

Summary
Synapses are crucial to brain function and frequent disease targets, but current analysis methods cannot report on individual synaptic components in situ or present barriers to widespread adoption. SEQUIN was developed to address this challenge. SEQUIN utilizes a widely available super-resolution platform in tandem with image processing and analysis to quantify synaptic loci over large regions of brain and characterize their molecular and nanostructural properties at the individual and population level. This protocol describes quantification of synaptic loci using SEQUIN.
For additional details on the use and execution of this protocol, please refer to Sauerbeck et al. (2020).
Subject areas: Microscopy, Neuroscience
Graphical Abstract
Highlights
-
•
SEQUIN permits rapid quantification and fine characterization of synaptic loci in situ
-
•
Accessible super-resolution microscopy is used to image individual synaptic elements
-
•
Synaptic loci are identified via characteristic pre-to-postsynaptic separations in 3D
-
•
SEQUIN is suitable for synaptic analysis over large sample numbers or regions of brain
Synapses are crucial to brain function and frequent disease targets, but current analysis methods cannot report on individual synaptic components in situ or present barriers to widespread adoption. SEQUIN was developed to address this challenge. SEQUIN utilizes a widely available super-resolution platform in tandem with image processing and analysis to quantify synaptic loci over large regions of brain and characterize their molecular and nanostructural properties at the individual and population level. This protocol describes quantification of synaptic loci using SEQUIN.
Before you begin
Preparation of 30% sucrose buffer
Timing: 15 min
30% sucrose in PBS with 0.02% sodium azide
-
1.
For 1 L, dissolve 300 g of sucrose in ∼300 mL distilled water.
-
2.
Add 100 mL of 10× PBS.
-
3.
Adjust final volume to 1 L with distilled water.
-
4.
Add 2 mL of 10% sodium azide to mixture.
-
5.
Mix well before use.
Caution: Sodium azide is toxic. Use appropriate personal protective equipment when preparing and using solution.
Preparation of cryoprotectant
30% ethylene glycol/30% sucrose in PBS with 0.02% sodium azide
-
6.
For 1 L, combine 300 mL ethylene glycol, 100 mL 10× PBS, and 200 mL distilled water.
-
7.
Dissolve 300 g sucrose in solution.
-
8.
Adjust final volume to 1 L with distilled water.
-
9.
Add 2 mL of 10% sodium azide to mixture.
-
10.
Mix well before use. Store at −20°C for up to 6 months.
Caution: Sodium azide is toxic. Use appropriate personal protective equipment when preparing and using solution.
Tissue preparation
-
11.Perfusion-fix tissue.
-
a.Anesthetize animals using 5% isoflurane.
-
b.Transcardially perfuse with ice cold PBS at a rate of ∼5 mL/min until the liver clears.
-
c.Switch to fresh, ice cold 4% PFA in PBS pH 7.6 (made day of perfusion). Perfuse with PFA for 3 min at the same rate. Following successful perfusion, body will be very stiff, and vasculature will no longer be visible in brain.
-
d.Decapitate mouse, open the skull, and carefully extract the brain.
-
e.Store in 20 mL 4% PFA for 16–24 h at 4°C.
-
a.
-
12.Sucrose tissue.
-
a.Dunk tissue in 1× PBS to remove excess 4% PFA.
-
b.Transfer to 20 mL 30% sucrose buffer.
-
c.Store in 30% sucrose buffer at 4°C at least 48 h (ensure brain sinks to bottom of sucrose buffer before continuing).
-
a.
Pause point: Fixed brains can be stored in 30% sucrose (with azide) for at least several days to a week at 4°C
-
13.Section tissue.
-
a.Freeze tissue to microtome stage using PBS and section tissue at 50 μm
-
b.Store in cryoprotectant buffer at −20°C until immunolabeling.
-
a.
Optional: Labeling protocol below is described for free-floating tissue sections. 15 μm slide-mounted cryostat sections can be used with slight modifications.
Preparation of blocking buffer (20% normal goat serum in 1× PBS)
-
14.
Completely thaw and thoroughly mix stock NGS. (NOTE: blocking buffer is prepared fresh day of use.)
-
15.
Combine NGS and 1× PBS in a ratio of 1:4.
-
16.
Vortex well.
-
17.
Centrifuge mixture at 10,000–17,000 × g for 5 min.
-
18.
Avoid pellet when pipetting blocking buffer.
Preparation of antibody buffer (10% normal goat serum, 0.3% Triton X-100 in 1× PBS)
-
19.
Completely thaw and thoroughly mix stock NGS. (NOTE: antibody buffer is prepared fresh day of use.)
-
20.
Combine NGS and 1× PBS in a ratio of 1:9.
-
21.
Add Triton X-100 in a ratio of 3:1,000.
-
22.
Vortex well.
-
23.
Centrifuge mixture at 10,000–17,000 × g for 5 min.
-
24.
Avoid pellet when pipetting antibody buffer.
Optional: 10% NGS can also be attained by diluting blocking buffer 1:1 with 1× PBS and then adding Triton X-100.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-PSD-95 (1:200) | Invitrogen | Cat# 51-6900, RRID:AB_2533914 |
| Guinea pig anti-Synapsin 1/2 (1:500) | Synaptic Systems | Cat# 106004, RRID:AB_1106784 |
| Mouse anti-Synapsin 1 (1:200) | Invitrogen | Cat# MA5-31919, RRID:AB_2787542 |
| Goat anti-rabbit IgG-Alexa 594+ (1:200) | Invitrogen | Cat# A32740, RRID:AB_2762824 |
| Goat anti-rabbit Alexa 488 (1:200) | Invitrogen | Cat# A11034, RRID:AB_2576217 |
| Goat anti-guinea pig IgG-Alexa 488 (1:200) | Invitrogen | Cat# A11073, RRID:AB_2534117 |
| Goat anti-mouse IgG-Alexa 488+ (1:200) | Invitrogen | Cat# A32723, RRID:AB_2633275 |
| Chemicals, peptides, and recombinant proteins | ||
| 10× phosphate buffered saline | Corning | Cat# 46-013-CM |
| Normal goat serum | Vector | Cat# S-1000, RRID:AB_2336615 |
| Mowiol 488 | Electron Microscopy Services | Cat# 17977-150 |
| Antifade 300 | Electron Microscopy Services | Cat# 17977-25 |
| 1.5 H coverglass | Marienfeld | Cat# 0107242 |
| Experimental models: organisms/strains | ||
| Mouse: C57/BL6J | The Jackson Laboratory | Cat# 000664 RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| Zen Digital Imaging for Light Microscopy | Zeiss | RRID:SCR_013672 |
| Imaris | Bitplane | RRID:SCR_007370 |
| Python | Python Software Foundation | RRID:SCR_008394 |
| MATLAB | MathWorks | RRID:SCR_001622 |
| SEQUIN MATLAB Code | This report | https://github.com/KummerLab/SEQUIN |
| Other | ||
| Zeiss LSM 880 Fast Airyscan | Zeiss | N/A |
| Z-Piezo | Zeiss | N/A |
| 63× oil objective | Zeiss | Cat# 420782-9900-000 |
Step-by-step method details
Immunofluorescent labeling
Immunofluorescent labeling of synaptic proteins allows for visualization and identification of synaptic loci based on measured separation distances between pre- and postsynaptic markers. This protocol demonstrates dual-antibody fluorescent labeling of 50 μm free-floating sections using antibodies against the presynaptic marker synapsin and the postsynaptic marker PSD-95. Below we present two possible anti-synapsin antibodies that can be used. Guinea pig anti-synapsin 1/2 is a convenient choice to avoid species cross-reactivity. Mouse anti-synapsin 1 is a suitable option as well that limits lot-to-lot variability. NOTE: SEQUIN has been validated in female and male mice 8 weeks-18 months of age.
-
1)Rinse sections three times in 1× PBS to remove cryoprotectant.
-
a)Fill 6 well plate with PBS (∼12 mL/well) and add netwell inserts.
-
b)Fill 1 L beaker with distilled water.
-
c)Using a clean paintbrush, carefully move sections to netwells. Ensure sections are fully submerged.
-
d)Rinse on shaker at low speed for 5 min.
-
e)Fill another 6 well plate with PBS and move netwells from first plate to second.
-
f)Rinse on shaker at low speed for 5 min.
-
g)Repeat steps 1e and 1f for 3 washes total.
-
a)
-
2)Block in 20% Normal Goat Serum (NGS) blocking buffer to prevent non-specific antibody binding (see “Before you begin”).
-
a)Add blocking buffer to wells of a separate plate: ∼500 μL/well for a 24 well plate (up to 4 sections/well); ∼750 μL/well for a 12 well plate (up to 8 sections/well).
-
b)Using a clean paintbrush, carefully move sections from netwells with PBS to plates with blocking buffer.
-
c)Cover and incubate at 20°C–24°C (room temperature) on shaker at low speed for 1 h.
-
a)
-
3)Prepare primary antibody solutions while sections are incubating in blocking buffer.
 CRITICAL: The steps below describe a method of preparing these solutions using “master mixes” of various primary antibodies to avoid well-to-well variation in antibody concentration resulting from pipetting error. Allow ∼500 μL/well for a 24 well plate (up to 4 sections/well) or ∼750 μL/well for a 12 well plate (up to 8 sections/well).
CRITICAL: The steps below describe a method of preparing these solutions using “master mixes” of various primary antibodies to avoid well-to-well variation in antibody concentration resulting from pipetting error. Allow ∼500 μL/well for a 24 well plate (up to 4 sections/well) or ∼750 μL/well for a 12 well plate (up to 8 sections/well).-
a)Prepare appropriate volumes of antibody “master mixes”: Master mixes are separate, 2× working solutions of each primary antibody (anti-PSD-95 or anti-synapsin) prepared in antibody buffer (see “Before you begin”). When mixed 1:1 with a master mix of the other primary antibody (or with an equivalent volume of antibody buffer for the no-primary or channel alignment controls), the final working concentration is 1×. For experiments entailing >2 primary antibodies, master mixes will need to be further concentrated (e.g., 3× for 3 antibodies). Mix well.
-
b)Centrifuge mixture at 10,000–17,000 × g for 5 min.
-
c)Avoid pellet when pipetting antibody.
-
d)Combine master mixes to create final experimental (anti-PSD-95 master mix + anti-synapsin master mix 1:1), channel alignment control (anti-PSD-95 master mix + antibody buffer 1:1), and no-primary control (either anti-PSD-95 master mix OR anti-synapsin master mix + antibody buffer 1:1) antibody solutions.
-
a)
-
4)Incubate sections for 16–24 h in these antibody labeling solutions at 20°C–24°C (room temperature) on a shaker.
-
a)Using a paintbrush, carefully transfer tissue from blocking buffer plate to primary antibody plate. Ensure sections are submerged and mobile.
-
b)Fill empty wells with distilled water, cover.
-
c)Incubate for 16–24 h at 20°C–24°C (room temperature) on shaker at a low speed.
-
a)
-
5)
Rinse three times for 5 min each in 1× PBS to remove unbound primary antibody by repeating step 1.
-
6)Prepare secondary antibodies.
-
a)Calculate total volume of each antibody mixture to be used (same per-well volumes as for primary antibody labeling). Creation of master mixes is unnecessary for secondary antibodies.
-
b)Prepare fresh antibody buffer (see “Before you begin”).
-
c)Add appropriate volumes of antibodies to buffer.
-
d)Mix well.
-
e)Centrifuge mixture at 10,000–17,000 × g for 5 min.
-
f)Avoid pellet when pipetting antibody.
-
a)
-
7)Incubate sections in secondary antibody (NOTE: although modern fluorophores are relatively photostable, it is advisable to protect solutions and tissue labeled with fluorophores from prolonged or bright illumination).
-
a)Plate secondary antibodies.
-
b)Transfer tissue from PBS to secondary antibody. Ensure sections are submerged and able to move freely.
-
c)Incubate on shaker at slow speed for 4 h at 20°C–24°C (room temperature). Continue to steps 8 and 9 while incubating.
-
a)
-
8)Prepare mounting media.
-
a)Calculate total volume required (150 μL/slide).
-
b)Mix AF300 and MWL-488 in 1:9 ratio.
-
c)Vortex well.
-
d)Centrifuge until bubbles are gone.
-
a)
-
9)
Clean 1.5H coverslips with distilled water, then 100% ethanol, and allow to dry in a warmer at 60°C.
-
10)
After incubation in secondary antibodies, rinse sections three times in 1× PBS per step 1
-
11)Mount tissue and coverslip.
-
a)Mount free-floating sections in 1× PBS. The maximum number of sections mounted per slide is dependent on the XY range of the available microscope stage.
-
b)Allow to dry in flat, dark location.
-
c)Immediately after tissue has completely dried and adheres to the slide (∼5–10 min), dunk slide in distilled water 5× quickly to remove salt residue.
-
d)Allow to dry again in dark, flat location (∼2–10 min).
-
e)Apply 150 μL mountant prepared above per slide by distributing among each section on the slide. Larger mountant volumes may be necessary if working with large numbers of sections.
-
f)Slowly apply coverslip to avoid bubbles and apply gentle pressure to expel excess mountant.
-
g)Store in dark, flat place at 20°C–24°C (room temperature) for mountant to cure.
-
a)
Optional: Between steps 9 and 10, DAPI may be applied to visualize nuclei. To do so, rinse sections once for 5 min in PBS, incubate in 1:50,000 DAPI in PBS at 20°C–24°C (room temperature) in the same volume used for primary and secondary antibodies for 10–15 min, and then return sections to netwells and continue with step 10.
Optional: label with other/alternate markers (for primary synaptic analysis or subset analysis).
Optional: While above protocol is designed for free-float microtome-cut sections, a modified version of this protocol is also effective using 15 μm cryostat-cut sections. For this variation, tissue is mounted on slides prior to immunolabeling.
Image acquisition
The below protocol describes a typical acquisition suitable for SEQUIN analysis. Other parameters may suffice or be necessary for certain experimental aims.
-
12)Set up microscope.
-
a)Turn on Zeiss Airyscan microscope.
-
b)Turn on light source.
-
c)Select 63× 1.4 NA oil objective on the microscope display.
-
d)Clean objective with lens cleaner.
-
e)Mount Z-Piezo, a slide holder allowing for greater precision in Z translation.
-
f)Apply Zeiss 518F immersion oil to objective.
-
a)
-
13)Configure software.
-
a)Open the Zen Black software, click “start system.”
-
i)While software loads, ensure slide is clean. (Note: use distilled water and lens cleaner to clean the cover glass.)
-
ii)Insert slide in Z-Piezo and tighten knobs securing slide until finger-tight. Slide should be firmly immobilized. If performing long/unsupervised scans slide should be first loosely placed in the piezo and then firmly secured with dental wax.
-
i)
-
b)Navigate to “Maintain” tab.
-
i)Confirm objective listed in the software matches objective shown in the freestanding microscope control box.
-
ii)In the “System Options” box, navigate to the “Hardware” tab and ensure “Extended Z-Piezo” is unchecked.
-
iii)Navigate to the Airyscan menu within this tab and ensure “Adjust in continuous scans” and “Weak or sparse samples” are checked and “Adjust in slow time series” is unchecked.
-
i)
-
a)
-
14)Set experimental parameters.
-
a)Move to “Acquisitions” tab.
-
i)Choose “8-bit” bit depth.
-
ii)Choose “bidirectional” scan mode.
-
iii)Use a zoom of greater or equal to 1.8× to facilitate post-acquisition Airyscan processing.
-
iv)Choose “Line” scan mode.
-
v)Design appropriate light paths to image the fluorophores being used.
-
(a)For each light path, select the appropriate laser to be used. For the fluorophores applied in this protocol, the 488 and 561 nm laser lines are used.
-
(b)Open pinhole to maximum size.
-
(c)Set gain to 800.
-
(d)Configure the light path. For the above fluorophores on the Airyscan system, the 488/561 main beam splitter is one appropriate option. Set the secondary beam splitter to “Plate” and the emission filter to “BP 420–480 + BP 495–620” (these emission filters allow for line scan imaging of both fluorophores).
-
(a)
-
vi)Click “Optimal” to set XY pixel resolution of all channels selected. Note that all channels being imaged should be selected before clicking “Optimal.”
-
i)
-
a)
-
15)
Navigate to region of interest (ROI) using epifluorescence light source and oculars (“Locate” tab in Zen Black).
-
16)Set up imaging Z-stack.
-
a)Ensure “Z-stack” is enabled in the top left corner of the “Acquisitions” tab.
-
b)In the “Z-stack” window, Check “Use Piezo.”
-
c)Set Z-stack configuration to “First-Last” mode.
-
d)Select one of the laser channels and click “Live” in the top left panel of the “Acquisitions” tab.
-
e)Using Z-Piezo control knob, find the cut edge of the tissue at the cover glass. (NOTE: it may help to “Best fit” the display parameters to visualize signal.)
-
f)Find the most superficial Z position that avoids irregularity at the cut surface (the first optical section in which the entire field is intact) and choose “set first” in the Z-stack control windows.
-
g)Rotate the piezo focus control to find the deepest tissue section that avoids surface irregularity and select “set last.”
-
h)Navigate to the center of the stack (click “c” on the Z-stack diagram in the “Z-stack” window).
-
i)Within the Z-stack window, set the step size (“Interval”) to 120 nm to modestly oversample in Z, and set the number of optical sections (“Slices”) to 35. Note that other stack thicknesses (number of optical sections) or step sizes (Z intervals) may be appropriate for a given experiment, though increasing step size will lead to some reduction in Z-resolution and puncta detection (with the advantage of faster scan speeds).
-
j)Scroll through the entirety of the now shorter stack (4.08 μm centered in the section) and ensure qualitative suitability of all optical sections (NOTE: see “Troubleshooting” section for examples of problematic optical sections).
-
k)Turn off “Live” mode.
-
a)
-
17)Align Airyscan detector.
-
a)Turn on “Continuous mode.”
-
b)While continuous mode is running, open the Airyscan view on the display window. A display of the 32 detectors is depicted. The brightest detector should be the central element. If not, give the scope several moments to auto-adjust. Do not proceed until the brightest detector is centered.
-
a)
-
18)Set up laser ramping.
-
a)Check the “Z-correction” box in the Z-stack window.
-
b)Select the first channel to laser ramp for and start “Continuous” mode.
-
c)Move to the deepest position of the Z-stack.
-
d)Change the laser intensity of the selected channel until the lower 1/3 of the dynamic range of pixel intensity is occupied, as represented by the histogram in the “Display” tab.
-
e)Move to the first position in the Z-stack and find the laser intensity that best replicates the signal acquired at the deepest position.
-
f)Repeat this for all channels.
-
g)When desired laser ramp is set for all channels, move back to the most superficial position, select all channels, and enter the laser intensity for the first position for each. Then, click “Add position” in the Z-stack window.
-
h)Move to the last position and repeat.
-
a)
-
19)
Ensure acquisition speed is set to maximum.
-
20)
Now, select “begin experiment” to begin the scan (duration is approximately 5 min for above parameters).
-
21)
When image acquisition is complete, save image. Before proceeding with further imaging, Airyscan process the image (see Image Processing below) and check the orthogonal view to ensure consistent Z intensity (see Troubleshooting). If brightness is not consistent, move to a different field and reset the laser ramp.
Optional: Zoom can be varied to any value greater than 1.8×. Increasing zoom will decrease scan time but yield a smaller field and therefore smaller sample size.
Optional: While this protocol discusses imaging synaptic markers that emit in the green and red portions of the visible light spectrum, SEQUIN can be completed with alternate or additional fluorophores. However, settings for lasers, filter sets, and scanning mode must be adjusted.
Optional: Using microscope settings not discussed in this protocol, large automated scans can be set up to cover many contiguous or non-contiguous ROIs, as described in Sauerbeck et al., 2020.
Image pre-processing
SEQUIN analysis post-acquisition consists of image pre-processing, puncta detection, and pre-to-postsynaptic puncta separation analysis (Figure 1). Image pre-processing consists of Airyscan processing (pixel reassignment and hardware-enabled deconvolution) and channel alignment (channel shifting to correct for chromatic aberration) (Figure 2).
-
22)Airyscan Processing.
-
a)Select “Processing” tab within Zen Black and choose “Airyscan Process” from dropdown menu.
-
b)With the image of interest open, choose “Select Image.”
-
c)Set Airyscan strength to 6. Note: this value was determined by automatically determining the Airyscan strength for many images in Zeiss Zen. The lowest auto-determined Airyscan strength tended to hover near 6. It is reasonable to similarly empirically determine this value at the outset for a given system and experiment, then apply it to the entire dataset.
-
d)Choose “3D” option.
-
e)Click “Apply.”
-
f)Save new image as CZI.
-
a)
-
23)Channel Alignment of Airyscan Processed images (Figure 2).
-
a)Find channel alignment value
-
i)Select “Processing” tab within Zen Black.
-
ii)Choose “Channel Align” from dropdown menu in left sidebar.
-
iii)Open previously acquired and Airyscan Processed channel alignment image and choose “Select Image” in the left sidebar.
-
iv)Unselect “Fit.”
-
v)Offset one channel in Y such that channels are no longer overlapping to facilitate visualization of chromatic aberration.
-
vi)Click “Apply” to create a new image.
-
vii)View the orthogonal projection of this image and focus on one dual-labeled punctum. Manually shift the Z value for one of the channels until the channel images are fully aligned. Record the Z value offset.
-
viii)Once image has been aligned, confirm suitability of adjustment value on multiple puncta throughout the image.
-
ix)While XY correction is generally unnecessary when using the recommended 63× oil objective and filter sets, other combinations may require XY channel alignment. It is advisable to empirically determine degree of shift for a given system and light path.
-
i)
-
b)Channel align experimental images.
-
i)While in “Processing” tab, choose “Channel Alignment” and open and select experimental image to be channel aligned.
-
ii)Unselect “Fit.”
-
iii)Input the recorded Z offset and save the new CZI.
-
i)
-
a)
Optional: Airyscan processing can be batched. To do so, click “Run Batch” in the bottom of the window and select the files to be processed. Ensure “Auto” is unselected, strength is set to 6.0, and “3D” is selected.
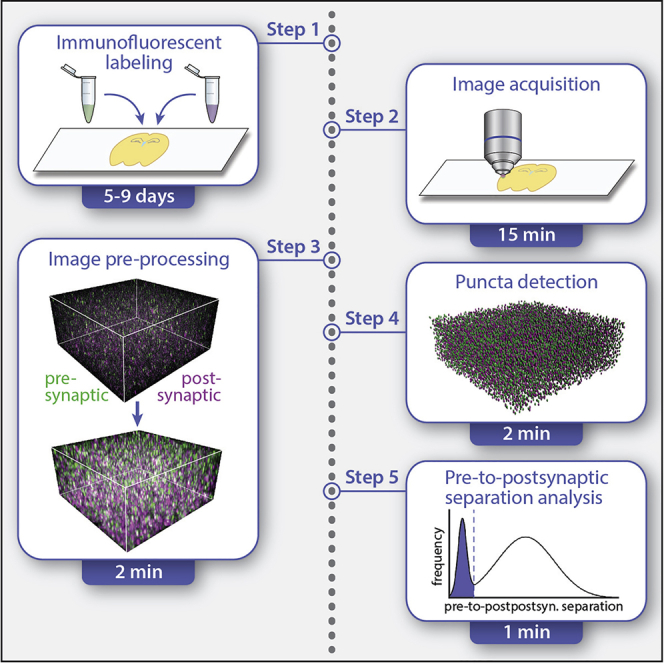
Figure 1.

SEQUIN workflow
Overview of the SEQUIN data analysis workflow. Software associated with each step is listed in blue. Figure adapted with permission from Sauerbeck et al. (2020).
Figure 2.

Image pre-processing
(A) Single detector (central detector) image prior to Airyscan processing (above) and the Airyscan processed result (below).
(B) Channel alignment image shown with orthogonal views before (upper left) and after (lower left) adjustment. Panels to right depict process for determining and correcting chromatic aberration in Z, involving a temporary transposition in Y.
Puncta detection
Using Imaris, puncta are detected in the three-dimensional Airyscan processed, channel-aligned image. This will yield X, Y, and Z coordinates of the centroid of each puncta as well as metadata such as average intensity of each punctum. Although a minimal amount of coding experience is required, a SEQUIN Python code that accomplishes the tasks below is also available (https://github.com/KummerLab/SEQUIN) if Imaris software is not.
-
24)Place all Airyscan processed and channel-aligned images in a single folder.
-
a)Note all images must have the same Z dimensions.
-
a)
-
25)Select images in Imaris (below steps describe spot creation using Imaris v9.5, but other versions are suitable with minor adaptations).
-
a)Open Imaris.
-
b)Click “Observe Folder” and navigate to folder of CZI images.
-
c)Choose “Select Folder;” folder will be added to list of “Observed Folders.”
-
d)Open folder in Imaris (double-click).
-
e)Select all images in the folder, right click, and select “convert to IMS.”
-
a)
-
26)Create spots (Figure 3).
-
a)Select “New Batch” in top menu.
-
b)When a preview of the image appears, select “Next” to begin object creation.
-
c)To create spots, click “Spots” button. Create spots for each channel for analysis.
-
d)Set parameters for channel, size, and boundaries of spot creation in the “Create” window.
-
i)“Algorithm Settings”: leave all unchecked. Advance to next screen.
-
ii)“Source Channel”: choose the source channel for spots detection.
-
iii)Under “Spot Detection” (same screen).
-
(1)Check “Model PSF-elongation” along Z-axis and “Background Subtraction.”
-
(2)“Estimated XY Diameter” for punctum is generally set to 0.2 μM.
-
(a)Indicated X,Y, and Z puncta dimensions are appropriate for the indicated imaging parameters including the 63× 1.4 NA objective, but may require modification for alternate set-ups or objectives.
-
(a)
-
(3)“Estimated Z Diameter” for punctum is generally set to 0.6 μM.
-
(4)Navigate to the next screen.
-
(1)
-
iv)“Classify Spots”: perform edge filtering (exclude spots not entirely contained within image boundaries).
-
(1)Create two filters by clicking the plus sign twice.
-
(2)Highlight one filter and change it to “Distance to Image Border XY Img=1” above automatic threshold.
-
(a)Set the leftmost value of this filter to 0.1 μm (half XY diameter from above) and ensure rightmost value is not activated.
-
(a)
-
(3)Change the second filter to “Position Z.”
-
(a)Set the left and right values so they are 0.3 μm (half Z diameter from above) from the extremes. (ex: an image that ranged from 0.06 to 4.14 in Z would be filtered to between 0.36 to 3.84).
-
(a)
-
(4)Click the green arrow to complete the setup process for this analysis.
-
(1)
-
v)Repeat step 26 for all channels in need of puncta detection.
-
i)
-
a)
-
27)
When all spots have been created and edited, choose the “OK” button in the Objects window.
-
28)
Name the batch.
-
29)
Right click the new batch and select “Run Batch” to run on all images in the folder. After the batch finishes, export the results in the CSV file format.
Figure 3.

Puncta detection in tissue
Three-dimensional image taken in mouse cortex labeled with PSD-95 and Synapsin. Panels to right show puncta detection and rendering using Imaris, with appropriate filtering applied to remove puncta at the edges of the image.
Pre-to-postsynaptic puncta separation analysis
Using MATLAB, Euclidian distances between the centroids of pre- and postsynaptic puncta are measured for each pre- or postsynaptic punctum in 3 dimensions, yielding raw as well as binned separation data (sorted by separation distance). One marker, which can be pre- or postsynaptic, is used as the reference dataset, and the nearest neighbor from the other dataset will be matched to each puncta in the referenced set (while either pre- or postsynaptic puncta can serve as the reference dataset, all examples shown reference PSD-95). All needed code is available on GitHub (see Key resources table). The Pre-to-Postsynaptic Puncta Separation Analysis utilizes two MATLAB codes (“SEQUINv3.m” and “compileExceldocs_SEQUINv3.m”), each with an associated txt file that specifies their parameters (“Analysis parameters.txt” and “Compile Excel Docs parameters.txt,” respectively). The general protocol involves specifying the parameters and running the analysis with SEQUINv3.m, then compiling the data with compileExceldocs_SEQUINv3.m with its user-specified parameters.
In many cases, only the top 20% brightest pre- and postsynaptic puncta are analyzed. Spots of lower intensity contain fewer puncta pairs consistent with synapses, though this is dependent on antibody and marker choice and should be experimentally determined (Sauerbeck et al., 2020). We recommend puncta separation analysis is performed on a system containing at minimum a multicore processor and 8 GB RAM.
-
30)
Download “SEQUINv3.m,” “Analysis parameters.txt,” “compileExceldocs_SEQUINv3.m,” and “Compile Excel Docs parameters.txt” from https://github.com/KummerLab/SEQUIN. The “.m” files contain the necessary MATLAB code, and the “.txt” file contains the user-specified parameters necessary to run each code.
-
31)Analyze separation distances.
-
a)Open MATLAB.
-
b)Within the MATLAB “Current Folder” window, navigate to the folder containing the SEQUINv3 code.
-
c)Edit the “Analysis parameters.txt” file to reflect all needed parameters. Note that additional instructions are found within the txt file.
-
i)Whichever spot data is listed as “Spot1” in the user-updated “Analysis parameters.txt” document will be used as the referenced dataset for nearest neighbor analysis.
-
i)
-
d)Paste all the text of the user-updated “Analysis parameter.txt” file into MATLAB’s command window and press “enter.” This will call the SEQUINv3 script to analyze the data, which will appear in a newly created “data output” folder.
-
a)
-
32)Compile Pre-to-Postsynaptic Puncta Separation Analysis data into one workbook.
-
a)Within the MATLAB “Current Folder” window, navigate to the folder containing the compileExceldocs_SEQUINv3.m code (if this differs from the location of the SEQUINv3.m code).
-
b)Edit the “Compile Excel Docs parameters.txt” file to reflect all needed parameters. Note that additional instructions are found within the txt file.
-
c)Paste all the text of the user-updated “Compile Excel Docs parameters.txt” file into MATLAB’s command window and press “enter.” This will call the compileExceldocs_SEQUINv3 script to compile all Pre-to-Postsynaptic Puncta Separation Analysis outputs of the SEQUINv3 code into a single file into the newly created “compileddata” folder.
-
a)
Data representation (optional)
-
33)Retrieve generated pre-to-postsynaptic separation distances.
-
a)The Nearest Neighbor code will create a “data output” folder where the input Imaris data was located. The “compile Excel docs” code will create a “compileddata” folder inside of “data output.”
-
b)The tab “Normal Frequency” within the Excel doc will contain frequency distributions of images sorted into bins of 20 nm. Each column represents a different image.
-
a)
-
34)Graph data.
-
a)Plot the frequency distribution data found on the “Normal Frequency” tab in the Excel doc against a geometric series “10 nm, 30 nm, 50 nm, […]”. This series represents the center of the histogram bins. All graphs shown and subsequent quantifications were modeled using GraphPad Prism, but any statistical software package can be used.
-
a)
Expected outcomes
When the technique is performed as described in healthy brain tissue, one expects to see a bimodal histogram of pre-to-postsynaptic separation distances. The first peak, typically found at 150–200 nm, represents pre-postsynaptic marker pairs separated by distances ultrastructurally consistent with synapses. The second peak, found at longer separation distances, represents non-synaptic associations in the volume (Figure 4). The area under the first peak (quantified as described below) corresponds to the quantity of synaptic loci within the imaged volume. In the setting of certain forms of acute injury (Sauerbeck et al., 2020), neurodegenerative processes (Sauerbeck et al., 2020; Terry et al., 1991; Gratuze et al., 2020) or other processes that reduce synapse number, the area under this peak is expected to be lower compared to controls.
Figure 4.

Synaptic quantification strategy
Bimodal histogram indicating both the first and second peaks. Dashed line marks cutoff point, and shaded region indicates area to sum for synaptic quantification (NOTE: other quantification options may be used; see text and Figure 5).
Quantification and statistical analysis
To perform a basic quantification of synaptic loci, sum all puncta counts falling to the left of an experimentally determined cutoff (for example, < 250 nm pre-to-postsynaptic separation, or the trough between the first and second peak). Given variability between experimental models and conditions, this cutoff point may vary for different applications (Figure 4).
The early peak may be more completely “disentangled” from the broad second peak by incorporating “flipped” data into the analysis, or modeling the pre-to-postsynaptic separation histogram as a mixture of two Gaussians (Figure 5). For the “flipped” analysis, spots data for one of the marker channels (pre- or postsynaptic) are first reflected across the Y axis and then the nearest neighbor analysis is undertaken with the paired, unaltered marker channel coordinates (e.g., the PSD-95 spots coordinates are reflected and a separation analysis is performed with un-reflected synapsin spot coordinates). Plotting the flipped data (MATLAB, R, GraphPad Prism, etc.) should yield a unimodal distribution indicative only of random pairings. This data can be found in the generated spreadsheet in the tab “Flipped Frequency.” These random pairings can then be subtracted from the prior (unflipped) nearest neighbor analysis on a bin-wise basis. The results of this subtraction can be found in the sheet titled “Flip Subtraction Frequency.” Plotting the flip subtraction data should yield a sharp early peak with a minimal or negative second peak. Finally, the “Flip Sub Zeros Frequency” uses the same algorithm to remove random pairings as found in the “Flipped Frequency,” but subsequently zeros any biologically meaningless negative values.
Figure 5.

Additional synaptic quantification strategies
(A) Standard frequency distribution, overlaid with flipped distribution and flipped subtracted distribution.
(B) Gaussian curves fitted to the first and second peak of a standard frequency distribution.
2-Gaussian modeling can be performed with a number of graphing and analysis packages (MATLAB, R, GraphPad Prism, etc.). In this case the first of the 2 resulting Gaussians represents paired puncta consistent with synaptic loci, while the second (at a longer separation distance) represents random pairings.
Limitations
SEQUIN has only been validated in aldehyde-fixed tissue.
The quality of refractive index-matching currently limits the depth at which SEQUIN can be applied. With this protocol, SEQUIN analysis becomes challenging at depths greater than 50 μm from the cut surface.
Troubleshooting
Problem
The synapse peak is minimal or indistinguishable from the noise peak (Figure 6).
Figure 6.

Minimal-to-no synapse peak
Frequency distribution with little-to-no synapse peak.
Potential solution
Lack of a typical bimodal frequency distribution of pre-to-postsynaptic separations with a clear early peak and broader following peak could be due to a variety of problems occurring in the tissue preparation, labeling, and/or imaging steps. We present a strategy to isolate the problem. Working backward:
Quality check image analysis workflow: Repeat quantification on imaging data that has historically yielded expected results (or use sample data provided at https://github.com/KummerLab/SEQUIN) in tandem with experimental images. Repeat analysis of historical data should yield identical results.
Quality check imaging workflow: Compare previous and current quantification of the previously immunolabeled and validated sample (imaging validation sample; Table 1) that was imaged as a historical control. While quantification is not expected to be identical, an image from the same region using the same filter settings should yield a similar frequency distribution vs. historical data. Additional quality checks should include:
-
•
Puncta brightness should be stable throughout the Z stack (assuming uniform distribution of markers).
-
•
Image should be free of tissue artifacts or excessive autofluorescence.
-
•
Z stack should be centered in tissue.
Table 1.
List of immunolabeling and imaging controls
| Necessary controls | Function | Antibodies |
|---|---|---|
| No anti-PSD-95 | to ensure secondary antibody specificity and lack of spectral cross-talk | 10: Gp anti-Synapsin 1/2 OR Ms anti-Synapsin 1 20: Gt anti-Rb 594, Gt anti-Gp 488 OR Gt anti-Ms 488+ |
| No anti-synapsin | 10: Rb anti-PSD-95 20: Gt anti-Rb 594, Gt anti-Gp 488 OR Gt anti-Ms 488+ |
|
| Channel alignment | to measure and correct chromatic aberration post-acquisition | 10: Rb anti-PSD-95 20: Gt anti-Rb 594 AND Gt anti-Rb 488 |
| Previously validated tissuea (optional) | to differentiate tissue preparation from immunolabeling problems | 10: Rb anti-PSD-95, Gp anti-Synapsin 1/2 OR Ms anti-Synapsin 1 20: Gt anti-Rb 594, Gt anti-Gp 488 OR Gt anti-Ms 488+ |
| Previously validated slideb (optional) | to validate imaging and analysis workflow |
Rb, rabbit; PSD, postsynaptic density; Gt, goat; Gp, guinea pig; Ms, mouse.
Previously validated tissue is tissue that has previously been labeled, imaged, and analyzed and has yielded quantifiable results.
Previously validated slide is tissue that was previously labeled, imaged, and analyzed (using the same antibody combinations as the current experimental tissue) and yielded quantifiable results.
Quality check histological workflow: Examine no-primary controls to assess background levels and unexpected antibody cross-reactivity (primary or secondary). This protocol should yield very low background using the suggested fluorophores. Examine results of analysis of tissue control (Tissue validation control; Table 1) to rule out poor clearance or fixation of experimental tissue, or other potential problems with tissue harvesting and processing.
Problem
The frequency distribution is far-left shifted, with a small or nonexistent noise peak (Figure 7).
Figure 7.

Left shifted frequency distribution
Left shifted frequency distribution with minimal secondary peak, often indicative of antibody or fluorophore cross-talk.
Potential solution
This can occur with antibody or fluorophore cross-talk. To identify the issue:
-
•
Confirm correct filter sets were used to isolate target signal.
-
•
Confirm antibody specificity using no-primary controls.
-
•
Confirm correct immunolabeling techniques were implemented.
Problem
Puncta appear to “zigzag” through Z (Figure 8).
Figure 8.

XY shift
An excerpt from an orthogonal view of an image from an imaging session in which the stage shifted during imaging (bottom panel) compared to an image from a session with minimal shift (top panel).
Potential solution
This indicates an XY shift of the stage during image acquisition. This can be an indication that the stage and/or slide are not properly secured or that the system was not given enough time to warm prior to the start of imaging (thermal shift). Ensure proper installation of both components and allow at least 30 min from the time the system is started until the first experimental images are acquired.
Problem
Fluorescence intensity is highly variable through Z, which causes faulty puncta detection (Figure 9).
Figure 9.

Fluorescence intensity variation through Z
An image with varying fluorescence intensity through Z (bottom) compared to an image with stable fluorescence intensity (top).
Potential solution
First confirm the Z-stack is centered in the middle of the tissue and neither the top nor the bottom are approaching the tissue cut surface. Tissue at the cut surface is occasionally more intensely labeled than tissue deeper within the section. Improper laser ramping is another possible cause.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Terrance T. Kummer (kummert@wustl.edu).
Materials availability
This study did not generate new unique materials or reagents.
Data and code availability
The code utilized in the protocol and example datasets are available on GitHub [https://github.com/KummerLab/SEQUIN].
Acknowledgments
The authors wish to thank Dr. James Fitzpatrick and Matthew Curtis for imaging discussions. This study was supported by the BrightFocus and Brain Research Foundation, the McDonnell Center for Cellular and Molecular Neurobiology, and NIH I01BX005204 (all to T.T.K.). Experiments were performed in part through the Washington University Center for Cellular Imaging supported by Washington University School of Medicine, The Children’s Discovery Institute, St. Louis Children’s Hospital (CDI-CORE-2015-505), the Foundation for Barnes-Jewish Hospital (3770), and the NIH Office of Research Infrastructure Programs (OD021629).
Author contributions
S.R. collected the data for this report. S.R. wrote the manuscript with input from A.D.S. and T.T.K. A.D.S., T.T.K., and S.R. developed the SEQUIN technique. T.T.K. was responsible for overall project management.
Declaration of interests
The authors declare no competing interests.
References
- Sauerbeck A.D., Gangolli M., Reitz S.J., Salyards M.H., Kim S.H., Hemingway C., Gratuze M., Makkapati T., Kerschensteiner M., Holtzman D.M. SEQUIN multiscale imaging of mammalian central synapses reveals loss of synaptic connectivity resulting from diffuse traumatic brain injury. Neuron. 2020;107:1–17. doi: 10.1016/j.neuron.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Gratuze M., Leyns C.E.G., Sauerbeck A.D., St-Pierre M., Xiong M., Kim N., Serrano J.R., Tremblay M., Kummer T.T., Colonna M. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 2020;130:4954–4968. doi: 10.1172/JCI138179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The code utilized in the protocol and example datasets are available on GitHub [https://github.com/KummerLab/SEQUIN].