang et al., 2012; Lock et al., 2013). This may be due to inability of these compounds to inhibit KDM, highlighting the importance of the KDM-inhibitory activity of 17-DMAG in treating aRMS.
Figure 6.

17-DMAG suppresses tumor growth, inhibits tumor angiogenesis, and disrupts multiple oncogenic pathways
(A) Tumor growth curve of Rh30 in CB17 scid mice treated with vehicle (n = 7), 25mg/kg of 17-DMAG (n = 11) and 50mg/kg of 17-AAG (n = 7). Unpaired t test for comparison of tumor volumes of each group. ∗∗p < 0.01, ∗p < 0.05.
(B) Kaplan-Meier analysis of mouse survival treated with vehicle, 17-DMAG and 17-AAG. Log rank test for comparison of survival of each group. (C) RNA-seq read of FGFR4 and MYCN from 3 individual tumors of vehicle, 3 individual tumors of 17-DMAG and 2 individual tumors of 17-AAG.
(D) GSEA analyses of pathways downregulated by 17-DMAG.
(E) GSEA analyses of pathways downregulated by 17-DMAG.
(F) Hematoxylin and Eosin (H&E), cleaved caspase 3 (Cl-CASP3) and TUNEL immunohistochemistry staining of tumor tissue sections from vehicle and 17-DMAG treatment. (G) IHC scores of 3 different areas per section from 4 tumors (n = 12) in each group were compared with unpaired student t test. Data are represented as mean ± SEM.
See also Figures S4 and S5 and Table S3.
The anticancer activity of 17-DMAG is partly dependent on KDM4B
To test whether inhibition of KDM4B by 17-DMAG contributes to the anti-proliferative phenotype, we treated KDM4B-null Rh30 cells with 17-DMAG. Loss of KDM4B led to at least 3-fold greater resistance to 17-DMAG (Figures 7A–7C), suggesting that KDM4B, at least in part, is responsible for the anti-proliferative phenotype. Furthermore, we tested a selective KDM5 inhibitor KDM5-C70 (Tumber et al., 2017) and KDM6 inhibitor GSK-J4 (Kruidenier et al., 2012), both of which showed no anti-proliferative effect (Figures 7D and 7F) at the concentrations that hit the targets (Figures 7E and 7G). These data suggest that KDM4, but not other KDMs, are responsible for the anticancer activity of 17-DMAG.
Figure 7.

The anticancer activity of 17-DMAG is partly dependent on KDM4B
(A) Western blot shows KDM4B expression in Rh30 wildtype (Rh30-WT) and KDM4B knockout (Rh30-KDM4B KO) cells.
(B) Rh30-WT and Rh30-KDM4B KO cells are treated with 100nM of 17-DMAG or 500nM of 17-AAG for 5 days, followed by crystal violet staining.
(C) The intensity of cells stained by crystal violet in Figure 7B is quantified by ImageJ, which is then normalized to the DMSO control.
(D) Rh30 cells are treated with 2.5μM of GSK-J4 for 5 days, followed by crystal violet staining. The values indicate the intensity of cells stained by crystal violet quantified by ImageJ, which is normalized to the DMSO control.
(E) The whole cell lysates of Rh30 cells treated with GSK-J4 for 72 hr are assessed by Western blot with indicated antibodies.
(F) Rh30 cells are treated with different concentrations of KDM5-C70 for 5 days, followed by crystal violet staining. The values indicate the intensity of cells stained by crystal violet quantified by ImageJ, which is normalized to the DMSO control.
(G) The whole cell lysates of Rh30 cells treated with KDM5-C70 for 72 hr are assessed by Western blot with indicated antibodies.
Combination of 17-DMAG with conventional chemotherapy enhances therapeutic efficacy
In addition to targeting the PAX3-FOXO1 pathway and other key oncogenic pathways (Figure 6), GSEA analysis revealed that 17-DMAG also inhibits gene signatures involved in DNA repair and the cell cycle (Figures S5A–S5D). We therefore surmised that 17-DMAG may enhance the efficacy of standard chemotherapy agents which damage DNA (such as irinotecan, a topoisomerase I inhibitor which generates DNA single- and double-strand breaks) and that halt the cell cycle (such as vincristine, an inhibitor of microtubule formation in mitotic spindle progression). The combination of vincristine and irinotecan (VCR + IRN) has been used in multiple clinical trials for relapsed patients with sarcoma (clinicaltrials.gov). We hypothesized that the combination of 17-DMAG and VCR + IRN may achieve better efficacy than either approach alone. Indeed, combining 17-DMAG significantly improved the efficacy of VCR + IRN in two PAX3-FOXO1-positive RMS xenograft models implanted in NSG mice (Figures 8A–8F). In the Rh30 xenograft model, all mice had a complete response (CR) to the combination therapy while nearly all control mice had progressive disease when treated with 17-DMAG alone or chemotherapy alone (Figures 8A–8C). In the Rh41 xenograft model, combination therapy also showed a better response (11% stable disease, 22% partial response, and 67% CR) than monotherapy (Figures 7D and 7F).
Figure 8.

Combination of 17-DMAG with conventional chemotherapy enhances therapeutic efficacy
(A–C) Tumor growth curve of Rh30 in NSG mice treated with vehicle (n = 10), 17-DMAG (n = 12), VCR/IRN (n = 6), 17-DMAG/VCR/IRN (n = 11) for two weeks (A), Waterfall plot of response to treatment with 17-DMAG, VCR/IRN, and 17-DMAG/VCR/IRN (B), Summary of tumor response to treatment with 17-DMAG, VCR/IRN, and 17-DMAG/VCR/IRN (C). p value is determined by Wilcoxon Rank-Sum test between treatment groups VCR + IRN and 17-DMAG + VCR + IRN.
(D–F) Tumor growth curve of Rh41 in NSG mice treated with vehicle (n = 7), 17-DMAG (n = 8), VCR + IRN (n = 8), 17-DMAG + VCR + IRN (n = 9) for three weeks (D), Waterfall plot of response to the treatment with 17-DMAG, VCR + IRN, and 17-DMAG + VCR + IRN (E), Summary of tumor response to the treatment with 17-DMAG, VCR/IRN, and 17-DMAG/VCR/IRN (F). p value is determined by Wilcoxon Rank-Sum test between treatment groups VCR + IRN and 17-DMAG + VCR + IRN.
Discussion
Geldanamycin, and its analogs 17-AAG and 17-DMAG, are prototype Hsp90 inhibitors which bind the N-terminal pocket of Hsp90(Stebbins et al., 1997), and have potent anticancer activity. However, the mechanism of action of these ansamycins is not entirely clear. It has been reported that geldanamycin and its analogs have inhibitory activity and binding affinity to Hsp90 in the range of 0.3–10 μM (Chiosis et al., 2001; Panaretou et al., 1998; Roe et al., 1999), which is in contrast to the low nanomolar antiproliferative activity of the compounds in multiple cell lines in culture(Chiosis et al., 2001; Neckers, 2002; Panaretou et al., 1998; Schulte and Neckers, 1998). Three major mechanisms were proposed to interpret the discrepancy of the 100-fold greater potency in cell culture. The first theory is ansamycins bind to and inhibit an Hsp90 multiprotein complex with much higher affinity than to Hsp90 alone (Kamal et al., 2003). The second explanation is that the physicochemical properties of the ansamycins result in greater intracellular accumulation from cell culture media (Chiosis et al., 2001), leading to highly potent antiproliferative activity. The third possible reason may be due to geldanamycin's time-dependent, tight binding to Hsp90 (Gooljarsingh et al., 2006). However, in this study, we identified geldanamycin and 17-DMAG as potent KDM inhibitors using multiple orthogonal validation approaches (TR-FRET, high-content immunofluorescence, MALDI-FTICR mass spectrometry, ALPHAscreen and MST assay), demonstrating that these compounds are epigenetic modulators. Other chemotypes of Hsp90 inhibitors showed no direct KDM inhibition, indicating that geldanamycin has unique features being a dual inhibitor of KDM/Hsp90.
Rhabdomyosarcoma (RMS) is a devastating myogenic cancer in children, adolescents, and young adults (Hettmer et al., 2014; Xia et al., 2002). This soft tissue sarcoma is mainly classified into two histological subtypes, aRMS and eRMS. aRMS is more aggressive, with a higher rate of metastasis and a poorer prognosis (Hettmer et al., 2014; Skapek et al., 2013; Sorensen et al., 2002). aRMS is primarily driven by the pathognomonic fusion oncoprotein PAX3-FOXO1 (Barr, 2001; Linardic, 2008) or its variant PAX7-FOXO1. However, no effective molecules are available to target PAX3-FOXO1 and its transcriptional activity. It was previously shown that 17-DMAG, the geldanamycin analog, was more potent at inhibiting the tumor growth of PAX3-FOXO1-positive aRMS but not PAX3-FOXO1-negative eRMS (Smith et al., 2008); while two other specific and more potent Hsp90 inhibitors, AT13387 and Ganetespib, showed no effect on either aRMS or eRMS xenograft growth(Kang et al., 2012; Lock et al., 2013). Our study provides evidence that the first prototype Hsp90 inhibitor geldanamycin and its analog 17-DMAG are new KDM inhibitors, exerting dual inhibition of Hsp90 and histone demethylases, which may account for the discrepancies of in vivo efficacy against aRMS among these Hsp90 inhibitors. 17-DMAG disrupted the epigenetic landscape of PAX3-FOXO1 target genes by elevating the histone marks H3K9me3 and H3K36me3 and inhibited the expression of PAX3-FOXO1 targets such as FGFR4 and MYCN. Multiple oncogenic pathways including MYC, E2F1 and NOTCH were inhibited by 17-DMAG. Previous studies have shown that MYC, E2F1 and NOTCH pathways play an important role in RMS (Ignatius et al., 2017; Slemmons et al., 2017; Takahashi et al., 2004; Tonelli et al., 2012). Combination of 17-DMAG with conventional chemotherapy further enhanced efficacy. Our data provide a rationale for targeting PAX3-FOXO1-positive aRMS using chemotherapy in combination with KDM inhibitors. Nevertheless, other potential off-targeting molecules may also contribute to the therapeutic efficacy of 17-DMAG, as 17-AAG has been shown to bind to mitochondrial voltage-dependent anion channel and inhibits cell invasion(Xie et al., 2011), and ansamycins may also induce reactive oxygen species(Samuni et al., 2010). In the future, the optimization of 17-DMAG dosing in combination with chemotherapy might be key to find the optimal therapeutic window.
KDMs play an important role in carcinogenesis, metastasis and therapy resistance(D'Oto et al., 2016). Pharmacologically targeting KDM effectively inhibits tumor growth in multiple preclinical models(Grasso et al., 2015; Lochmann et al., 2018; Metzger et al., 2017; Pishas et al., 2018; Yang et al., 2017). KDM1 inhibitors have entered clinical trials in cancer treatment(Fang et al., 2019). We recently identified an antifungal drug ciclopirox as a JmjC-domain containing KDM inhibitor, which targets KDMs and suppresses neuroblastoma growth(Yang et al., 2017). Here, we further showed 17-DMAG inhibits RMS growth, providing a proof-of-concept that KDMs are a potential vulnerability of solid tumors driven by oncogenic transcription factors such as MYC and PAX3-FOXO1, whose activity may require epigenetic modifiers to facilitate their transformation activity. Considering that most oncogenic transcription factors are difficult to target directly as they usually do not bear druggable pockets, targeting such epigenetic modifiers may provide alternative options to inhibiting cancer drivers. The surprising discovery of geldanamycin and its analogs as KDM inhibitors suggests that cancers driven by oncogenic transcription factors may be more sensitive to 17-DMAG than other chemotypes of Hsp90 inhibitor, considering that solid tumors are hypoxic and multiple KDMs are hypoxia-inducible genes(Yang et al., 2018). While the chaperone dependency of oncoproteins is already known, dependency of PAX3-FOXO1 on Hsp90 for protein stability has not been previously reported. Our study shows that PAX3-FOXO1 physically interacts with Hsp90 and pharmacologic inhibition of Hsp90 induces proteasome-dependent degradation of PAX3-FOXO1, indicating that PAX3-FOXO1 is a new Hsp90 client. This may further enhance the effect of geldanamycin/17-DMAG on PAX3-FOXO1 function, as the heterochromatin mark, H3K9m3 was profoundly elevated at PAX3-FOXO1 downstream target genes. As chimeric transcription factors such as MLL-AF9 and MOZ-TIF2 play a critical role in cellular transformation, and need KDM4 for their function26 (Cheung et al., 2016), geldanamycin analogs might be suitable to target cancers driven by such oncofusion proteins, warranting further investigation using animal models.
Geldanamycin is a natural antibiotic isolated as the fermentation product of Streptomyces hygroscopicus, a bacterial strain widely distributed in nature, especially in the soil. Activity profiling of geldanamycin against KDMs showed that geldanamycin is more selective to inhibit human KDM4 and KDM5 members, two closest subfamilies in the KDM phylogenetic tree (Figure 3A). Why would a bacterium produce an antibiotic that is able to dually inhibit KDM and Hsp90? One possibility is that it may help bacteria more effectively defend surrounding competitors such as other bacterial strains or fungi. Although bacteria do not have homologs of KDM4 and KDM5, they do have JmjC domain containing proteins and Hsp90 homologs. However, based on phylogenetic analysis, fungi have homologs of KDM4 and KDM5 (Figure S6). 17-DMAG and 17-AAG, two derivatives of geldanamycin used in clinical application, have distinct selectivity against KDMs. While 17-DMAG showed a broader KDM inhibition to KDM3, KDM4, KDM5, and KDM6, 17-AAG seemed to be more selective to KDM4A and KDM4C. This might be the reason that 17-DMAG had a higher therapeutic efficacy than 17-AAG in suppressing tumor growth in aRMS xenograft models. Particularly, all three compounds showed very low activity to KDM1A (IC50 > 10μM), which is not a JmjC domain containing demethylase. Notably, bacteria do not have KDM1 homologs(Zhou and Ma, 2008). These data indicate that geldanamycin has an evolutionary selectivity to JmjC KDMs. While the structural basis for selectivity of geldanamycin and its analogs against KDM remains to be solved, the selectivity is probably determined by the differences of 17-position of benzoquinone ring (17-methoxy group of geldanamycin, 17-dimethylaminoethylamino of 17-DMAG, and 17-N-allylamino of 17-AAG). Based on structural studies, this position is highly solvent-exposed in the Hsp90-Geldanamycin crystal complex and is a poor candidate for additional Hsp90 contacts(Stebbins et al., 1997), suggesting that it is not essential to Hsp90 binding. However, whether geldanamycin could inhibit other related dioxygenases such as TETs, PHDs, and ALKB whose activity also depend on Fe2 and 2-oxoglutarate warrant further studies. Hsp90 has 3 families and warrants further studies to determine which one is important to RMS and whether depletion of Hsp90 could lead to same phenotype as KDM4B inhibition.
We noticed the IC50 values of geldanamycin were different among TR-FRET, HTRF, ALPHA screen, MALDI-FTICR and cell-based immunofluorescence assays. The TR-FRET IC50 value of 50.1 nM was only slightly different from the IC50 value of 84.4 nM observed in the HTRF assay. The TR-FRET assay and the HTRF assay were similarly formulated with slightly different acceptors (AF488-streptavidin in the TR-FRET assay vs AF647-streptavidin in the HTRF assay). The small difference in the acceptors was believed to contribute to the subtle IC50 value difference. While ALPHA assay is similar to TR-FRET, the KDM4B concentration and acceptor were different from our in-house TR-FRET, which may account for the IC50 difference. In the MALDI-FTICR assay, the substrate peptide concentration was 10 μM instead of 1.5 μM in the TR-FRET or HTRF, the higher concentration of the substrate peptide ultimately resulted an observed higher IC50 value of 1.24 μM. In the cell-based immunofluorescence assay, an IC50 value of 21.2 nM for geldanamycin was observed which agreed well with the TR-FRET or HTRF assay, although protein binding, cellular permeability or metabolism may all complicate geldanamycin's cellular activity at different ways. Taken together, we believe different assay formats with different mechanisms contribute to the discrepancies of IC50. Nevertheless, all these orthogonal methods validated geldanamycin as a KDM inhibitor.
In summary, we identified Hsp90 inhibitors, geldanamycin and its analog 17-DMAG, as potent KDM inhibitors. We also found that PAX3-FOXO1 is a Hsp90 client, which was destabilized by geldanamycin. Our findings support the concept that PAX3-FOXO1 creates an epigenetic dependency to KDMs and chaperone dependency to Hsp90, and thus dually targeting KDMs and Hsp90 is a potentially valuable therapeutic option for PAX3-FOXO1-driven aRMS (Figure 9).
Figure 9.

Mechanism of action of geldanamycin or 17-DMAG
PAX3-FOXO1 binds at enhancer regions enriched with PAX3 and E-box motifs(Cao et al., 2010), driving expression of the key oncogenes such as MYCN and FGFR4. KDM demethylates H3K9me3 at PAX3-FOXO1 target genes, maintaining the euchromatin status for facilitating active gene transcription mediated by PAX3-FOXO1. When targeted by 17-DMAG, KDM activity is inhibited, resulting in epigenetic reprograming of PAX3-FOXO1 target genes by inducing heterochromatin mark H3K9me3, and consequently disrupting the transcriptional activity of PAX3-FOXO1. In addition, as an Hsp90 inhibitor, 17-DMAG leads to destabilization of the Hsp90 client PAX3-FOXO1, further enhancing the suppression of PAX3-FOXO1 function. Thus, being a dual KDM/Hsp90 inhibitor, 17-DMAG has more potent antitumor effects to PAX3-FOXO1-positive aRMS than other Hsp90 inhibitors.
Limitations of the study
Our study has limitations in that geldanamycin/17-DMAG are not specific KDM4B inhibitors and further future experiments will be needed to clarify this and the mechanism of action.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jun Yang (Jun.Yang2@stjude.org).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon request pending completed Materials Transfer Agreement.
Data and code availability
The accession number for the RNA-seq and ChIP-sed data reported in this paper has been submitted to GEO, GSE151493 [ChIP-seq], GSE151514 [RNA-seq].
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151598&token=utkpiockhrglnkx
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was partly supported by American Cancer Society-Research Scholar (130421-RSG-17-071-01-TBG, J.Y.) and National Cancer Institute (R03CA212802-01A1, J.Y.; 1R01CA229739-01, J.Y.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
A.A.Z, S.S., W.L., J.L., J.F., A.A., B.C., J.B., M.K.Y., A.A.T, D.F., J.M., H.T., B.Y., S.D., Q.W., and Z.L. performed research. A.A.Z, A.S., D.C., S.S., H.J., W.L., J.L., A.A., R.L., M.K.Y., J.Y. P.B., Z.L. analyzed data. J.Y., A.M.D. T.C., S.W., and Z.R. designed the research. J.Y. wrote the paper with input from A.M, C.T., A.M.D.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101996.
Contributor Information
Taosheng Chen, Email: taosheng.chen@stjude.org.
Jun Yang, Email: jun.yang2@stjude.org.
Supplemental information
References
- Ambati S.R., Lopes E.C., Kosugi K., Mony U., Zehir A., Shah S.K., Taldone T., Moreira A.L., Meyers P.A., Chiosis G. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 2014;8:323–336. doi: 10.1016/j.molonc.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharzadeh S., Pique-Regi R., Sposto R., Wang H., Yang Y., Shimada H., Matthay K., Buckley J., Ortega A., Seeger R.C. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J. Natl. Cancer Inst. 2006;98:1193–1203. doi: 10.1093/jnci/djj330. [DOI] [PubMed] [Google Scholar]
- Barr F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- Butler L.M., Ferraldeschi R., Armstrong H.K., Centenera M.M., Workman P. Maximizing the therapeutic potential of HSP90 inhibitors. Mol. Cancer Res. 2015;13:1445–1451. doi: 10.1158/1541-7786.MCR-15-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Yu Y., Bilke S., Walker R.L., Mayeenuddin L.H., Azorsa D.O., Yang F., Pineda M., Helman L.J., Meltzer P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497–6508. doi: 10.1158/0008-5472.CAN-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., Liang H., Chen Y., Chen J., Wu Y. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Chen Y.K., Bonaldi T., Cuomo A., Del Rosario J.R., Hosfield D.J., Kanouni T., Kao S.C., Lai C., Lobo N.A., Matuszkiewicz J. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med. Chem. Lett. 2017;8:869–874. doi: 10.1021/acsmedchemlett.7b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Yuan Q., Vergnes L., Rong X., Youn J.Y., Li J., Yu Y., Liu W., Cai H., Lin J.D. KDM4B protects against obesity and metabolic dysfunction. Proc. Natl. Acad. Sci. U S A. 2018;115:E5566–E5575. doi: 10.1073/pnas.1721814115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N., Fung T.K., Zeisig B.B., Holmes K., Rane J.K., Mowen K.A., Finn M.G., Lenhard B., Chan L.C., So C.W. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell. 2016;29:32–48. doi: 10.1016/j.ccell.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P., Allis C.D., Wang G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiosis G., Rosen N., Sepp-Lorenzino L. LY294002-geldanamycin heterodimers as selective inhibitors of the PI3K and PI3K-related family. Bioorg. Med. Chem. Lett. 2001;11:909–913. doi: 10.1016/s0960-894x(01)00099-3. [DOI] [PubMed] [Google Scholar]
- Chu C.H., Wang L.Y., Hsu K.C., Chen C.C., Cheng H.H., Wang S.M., Wu C.M., Chen T.J., Li L.T., Liu R. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J. Med. Chem. 2014;57:5975–5985. doi: 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey K., Rogerson L., Ryan-Munden C., Alkharaif D., Stockley J., Heer R., Sahadevan K., O'Neill D., Jones D., Darby S. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41:4433–4446. doi: 10.1093/nar/gkt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Oto A., Tian Q.W., Davidoff A.M., Yang J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016;1:34–40. [PMC free article] [PubMed] [Google Scholar]
- Das P.P., Shao Z., Beyaz S., Apostolou E., Pinello L., Angeles A.L., O'Brien K., Atsma J.M., Fujiwara Y., Nguyen M. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell. 2014;53:32–48. doi: 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L., Chen Z., Lu J., Liang Y., Wang M., Roggero C.M., Zhang Q.J., Gao J., Fang Y., Cao J. Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res. 2019;47:11623–11636. doi: 10.1093/nar/gkz1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L., Rai G., Roggero C., Zhang Q.J., Wei Q., Ma S.H., Zhou Y., Santoyo J., Martinez E.D., Xiao G. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem. Biol. 2015;22:1185–1196. doi: 10.1016/j.chembiol.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N. Engl. J. Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fang Y., Liao G., Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 2019;12:129. doi: 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooljarsingh L.T., Fernandes C., Yan K., Zhang H., Grooms M., Johanson K., Sinnamon R.H., Kirkpatrick R.B., Kerrigan J., Lewis T. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc. Natl. Acad. Sci. U S A. 2006;103:7625–7630. doi: 10.1073/pnas.0602650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso C.S., Tang Y., Truffaux N., Berlow N.E., Liu L., Debily M.A., Quist M.J., Davis L.E., Huang E.C., Woo P.J. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015;21:827. doi: 10.1038/nm0715-827a. [DOI] [PubMed] [Google Scholar]
- Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder B.E., Yohe M.E., Chou H.C., Zhang X., Marques J., Wachtel M., Schaefer B., Sen N., Song Y., Gualtieri A. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–899. doi: 10.1158/2159-8290.CD-16-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Dai X., Laurent B., Zheng N., Gan W., Zhang J., Guo A., Yuan M., Liu P., Asara J.M. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019;21:226–237. doi: 10.1038/s41556-018-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm E.R., Moura M.B., Kelley E.E., Van Houten B., Shiva S., Singh S.V. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One. 2011;6:e23354. doi: 10.1371/journal.pone.0023354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmer S., Li Z., Billin A.N., Barr F.G., Cornelison D.D., Ehrlich A.R., Guttridge D.C., Hayes-Jordan A., Helman L.J., Houghton P.J. Rhabdomyosarcoma: current challenges and their implications for developing therapies. Cold Spring Harb Perspect. Med. 2014;4:a025650. doi: 10.1101/cshperspect.a025650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillringhaus L., Yue W.W., Rose N.R., Ng S.S., Gileadi C., Loenarz C., Bello S.H., Bray J.E., Schofield C.J., Oppermann U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojfeldt J.W., Agger K., Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- Huether R., Dong L., Chen X., Wu G., Parker M., Wei L., Ma J., Edmonson M.N., Hedlund E.K., Rusch M.C. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatius M.S., Hayes M.N., Lobbardi R., Chen E.Y., McCarthy K.M., Sreenivas P., Motala Z., Durbin A.D., Molodtsov A., Reeder S. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. 2017;19:2304–2318. doi: 10.1016/j.celrep.2017.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jez J.M., Chen J.C., Rastelli G., Stroud R.M., Santi D.V. Crystal structure and molecular modeling of 17-DMAG in complex with human Hsp90. Chem. Biol. 2003;10:361–368. doi: 10.1016/s1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- Kamal A., Thao L., Sensintaffar J., Zhang L., Boehm M.F., Fritz L.C., Burrows F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M.H., Reynolds C.P., Houghton P.J., Alexander D., Morton C.L., Kolb E.A., Gorlick R., Keir S.T., Carol H., Lock R. Initial testing (Stage 1) of AT13387, an HSP90 inhibitor, by the pediatric preclinical testing program. Pediatr. Blood Cancer. 2012;59:185–188. doi: 10.1002/pbc.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazu M., Saso K., Tong K.I., McQuire T., Goto K., Son D.O., Wakeham A., Miyagishi M., Mak T.W., Okada H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6:e17830. doi: 10.1371/journal.pone.0017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King O.N., Li X.S., Sakurai M., Kawamura A., Rose N.R., Ng S.S., Quinn A.M., Rai G., Mott B.T., Beswick P. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra S.M., Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kruidenier L., Chung C.W., Cheng Z., Liddle J., Che K., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardic C.M. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008;270:10–18. doi: 10.1016/j.canlet.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmann T.L., Powell K.M., Ham J., Floros K.V., Heisey D.A.R., Kurupi R.I.J., Calbert M.L., Ghotra M.S., Greninger P., Dozmorov M. Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Sci. Transl Med. 2018;10:eaao4680. doi: 10.1126/scitranslmed.aao4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R.B., Carol H., Maris J.M., Kang M.H., Reynolds C.P., Kolb E.A., Gorlick R., Keir S.T., Billups C.A., Kurmasheva R.T. Initial testing (stage 1) of ganetespib, an Hsp90 inhibitor, by the pediatric preclinical testing program. Pediatr. Blood Cancer. 2013;60:E42–E45. doi: 10.1002/pbc.24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C., Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Metzger E., Stepputtis S.S., Strietz J., Preca B.T., Urban S., Willmann D., Allen A., Zenk F., Iovino N., Bronsert P. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. 2017;77:5900–5912. doi: 10.1158/0008-5472.CAN-17-1754. [DOI] [PubMed] [Google Scholar]
- Mishra S., Van Rechem C., Pal S., Clarke T.L., Chakraborty D., Mahan S.D., Black J.C., Murphy S.E., Lawrence M.S., Daniels D.L. Cross-talk between lysine-modifying enzymes controls site-specific DNA amplifications. Cell. 2018;175:1716. doi: 10.1016/j.cell.2018.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L. Heat shock protein 90 inhibition by 17-allylamino-17- demethoxygeldanamycin: a novel therapeutic approach for treating hormone-refractory prostate cancer. Clin. Cancer Res. 2002;8:962–966. [PubMed] [Google Scholar]
- Panaretou B., Prodromou C., Roe S.M., O'Brien R., Ladbury J.E., Piper P.W., Pearl L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C., Brain J., Hu Y., Goodrich A., Kong L., Grayzel D., Pak R., Read M., Li S. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood. 2007;110:678–685. doi: 10.1182/blood-2006-10-054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishas K.I., Drenberg C.D., Taslim C., Theisen E.R., Johnson K.M., Saund R.S., Pop I.L., Crompton B.D., Lawlor E.R., Tirode F. Therapeutic targeting of KDM1A/LSD1 in ewing sarcoma with SP-2509 engages the endoplasmic reticulum stress response. Mol. Cancer Ther. 2018;17:1902–1916. doi: 10.1158/1535-7163.MCT-18-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards M.W., Law E.W., Rennalls L.P., Busacca S., O'Regan L., Fry A.M., Fennell D.A., Bayliss R. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc. Natl. Acad. Sci. U S A. 2014;111:5195–5200. doi: 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Paredes M., Esteller M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- Roe S.M., Prodromou C., O'Brien R., Ladbury J.E., Piper P.W., Pearl L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Samuni Y., Ishii H., Hyodo F., Samuni U., Krishna M.C., Goldstein S., Mitchell J.B. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med. 2010;48:1559–1563. doi: 10.1016/j.freeradbiomed.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T.W., Neckers L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- Seidel S.A., Dijkman P.M., Lea W.A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J.S., Srinivasan P., Baaske P., Simeonov A. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59:301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J.A., Kirkland J.G., Caswell-Jin J.L., Crabtree G.R., Curtis C. Chromatin regulators mediate anthracycline sensitivity in breast cancer. Nat. Med. 2019;25:1721–1727. doi: 10.1038/s41591-019-0638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A., Casero R.A., Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi Y., Whetstine J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Skapek S.X., Anderson J., Barr F.G., Bridge J.A., Gastier-Foster J.M., Parham D.M., Rudzinski E.R., Triche T., Hawkins D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children's oncology group report. Pediatr. Blood Cancer. 2013;60:1411–1417. doi: 10.1002/pbc.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slemmons K.K., Crose L.E.S., Riedel S., Sushnitha M., Belyea B., Linardic C.M. A novel notch-YAP circuit drives stemness and tumorigenesis in embryonal rhabdomyosarcoma. Mol. Cancer Res. 2017;15:1777–1791. doi: 10.1158/1541-7786.MCR-17-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.A., Morton C.L., Phelps D.A., Kolb E.A., Lock R., Carol H., Reynolds C.P., Maris J.M., Keir S.T., Wu J. Stage 1 testing and pharmacodynamic evaluation of the HSP90 inhibitor alvespimycin (17-DMAG, KOS-1022) by the pediatric preclinical testing program. Pediatr. Blood Cancer. 2008;51:34–41. doi: 10.1002/pbc.21508. [DOI] [PubMed] [Google Scholar]
- Sorensen P.H., Lynch J.C., Qualman S.J., Tirabosco R., Lim J.F., Maurer H.M., Bridge J.A., Crist W.M., Triche T.J., Barr F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J. Clin. Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- Stebbins C.E., Russo A.A., Schneider C., Rosen N., Hartl F.U., Pavletich N.P. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Oda Y., Kawaguchi K., Tamiya S., Yamamoto H., Suita S., Tsuneyoshi M. Altered expression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcoma. Mod. Pathol. 2004;17:660–669. doi: 10.1038/modpathol.3800101. [DOI] [PubMed] [Google Scholar]
- Tonelli R., McIntyre A., Camerin C., Walters Z.S., Di Leo K., Selfe J., Purgato S., Missiaglia E., Tortori A., Renshaw J. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012;18:796–807. doi: 10.1158/1078-0432.CCR-11-1981. [DOI] [PubMed] [Google Scholar]
- Tumber A., Nuzzi A., Hookway E.S., Hatch S.B., Velupillai S., Johansson C., Kawamura A., Savitsky P., Yapp C., Szykowska A. Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell Chem Biol. 2017;24:371–380. doi: 10.1016/j.chembiol.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Oguz G., Lee P.L., Bao Y., Wang P., Terp M.G., Ditzel H.J., Yu Q. KDM4B-regulated unfolded protein response as a therapeutic vulnerability in PTEN-deficient breast cancer. J. Exp. Med. 2018;215:2833–2849. doi: 10.1084/jem.20180439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L., Mimnaugh E.G., De Costa B., Myers C.E., Neckers L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C., Krieg A.J. KDM4B: a nail for every hammer? Genes (Basel) 2019;10:134. doi: 10.3390/genes10020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.J., Pressey J.G., Barr F.G. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol. Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- Xie Q., Wondergem R., Shen Y., Cavey G., Ke J., Thompson R., Bradley R., Daugherty-Holtrop J., Xu Y., Chen E. Benzoquinone ansamycin 17AAG binds to mitochondrial voltage-dependent anion channel and inhibits cell invasion. Proc. Natl. Acad. Sci. U S A. 2011;108:4105–4110. doi: 10.1073/pnas.1015181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., AlTahan A.M., Hu D., Wang Y., Cheng P.H., Morton C.L., Qu C., Nathwani A.C., Shohet J.M., Fotsis T. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J. Natl. Cancer Inst. 2015;107:djv080. doi: 10.1093/jnci/djv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Harris A.L., Davidoff A.M. Hypoxia and hormone-mediated pathways converge at the histone demethylase KDM4B in cancer. Int. J. Mol. Sci. 2018;19:240. doi: 10.3390/ijms19010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Jubb A.M., Pike L., Buffa F.M., Turley H., Baban D., Leek R., Gatter K.C., Ragoussis J., Harris A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010;70:6456–6466. doi: 10.1158/0008-5472.CAN-10-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Ledaki I., Turley H., Gatter K.C., Montero J.C., Li J.L., Harris A.L. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y Acad. Sci. 2009;1177:185–197. doi: 10.1111/j.1749-6632.2009.05027.x. [DOI] [PubMed] [Google Scholar]
- Yang J., Milasta S., Hu D., AlTahan A.M., Interiano R.B., Zhou J., Davidson J., Low J., Lin W., Bao J. Targeting histone demethylases in MYC-driven neuroblastomas with ciclopirox. Cancer Res. 2017;77:4626–4638. doi: 10.1158/0008-5472.CAN-16-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Fan Z., Yu B., Chang J., Al Hezaimi K., Zhou X., Park N.H., Wang C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11:50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L.C., McDonald D.W., Hendzel M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013;288:21376–21388. doi: 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhou X., Ma H. Evolutionary history of histone demethylase families: distinct evolutionary patterns suggest functional divergence. BMC Evol. Biol. 2008;8:294. doi: 10.1186/1471-2148-8-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq and ChIP-sed data reported in this paper has been submitted to GEO, GSE151493 [ChIP-seq], GSE151514 [RNA-seq].
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151598&token=utkpiockhrglnkx