

Summary
A FACS protocol is described that eliminates isolation and staining artifacts to allow accurate comparison between cell populations isolated from organs obtained from disparate mouse groups. This protocol was validated by characterizing the estrogen receptor positive cells within the mammary gland of transgenic mice with different genotypes at different stages of the estrous cycle. We include protocols necessary to batch stage animals within the cycle to proceed directly to FACS, which provides optimal RNA yields for RNA-seq.
For complete details on the use and execution of this protocol, please refer to Ludwik et al. (2020).
Subject areas: Cell biology, Cell isolation, Flow cytometry/mass cytometry, Genomics, Model organisms
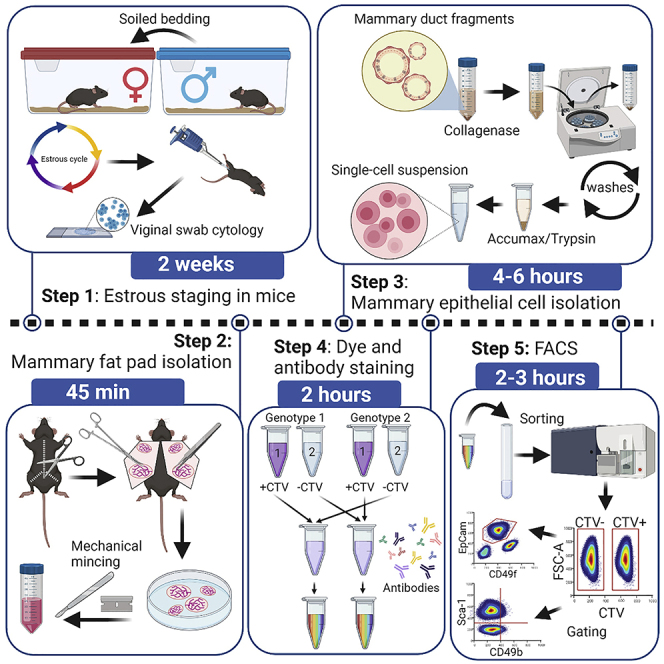
Graphical Abstract
Highlights
-
•
FACS protocol for direct comparison of cell populations from disparate mouse groups
-
•
Cells from varying genotypes or estrous stage are marked by dye and processed together
-
•
Protocol validated by analysis of mammary gland estrogen receptor positive cells
-
•
Procedure optimized to enhance RNA yields for RNA-seq
A FACS protocol is described that eliminates isolation and staining artifacts to allow accurate comparison between cell populations isolated from organs obtained from disparate mouse groups. This protocol was validated by characterizing the estrogen receptor positive cells within the mammary gland of transgenic mice with different genotypes at different stages of the estrous cycle. We include protocols necessary to batch stage animals within the cycle to proceed directly to FACS, which provides optimal RNA yields for RNA-seq.
Before you begin
Equipment sterilization
Timing: 2 h
-
1.
Sterilize surgical scissors, forceps and 70 μm nylon mesh. Cut the mesh into 1 inch squares.
Working solutions
-
2.Digestion media (prepare fresh): The combined mammary glands (eight from one mouse as the #1 pair are very small and hard to access) from one mouse requires 15 mL of digestion media.
-
a.Add 30 mg of Collagenase A powder (2 mg/mL) and dissolve in 15 mL DMEM/F12 media by vortexing and heating in a 37°C water bath. After the powder is completely dissolved filter sterilize using a 0.22 μm syringe filter.
-
b.Add 150 μL of sterile penicillin/streptomycin (100×).
-
c.Solution should be at 37°C prior to use.
-
a.
-
3.DNase solution (prepare fresh):
-
a.Dilute DNase (1,600 U/50 μL) 1:100 in DMEM/F12 to achieve 320 U/mL.
-
a.
-
4.Trypsin solution (prepare fresh):
-
a.Dilute Trypsin (10×) 1:2 in DMEM/F12.
-
a.
-
5.FACS buffer (filter-sterilized solution can be stored at −80°C for 6 months):
-
a.A mixture of 5% FBS in PBS.
-
a.
-
6.Collection media (filter-sterilized solution can be stored at 4°C for 2 weeks):
-
a.Dilute FBS 1:2 in DMEM/F12.
-
a.
-
7.If needed freezing media (filter-sterilized solution can be stored at −80°C for 6 months):
-
a.A mixture of 90% FBS and 10% DMSO.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin anti-CD140 | BioLegend | RRID:AB_11211998 |
| Biotin anti-CD31 | BioLegend | RRID:AB_312910 |
| Biotin anti-Ter-119 | BioLegend | RRID:AB_313704 |
| Biotin anti-CD45 | BioLegend | RRID:AB_312968 |
| Anti-Sca1-PerCP | BioLegend | RRID:AB_893618 |
| Anti-CD49b-APC/Cy7 | BioLegend | RRID:AB_313416 |
| Anti-EpCAM-APC | BioLegend | RRID:AB_1134105 |
| Anti-CD49f-PE/Cy7 | BioLegend | RRID:AB_2561704 |
| Rat IgG isotype control | Invitrogen | RRID:AB_2532969 |
| Chemicals, peptides, and recombinant proteins | ||
| Accumax | Millipore Sigma | SCR006 |
| Collagenase A | Millipore Sigma | 11088793001 |
| 100× penicillin/streptomycin | Thermo Fisher | 15140122 |
| DNase | Millipore Sigma | D4527-200KU |
| 10× Trypsin | Thermo Fisher | 15400054 |
| DMEM/F12 | Thermo Fisher | 11330032 |
| Brilliant Violet 510 Streptavidin | BioLegend | 405233 |
| Cell Trace Violet | Life Technologies | C34557 |
| Zombie Yellow | BioLegend | 423104 |
| Fetal bovine serum | R&D Systems | Cat # S11150 |
| Gibco PBS, pH 7.4 | Thermo Fisher Scientific | Cat # 10010023 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6JRSK2−/−(RSK2-KO) age range 6–12 weeks | Institut de Genetique et Biologie Moleculaire et Cellulaire, C.U. de Strasbourg, France | Andre Hanauer, PhD |
| Software and algorithms | ||
| LSM-FCS/ZEN | Carl Zeiss | N/A |
| GraphPad Prism 6.0a | GraphPad | N/A |
| BioRender | BioRender | https://biorender.com/ |
| Cytobank | Cytobank | https://cytobank.org/ |
| Other | ||
| Becton-Dickinson’s FACAria II | BD Biosciences | n/a |
| Eppendorf New Brunswick Galaxy 170R CO2 incubator | VWR | n/a |
| Eppendorf ThermoMixer | VWR | n/a |
| Thermo/IEC Centra CL2 benchtop centrifuge | Thermo Scientific | n/a |
| 236 Aerocarrier rotor | Thermo Scientific | n/a |
| Aerocarrier inserts for 236 Aerocarrier rotor for 2 × 15 mL Falcon/Corning Conical | Thermo Scientific | n/a |
| Eppendorf centrifuge 5424R | Fisher Scientific | n/a |
| Rotor FA-45-24-11 | Fisher Scientific | n/a |
| Hausser Scientific Levy hemacytometer | Fisher Scientific | Cat # 02-671-55A |
| WPI Iris scissors, 10 cm, supercut, straight | Fisher Scientific | Cat # 50-822-291 |
| Micro dissecting forceps serrated, half curved 1 mm tip, 4 inch | George Tiemann | Cat # 160-18 |
| Syringe filters, sterile | Fisher Scientific | Cat # 09720004 |
| 70 μm nylon mesh | Fisher Scientific | Cat # NC0446099 |
| Deposited data | ||
| RNA sequencing data | Ludwik et al. 2020 | GEO:GSE113323 |
Step-by-step method details
Estrous staging in mice
This step describes the process of batch staging female mice during their estrous cycle, which has been described for setting up timed pregnancies (https://www.jax.org/news-and-insights/jax-blog/2014/september/six-steps-for-setting-up-timed-pregnant-mice#, 2014). Having multiple animals available at the same stage of the estrous cycle to proceed directly to FACS sorting improves isolated RNA yield for RNA-seq analysis.
Note: We have found that using freshly purified cells for RNA-seq increases RNA yield (Figure 1A). This goal can be achieved by analyzing the estrous stage and performing the mammary gland isolation by 9 a.m. Mammary cell isolation and antibody/dye staining can then be completed by 3 p.m. The FACS sorting will take an additional 3 h. Alternatively, if RNA-seq is not going to be performed intact mammary fat pads can be frozen at −80°C in a mixture of 90% FBS and 10% DMSO. We have found that cryopreservation of the mammary fat pad followed by single cell isolation does not increase the percentage of dead cells compared to using fresh tissue (Figure 1B).
Figure 1.

Analysis of isolated mammary epithelial cells isolated from either fresh or frozen glands
(A) Total RNA yield from the Sca+CD49− population (median ± quartile, each point represents isolation from eight glands obtained from an individual mouse) p = 0.0014.
(B) CTV staining was used for fresh versus frozen comparison and the lineage Abs were not used for staining. The gating was: FSC-A /SSC-A (p1), FSC-H /-A (p2), FSC-A/ CTV (fresh; frozen), FSC-A/ZY. The ZY+ population was selected to determine the percentage of dead cells (median ± quartile, each point represents isolation of eight glands from an individual mouse).
On average a mouse estrous cycle will last 5–6 days with 1 or 2 days in each stage.
Note: Mice caged in isolation tend to cycle more rapidly than mice in larger social groups (Cora et al., 2015).
The stages are defined by vaginal cytology and are comprised of proestrus, estrus, metestrus, and diestrus.
-
1.Synching setup
-
a.Weigh and randomize 4–5 females in cages with clean bedding for 2 days prior to step 1b.
-
b.From a male cage transfer one tablespoon of urine-saturated bedding from the corner of the cage primarily used as a toilet to the middle of the female cage. Male pheromones will promote regular cycling in the females.
-
a.
-
2.Vaginal cytology
-
a.Begin analysis of vaginal cytology in the morning 24–36 h after adding the male bedding.
-
i.Restrain the mouse by scruffing the skin on the dorsal side of the mouse between the shoulders. Rotate your wrist with the mouse in a supine ventral posterior position.
-
ii.Lavage the vagina using 10 μL of sterile PBS twice and then collect the third flush containing the vaginal cells into a labeled microcentrifuge tube.
 CRITICAL: Try to keep the mice relaxed during the process. Remove any excreted urine on the animal’s belly using a sterile dry towel. The pipet tip is placed against the vulva and the ejection of liquid is sufficient to wash out the vagina. Flushes should be collected at the same time each day. Mice which arrive at the desired stage after completion of a full successful cycle are considered regularly cycling. Exclude mice with irregular cycles or restart the process after a several day rest period in which the mice are not being handled.
CRITICAL: Try to keep the mice relaxed during the process. Remove any excreted urine on the animal’s belly using a sterile dry towel. The pipet tip is placed against the vulva and the ejection of liquid is sufficient to wash out the vagina. Flushes should be collected at the same time each day. Mice which arrive at the desired stage after completion of a full successful cycle are considered regularly cycling. Exclude mice with irregular cycles or restart the process after a several day rest period in which the mice are not being handled. -
iii.Repeat steps 2a, i and ii for each mouse.
-
i.
-
b.Analyze the vaginal cytology, which is based on the ratio of cell types (Figure 2) (Byers et al., 2012; Cora et al., 2015).Note: The vaginal cytology of different mice can vary at each stage, but once a full cycle is observed, the relative makeup is consistent for one individual. Taking notes of each day or saving images can aid in determining characteristics of an individual mouse. Proestrus (PE) is marked by low to moderate cellularity and the predominant cell type is small, nucleated epithelial cells. Estrus (E) is marked by high cellularity and may have a mix of epithelial phenotypes, but the predominant phenotype will be large, anucleate cells referred to as cornified epithelium. Often these will appear as sheets of > 10 cells. Metestrus (ME) is also marked by high cellularity and appears as a mix of neutrophils, which appear as small, highly circular cells, and epithelial cells. The epithelial portion in ME often matches that of estrus and is usually a mix of epithelial phenotypes. When mice are caged in groups of 4 to 5 females, diestrus (DE) usually extends over 2 days. The first day is marked by high cellularity predominantly made up of neutrophils. The epithelial portion can vary widely. The second day of DE usually has low cellularity, and the relative makeup will vary between mice.
-
i.If the dilution appears cloudy, dilute each of the vaginal flushes ∼1:5 or ∼1:10 with PBS. If the dilution remains cloudy then dilute further.
-
ii.Pipet ∼10 μL of the dilutions onto a microscope smear and image at 10× magnification.
-
i.
-
a.
Figure 2.

Vaginal cytology for determination of estrous stage
(A) Typical makeup of cell type and relative number from mice in proestrus (PE), estrus (E), metestrus (ME), and diestrus (DE) from different mice.
(B) Comparison of a normal cycle and an example of an aberrant cycle, wherein the mouse appeared to alternate between proestrus and diestrus with few to no lavages showing typically cytology for estrus or metestrus.
(C) Vaginal lavages from the mouse with an aberrant cycle in (B) showed several days (D3–D5) in diestrus. Scale bars, 100 μm.
Mouse mammary fat pad isolation
This step involves removal of the mouse mammary fat pads for various downstream analyses.
-
3.Removal of glands
-
a.Spray the ventral surface of the euthanized mouse with 70% ethanol and wipe with gauze.
-
b.Starting from the bottom of the abdomen create a Y-shaped incision along the ventral side and then proceed to the thorax. Take care not to pierce the peritoneal cavity as the mammary fat pad lies between the skin and the peritoneum.
-
c.Create additional incisions radiating from the central cut toward each of the four limbs.
-
d.Using forceps and scissors separate the skin fascia from the abdominal wall and expose the underlying mammary fat pads (Figure 3 and Methods Videos S1 and S2).
-
e.Remove any surrounding tissue from the mammary fat pad as hair and extraneous tissue will interfere with the single cell isolation. In particular the thin layer of muscle overlying thoracic glands 2 and 3 should be removed. Remove the lymph node in the number 4 gland.
-
f.Separately pinch each fat pad with forceps and gently pull away from skin. While pulling gently on the gland with forceps use scissors to separate the gland from the underlying connective tissue. Initially focus on the edge of the fat pad. Mammary fat pad pairs 2 and 3, and 4 and 5, are connected and removed together.
-
a.
-
4.Preparation for downstream analysis
-
a.To generate formalin-fixed paraffin embedded blocks place the tissue in formalin for 48 h and transfer to 70% ethanol for long term storage at 4°C followed by dehydration and embedding.
-
b.To cryopreserve the tissue place the glands of one mouse in 6–10 mL of premade freezing media (enough to submerge all glands) . For short term storage (< 1 month) keep at −80°C and for longer term storage (> 1 month) in liquid N2. Cryopreservation provides the ability to collect sufficient samples at a particular stage of the estrous cycle or a particular genotype before proceeding to the described FACS, which uses viable cells. To use cryopreserved samples for FACS rapidly thaw at 37°C with gentle swirling until the media is liquid with a few remaining ice crystals. The glands are easily removed from the media using forceps.
-
c.For immediate cell isolation following dissection place the glands in PBS and proceed.
-
a.
Figure 3.

Mouse mammary gland dissection
(A) Euthanized mouse (left) in which the mammary glands were quickly exposed and the mammary glands removed (right). A bilateral incision through the skin exposed the thoracic mammary glands (upper arrow) and abdominal glands (lower arrow). Scale bar, 1 cm.
(B) Thoracic mammary glands are located under muscle tissue which should be avoided in the mammary gland collection. Muscle tissue is indicated by forceps (lower image is zoom).
Mammary cell isolation
This step involves obtaining single cells from the isolated mammary fat pads. If necessary for downstream application the protocol should be performed in sterile and/or RNase-free conditions.
Note: When working with more than six samples we recommend two operators as there is not sufficient time between samples for proper processing.
-
5.Isolation of mammary cells
-
a.Initial digestion of mammary glands
-
i.Excise each of the mammary glands and place together in a 6 cm dish with 1 mL DMEM/F12.
-
ii.Move gland into a clean dry dish using forceps and with scissors or a scalpel chop the gland finely (takes ∼5–10 min per sample) so that it resembles a homogenous sludge with no large tissue pieces (Figure 4A).
-
iii.Transfer the homogenized tissue into a 50 mL conical containing 14 mL of pre-warmed digestion media.
-
iv.Place the tube in the incubator at 37°C in 5% CO2. The tube cap should be on loosely to allow for gas exchange.
-
v.Depending on the available time and labor resources the following approaches are recommended.
Minimal effort
Total digestion time:2–3hLabor intensive
Total digestion time: 1–1.5hEvery 20–30 min remove tube from incubator. Every 15 min remove tube from incubator. Resuspend the tissue in the digestion mixture by gentle swirling of the tube. Pipet each tube 10–15 times using a 10 mL serological pipet Note: Digestion times are dependent on how frequently the digestion is mixed. We advise against using a shaker or thermomixers as we have found that the repeated swirling causes the tissue to clump and hinders mammary cell isolation.CRITICAL: Long strands of fibrous material frequently appear during the digestion process. Isolated mammary cells attach to these fibers and interfere with pelleting during centrifugation. To evaluate whether fibers need to be removed monitor whether the fibers are increasing in size. It is important to remove the fibers as soon as they appear during the digestion process. Removal is performed by using a P20 and drawing the fibers into the pipet tip with as little liquid as possible. -
vi.Repeat steps 5a, iv-v until the digestion is complete: a fatty layer should be visible on top when the tube is undisturbed and there are no visible tissue fragments observed upon swirling the sample (Figure 4B).
-
i.
-
b.Epithelial cell clump isolation.Note: All centrifugation steps require a swinging bucket rotor in step 5b.
-
i.Pellet digested material at 150 × g for 5 min and gently aspirate the supernatant. Avoid using aspirators and do not completely remove all the liquid as the pellets are loosely adhered.
-
ii.Resuspend the pellet in 5 mL of DNase I solution and incubate at 37°C in 5% CO2 for 3–5 min. This step should remove any remaining fibrous clumps. If large fibers remain, they should be removed manually. See Critical at step 5a, v for removal of clumps (Figure 4B).
-
iii.At the end of the digestion time add 500 μL of FBS, mix gently at this stage and then transfer to a 15 mL conical tube.
-
iv.Pellet cell suspension at 150 × g for 10 min.
-
v.Carefully aspirate the supernatant and resuspend the pellet in 1 mL of PBS by gentle pipetting up and down ∼10–20 times using a P1000 pipet.
-
vi.Add 9 mL of PBS and pellet at 350 × g for 20s.
-
vii.Repeat steps 5b, v and vi twice using a 10 mL serological pipet instead of a P1000 to keep the cells as clusters so they spin down efficiently.CRITICAL: The number of mammary epithelial cells will decrease with every wash step. We strongly recommend monitoring the sample after each wash. After the pellet is resuspendend in 1 mL PBS remove 10 μL of the cell suspension and place as a single drop on a glass slide. Observe under a microscope (Figure 4C).
-
i.
-
c.Single cell isolation.Note: All centrifugation steps require a table top centrifuge in step 5c
-
i.After the last PBS wash, aspirate the supernatant and resuspend the pellet in 1 mL of Accumax by gently pipetting 20 times using a P1000 pipet.
-
ii.Bring up the Accumax volume to 3 mL and divide the sample equally into three 1.5 mL centrifuge tubes.Note: We recommend a total of 3 mL per mouse because if the cell density is too high clumps will form during the digestion step, which will reduce the epithelial cell yield.
-
iii.Incubate in a Thermomixer at 37°C for 10 min at 800 rpm. After every 2 min pipet the suspension with a P200 to break up clumps that may form. Large fibrous aggregates can be removed using a P200 as described.CRITICAL: Do not swirl the samples from steps 5c, iii onward as it will promote cell aggregation.
-
iv.Remove the tubes from the Thermomixer and pellet the cells at 500 × g for 5 min.
-
v.Carefully remove the supernatant and recombine the samples from the multiple tubes using a total of 0.5 mL Trypsin solution.
-
vi.Incubate at 37°C in 5% CO2 for 3–5 min.Note: Determine the proportion of single cells by taking 10 μL and observing under the microscope. At this stage 90% of the suspension should be single cells.
-
vii.Add 0.5 mL of FBS to quench the trypsin and gently mix by pipetting.
-
viii.Pellet the cells at 500 × g for 5 min.
-
ix.Carefully aspirate the supernatant and resuspend the pellet in 1 mL of PBS by gentle pipetting up and down ∼10–20 times using a P1000 pipet.
-
x.The suspension is taken up into a 1 mL syringe and passed through a 70 μm mesh into a new 1.5ml Eppendorf tube.Note: To construct the home made filter (Figure 5) take a 1 mL syringe with needle. Discard the needle and keep the needle cap. Cut off the bottom of the needle cap just under the part where it narrows. Sterilize cap in 70% ethanol. With sterile tweezers insert one sterile nylon mesh square into the wide portion of the needle cap. Use syringe filled with cell suspension to push the mesh into place. Pass the cell suspension through the mesh. We found that commercially available cell filters resulted in substantial cell loss.
-
xi.Take a 10 μL aliquot of the filtered cell suspension and count the cells using a hemocytometer. Expected total yield from eight mammary glands obtained from one mouse is ∼4 × 106.
-
xii.Place cell suspension on ice until ready for further processing.
-
i.
-
a.
Figure 4.

Mouse mammary gland digestion and mammary epithelial cell isolation
(A) Minced mammary tissue is chopped to a sludge-like consistency to aid the digestion process.
(B) Minced mammary tissue is placed in digestion media (left panel). Following incubation, the mammary tissue digest is a translucent solution with no large tissue fragments (middle panel). Large fibrous clumps may persist in the digestion. These fibrous pieces can be manually removed using a pipette (right panel).
(C) Isolated mammary cells visualized using a light microscope. Following mammary gland digestion the sample consists of large clumps of epithelial cells and contaminating cell types such as muscle tissue and fibrous tissue (left panel, arrows). Differential centrifugation allows the purification of mammary epithelial clumps (middle panel). The epithelial clumps can be further digested to single cells for FACS (right panel). Scale bar, 0.25 mm.
Figure 5.

Obtaining isolated mouse mammary epithelial cells by filtration
(A) Schematic of homemade filter device.
(B) The end of the plastic syringe cap is used to clamp the nylon mesh (70 μm pore) to the syringe. The syringe needle has been discarded. The cap can be soaked in 70% ethanol after cutting and dried before proceeding to the next step.
(C) Physical assembly of the sterile cell syringe filter.
Antibody and dye optimization
This step describes the dye and antibody titration and controls necessary to perform the mixed FACS. The cell type for the optimization should be the same as that used for the actual experiment.
Note: Total mammary cell yield/mouse based on using the paired glands 2–5 is ∼4 × 106 cells. Each antibody optimization will require ∼1 × 105 cells. Assuming one negative control and testing 4 concentrations for each antibody, the mammary glands from one mouse should be sufficient for testing all of the 8 antibodies/dyes used in this FACS protocol.
Note: We recommend optimizing the viability dye, i.e., Zombie Yellow, first, as it should be included for the antibody titration.
-
6.Dye staining (perform first)
-
a.For CellTrace Violet (CTV) and Zombie Yellow (ZY) optimization the cells remain in PBS.
-
b.For each dye, 4 concentrations should be tested as described:
-
i.Negative control where no dye/antibody is added.
-
ii.Recommend by manufacturer
-
iii.1:5 dilution of ii
-
iv.1:10 dilution of ii
-
v.1:100 dilution of ii
-
i.
-
c.Transfer 1 × 105 cells into each of five 1.5 mL microcentrifuge tubes labeled as indicated in b.Note: The recommended cell concentration is 1 × 105 cells in 50 μL. We recommend not using volumes smaller than 50 μL. Manufacturer recommendations frequently suggest 1 × 106 cells in 100 μL. This concentration can be very difficult to achieve from in vivo samples and we have found the staining efficiency is flexible in terms of the cell concentrations, as long as the antibody/dye concentrations remain constant. This approach simplifies scaling of the staining protocol from optimization to the experimental stage.
-
d.Adjust volume to 50 μL if necessary.
-
e.Add dyes at the calculated concentrations.
-
f.Incubate at 21°C–23°C in the dark for 20–30 min.
-
g.Add 1 mL FACS buffer to each tube and centrifuge at 350 × g for 5 min.
-
h.Remove the supernatant and resuspend in 1 mL FACS buffer.
-
i.Centrifuge at 350 × g for 5 min.
-
j.Remove the supernatant and resuspend in 250 μL to 1 mL of FACS buffer.Note: Consult with the FACS facility or operator regarding the preferred volumes.
-
k.Store on wet ice in the dark until ready to analyze.
-
a.
-
7.Antibody staining.
-
a.For antibody optimization the cells need to be resuspended in PBS supplemented with 0.5%–1% BSA.
-
b.For each antibody, 4 concentrations should be tested as described:
-
i.Negative control where no dye/antibody is added.
-
ii.Recommend by manufacturer
-
iii.1:5 dilution of ii
-
iv.1:10 dilution of ii
-
v.1:100 dilution of ii
-
i.
-
c.Transfer 1 × 105 cells into each of five 1.5 mL microcentrifuge tubes labeled as indicated in b. See Note at step 6c for cell concentration.
-
d.Adjust volume to 50 μL if necessary.
-
e.Perform the following steps:
-
i.Block by adding 0.5 μg of IgG
-
ii.Incubate at 21°C–23°C for 10 min.
-
iii.Add conjugated primary Ab at the calculated concentrationsNote: For indirect staining, titration is necessary for both the primary and the secondary antibody. In the case of a streptavidin conjugated primary and a biotin secondary, we recommend choosing a single concentration of the primary, for example that based on manufacturer instructions or a publication, and then titrating the secondary antibody as in step 7b. Using the optimal concentration of the secondary then proceed to titrate the primary.
-
i.
-
f.Incubate on wet ice in the dark for 20–30 min.
-
g.Add 1 mL FACS buffer to each tube and centrifuge at 350 × g for 5 min.
-
h.Remove the supernatant and resuspend in 1 mL FACS buffer.
-
i.Centrifuge at 350 × g for 5 min.
-
j.Remove the supernatant and resuspend in 250 μL to 1 mL of FACS buffer.Note: Consult with the FACS facility or operator regarding the preferred volumes.
-
k.Store on wet ice in the dark until ready to analyze.
-
a.
FACS
For FACS analysis, cells of different genotypes are stained with antibodies and analyzed separately. However, this approach results in artifacts such as unequal antibody staining, especially if a large number of samples are processed. To overcome this challenge we permanently mark cells of one genotype using CellTraceViolet (CTV) and mix the two samples for the antibody staining. The following protocol describes the staining of cells obtained from two animals that are being compared: genotype 1 (G1) and genotype 2 (G2). The described protocol is for the isolation of viable cells.
Figure 6.

Example of sample setup for two way comparison
-
8.CellTrace Violet (CTV) and Zombie Yellow (ZY) staining.CRITICAL: CTV and ZY staining must be done in the absence of BSA or FBS according to the manufacturer’s recommendation.
-
a.For your control samples aliquot ∼1 × 105 cells in PBS into each of 12 microfuge tubes and if necessary adjust the volume to 150 μL. Label the tubes 1 through 12, which are compensation controls (Figure 6). See Note at step 6c on volume.
-
b.For your experimental samples aliquot ∼2 × 106 from each genotype into two microfuge tubes and if necessary adjust the volume to 150 μL. Label: tube #13 as 13-G1_1, tube #14 as 14-G1_2, tube #15 as 15-G2_1, and tube #16 as 16-G2_2 (Figure 6).
-
c.This series of steps refers to tubes indicated in Figure 6.
-
i.Add ZY to tubes 2, 4, and 10–16.
-
ii.Add CTV to tubes 5, 10–13, and 15.
-
iii.Mix by gently flicking the tubes with your finger.
-
iv.Incubate at 21°C–23°C for 20–30 min.
-
v.Add 1,350 μL of FACS buffer to each tube and centrifuge at 350 × g for 5 min.
-
vi.Carefully remove the supernatant and resuspend the cells in tubes 1–12 in 150 μL of FACS buffer and tubes 13–16 in 75 μL of FACS buffer.
-
vii.Combine tube 13 (13-G1_1_CTV+) with tube 16 (16-G2_2_CTV-) (EXP 1; Figure 6) and tube 14 (14-G1_2-CTV-) with tube 15 (15-G2_1-CTV+) (EXP 2; Figure 6). In this manner comparisons between the genotypes are carried out twice.Note: It is advisable to limit the number of comparisons to two per genotype, which is dictated by the requirement for at least ∼1 × 105 cells per tube and technical considerations. With a higher number of comparisons the delay in handling the collection tubes during the sort can lead to loss of cell viability. As an example, Figure 7 shows the comparisons that could be set up between multiple different genotypes/conditions. Briefly, for comparing cells from three animals, the experimental samples from each genotype are divided in two equal parts. One portion is stained with CTV, and the other one is not. For the antibody staining, the samples are combined.
-
i.
-
a.
-
9.Antibody staining. This series of steps refers to Figure 6.Note: Depending on the cell sorters available, the setup of the dyes used might differ (Lambert, 2019). The protocol provided was developed with the Becton-Dickinson’s FACAria II, equipped with 3 lasers and standard (5-2-2) optical filter configuration (Table 1). Zombie Yellow marks dead cells. All lineage antibodies (anti-CD140, anti-CD31, anti-Ter-119 and anti-CD45) are tagged with biotin and then stained with streptavidin conjugated Brilliant Violet510 (BV510). BV510 spectrum partially overlaps with Zombie Yellow and allows the simultaneous gating of dead and lineage cells, creating a dump channel. The choice of having BV510 and Zombie Yellow to be in the same dump channel is specific to our sorter and filter setup (Table 1) and may not be applicable for other sorters. Alternatively, pairing BV570 with Zombie Yellow or BV510 with Zombie Aqua will have perfect spectra overlaps and allow for the simultaneous gating of dead and lineage cells in the dump channel.Antibodies were selected based on Shehata et al (Shehata et al., 2012) and Pasic et al (Pasic et al., 2011). Epithelial cell adhesion molecule (EpCAM) and integrin alpha 6 (CD49f) antibodies are used to identify mammary epithelial cells. Luminal epithelial cells are strongly positive for EpCAM (high) and moderately positive for CD49f (med), whereas basal epithelial cells are strongly positive for CD49f (high) and only slightly positive for EpCAM (low). The remaining cells, which are predominately fibroblasts, are negative for both markers. The luminal epithelial population can be further fractionated using stem cell antigen-1 (Sca1) and integrin alpha 2 (CD49b), resulting in four populations: Sca1+CD49b-, Sca1-CD49b+ Sca1+CD49b+ and Sca1-CD49b-. The Sca1+CD49b- population primarily consists of estrogen receptor alpha cells, the Sca1-CD49b+ and Sca1+CD49b+populations are luminal progenitors and Sca1-CD49b- are currently undefined.Note: FMO controls are primarily used for antibodies that do not form distinct populations or antibodies that you have not used previously. FMO controls are extremely important when multiplexing to ensure that the spectral overlap artifacts are removed and analysis gates for FACS are set correctly.
-
a.Add 1.5 μg of IgG to each tube.
-
b.Incubate at 21°C–23°C for 10 min.
-
c.Add the biotin anti-CD140, biotin anti-CD31, biotin anti-Ter-119, biotin anti-CD45 to tubes 3, 4, 10–12, 13+16, 14+15.
-
d.Incubate at 21°C–23°C for 20–30 min.
-
e.Add anti-EpCAM-APC to tubes 6, 10–12, 13+16, 14+15.
-
f.Add anti-CD49f-PE/Cy7 to tubes 7, 10–12, 13+16, 14+15.
-
g.Add anti-Sca1-FITC to tubes 8, 11–12, 13+16, 14+15.
-
h.Add anti-CD49b-APC/Cy7 to tubes 9, 10, 12, 13+16, 14+15.
-
i.Add streptavidin-Brilliant Violet 570 to tubes 3, 4, 10–12, 13+16, 14+15.
-
j.Incubate on wet ice for 20–30 min.
-
k.Add 1 mL FACS buffer to each tube and centrifuge at 350 × g for 5 min.
-
l.Carefully remove the supernatant and resuspend in 1 mL of FACS buffer.
-
m.Centrifuge at 350 × g for 5 min.
-
n.Carefully remove supernatant and resuspend in 250–1,000 μL of FACS buffer.Note: Consult with the FACS facility or operator regarding the preferred volumes.
-
o.Transfer to 5ml round bottom polystyrene tubes.
-
p.Store on wet ice in the dark until ready for sorter.
-
a.
-
10.Gating Strategy
-
a.Analyze negative control (tube 1) to set baselines for all channels.
-
b.Analyze compensation controls (tubes 2–9) to select appropriate laser power and outputs for each of the dyes.Note: Fluorescence compensation is performed to correct for spectral overlap when using multiple fluorophores. To correct for overlap single stained samples are required (tubes 2, 3, 5–9). Additionally, a tube containing a mix of stains from tubes 2 and 3 (tube 4) should be added to compensate for the dump channel (see Note at step 9 on antibody choice). The laser output and channel compensation settings are specific to the instrument used so that the gates are correctly established. Be prepared to re-run the samples after the adjustments are made.
-
c.Analyze FMO-Sca-1 (tube 10) and begin gating using the following order.
-
i.Use the Forward SCatter-Area(FSC-A) versus Side SCatter Area (SSC-A) parameters and choose a cell population with uniform SSC-A across the range of FSC-A (p1) (Figure 8A).
-
ii.Use the FSC-Height (FSC-H) versus FSC-A to reject doublets and aggregates (p2) (Figure 8B).
-
iii.Select FSC-A versus ZY+Lineage (appear in same channel; See Note at step 9 on antibody choice) to identify live Lineage- cells that are ZY- (Figure 8C).
-
iv.Select EpCAM versus CD49F to identify the luminal cells, which are EpCAM(high)CD49f(med) (Figure 8D).
-
v.Select Sca-1 versus CD49b with a four way gate to obtain 0% of cells in the upper quadrants (Figure 8E left panel).
-
i.
-
d.Analyze FMO-CD49b (tube 11) and select Sca-1 versus CD49b with a four way gate from step 10c, v to obtain 0% of cells in the right-side quadrants (Figure 8E right panel).
-
e.Analyze tube 12 and keep the gates from the above steps and determine whether quadrants from step 10c, v and step 10d are now populated with cells.Note: Tube 12, which is made up of an equal mixture of all genotypes/conditions being evaluated with all dyes/antibodies establishes the accuracy of the gates.
-
f.Insert CTV gate prior to 4-way gate: select FSC-A versus CTV and plot luminal population from step 10c, iv; choose CTV- and CTV+ populations (Figure 8F).Note: In our analysis we used CTV to subset the luminal cells from the different genotypes (Figures 8F and 8G). However, comparison between genotypes by using CTV gates can be carried out at any point of the FACS analysis. For example, applying the CTV gate to the LiveLin- cells shows the relative proportions of luminal and basal cells between the two genotypes (Figure 9).
-
g.Clone the 4-way gate from step 10d and plot CTV+ population on one of the graphs and CTV- on the other (Figure 8G).
-
h.Switch to the combined tube EXP1 (Figure 6), and select the subpopulation of interest e.g., CTV+Sca+CD49b− and CTV- Sca+CD49b− to initiate sorting.Note: Consult with the FACS facility or operator regarding sorting parameters and available collection set-ups. In the above experiments cells were sorted on FACS Aria II at 30psi using 100 μm nozzle. If possible, maintain the collected cells at 4°C during the sort.
-
i.The cells are collected in 500 μL of collection media.Note: Sheath fluid to media ratio in the collection tubes should be 1:1 to maintain cell viability. Therefore, when the volume in the collection tubes reaches ∼1 mL, but the sort is not complete, replace the collection tube with a new tube containing 500 μL of collection media. After the sort is complete spin down the cells and combine the tubes containing the same populations.
-
j.Repeat steps 10h and 10i with combined tube EXP2 (Figure 6).
-
a.
Figure 7.

Schematic illustrating the experimental set up to perform comparisons between multiple different genotypes/conditions
Table 1.
Experimental set up for FACS
| Laser | Detector | Mirror | Filter | Range (nm) | Fluorophore used |
|---|---|---|---|---|---|
| Blue 488 | B | 655LP | 695/40 | 675-715 | PerCP |
| Blue 488 | A | 735LP | 780/60 | 750-810 | PE-Cy7 |
| Red 633 | B | 660/20 | 650-670 | APC | |
| Red 633 | A | 735LP | 780/60 | 750-810 | APC-Cy7 |
| Violet 405 | B | 450/40 | 430-470 | CTV | |
| Violet 405 | A | 502LP | 530/30 | 515-545 | ZY/BV510 |
Figure 8.

Gating strategy for flow cytometry analysis and sorting of mouse mammary epithelium
(A) Cells were gated for forward (FCS-A) and side (SSC-A) scatter to remove debris and population p1 was selected.
(B and C) (B) P1 from (A) was plotted on FSC-H/A and single cells (p2) were selected and then gated (C) to remove dead cells (Zombie Yellow positive) and Lineage+ (Cd140a+; CD31+; Ter-119+; and CD45+) cells (Live lineage-).
(D) Live cells from (C) were plotted on EpCam and CD49f and the luminal population (EpCamhigh CD49fmedium) was selected.
(E and F) (E) Luminal cells from FMO controls (tube 10: FMO-Sca1; tube 11: FMO-CD49b) were plotted on Sca-1/CD49b to determine the 4-way gates. Gates from (A)–(D) were reapplied to tube 12, and (F) luminal cells from (D) were separated into CTV+ and CTV- populations.
(G) 4-way gates from (E) were reapplied to CTV+ and CTV- cells from (F) to perform 4-way split.
Figure 9.

Comparison of the luminal population from the CTV+ and CTV- populations
Live lineage- was obtained as described in Figure 7 and separated into CTV+ and CTV- populations, which were plotted on EpCam and CD49f.
Expected outcomes
The optimal antibody concentration is based on the dilution that provides the maximum separation between the positive and negative populations. A helpful tool for determination of optimal antibody concentration is staining index (SI), which is defined as the ratio of the separation between the positive and negative population divided by two times the standard deviation of the negative population: SI=(MFIpos-MFIneg)/2SDneg; MFI-mean fluorescence intensity, SD standard deviation. Optimal antibody concentration is the lowest concentration that results in the highest value of SI. Ideally the positive population should be clearly distinguishable from the negative population. However, this result might not always be the case. For example, Figure 8E shows staining of a population that is not very uniform and does not produce clearly separate populations. Instead a shift of the whole population is observed. In this case SI calculation might not be possible. Rather, the expected size of the positive population and its separation from the negative population can be based on published literature and from antibody/dye manufacturer instructions. However, when the antibody is poorly characterized or evaluated on a previously untested cell type, we recommend using a positive control. Gene expression and proteomics data are currently available for many cultured cell lines, which should allow the user to easily identify cells that do or do not express the antigen of interest as positive and negative controls, respectively.
We have found that keeping the antibody concentration and volume constant resulted in similar staining efficiencies regardless of the cell concentration. See Note at step 6c on volume. If antibody depletion is a concern then the optimization protocol can be performed with increasing cell numbers and keeping the concentration and volume constant. Alternatively, the sorting settings, e.g., laser output, sensitivity, can be reapplied to each experiment to ensure consistency of staining.
Epithelial cell isolation from paired glands 2–5 from a single mouse is expected to yield ∼4 × 106 cells. Accounting for cells needed for controls and based on gate percentages from FACS, there will be ∼0.4–0.8 × 106 luminal cells, and of those ∼50% will be Sca1+CD49b−, resulting in ∼2 × 105–4 × 105 cells. In our experience , the users should assume that the worst case recovery rate in FACS will be about 50%–60%, due to limitations in sort precision, sort efficiency, and target frequency. Therefore, the total number of recovered Sca1+CD49b− cells will be 1–2 × 105. This limitation should be especially considered for recovery of less abundant populations, i.e., Sca1−CD49b+ which consist of only ∼10% luminal cells. (Figure 8).
Quantification and statistical analysis
Despite the fact that thousands of cells are analyzed at the same time in a FACS experiment, the statistical comparison between the two conditions must be performed based on biological replicates, i.e., per animal. In a classical FACS protocol, each sample (corresponding to one animal) is analyzed separately. Therefore, to obtain sufficient power to observe statistically significant differences between two animal populations researchers frequently require large number of animals. In our approach, antibody staining and FACS analysis of two samples are performed simultaneously, eliminating artifacts. Our method introduces additional technical replicates which increases quality control and reduces sample variation.
Limitations
The number of animals that can be processed at the same time is ∼6 animals/ person. The number of comparisons is limited by the cell yield because accurate FACS analysis requires 100,000 cells total. For a good statistical analysis you need to exceed 20,000 cells per given gate.
Troubleshooting
Problem 1
Low yield (step 5c, xi).
Potential solution
Incomplete digestion in step 5a can be improved by increasing the digestion time and if not done so already by introducing mixing. See Note at step 5a, v.
Cells clumping can be reduced by removing fibrous material. See Critical at step 5a, v.
Be gentle when pipetting during washing steps, which can lead to dissociation of clumps to single cells. Single cells are not recovered using the provided centrifugation conditions. We advise against changing the centrifugation conditions because increasing the speed, g force, or time will bring down unwanted debris as well as single cells. See Critical at step 5b, vii.
Prolonged exposure to trypsin (>15min) can lead to increased cell death. Pipette the suspension to improve single cell generation.
Problem 2
No signal/low signal (step 10b–j).
Potential solution
Reason 1: Antigens could be removed by excessive exposure to trypsin. Solution: TrypLE could be used as an alternative.
Reason 2: A dim fluorophore used in combination with a low expressing marker. Solution: Identify a brighter fluorophore to use.
Reason 3: Precipitation of antibody could occur with expired antibodies or antibodies not stored according to the manufacturer’s instructions. Solution: Replace old antibodies with new ones.
Reason 4: Antibody/dye could have accidentally been left out or the wrong concentration added. Solution: Confirm dilutions and if correct, repeat the optimization of the antibody/dye using a higher concentration.
Reason 5: The presence of exogenous protein reduces the efficiency of labeling for ZY and CellTrace dyes, which is due to the binding of the dye to free amine groups (Tario et al., 2018). Solution: It is highly recommended that the buffer does not contain proteins when using dyes. However, when protein is necessary to maintain viability, use the minimum necessary, and perform the optimization in the presence of the exogenous protein. Reduced staining is to be expected when proteins (or other sources of free amines) are present in the buffer.
Reason 6: Antigen is not present. Solution: Verify that the expected antigen is expressed on the analyzed cells by RNA-seq, PCR, or mass spectroscopy if this information is available.
Reason 7: Cells are lost during the centrifugation. Solution: An aliquot of the cell suspension could be analyzed after the centrifugation steps.
Reason 8: Poor antibody avidity. Solution: Use a cell line expressing the antigen of interest to validate the efficacy of the antibody or alternatively, try a different clone of the antibody.
Reason 9: Overcorrection for autofluorescence caused by decreasing the laser power and detector sensitivity. Solution: A different fluorophore should be used.
Problem 3
High background (step 10b–j).
Potential solution
Reason 1: Antibody/dye is not sufficiently diluted. Solution: Repeat the optimization with lower antibody/dye concentrations.
Reason 2: Non-specific staining of the primary antibody. Solution: Include an isotype control antibody in the titration process. Make sure that the isotype control matches the immunoglobulin type and heavy chain type of the primary antibody being titrated.
Reason 3: Dead cells can stain non-specifically. Solution: We recommend using viability dye to exclude dead cells (see Antibody and dye optimization).
Reason 4: The blocking could be insufficient. Solution: There are many other blocking reagents available such as different types of Fc-blocks. We recommend following the manufacturer instructions. Blocking procedures are frequently similar, i.e., TruStain FcX (BioLegend) is used at 0.5 μg in 50 μL volume and incubated at 21°C–23°C for 5–10 min prior to antibody staining.
Reason 5: APC-Cy7 and APC-H7 tandems can be metabolized to APC, which changes their spectral properties. Solution: If your cells are very metabolically active the cells should be fixed after staining. ZY and CTV are fixable dyes (Le Roy et al., 2009).
Problem 4
Unexpected FACS profiles (step 10b–j).
Potential solution
Reason 1: Compensation issues. Solution: Consult with a FACS operator or facility; excessive compensation can lead to unexpected FACS profiles; consider changing the fluorophores.
Reason 2: Tandem-dye degradation: tandem-dyes (i.e., APC-Cy7) can be degraded by metabolically active cells. Solution: fix the cells (https://www.biolegend.com/tandem_dyes; Le Roy et al., 2009) if live cells are not required or switch dyes to more stable alternatives.
Reason 3: Incorrect staining buffer. Solution: For dyes, make sure that the staining buffer did not contain protein when using dyes.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deborah Lannigan (deborah.lannigan@vumc.org).
Materials availability
This study did not generate any new reagents.
Data and code availability
The accession number for the RNA sequencing reported in the manuscript using this protocol (Ludwik et al. Cell Reports, 32:10793) is GEO:GSE113323
Acknowledgments
This work was supported by NIH grant DK113423 (D.A.L.) and CA213201 (D.A.L). The authors would like to thank Catherine E. Alford at the FACS core, Nashville VA Medical for outstanding technical assistance. The graphical abstract was created with BioRender.com.
Author contributions
K.A.L. conceived the FACS approach, and Z.M.S. and K.A.L developed the staging and mammary epithelial isolation protocols. K.A.L. and Z.M.S. performed the experiments with assistance from E.B.W. K.A.L. analyzed the FACS data. D.A.L. conceived the project and aided in experimental design and data analysis. K.A.L., Z.M.S., and D.A.L. wrote the manuscript with editorial assistance from E.B.W.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2020.100270.
References
- Byers S.L., Wiles M.V., Dunn S.L., Taft R.A. Mouse estrous cycle identification tool and images. PLoS One. 2012;7:e35538. doi: 10.1371/journal.pone.0035538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cora M.C., Kooistra L., Travlos G. Vaginal cytology of the laboratory rat and mouse: review and criteria for the staging of the estrous cycle using stained vaginal smears. Toxicol. Pathol. 2015;43:776–793. doi: 10.1177/0192623315570339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert T.J. FPbase: a community-editable fluorescent protein database. Nat. Methods. 2019;16:277–278. doi: 10.1038/s41592-019-0352-8. [DOI] [PubMed] [Google Scholar]
- Le Roy C., Varin-Blank N., Ajchenbaum-Cymbalista F., Letestu R. Flow cytometry APC-tandem dyes are degraded through a cell-dependent mechanism. Cytometry A. 2009;75:882–890. doi: 10.1002/cyto.a.20774. [DOI] [PubMed] [Google Scholar]
- Ludwik K.A., Sandusky Z.M., Stauffer K.M., Li Y., Boyd K.L., O'Doherty G.A., Stricker T.P., Lannigan D.A. RSK2 maintains adult estrogen homeostasis by inhibiting ERK1/2-mediated degradation of estrogen receptor alpha. Cell Rep. 2020;32:107931. doi: 10.1016/j.celrep.2020.107931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasic L., Eisinger-Mathason T.S., Velayudhan B.T., Moskaluk C.A., Brenin D.R., Macara I.G., Lannigan D.A. Sustained activation of the HER1-ERK1/2-RSK signaling pathway controls myoepithelial cell fate in human mammary tissue. Genes Dev. 2011;25:1641–1653. doi: 10.1101/gad.2025611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M., Teschendorff A., Sharp G., Novcic N., Russell A., Avril S., Prater M., Eirew P., Caldas C., Watson C.J. Phenotypic and functional characterization of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012;14:R134. doi: 10.1186/bcr3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tario J.D., Jr., Conway A.N., Muirhead K.A., Wallace P.K. Monitoring cell proliferation by dye dilution: considerations for probe selection. Methods Mol. Biol. 2018;1678:249–299. doi: 10.1007/978-1-4939-7346-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing reported in the manuscript using this protocol (Ludwik et al. Cell Reports, 32:10793) is GEO:GSE113323