

Abstract
Acute graft-versus-host disease (aGVHD) remains a major complication of allogeneic hematopoietic cell transplantation (HCT). CD146 and CCR5 are proteins that mark activated T helper 17 (Th17) cells. The Th17 cell phenotype is promoted by the interaction of the receptor ICOS on T cells with ICOS ligand (ICOSL) on dendritic cells (DCs). We performed multiparametric flow cytometry in a cohort of 156 HCT recipients and conducted experiments with aGVHD murine models to understand the role of ICOSL+ DCs. We observed an increased frequency of ICOSL+ plasmacytoid DCs, correlating with CD146+CCR5+ T cell frequencies, in the 64 HCT recipients with gastrointestinal aGVHD. In murine models, donor bone marrow cells from ICOSL-deficient mice compared to those from wild-type mice reduced aGVHD-related mortality. Reduced aGVHD resulted from lower intestinal infiltration of pDCs and pathogenic Th17 cells. We transplanted activated human ICOSL+ pDCs along with human peripheral blood mono-nuclear cells into immunocompromised mice and observed infiltration of intestinal CD146+CCR5+ T cells. We found that prophylactic administration of a dual human ICOS/CD28 antagonist (ALPN-101) prevented aGVHD in this model better than did the clinically approved belatacept (CTLA-4-Fc), which binds CD80 (B7–1) and CD86 (B7–2) and interferes with the CD28 T cell costimulatory pathway. When started at onset of aGVHD signs, ALPN-101 treatment alleviated symptoms of ongoing aGVHD and improved survival while preserving antitumoral cytotoxicity. Our data identified ICOSL+-pDCs as an aGVHD biomarker and suggest that coinhibition of the ICOSL/ICOS and B7/CD28 axes with one biologic drug may represent a therapeutic opportunity to prevent or treat aGVHD.
INTRODUCTION
Allogeneic hematopoietic cell transplantation (HCT) remains the only curative therapy for many patients with hematologic malignancies and bone marrow (BM) failure states. However, wider application of HCT is limited by the morbidity associated with the procedure, particularly the acute graft-versus-host disease (aGVHD) that affects as many as 50% of HCT recipients (1). Gastrointestinal GVHD (GI-GVHD) is the most fatal type of aGVHD (2). The development of plasma aGVHD biomarkers has increased interest in finding additional biomarkers that can provide meaningful information early in the course of HCT and that can be drug-targeted (3–5). In contrast, few aGVHD cellular markers have been identified and validated in large cohorts. However, such cellular markers are likely present on immune cell subsets and may represent suitable therapeutic targets. Using state-of-the-art quantitative proteomics followed by flow cytometry, we identified and validated, in a cohort of 254 patients, a CD4+CD146+CCR5+ T cell population that is increased in the blood of GI-GVHD patients compared to controls without GVHD or to patients with GVHD of other target organs (6). This T cell population produced interleukin-17 (IL-17) and interferon-γ (IFNγ) and was activated by the receptor inducible costimulator (ICOS), a critical costimulatory molecule for the development of often pathogenic T helper 17 (Th17) cells (6). This T cell population was also found in patients with chronic GVHD, and the cells displayed similar Th1- and Th17-like characteristics (7).
The CD28/cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) pathway is the prototypic cosignaling pathway in T cells, with CTLA-4 coinhibition acting to counterbalance CD28 costimulation as they bind the same receptors (CD80 and CD86). A recombinant fusion protein, abatacept (CTLA-4-Fc), is an approved drug that comprises the extracellular domain of human CTLA-4 fused with a fragment of the Fc portion of human immunoglobulin G1 (IgG1) and mediates immunosuppressive effects by binding CD80 and CD86 and blocking their interactions with CD28/CTLA-4. Abatacept is approved for the treatment of rheumatoid arthritis, whereas a second-generation CTLA-4-Fc (belatacept) is approved for prophylaxis of organ rejection in renal transplant recipients (8). However, while most human T cells express CD28 constitutively, it is known that various factors like age, inflammation, viral infection, or transplant can drive the generation of CD28null memory T cells that retain effector function and are resistant to belatacept treatment; these T cells may use other costimulatory pathways (9).
Here, we reasoned that ICOS ligand (ICOSL) present on dendritic cells (DCs) that prime T cell responses may provide the key stimulus for generating pathogenic IL-17–producing, CD4+CD146+CCR5+ T cells. Using a subset of the previously described cohort of patients with GVHD, we determined that ICOSL was present on plasmacytoid DCs (pDCs) in human peripheral blood and the frequency of these cells was increased in patients with GI-GVHD. Because this marker was both correlated to GI-GVHD and appeared implicated in its pathophysiology, ICOSL could not only serve as a biomarker to guide clinical trials, but the ICOSL/ICOS pathway could potentially be therapeutically targeted. Therefore, we explored the importance of modulating the ICOSL-ICOS pathway in aGVHD by evaluating ICOSL-knockout (KO) mice and investigated the impact of treatment with a dual CD28 and ICOS antagonist in humanized mouse models of aGVHD.
RESULTS
ICOSL+ pDC frequencies correlate with CD4+CD146+CCR5+ T cells and survival in patients with GI-GVHD
ICOS binds its ligand, ICOSL, which is expressed on DCs that prime naïve T cells to initiate immune responses (10). This prompted us to examine whether ICOSL+ DCs contributed to the activation of CD4+CD146+CCR5+ T cells in GI-GVHD; we examined DCs from a subset of 156 HCT patients from the larger cohort of 234 HCT patients previously published (6). We analyzed ICOSL on the two circulating DC subsets that can be identified in human peripheral blood: Lineage (Lin) −HLA-DR+CD11c+ conventional DCs (cDCs) and Lin−HLA-DR+CD123+ pDCs (fig. S1). Peripheral blood mono-nuclear cells (PBMCs) were collected weekly during the first 4 weeks and then monthly after allogenic HCT. PBMCs were also collected at the development of key clinical events (symptoms of GVHD, skin rash, or non-GVHD enteritis) (see table S1 for median day) and at matched median time points after HCT for patients without aGVHD (median day 29 after HCT). All patients received pharmacologic GVHD prophylaxis with a least two agents, including a calcineurin inhibitor (tacrolimus in >90% of the cases). Patient characteristics and GVHD prophylaxis are summarized in table S1.
We found a higher frequency of ICOSL+ pDCs in patients with GI-GVHD (24.1%) compared to patients without GVHD (4.6%) or non-GVHD enteritis (5.7%) or with skin GVHD (5.6%) (P < 0.0001 for GI-GVHD compared to the rest of patients with allogenic HCT) (Fig. 1, A and B). There were no differences in the frequencies of total DCs or cDCs populations in these samples from the patients (fig. S2). Moreover, the frequency of ICOSL+ cDCs was not different between groups (fig. S3). Because lower GI-GVHD is more prevalent and severe than upper GI-GVHD (2), we also compared ICOSL+ pDCs in patients with upper and lower GI-GVHD. We found no difference in ICOSL+ pDC frequencies between patients with upper and lower GI-GVHD (fig. S4).
Fig. 1. ICOSL+ pDCs in patients after allogeneic HCT.
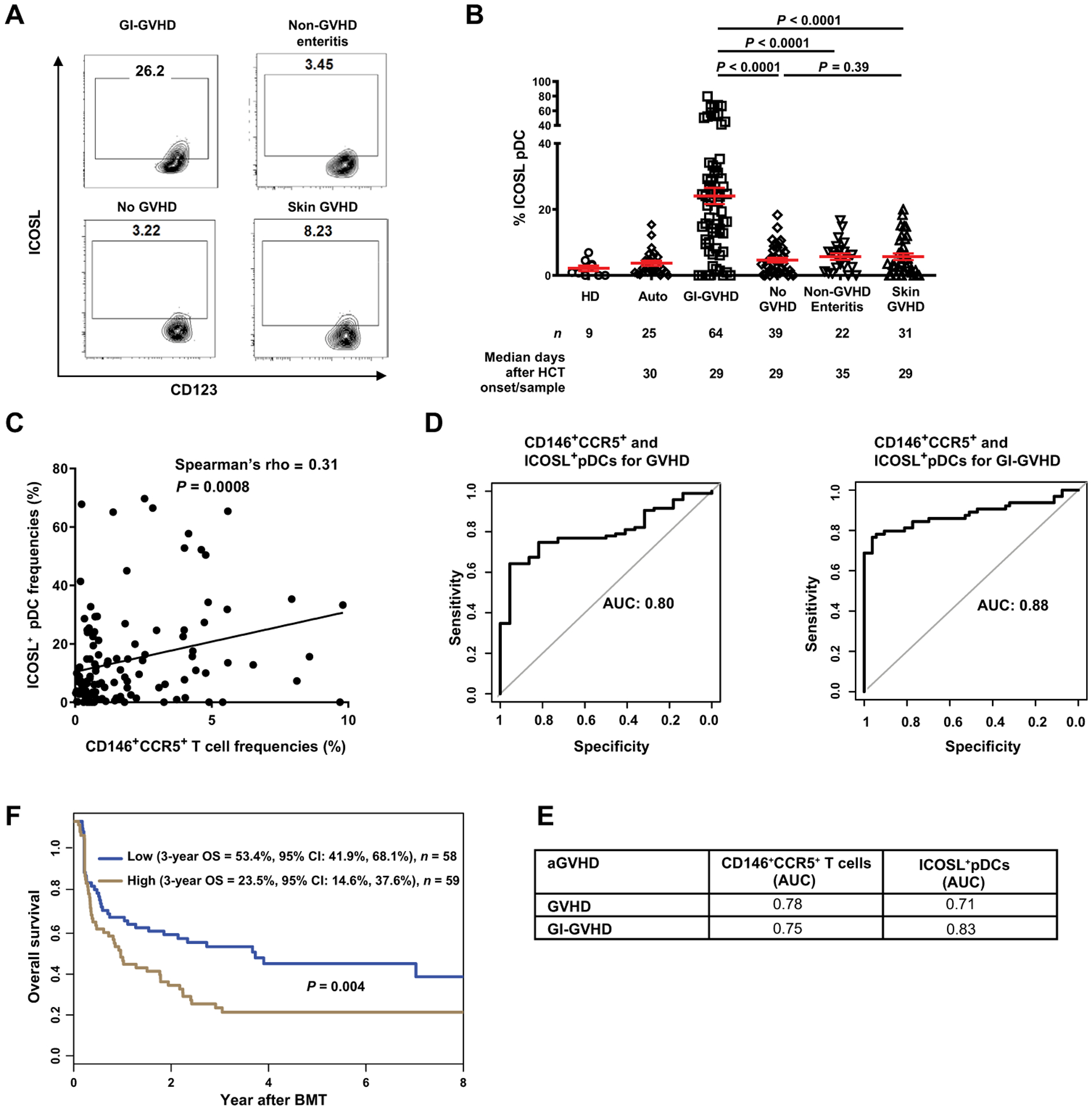
(A) Representative plots showing percentage of cells positive for ICOSL and CD123 in samples from patients without GVHD or with GI-GVHD, non-GVHD enteritis, or skin GVHD. (B) ICOSL+CD123+CD11c−HLA-DR+Lin− pDC frequencies in healthy donors (HDs) and in autologous transplant (Auto) or allogeneic patients. Number of patients (n) and median days after HCT onset of signs/sample collection are shown below the graphs. The data are shown as mean ± SEM, unpaired t test. (C) The correlation between ICOSL+CD123+CD11c−HLA-DR+Lin− pDCs and CD146+CCR5+CD4+ T cells frequencies in patients with GVHD (n = 95) and non-GVHD enteritis (n = 22) (total n = 117) using Spearman’s correlation. (D) Receiver operating characteristic curves of ICOSL+ pDCs and CD146+CCR5+ T cells in GVHD and GI-GVHD. (E) AUCs calculated from the curves in fig. S5 (B to E). (F) Three-year overall survival (OS) in allogeneic HCT patients with symptoms [GI-GVHD (n = 64), non-GVHD enteritis (n = 22), and skin GVHD (n = 31)] divided by low and high ICOSL+ pDC frequencies. High-risk group is shown in brown (ICOSL+ pDC frequency ≥ 8.23%, n = 59); the low-risk group is shown in blue (n = 58). Statistical significance was calculated for the overall curve by log-rank test. CI, confidence interval.
In a diagnostic test, the use of several biomarkers simultaneously may increase predictability, diagnostic performance, or specificity. Thus, we analyzed the correlation between CD4+CD146+CCR5+ T cells and ICOSL+ pDC frequencies collected at the same time points from all patients in this cohort and analyzed on the same day. We found a positive correlation with all cells in patients with clinical signs (Fig. 1C), as well as with cells from patients with GI-GVHD only (fig.S5A). To assess the sensitivity and specificity of using either CD146+CCR5+ T cells and ICOSL+ pDC frequencies or the combination as a predictor of GVHD, we determined the receiving operating characteristic (ROC) area under the curve (AUC) for each. We found that combining CD146+CCR5+ T cells and ICOSL+ pDC frequencies in patients with GVHD or GI-GVHD (Fig. 1D) resulted in higher AUC values, indicating better predictive ability, compared to those with ICOSL+ pDCs or CD146+CCR5+ T cells alone in GVHD and GI-GVHD (Fig. 1, D and E, and fig. S5, B to E).
We analyzed the impact of ICOSL+ pDC frequency on the overall survival in all patients with symptoms (GI-GVHD, non-GVHD enteritis, and skin GVHD). We used the median ICOSL+ pDC frequency in these patients (8.23%) as a threshold to assess the risk of mortality from the time of HCT to 8 years after HCT (Fig. 1G). Patients with a high frequency of ICOSL+ pDCs had a significantly lower 3-year overall survival compared with those who had a low frequency of these cells (P = 0.004). Causes of mortality in patients with GVHD symptoms are provided in table S2. Among the patients with GI-GVHD, there was no significant difference in overall survival when divided into groups according to ICOSL+ pDC frequencies (fig S5F, P = 0.87). This is consistent with our finding of high ICOSL+ pDC frequencies in patients with GI-GVHD (Fig. 1B) and the usual low survival rates associated with GI-GVHD (2). These findings suggested that ICOSL+ pDC frequencies could be used as a potential additional biomarker with CD146+CCR5+ T cells in GI-GVHD. Furthermore, the results are consistent with the hypothesis that ICOSL+ pDCs represent the inducers of the Th17-type CD4+CD146+CCR5+ T cell population in patients with GI-GVHD.
ICOSL−/− donor BM prevents aGVHD and reduces the frequency of intestinal pDCs and pathogenic Th17 cells in two experimental HCT models
The growth factor Feline McDonough Sarcoma (fms)-like tyrosine kinase 3 ligand (hFLT3L for human and mFLT3L for mouse) is necessary for the development and differentiation of DCs, and the transcription factor, signal transducer and activator of transcription 3 (STAT3), is required for the development of FLT3L-dependent common DC precursors and their progeny in mice (11, 12). In mice, pDCs are defined as CD11b−CD11c+B220+CD103+, whereas cDCs are CD11b+CD11c+B220−CD103+. Conditional KO of STAT3 in CD11c+ cells has no impact on the generation of cDCs but decreases pDC generation (13). The role of pDCs in aGVHD is controversial, because these have been reported to have either tolerogenic or disease-promoting properties, depending on the murine model (14–16). Thus, additional studies of their function in aGVHD are necessary before a therapeutic approach based on this mechanism can be contemplated. On the basis of the data from our patient cohort, we hypothesized that absence of ICOSL signaling in donor BM-derived DCs would protect against aGVHD by impairing the activation of pDCs dependent on FLT3L, STAT3, or both pathways.
We used a major experimental mismatch HCT model in which BALB/c, H-2d mice were the recipients of T cells and BM cells from C57BL/6, H-2b mice (C57BL/6, H-2b ➔ BALB/c, H-2d). Although all recipient mice received T cells from wild-type mice, the BM was from wild-type, ICOSL−/−, or STAT3 conditionally deficient (CD11cCreSTAT3fl/fl, hereafter referred to as STAT3−/−) mice. We found that recipients of ICOSL-deficient BM had lower GVHD severity scores and longer survival compared to recipients of wild-type BM or STAT3−/− BM (Fig. 2, A and B). Previously, FLT3L treatment of murine donors did not affect GVHD, whereas FLT3L treatment of recipients after HCT accelerated GVHD lethality (17). Therefore, we assessed FLT3L concentrations in the serum of the recipient mice. We found a significant decrease of FLT3L concentration in serum collected at day 3 after HCT from ICOSL−/− BM recipients compared to mice that received wild-type BM in this model (P = 0.01, Fig. 2C). To confirm that these results were not specific to these mouse strains, we used a haploidentical (C57BL/6, H-2b ➔ B6D2F1, H-2bxd) experimental model and observed decreased serum FLT3L concentrations at days 7, 14, and 21 after HCT (Fig. 2D).
Fig. 2. aGVHD severity and mortality in ICOSL−/− BM and ICOSL+ pDCs recipients in both a major mismatch murine model and a human PBMC-NSG GVHD model.
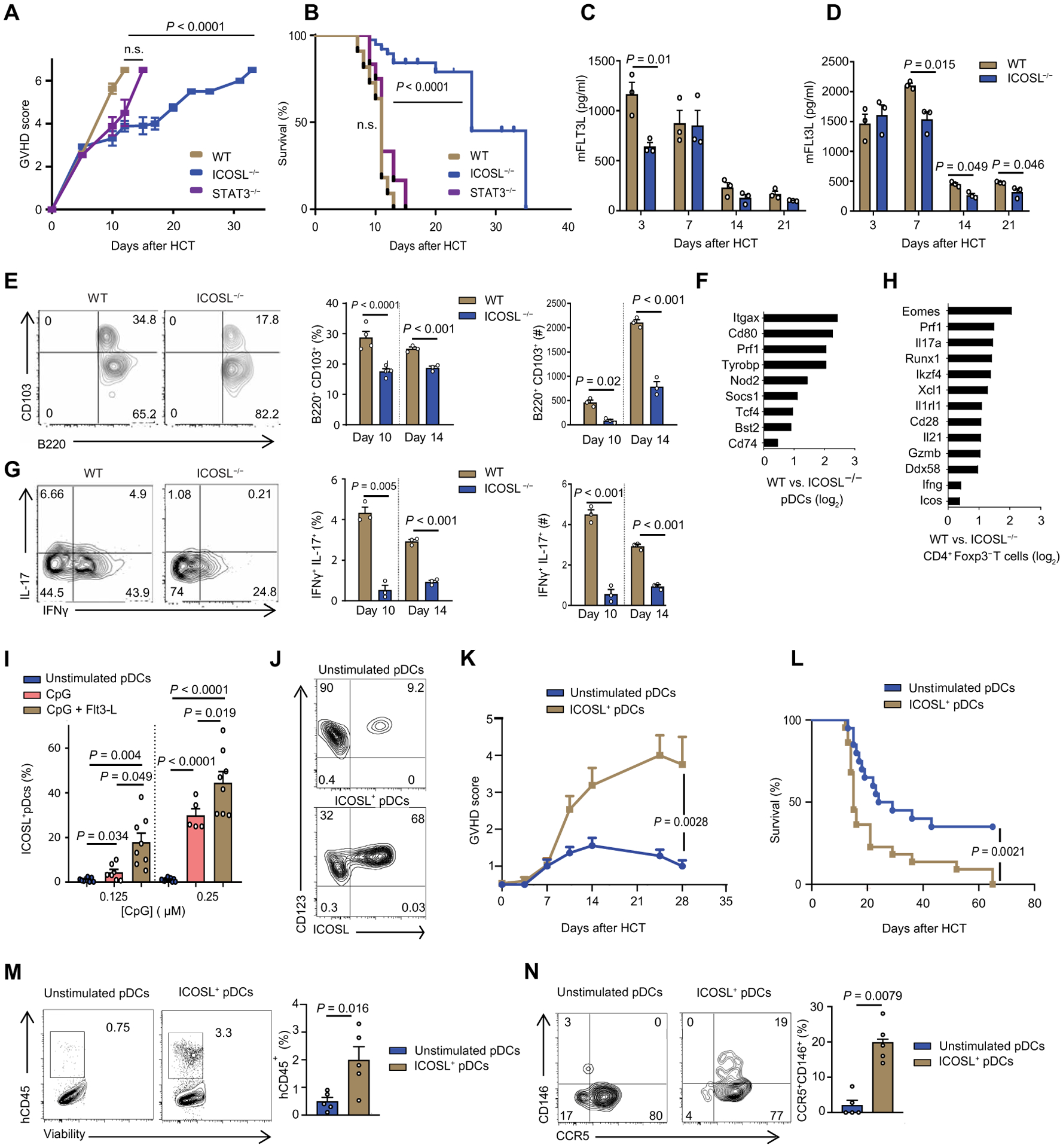
(A and B) Clinical GVHD score (A) and survival curve (B): B6 wild-type (WT) (n = 15), B6 ICOSL−/− (n = 15), or B6 CD11cCreSTAT3fl/fl (n = 15). BALB/c mice were irradiated with 900 cGy and then injected intravenously with 1 × 106 of WT B6 T cells and 5 × 106 of BM from WT, ICOSL−/−, or CD11cCreSTAT3fl/fl B6 mice for allogeneic transplant. n.s., not significant. (C) Kinetics of serum concentrations of murine FLT3L in BALB/c recipient mice at the indicated days after allogeneic HCT WT or ICOSL−/− BM into BALB/c (n = 3 each group). (D) Kinetics of serum concentrations of murine FLT3L in the haploidentical model (WT or ICOSL−/− B6 ➔ BALB/c) at the indicated days after allogeneic HCT (n = 3 each group). (E) CD11b−CD11c+CD103+B220+ pDCs from the gut of recipient mice injected with WT or ICOSL−/− BM analyzed at days 10 and 14 after HCT (n = 3). Representative flow cytometry and percent positive and absolute number of cells are shown. (F) Transcriptome analysis comparing sorted intestinal pDCs from recipient mice injected with WT or ICOSL−/− BM at day 10 after HCT. Data are shown as ratio of fold change between sorted WT and ICOSL−/− pDCs log2-transformed. (G) Representative plots of Th17 positive for IL-17 or IFNγ or both in the gut of mice that received WT or ICOSL−/− BM, analyzed at day 10 after HCT (n = 3). Representative flow cytometry and percent double positive and absolute number of cells are shown. (H) Transcriptome analysis comparing sorted intestinal CD4+ Foxp3− T cells from WT and ICOSL−/− BM recipient B6 mice at day 10 after HCT. Data are shown as ratio of fold change between sorted WT and ICOSL−/− CD4+ T cells log2-transformed. (I) Percentage of human pDCs positive for ICOSL after stimulation with 0.125 and 0.25 μM CpG or with Flt3-L (200 ng/ml) overnight. pDCs were enriched from human PBMCs. (J) Representative flow cytometry for the experiment in (I), showing percentage of human pDCs positive for ICOSL. Cells were either unstimulated or exposed to both CpG (0.25 μM) and Ftl3-L (200 ng/ml) overnight. (K to N) GVHD score (K), survival (L), intestinal hCD45 infiltration (M), and intestinal CD146+CCR5+ T cells (N) in NSG mice receiving 50,000 of stimulated or unstimulated pDCs with 5 × 106 PMBCs depleted of pDCs (n = 10). In (A), (C) to (E), (G), (I), (K), (M), and (N), data are shown as mean ± SEM. In (C) to (E), (G), and (K) to (N), statistical significance was determined by unpaired t test; in (A) and (I), by ANOVA with Bonferroni’s correction; in (B) and (L), by Mantel-Cox log-rank test.
We also examined the intestines of the transplanted mice at days 10 and 14 after HCT for infiltration of pDCs and compared the ICOSL−/− BM recipients to the recipients of wild-type BM. Recipients of ICOSL−/− BM showed a decrease in infiltration of intestinal pDCs compared to the wild-type BM recipients at both time points (Fig. 2E). The frequencies and absolute numbers of splenic pDCs were also significantly decreased in recipients of ICOSL−/− BM versus wild-type BM recipients (P < 0.001, fig. S6). We performed transcriptome analysis of sorted intestinal pDCs (purity, >95%) from recipients of either wild-type or ICOSL−/− BM at day 14 after HCT. Compared to the intestinal pDCs from recipients of ICOSL−/− BM, intestinal pDCs from recipients of wild-type BM had greater expression of genes encoding key molecules essential for pDCs development (such as Itgax, Socs1, Tcf4, and Bst2) and for costimulation (such as CD80 and CD74) and of molecules essential for pDCs function (such as Tyrobp and Nod2) (Fig. 2F and data file S1).
In parallel, we examined infiltrating intestinal T cells at days 10 and 14 after HCT. In contrast to human T cells, CD146 is not present on murine T cells (6); therefore, we detected Th17 cells using IL-17 and IFNγ expression by infiltrating T cells in the intestine, or quantified RORγt+CXCR3+ Th17 cells in the spleen. We found lower frequencies of infiltrating intestinal T cells coexpressing IL-17 and IFNγ cells in mice receiving ICOSL−/− BM compared to the recipients of wild-type BM (Fig. 2G). The frequencies of splenic RORγt+CXCR3+ Th17 cells were also decreased in recipients of ICOSL−/− BM versus wild-type BM recipients, and absolute counts of this population followed the same trend (fig. S7). Transcriptome analysis of sorted CD4+ Foxp3− effector T cells (Teffs) (purity, >95%) from the recipients of ICOSL−/− or wild-type BM at day 14 after HCT showed decreases in the expression of T cell activation markers such as Prf1, Eomes, Il17a, CD28, Gzmb, Ifng, and Icos (Fig. 2H and data file S2). We performed similar analyses in the haploidentical experimental model and on days 10 and 14 observed similar decreases in frequencies of intestinal pDCs (fig. S8) and in IFNγ+IL-17+ T cells (fig. S9) from recipients of ICOSL−/− BM compared to recipients of wild-type BM.
To investigate the mechanistic role of human ICOSL+ pDCs in the development of intestinal CD146+CCR5+ T cells, we used a human-to-mouse aGVHD model in which human PBMCs (huPBMCs) are injected into immune-deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (huPBMCs ➔ NSG) (18). Although the use of humanized models to evaluate aGVHD is controversial, they are currently the best small animal tool to evaluate human therapeutics, and some studies have suggested that human T cells are xenoreactive with mouse major histocompatibility complex (MHC) antigens on host cells (19, 20). Either 0.125 or 0.25 μM cytosine-phosphate-guanosine (CpG) increased the percentage of pDCs positive for ICOSL on pDCs, and the addition of FLT3L (200 ng/ml) further increased the proportion of ICOSL+ cells (Fig. 2I). Thus, we used CpG (0.25 μM) and FLT3L (200 ng/ml) for the subsequent experiments. The enriched human pDCs [stimulated or not with CpG (0.25 μM) and FLT3L (200 ng/ml)] were analyzed for the percent ICOSL+ (Fig. 2J). After verification of the ICOSL+ frequency, the stimulated or unstimulated pDCs were mixed with pDC-depleted PMBCs from the same donor and transplanted into NSG mice (fig. S10, A to C). More severe GVHD and lower survival was observed in mice receiving the ICOSL+ pDCs compared to the mice receiving unstimulated pDCs (Fig. 2, K and L). Frequencies of human hematopoietic cells (CD45+), human Th17 cells (CD146+CCR5+), and human pDCs (Lin−HLA-DR+CD123+CD11c−) were increased in the spleen of NSG mice receiving ICOSL+ pDCs compared to those in mice receiving unstimulated pDCs (fig. S11). Furthermore, infiltration of human CD45+ cells (Fig. 2M) and human Th17 cells (Fig. 2N) into the intestines was increased in recipients of the stimulated ICOSL+ pDCs compared to the recipients of unstimulated pDCs.
ALPN-101, a dual human ICOS/CD28 inhibitor, potently suppresses T cell responses
With data supporting a role of the ICOSL/ICOS pathway in aGVHD, we evaluated the translational potential of interfering with this pathway in humanized mouse models of aGVHD using the dual CD28 and ICOS antagonist ALPN-101, which was generated using the variant immunoglobulin domain (vIgD) platform (Fig. 3A and fig. S12) (21). The therapeutic candidate ALPN-101 is a dimer of the platform-matured ICOSL vIgD domain fused to a modified IgG1 that does not bind FcγR (i.e., CD16a, CD32, or CD64) or complement component 1q (C1q) but does bind FcRn. Flow cytometry–based characterization of ALPN-101 confirmed its high-affinity binding to ICOS, CD28, and CTLA-4 (which is highly homologous to CD28), with similar half-maximal effective concentration (EC50) values of ~650 pM for all three targets (fig. S13A). ALPN-101 also bound mouse ICOS (EC50 of 1153 pM), CD28 (EC50 of 1428 pM), and CTLA-4 (EC50 of 1112 pM) (data not shown), nearly twice those calculated for the human receptors. Biolayer interferometry (BLI) measurements of the affinity of ALPN-101 for soluble versions of each of its targets yielded average KD values of 306 pM for ICOS, 1257 pM for CD28, and 471 pM for CTLA-4 (table S3). The discrepancy between the flow-based and BLI affinity measurements (EC50s) may reflect both the different forms of the targets (soluble versus membrane bound) or that CD28 forms a dimer in vivo but the recombinant protein used in the BLI assessments was a monomer. Although ALPN-101 can bind to CTLA-4, the binding to CD28 is functionally dominant, as indicated by its immunosuppressive activity.
Fig. 3. ALPN-101 suppresses activated T cell expansion in the human PBMC-NSG GVHD model.
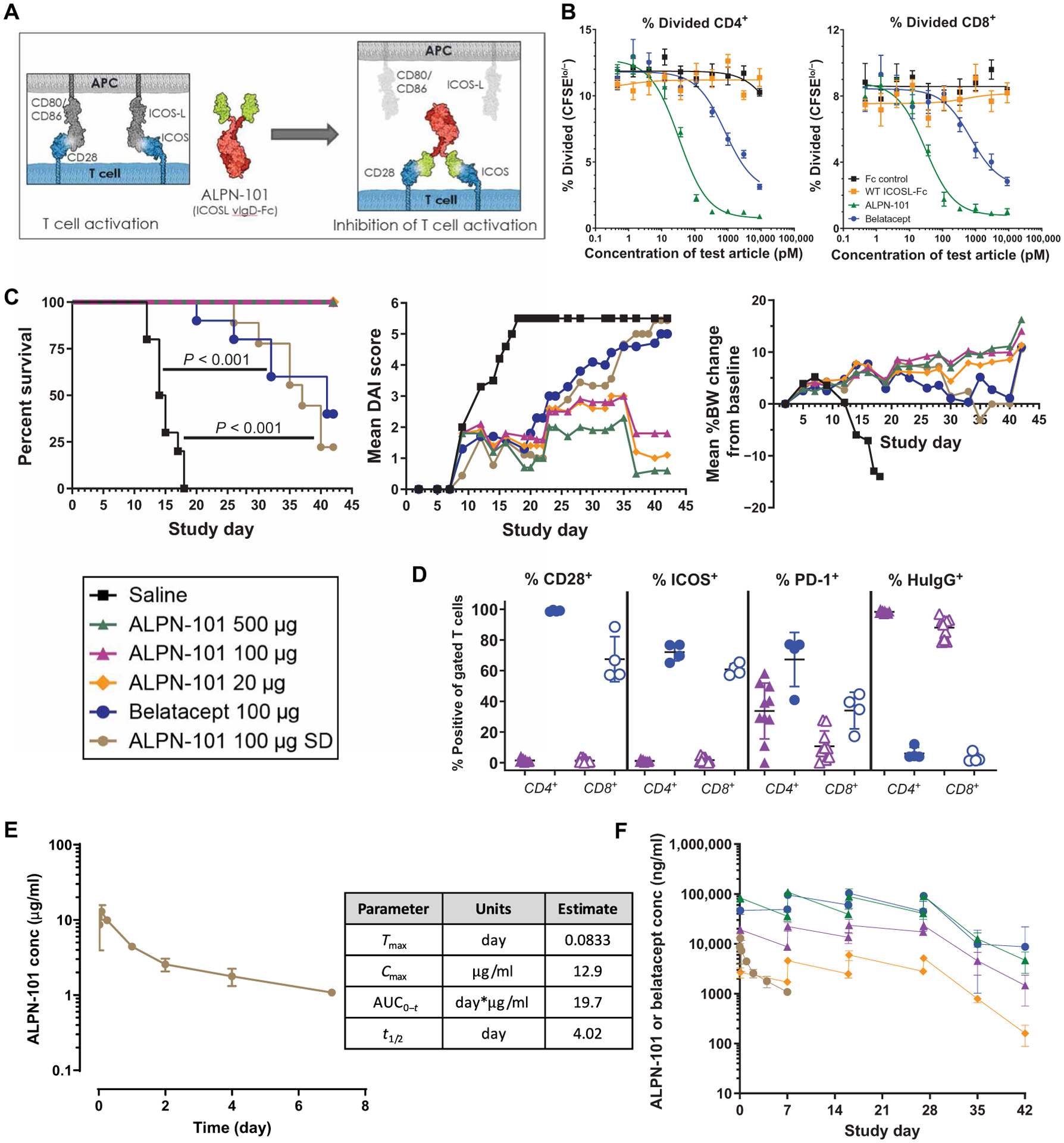
(A) Diagram of the structure of ALPN-101 and its mechanism of action. ALPN-101 was generated on the vIgD yeast display platform (21) and comprises two ICOSL “vIgD” domains (green) fused to a dimeric Fc tail (red) engineered to lack appreciable FcγR (CD16a, CD32, or CD64) or complement (C1q) binding while retaining FcRn binding. (B) Proliferation of CD4+ and CD8+ T cells was determined by quantifying the percentage of carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled cells remaining over time. As cells divide, CFSE signal decreases, and the percent CFSElo/− cells is used to assess the fraction of divided cells. Data are representative of at least six experiments with different donor pairs. (C) Survival, disease activity index (DAI) scores, and body weight (BW) of NSG mice x-ray irradiated (100 cGy) and administered 10 mg of human γ globulin subcutaneously on day −1 and then transplanted intravenously with 10 × 106 human PBMCs on day 0 and treated intraperitoneally with saline 3× per week (TIW) for 4 weeks (TIW × 4; days 0 to 28); 20, 100, or 500 μg of ALPN-101 TIW × 4; 100 μg of belatacept TIW × 4; or 100 μg of ALPN-101 single dose (SD) on day 0. On day 42, human CD45+ cells in blood were characterized by flow cytometry. For DAI analysis, the last observations or scores were carried forward for mice that were euthanized before the end of the study. For BW analysis, mice were weighed daily. See table S5 for statistical differences among groups. (D) Analysis of terminal blood collected from euthanized mice by flow cytometry for the 100 μg TIW × 4 ALPN-101 and belatacept treatment groups from the study described in (C). The % CD28+, ICOS+, PD-1+, or anti-human IgG-binding cells of gated CD4+ (filled) and CD8+ (open) from mice administered ALPN-101 (purple) or belatacept (blue). ALPN-101 blocks detection of CD28 and ICOS, and the anti-human IgG reagent binds to the Fc of cell-bound ALPN-101. Very few myeloid or B cells remained, so binding of belatacept to CD80/CD86 on APC was not detected. (E) Concentration (conc) of ALPN-101 in the serum of mice receiving a single dose of 100 μg of ALPN-101 in the GVHD model described in (C). Serum samples from three mice per group per time point after a single intraperitoneal injection of ALPN-101 were evaluated for drug concentrations by plate-based ELISA. (F) Concentrations of ALPN-101 and belatacept in the serum of mice receiving the indicated repeated doses (RD, TIW × 4) of the test articles in the GVHD model described in (C). Concentrations of ALPN-101 or belatacept were measured by ELISA in serum samples collected 2 hours post-dose on D0 (first dose), pre-dose, and 2 hours post-dose on D7, D16, and D27 (4th, 8th, and 12th doses) and D35 and D42 (8 and 15 days after the last dose). In (B), data are shown as mean ± SEM. In (C) (left), statistical significance was determined by Mantel-Cox log-rank test; in (C) (middle), by two-way repeated measures ANOVA for treatment effect; in (C) (right), by one-way ANOVA with Bonferroni correction. In (D), statistical significance was determined by unpaired t test.
We demonstrated the ability of ALPN-101 to inhibit both the ICOS and CD28 pathways using a human Jurkat T cell line with endogenous CD28 and expressing an IL-2-luciferase reporter gene (stimulated by CD28 signaling), and a transfected chimeric ICOS/CD28 molecule (the ICOS extracellular domain fused to the intracellular tail of CD28) (fig. S13B). ALPN-101 inhibited ICOS-mediated signaling at least as well as the positive control anti-ICOSL monoclonal antibody (mAb) and inhibited CD28-mediated costimulation more potently than belatacept and abatacept, two drugs targeting the CD80/CD86-CD28 pathway that are currently used clinically (fig. S13C). To evaluate the effects of ALPN-101 on the proliferation of primary CD4+ and CD8+ Teffs, a human mixed lymphocyte reaction (MLR) was performed in the presence of ALPN-101, belatacept, wild-type ICOSL-Fc, or Fc control. ALPN-101 decreased the percentage of proliferating CD4+ and CD8+ Teffs (Fig. 3B) and inhibited the production of Teff-derived cytokines—IFNγ, IL-2, IL-17A, IL-13, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor–α (TNFα)—in a dose-dependent manner (fig. S13D). These data confirmed that ALPN-101 is a potent dual human CD28 and ICOS inhibitor and showed that ALPN-101 is a more potent inhibitor of the CD28 pathway in vitro than belatacept or abatacept.
Dual ICOS/CD28 inhibition with ALPN-101 prevents xenogeneic aGVHD in models with immunodeficient mice transplanted with human PBMCs
ALPN-101 was evaluated in vivo in the huPBMCs ➔ NSG model. NSG mice were irradiated and transplanted with human PBMCs and then treated with a single dose of 100 μg of ALPN-101 at day 0; repeat doses of 20, 100, or 500 μg of ALPN-101; or repeat doses of 100 μg of belatacept (three times per week, for 4 weeks, 12 doses) (table S4). Mice were monitored for clinical signs of GVHD for 42 days and T cell counts were determined for surviving mice on day 42 (table S5A). We found that 12 injections of ALPN-101 at any of the three dose levels tested prevented the development of xenogeneic aGVHD compared to the saline-treated recipient mice (100% survival versus 0%, P < 0.001). Single-dose (100 μg) administration of ALPN-101 resulted in similar protection from xenogeneic aGVHD as repeat dosing of 100 μg of belatacept, with 25% of mice that received the single dose of ALPN-101 surviving compared to 40% of those receiving belatacept (Fig. 3C, left). The survival of mice in the groups for single-dose ALPN-101 or repeated doses of belatacept was both significantly improved compared to that of the group receiving saline (P < 0.001, table S5B). Consistent with the observed survival advantage, repeat dosing with ALPN-101 at all dose levels significantly reduced the disease activity index (DAI) score compared to animals treated with saline (P < 0.0001) or belatacept (P = 0.002 for 500-μg group; P = 0.011 for the 20-μg group) (Fig. 3C, middle, and table S5C). Compared to saline treatment, repeated-dose treatment with belatacept or single-dose treatment with ALPN-101 treatment significantly reduced DAI scores (P = 0.0086 for belatacept and P < 0.0001 for single-dose ALPN-101), although these treatments were not significantly different from each other (P = 0.3625) (Fig. 3C, middle, and table S5C). Also consistent with better overall health, the body weight (BW) of the mice receiving any of the repeated dose treatments with ALPN-101 increased over the course of the experiment, whereas the mice receiving repeated doses of belatacept or a single-dose treatment with ALPN-101 maintained their BW (Fig. 3C, right, and table S5D).
We performed flow cytometric analysis of blood isolated from all surviving mice 2 weeks after the last treatment with repeat doses of 100 μg of ALPN-101 or belatacept (day 42), and enumerated and characterized the remaining human T cells (table S5A). The cells were labeled with antibodies recognizing CD28, ICOS, or PD-1, or with a reagent that recognizes human IgG to detect the Fc tail of ALPN-101 bound to the T cells, to estimate target saturation. ALPN-101 blocks the binding of the flow antibodies to CD28 and ICOS, so the absence of signal for these two targets independently confirms the presence of ALPN-101 bound to the T cells (Fig. 3D). In mice treated with belatacept, CD28+ and ICOS+ cells were detectable, but human Ig staining was not present on the T cells, since belatacept binds to CD80 and CD86 on antigen-presenting cells (APCs) (Fig. 3D). The fraction of surviving human T cells positive for ICOS (~60 and 70% in the CD8+ and CD4+ T cells, respectively) was high in the mice treated with belatacept, reflecting their high state of activation (Fig. 3D). In addition to persistently blocking CD28 and ICOS, ALPN-101 reduced the fraction of PD-1+ activated CD4+ and CD8+ T cells significantly more so than did belatacept (P = 0.0085 for CD4+ and P = 0.0028 for CD8+, by unpaired parametric t test) (Fig. 3D).
Quantification of inflammatory cytokines released by the transplanted human cells in serum collected at termination (as the mice succumbed to disease or on day 42, the last day of the study) showed that ALPN-101 at all repeated doses reduced the concentration of serum IFNγ and TNFα and other key cytokines more effectively than repeat dose belatacept or saline, whereas a single dose of ALPN-101 had no protective effect (fig. S14 and table S6). Serum concentrations of ALPN-101 were measured for the first week after a single intraperitoneal dose of 100 μg of ALPN-101 on day 0 in the aGVHD model. Tmax was observed at 2 hours after the injection, and the serum half-life was ~4 days (Fig. 3E). The extent of target saturation noted in the repeated dose groups suggested that ALPN-101 exerts its pharmacodynamic effects longer than is reflected by its concentration in circulation (Fig. 3D). After repeat administration, ALPN-101 exposure increased in a dose-dependent fashion, although mean concentrations of belatacept after a 100-μg dose were higher than those after a 100-μg ALPN-101 dose at all evaluated time points (Fig. 3F).
In a second study, we specifically tracked the increase in ICOS+ T cells over time in the huPBMCs ➔ NSG model and compared the efficacy of single versus repeated dosing with 100 μg of ALPN-101 or belatacept (Fig. 4, A to C, and table S4). As we observed in the first study (Fig. 3, C to F), repeated dosing with ALPN-101 provided strong protection against xenogeneic aGVHD and a single dose of ALPN-101 protected against disease as effectively as repeated doses of belatacept (Fig. 4A and table S7). However, a single dose of belatacept conferred essentially no protection, yielding survival rates comparable to those in the saline control group (table S7). We conducted flow cytometric analyses to track the percentage of ICOS+ and CD28+ human Teffs in peripheral blood over time. In the groups treated with saline or a single dose of belatacept, the fraction of human Teffs positive for ICOS increased from <10% on day 0 to ~80% by days 12 to 15 when the mice succumbed to disease. Human Teffs in the mice treated with repeated doses of belatacept also exhibited an increase in the percentage of ICOS+ CD4+ human Teffs, although this population was lower in the mice that survived longer (Fig. 4B, left). In contrast, the fraction of transferred human CD4+ Teffs positive for CD28 remained >95% in saline- or belatacept-treated mice throughout the study (Fig. 4B, right). The percentage of ICOS+ CD4+ or CD8+ human Teffs (Fig. 4C, left, and table S8) in terminal blood correlated with the final DAI scores for each mouse (r2 = 0.49), whereas the CD28+ population of human CD4+ or CD8+ Teffs did not (Fig. 4C, right, and table S8). ALPN-101 blocks the detection of CD28 and ICOS by the flow cytometry antibodies, so data from those groups were not included in the correlation calculations. However, as the ALPN-101 concentrations in the repeated dose group declined between the last dose on day 28 and the final day of the study (day 55), CD28 became unmasked and partially detectable (Fig. 4C, right). In contrast, the percentage of ICOS+ T cells remained low in those mice, possibly reflecting better inhibition of T cell activation compared to single dose treatment with belatacept or ALPN-101 (Fig. 4C, left). These data suggested that ICOS+ Teffs are playing an important role in the pathogenesis of aGVHD.
Fig. 4. ICOS expression on activated T cells in the human PBMCs-NSG GVHD model correlates with disease severity, and the suppressive effects of ALPN-101 are not altered by CsA.
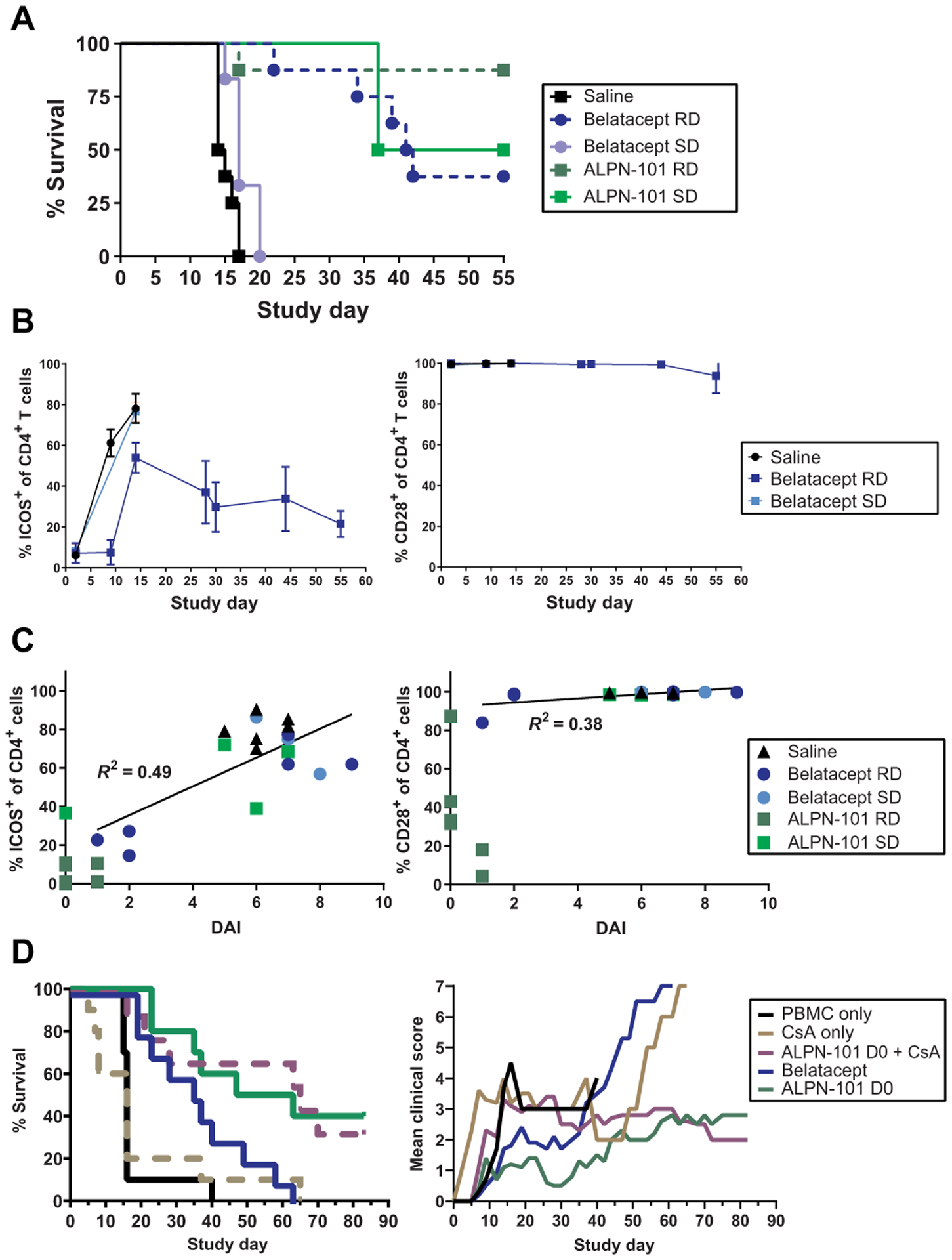
(A) Survival of mice receiving repeated doses (RD) or a single dose (SD) of the indicated test articles. NSG mice x-ray irradiated (100 cGy) and administered 10 mg of human γ globulin subcutaneously on day −1 and then transplanted intravenously with 10 × 106 human PBMCs on day 0 were treated intraperitoneally with saline; 100 μg of ALPN-101 TIW × 4 (days 0 to 28) or once on day 0; or 100 μg of belatacept TIW × 4 (days 0 to 28) or once on day 0. (B) Blood from each mouse treated with saline or a single dose (SD) or repeat doses (RDs) of belatacept or ALPN-101 [as described in (A)] was evaluated by flow cytometry for percent cells positive for ICOS or CD28 in the population of human CD4+ cells. Because ALPN-101 blocks the binding of anti-ICOS and anti-CD28 antibodies used for flow cytometry, data from the ALPN-101 groups are omitted. (C) Percent ICOS+ or CD28+ human CD4+ cells are plotted versus the terminal DAI score for each mouse as described in (A). Linear regression curves were calculated and correlation coefficients (R2) are indicated for each dataset. Because ALPN-101 blocks the binding of the antibodies used for flow cytometry, data from the ALPN-101 groups are shown on the graphs but were omitted from the correlation analyses. See table S7 for additional correlation analyses. (D) Effect of CsA on ALPN-101 therapy. Percent survival and clinical scores for NSG mice x-ray irradiated (100 cGy) and administered 10 mg of human γ globulin SC on day −1, then transplanted intravenously with 10 × 106 human PBMCs on day 0 (D0) and treated intraperitoneally with saline daily from day −1 to 13, then TIW through day 28; CsA 20 mg/kg daily from day −1 to 13, then TIW through day 28; 500 μg of ALPN-101 once on day 0; combination of 500 μg of ALPN-101 once on day 0 plus CsA 20 mg/kg daily from day −1 to 13, then TIW through day 28; or 75 μg of belatacept TIW × 4. In (A) (left), statistical significance in survival proportions between groups was determined by Mantel-Cox log-rank test. In (B), data are shown as mean ± SEM. In (D) (left), statistical significance in survival proportions between groups was determined by Mantel-Cox and Gehan-Breslow-Wilcoxon log-rank tests. In (D) (right), statistical significance was determined by two-way repeated measures ANOVA for treatment effect.
To dissect the relative contributions of CD28 and ICOS blockade, we produced and tested two Fc fusion proteins that blocked either CD28 or ICOS (fig. S15A). We compared their activity to that of ALPN-101 in vitro and in vivo. ALPN-101 bound to Jurkat cells expressing endogenous CD28 and transfected chimeric ICOS-CD28 molecules with EC50 values ~8 times lower than that of the anti-ICOS fusion protein and ~600 times lower than that of the anti-CD28 fusion protein (fig. S15B). We developed a cell-based assay to evaluate the relative contributions of CD80, CD86, and ICOSL, expressed singly or together by artificial APC, in stimulating the transfected ICOS-CD28–expressing Jurkat cells (fig. S16A). ALPN-101 consistently inhibited stimulation with IC50 values ~3- to 250-fold lower than those for the anti-CD28 or anti-ICOS fusion proteins alone and ~3- to 64-fold lower than both reagents combined (fig. S16B).
In the huPBMCs ➔ NSG model, treatment of mice with ALPN-101, anti-CD28, anti-ICOS, or a combination of anti-CD28 and anti-ICOS showed that disruption of the CD28 costimulation pathway with ALPN-101 or anti-CD28 conferred similar survival protection (100% of mice surviving), whereas single blockade with anti-ICOS reduced aGVHD and enhanced survival compared to Fc control–treated mice (P = 0.0004 for survival and P < 0.0001 for DAI) (fig. S17). These findings are consistent with the hypothesis that CD28 plays a predominant role during early, naïve T cell activation, and ICOS plays a later role in activated T cells, both of which are blocked by ALPN-101.
Prophylactic regimen with ALPN-101 extends survival and reduces human DCs and CD4+CD146+CCR5+ T cell populations in target organs in xenogeneic aGVHD model
We investigated whether a prophylactic regimen with a single 500-μg dose of ALPN-101 alone or in combination with the immunosuppressant cyclosporine A (CsA; 20 mg/kg daily from days −1 to 13, then three times weekly for 4 weeks) compared favorably to currently used prophylaxis with belatacept or CsA alone. We tested this in a slightly modified xenogeneic aGVHD model, in which the mice were monitored through 85 days after HCT (table S4). We found that ALPN-101 extended survival to 50% and ALPN-101 plus CsA extended the survival to 40% by day 85; in contrast, no mice survived to day 85 in the saline, CsA alone, or repeated dose belatacept treatment groups (Fig. 4D and table S9). The survival protection provided by each treatment was similarly reflected in the mean percent change in BW (fig. S18A) and mean clinical scores (Fig. 4D) for each group.
We also evaluated the effects of these treatments on the human T cell populations in the recipient mice. Both ALPN-101 and ALPN-101 plus CsA reduced the frequencies of total human CD4+ Teffs, proliferating Ki67+ T cells, central memory CD27+, effector memory CD27−, and CD27−PD-1+ cells (fig. S18B). Similar effects were observed for the CD8+ Teffs (fig. S18C). Regulatory T cells (Tregs; CD4+CD25+Foxp3+) were not detected in the blood in any of the treatment groups (fig. S18, B and C). In parallel, we evaluated the effect of ALPN-101 and belatacept on human Treg proliferation and suppressive function in vitro. We found that although ALPN-101 and belatacept both reduced the proliferation of Treg (Fig. 5A) and Teff (Fig. 5B), the relative ratios of these cells were maintained. Consequently, the impact of ALPN-101 or belatacept on Treg suppressive activity was negligible (Fig. 5C). These findings suggested that ALPN-101 inhibits CD4+ and CD8+ Teff function while preserving Treg function in vitro. In vivo, both a single dose at the time of transplantation and repeated doses of ALPN-101 prevent the development of xenogeneic aGVHD, by decreasing Teff proliferation and activation (Figs. 3, C and D, and 4, A and D, and fig. S18, B and C).
Fig. 5. Impact of ALPN-101 on Teff and Treg proliferation and on huDCs and huCD4+CD146+CCR5+ T cell populations in target organs.
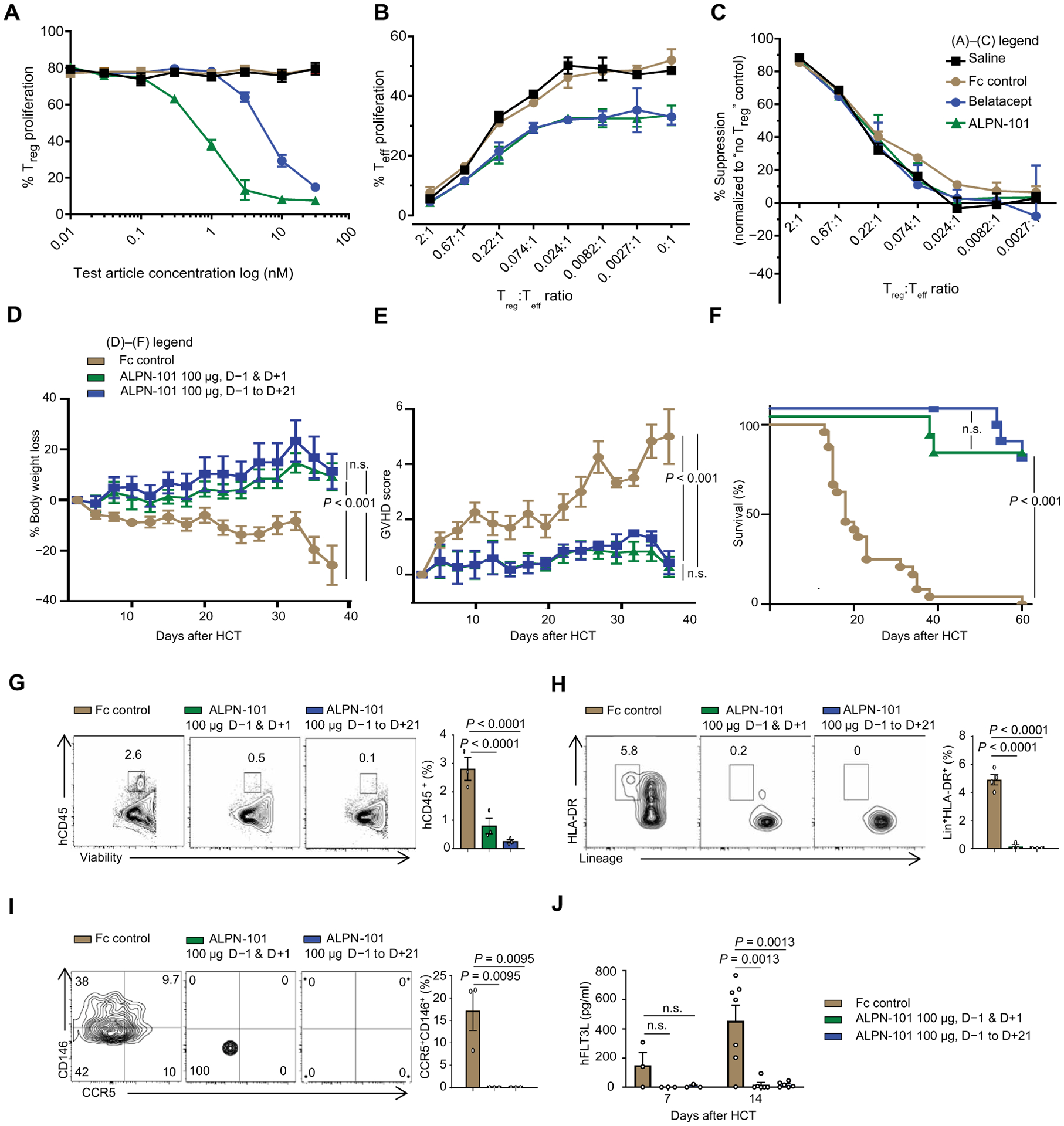
(A to C) Effect of ALPN-101 on T cell proliferation. Human Teffs and Tregs were stained with antibodies recognizing FoxP3, CD4, PD-1, CD25, CD28, CD127, ICOS, PD-L1, and Helios to confirm their surface phenotype before culture. In (A), enriched Tregs were labeled with CellTrace Violet (CTV) and cultured with soluble anti-CD3 antibody, recombinant IL-2 (rIL-2), and K562 APCs (transfected with CD80 and treated with mitomycin C) in medium with saline (black) or with various concentrations of the test molecules: Fc control (brown), belatacept (blue), or ALPN-101 (green). (A) After 3 days, Treg proliferation was assessed by CTV dilution by flow cytometry. Statistical differences between Fc control and the other test molecules were determined by an unpaired t test. The ALPN-101 treatment effect is significantly different from the Fc control group (P = 0.0037 at 1 nM, P = 0.0034 at 3 nM, and P = 0.0003 at 30 nM by unpaired t test) and also significantly different from the belatacept group (P = 0.004 at 1 nM and P = 0.007 at 3 nM). (B) Tregs labeled with CTV were mixed at the indicated ratios with Teff labeled with CFSE and cultured with mitomycin C–treated, CD80low K562 APCs, and soluble anti-CD3 antibody in medium containing added saline or 30 nM of each test molecule. After 4 days, Teff proliferation was assessed by CFSE dilution by flow cytometry. The ALPN-101 treatment effect is significantly different from the Fc control group at all Treg:Teff ratios except 2:1 (P = 0.0001 at 0.67:1; P = 0.0345 at 0.22:1; P = 0.0158 at 0.074:1; P = 0.0313 at 0.024:1; P = 0.0196 at 0.0082:1, P = 0.0187 at 0.0027:1, and P = 0.0342 at 0:1, by unpaired t test). (C) Specific Treg suppression activity for each culture condition was determined (see Materials and Methods). Concentrations are the same as in (B). Data are presented normalized to Teff activity in the absence of Tregs. (D to F) Effect of ALPN-101 on health [BW (D) and GVHD severity score (E)] and survival (F) of the human PBMC-NSG GVHD model. NSG mice were irradiated at 300 cGy at day −1 and then transplanted with 3.5 × 106 human PBMCs at day +1. Mice were treated every other day with Fc control (brown) or 100 μg of ALPN-101 from days −1 to +21 (12 doses total; blue) or 100 μg of ALPN-101 on days −1 and +1 (2 doses total; green). n = 10 mice in all groups. (G to I) Human hematopoietic cell engraftment (G), Lin−HLA-DR+ total DCs (H), and CD4+CD146+CCR5+ T cells (I) in the GI tract of NSG recipient mice. Five mice from (D) to (F) were analyzed at day 14 after HCT comparing Fc control and ALPN-101–treated groups. (J) Concentration of human FLT3L in plasma from NSG mice from (D) to (F) at days 7 and 14 after HCT (n = 3 mice per time point). Data are shown as mean ± SEM, except for survival curves and representative flow cytometry. In (A) to (C), statistical significance was determined by unpaired t test, and in (D), (E), and (G) to (J), it was determined by ANOVA with Bonferroni’s correction. Log-rank test (Mantel-Cox) was used for survival analysis in (F).
To further evaluate the prophylactic effects of ALPN-101 in the xenogeneic NSG model and correlate aGVHD with phenotypes of human DCs and CD4+CD146+CCR5+ T cell populations that infiltrated the intestines, we performed another study (table S4) using a modified PBMCs ➔ NSG model with NSG mice subjected to 300 centigray (cGy) dose of irradiation followed by injection with 3.5 × 106 PBMCs (22). In this model, mice received 10 mg of huIgG subcutaneously followed by a prophylactic dosing schedule from days −1 to +21 with 100 μg of ALPN-101 every other day (11 doses total) or a peri-transplant regimen with just two doses of 100 μg of ALPN-101 on days −1 and +1. The control group received 100 μg of Fc control from days −1 to +21. We found that NSG mice treated with ALPN-101 using either the full or peri-transplant dose regimens were protected from xenogeneic aGVHD as measured by BW loss (Fig. 5D) and aGVHD clinical score (Fig. 5E). Furthermore, survival was also improved (Fig. 5F) with most mice receiving either treatment surviving to day 60, whereas only one of the control mice survived beyond day 40.
Analysis of the GI tract, which is a key target organ in this NSG model (6, 23), showed reduced intestinal infiltration of human CD45+ cells (Fig. 5G), human DCs (Lin−HLA-DR+) (Fig. 5H), and human Th17 cells (CD146+CCR5+) (Fig. 5I). Similarly, we found decreased infiltration of human CD45+ cells, total DCs, as well as pDCs, ICOSL+ pDCs, and CD146+CCR5+ T cells in the spleens of the ALPN-101–treated mice compared to the Fc-treated control mice (fig. S19). Lower serum FLT3L concentrations were also detected in the ALPN-101–treated mice compared to that in the Fc-treated controls (Fig. 5J).
Last, we used a more aggressive model. NSG mice received a higher dose of irradiation (350 cGy), which was followed by injection with 5 × 106 PBMCs after receiving 10 mg of huIgG subcutaneously (table S4). We compared treatment using a prophylactic schedule from days −1 to +21 with 100 μg of ALPN-101 every other day (12 doses total) or a peri-transplant regimen with just two doses of 100 μg of ALPN-101 on days −1 and +1, versus 100 μg of Fc control comparator from days −1 to +21. By day 60, the ALPN-101–treated mice had gained weight (fig. S20A). Compared to the Fc control–treated mice, the mice treated with either ALPN-101 regimen had lower GVHD clinical scores throughout the study (fig. S20B) and improved survival (fig. S20C). These data suggested that administration of ALPN-101 before allogeneic HCT protects against the development of xenogeneic aGVHD and that ALPN-101 might serve as a new prophylactic treatment for aGVHD.
ALPN-101 administered in a therapeutic dosing regimen starting at onset of aGVHD signs protects against disease and inhibits pathogenic human DCs and CD4+CD146+CCR5+ T cell populations
Because therapeutic treatment of aGVHD after allogeneic HCT is a greater challenge than therapeutic prophylaxis, we investigated whether ALPN-101 effectively treated aGVHD. To test this, we used the aggressive PBMCs ➔ NSG model with NSG mice irradiated with 350 cGy followed by 10 mg of subcutaneous huIgG and 5 × 106 PBMCs (table S4). The mice received either 100 μg of Fc control or 20 μg or 100 μg of ALPN-101, which was initiated at the onset of aGVHD signs and continued every other day from days +7 to +14 (five doses total). The mice were monitored through 120 days after HCT.
We observed reduced percentages of BW loss (Fig. 6A) and aGVHD clinical scores (Fig. 6B), as well as extended survival (Fig. 6C), in the two groups of ALPN-101–treated mice as compared to the Fc control–treated group. Analysis of the human immune cells infiltrating the GI tract of the recipient mice revealed the presence of a higher frequency of human hematopoietic cells (CD45+) (Fig. 6D), human DCs (Lin−HLA−DR+) (Fig. 6E), and Th17 cells (CD146+CCR5+) (Fig. 6F) in the Fc control–treated group, and these human cells were almost undetectable in the intestines of the ALPN-101–treated mice. Lower serum FLT3L concentrations were found in the ALPN-101–treated groups as compared to the Fc control–treated group (Fig. 6G).
Fig. 6. Treatment with ALPN-101 at onset of xenogeneic aGVHD and huPBMC-NSG model with human leukemia cells.
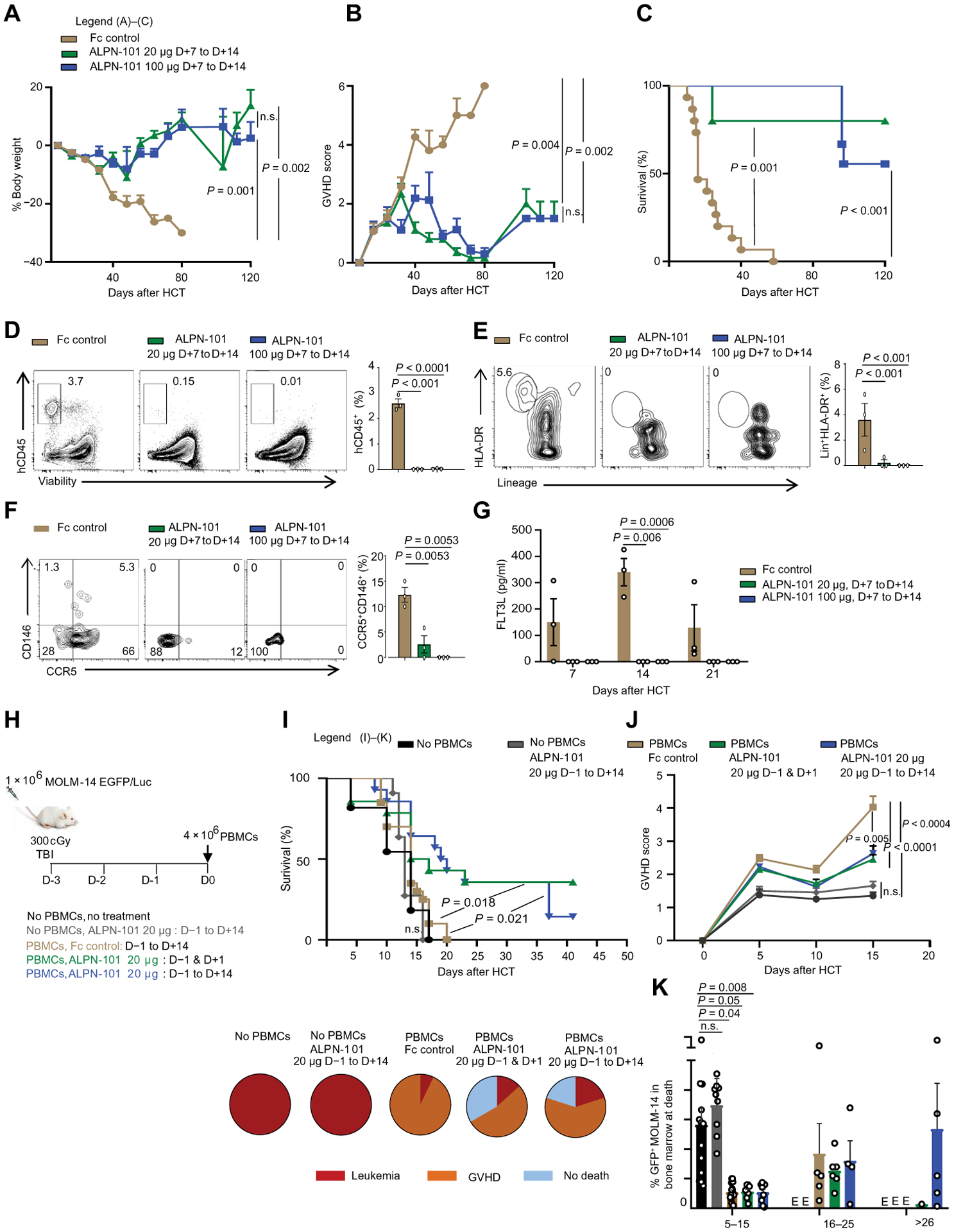
(A to C) Effect of ALPN-101 on health [BW (A) and GVHD severity score (B)] and survival (C) of the human PBMC-NSG GVHD model. NSG mice were irradiated at 350 cGy at day −1 and then transplanted with 5 × 106 human PBMCs at day +1. Mice were treated with either Fc control every other day from days +7 to +14 (n = 9), or 20 μg of ALPN-101 every other day from days +7 to +14 (n = 9), or 100 μg of ALPN-101 (n = 15) every other day from days +7 to +14. (D to F) Human hematopoietic cell engraftment (D), Lin−HLA-DR+ DC frequencies (E), and CD4+CD146+CCR5+ T cell frequencies (F) in the GI tract of NSG recipient mice. Three NSG mice from each group in (A) to (C) were analyzed at day 14 after HCT, comparing the Fc control and ALPN-101–treated groups. (G) Concentration of human FLT3L in plasma from three NSG mice from (A) to (C) at the indicated times after HCT. (H) Study design for evaluation of ALPN-101 GVHD prophylaxis in mice with leukemia. NSG mice were irradiated at 300 cGy and injected with 1 × 106 human leukemic cells of the line MOLM-14-EGFP on day −3 and then transplanted with 4 × 106 human PBMCs on day 0 (day of PBMC transplant). Two groups of mice did not receive PBMCs, while one did not receive any treatment, the other group was treated with ALPN-101 from days −1 to +14 (n = 15); the other mice all received PBMCs and were treated with either ALPN-101 only from days −1 to +14 (n = 20), Fc control every other day from days −1 to +14 (n = 20), 20 μg of ALPN-101 on day −1 and +1 (n = 20), or 20 μg of ALPN-101 every other day from days −1 to +14 (n = 20). (I) Survival of leukemia-recipient mice. Pie charts represent the percentage of surviving mice or cause of death for each group. (J) Clinical GVHD score in the three groups of leukemia-recipient mice receiving PBMCs. (K) Percentage of GFP+MOLM-14 leukemic cells in the BM of mice at death. In (A), (B), (D) to (G), and (I) to (K), data are shown as mean ± or + SEM. In (A), (B), (D) to (G), and (I) to (K), statistical significance was determined by ANOVA with Bonferroni’s correction. Kaplan-Meier method using the log-rank test for comparison was used for survival analysis in (C).
Prophylactic regimen with ALPN-101 in mice with ongoing acute myeloid leukemia provides protection from aGVHD without compromising antitumor treatment
Our results suggested that ALPN-101 can be used in a prophylactic or therapeutic regimen to prevent aGVHD. Because it is also important for an effective therapy to maintain antitumoral activity while reducing aGVHD, we assessed the impact of ALPN-101 on T cell responses. We evaluated the impact of ALPN-101 in mice with established acute myeloid leukemia (AML) using human MOLM-14 AML cells engineered to express enhanced green fluorescent protein (EGFP) so the leukemia cells could be tracked. NSG mice were irradiated at 300 cGy and injected with 1 × 106 MOLM-14-EGFP cells 3 days before the transplant to allow time for the human MOLM-14 AML cells to engraft (table S4). Three days after implantation of the MOLM-14 cells, the mice were injected with 4 × 106 human PBMCs. One group of mice did not receive PBMCs and served as a leukemia control group. The mice that received human PBMCs were injected subcutaneously with 10 mg of huIgG on day −1, then injected intravenously with the PBMCs on day 0. Treatment regimens were 20 μg of Fc control protein every other day from days −1 to +14, or 20 μg of ALPN-101 on days −1 and +1, or every other day from days −1 to +14 after HCT (Fig. 6H). As expected, all mice that did not receive any PBMCs died of leukemia, whereas all Fc control–treated mice except two died of aGVHD (Fig. 6I). Cause of death was inferred from GVHD clinical scores (high in the Fc control group) (Fig. 6J) and the percentage of GFP+ MOLM14 cells in the BM (high for the no-PBMC leukemia control group) (Fig. 6K). ALPN-101–treated mice showed increased survival compared to the “no PBMCs, no treatment” group, the “no PBMCs, ALPN-101” treatment group, and the “PBMCs/Fc control” treatment group (Fig. 6I). For the mice that died in these groups, the cause of death was a mix of aGVHD (although delayed) and relapse for the later death (Fig. 6, I, to K). These data suggested that ALPN-101 may preserve allogeneic anti-leukemic activity while inhibiting aGVHD.
DISCUSSION
A previous study from our group identified a CD146+CCR5+ T cell population up-regulated in patients with GI-GVHD and correlated with mortality, as compared to patients without GVHD or to patients with non-GVHD enteritis or skin GVHD (6). These cells were shown to be Th17-prone and induced by ICOS (6). Therefore, our first aim in the current study was to determine whether ICOSL was increased on APCs (especially DCs) in the patients and whether ICOSL+ DCs might enable increased accuracy of the risk stratification and more stringent monitoring of patients at risk for aGVHD. A high frequency of ICOSL+ pDCs was detected in patients with GI-GVHD compared to patients without GVHD or with non-GVHD enteritis or skin GVHD from the same cohort of patients we analyzed previously (11). ICOSL+ pDC frequencies were positively correlated with the frequencies of CD146+CCR5+ T cells. Further analysis of the patient cohort data demonstrated that high ICOSL expression was also associated with lower overall survival. These data suggest that early quantification of ICOSL+ pDC frequency may allow for identification of patients at risk of GI-GVHD and thus enable preemptive intervention; however, evaluation of additional independent cohorts will be required for a generalizable definition of high GI-GVHD risk.
The second aim of this study was to mechanistically understand the role of ICOSL+ pDCs in the polarization of Th17 cells in aGVHD murine models. Our analysis did not reveal whether ICOSL+ pDCs are derived from the host or the donor in recipients of allogeneic HCT. Because of the limitations of peripheral blood collection volumes and because circulating DCs are rare in the peripheral blood, we were unable to explore this issue further. However, a previous study has demonstrated that about 80% of circulating DCs were of donor origin 14 days after transplantation and increased to 95% by 56 days after transplantation (24). Therefore, it is likely that in our studies using blood samples from patients between 11 days and 92 days after transplantation, ICOSL+ pDCs were primarily of donor origin. Studies in mice indicated that while host APCs including recipient nonhematopoietic APCs (25, 26) play a major role in GI-GVHD, donor APCs can also enhance GVHD reactions (27). In addition, donor BM pDCs but not mature pDCs contained within stem cell grafts attenuate aGVHD in murine models (15). In humans, several studies have shown an important role of donor DCs, particularly nonactivated pDCs that are tolerogenic and improve survival (28, 29). Practically, recipient’s DCs are extremely difficult to obtain as they require biopsies only available in limited amount. On the basis of our patients’ data, we hypothesized that donor ICOSL+ pDCs were inducers of GI-GVHD. In the current study, absence of ICOSL in donor BM reduced the severity of aGVHD and the resulting mortality in recipients of major mismatch and haploidentical HCT models. Lower aGVHD and better survival were associated with lower frequencies of pDCs and pathogenic Th17+IFNγ+ T cells in the intestine and in the spleen. Transcripts of genes associated with key functions of pDCs and with T cell activation in the recipients of ICOSL−/− BM were also down-regulated in sorted intestinal pDCs and CD4+Foxp3− T cells, respectively.
We hypothesized that absence of ICOSL signaling in donor DCs would protect against aGVHD through STAT3, FLT3L, or both. FLT3L is a cytokine expressed by all tissues in mice and humans that is essential for the development, differentiation, and homeostasis of pDC (12, 30). STAT3 is required for FLT3L-dependent DC differentiation in mice (11), although FLT3L can also signal through other distinct and/or overlapping pathways such as PI3K/Akt/mTOR (31). In the current study, STAT3 deficiency in the donor BM had no effect on aGVHD severity in the recipient mice. However, in the recipient mice that received ICOSL KO BM (and thus no ICOSL+ pDCs to activate and expand pathogenic donor T cells), serum mFLT3L concentrations were markedly decreased. Although the source of mFLT3L in our HCT model is unclear, we theorize that because its concentration declines early after HCT, it is likely produced by stromal cells in the recipient and induced during the cytokine storm that occurs just before the onset of GVHD (32). The exact mechanism by which ICOSL deficiency in the donor BM leads to a reduction in FLT3L secretion by the recipient stromal cells remains a focus of active investigation.
Our study sheds light on the controversial role of pDCs during alloreactivity. Informed by patients’ data, we have shown that ICOSL-expressing donor pDCs induce GI-GVHD, which seems to be mediated by FLT3L but not STAT3. Thus, we further investigated the roles of human pDCs and FLT3L in the development of intestinal CD146+CCR5+ T cells. Activation of pDCs by the combination of CpG and FLT3L led to high expression of ICOSL on their surface. For translational purposes, we then used the human PBMCs ➔ NSG model, mainly because CD146 is expressed at an extremely low level on murine T cells compared to human T cells and has not been detectable in murine GVHD models (6, 33). The use of humanized models to evaluate aGVHD is contentious, particularly due to the absence of human APCs in the recipient mice (19). However, aGVHD can be reduced by MHC class I or class II deficiency in the host mice, indicating that human T cells are xenoreactive with mouse MHC antigens on NSG host cells (19). Furthermore, an enhanced “graft-versus-tumor/leukemia (GVT/L)” effect was shown in NSG-HLA-A2 mice (20). These humanized models are currently the best option to test GVT/L or antitumoral activity of human antibodies. Comparing transplantation of ICOSL+ pDCs and unstimulated pDCs along with PBMCs, only ICOSL+ pDCs prompted intestinal CD146+CCR5+ T cell infiltration, resulting in severe aGVHD and death.
Our third aim was to target the ICOS/ICOSL pathway for the prevention and treatment of aGVHD in humanized xenogeneic models. Both CD28 and ICOS costimulatory pathways appear to promote aGVHD (34). Currently approved biologic therapies are limited to CD28/B7 pathway inhibition, potentially permitting the escape of activated ICOS+ T cells (35, 36). ALPN-101 is a dual CD28 and ICOS T cell costimulation pathway inhibitor that can thus target ICOS+ cells that may escape CD28-only blockade. Our data show that ALPN-101 consistently demonstrates superior inhibition of T cell proliferation and cytokine production in MLRs and other cell-based assays compared to CD28 or ICOS single pathway blockade. Repeat dosing with ALPN-101 can completely protect mice from disease in a humanized PBMCs ➔ NSG model, and a single equimolar dose of ALPN-101 provides similar protection as repeat doses of belatacept, whereas a single dose of belatacept confers no protection from disease. After a single 100-μg dose of ALPN-101 in the xenogeneic model, Tmax was observed at 2 hours after dose, and the serum half-life was ~4 days. Mean concentrations after a 100-μg belatacept dose were substantially higher than after a 100-μg ALPN-101 dose at all evaluated time points, while target saturation of the human T cells by ALPN-101 remained high even >2 weeks after the final dose. These observations suggest tight target binding of ALPN-101 in vivo that is consistent with its superior potency and pharmacodynamic effects. ALPN-101 also potently inhibited inflammatory cytokine secretion and Teff expansion in xenogeneic models. The human PBMCs ➔ NSG model reproduced the increase of ICOS expression on activated human T cells in aGVHD over time, and the increase of circulating ICOS+ T cells in the model correlated with disease activity, whereas clinical scores did not correlate with CD28+ or PD1+ expression. Thus, one mechanism of action of ALPN-101 may be to inhibit the emergence of activated ICOS+CD28+ pathogenic T cells that escape CD28 single pathway blockade. Both ALPN-101 and belatacept preserved Treg function in vitro [(37) and this study]. Although belatacept reportedly blocks Treg function in vivo in a skin graft model through inhibition of the CTLA-4 pathway (37), we did not directly assess the impact of either treatment on Treg function in vivo.
Although previous studies in murine models have shown that inhibition of the ICOS and CD28 pathways with both anti-ICOS and anti-CD28 neutralizing antibodies is required for effective prevention of aGVHD (34, 38), as we have also confirmed here, this approach has not yet been implemented in the clinic. Thus, dual antagonism of ICOS and CD28 via one biologic such as ALPN-101 may be particularly effective as prophylaxis and/or therapeutic treatment for GVHD and other acute and chronic T cell–mediated inflammatory diseases. The clinically approved therapeutic protein abatacept, inhibiting solely the CD28 pathway, has shown some efficacy in patients with steroid-refractory aGVHD (39). A multi-center phase 2 trial (NCT01743131) with abatacept combined with calcineurin inhibitors and methotrexate for aGVHD prophylaxis is ongoing. Belatacept, which includes two point mutations resulting in an increased affinity for the CD28 ligands CD80 and CD86 relative to abatacept (wild-type CTLA-4-Fc), is approved for prevention of renal allograft rejection and is currently being evaluated in clinical trials to prevent organ transplantation rejection. However, it has not been evaluated for aGVHD because of mixed results in preclinical murine and canine models of HCT, as well as a lack of efficacy in a recently published nonhuman primate HCT model (40). Dual inhibition of CD28 using FR104, an antagonistic CD28-specific pegylated-Fab′, and the mTOR pathway inhibitor sirolimus resulted in enhanced control of Teff proliferation and activation and decreased aGVHD compared with the use of CTLA-4-Fc or CTLA-4-Fc/sirolimus in a nonhuman primate HCT model (40). Because our findings suggest that dual ICOS/CD28 inhibition with ALPN-101 dampens T cell activation and aGVHD better than abatacept and belatacept, a clinical trial with ALPN-101 in healthy volunteers was recently completed (NCT03748836), enabling development in multiple autoimmune and/or inflammatory conditions.
Using humanized xenogeneic mouse models with different doses of radiation and altering the numbers of PBMCs injected, we showed that prophylactic treatment with ALPN-101 decreased weight loss and aGVHD disease activity, increased survival compared to the Fc control group, and was better than several current standards of care such as CsA alone or in combination with belatacept. We then explored the frequencies of human Lin−HLA-DR+ pDCs and CD4+CD146+CCR5+ T cell populations, which were both decreased in the ALPN-101–treated groups as compared to the Fc control–treated mice. ALPN-101 could also treat established xenogeneic aGVHD, and ALPN-101 did not interfere with the antitumoral effect mediated by PBMCs. These results suggest that with an appropriate dosing regimen in patients, ALPN-101 may preserve allogeneic anti-leukemic activity while inhibiting aGVHD.
Our findings have certain limitations. The mechanism of FLT3L-mediated induction of pDCs is not well-characterized. In addition, because of the low frequency of pDCs in the peripheral blood, it has not been possible to extensively characterize the infiltrating pDCs.
We conclude that early quantification of ICOSL+ pDC frequency in HCT patients may allow for identification of individuals at risk of GI-GVHD and thus enable preemptive intervention. The use of ICOSL−/− donor BM and/or therapeutic blockade of ICOS/CD28 alleviates aGVHD, decreases pDC frequency, and decreases pathogenic Th17 (murine) and CD146+CCR5+ (human) cell expansion via FLT3L but not STAT3. ALPN-101 is a dual ICOS/CD28 T cell antagonist capable of inhibiting lethal inflammatory processes even with a single dose, with potent efficacy as compared to continuous CD28/CTLA-4-B7 or ICOS-ICOSL single pathway inhibition. This efficacy is achieved despite lower PK exposure of a single dose and is probably attributable to suppression of ICOS+ T cells that may otherwise escape single pathway blockade. In vivo preclinical studies of ALPN-101 showed efficacy for the prevention and treatment of aGVHD, while preserving anti-leukemic activity in the humanized aGVHD model. ALPN-101 is thus a promising therapeutic candidate for GVHD, and clinical trials are needed to explore its therapeutic potential in GVHD and other inflammatory diseases.
MATERIALS AND METHODS
Study design
This study was designed to investigate the role of ICOSL-expressing pDCs in the induction of intestinal CD146+CCR5+ T cells during the development of aGVHD. In the first phase of this study, we analyzed the expression profile of ICOSL on the pDCs (Lin−HLA-DR+CD123−CD11c+) of HCT patients (64 with GI-GVHD, 39 without GVHD, 22 with non-GVHD enteritis, and 31 with skin GVHD). In the second part, we used both an aGVHD major mismatch HCT model (C57BL/6, H2b → BALB/c, H-2d) and a translational in vivo GVHD model using the human PBMCs → NSG model to characterize intestinal CD146+CCR5+ T cells and pDCs during aGVHD. Last, we assessed the therapeutic potential of ALPN-101, a dual ICOS/CD28 inhibitor, in the prevention and treatment of aGVHD. Details regarding sample sizes and statistical analyses can be found in the legends and statistics section in Supplementary Materials and Methods. Power calculation for survival in our typical HCT/leukemia experiments (GVHD, GVT, and leukemia) studies was as follows: if p1 denotes mortality at the end of observation period (usually 60 days) in the animals treated and p2 in the wild-type animals, then the appropriate number of animals needed to detect >40% survival difference with 80% power with a type I error rate is 10. Survival of the animals was based on the (i) maximum GVHD score of 6 or (ii) maximum percentage of circulating GFP-labeled leukemic cells of 20%. The mice were monitored daily to check their health, blood was collected weekly to assess GFP-labeled leukemic cells, and euthanasia was performed according to the approved protocols at the Indiana University School of Medicine, University of Minnesota, the Jackson Laboratory (JAX), and Alpine Immune Sciences Institutional Animal Care and Use Committee (IACUC protocol 18033, 1805–35891A, and AUS #18003). Randomization of subjects and blinded assessments of outcomes were not formally implemented in these experiments.
Patients and samples
All patients or their legal guardians provided written informed consent, and the collection of samples for studying after HCT complications was approved by the institutional review board of the University of Michigan. Heparinized PBMCs were collected weekly during the first 4 weeks and then monthly after allo-HCT. PBMCs were also collected at the development of key clinical events (symptoms of GVHD or non-GVHD enteritis or skin rash). Samples were not drawn if methylprednisolone at a dose higher than 1 mg/kg was administered 48 hours or more before sample collection. PBMCs were isolated by density gradient separation using Ficoll and stored frozen at −120°C in liquid nitrogen. All patients received a T-replete graft and GVHD prophylaxis (details in table S1).
Multicolor flow cytometry for patients
Cell surface markers on frozen PBMCs were analyzed as follows: panels for T cells and DCs were run per batch of three to four patients, three times a week using the same instrument for the whole cohort the same day by the same operator. If viability >80% was reached and if there were >500,000 cells, then both flow panels were run; if there were less than 500,000 cells, then only the T cell panel was run. The T cell panel has been published (6); for the DC panel, we used human antibodies to lineage markers as a cocktail, including antibodies recognizing HLA-DR (clone LN3), CD123 (clone 6H6), CD11c (clone 3.9), ICOSL (clone MIH12), CD86 (clone IT2.2), CD4 (clone OKT4), CD8 (clone SK1), CD146 (clone P1H12), CCR5 (clone 2D7), and CD45 (clone 2D1). Antibodies were from BD Biosciences.
Animal study approval and mice
Animal experiments and euthanasia protocols, including all endpoint analyses such as blood and tissue collection, performed in these studies were approved by the Indiana University School of Medicine, University of Minnesota, JAX, and Alpine Immune Sciences IACUC (IACUC protocol 18033, 1805–35891A, and AUS #18003). C57BL/6 (B6, H-2b, CD45.2+), BALB/c (H-2d), B6D2F1 (H-2b), ICOSL−/− C57BL/6 (B6, H-2b, CD45.2+), and immunodeficient NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were purchased from JAX. For some studies, NSG mice purchased from JAX were bred and provided by the In Vivo Therapeutics Core at the Indiana University Simon Cancer Center (see table S4).
Injection of ICOSL+ pDCs in vivo
Immunocompromised NSG mice received 350 cGy total body irradiation (137Cs source) on day −1. On day 0, mice were injected intravenously with 5 × 106 of pDC-depleted PBMCs with either 5 × 104 CpG + FLT3L stimulated pDCs or unstimulated pDCs.
Induction and assessment of aGVHD in murine allogeneic HCT models
Mice underwent allogeneic HCT as previously described (23). Briefly, BALB/c and B6D2F1 received 900 and 1000 cGy total body irradiation (137Cs source), respectively, on day −1. Recipient mice were injected intravenously with T cell–depleted (TCD) BM cells (5 × 106) plus splenic T cells (2 × 106 for B6D2F1 or 1 × 106 for BALB/c mice) from allogeneic donors at day 0. T cells from donor mice were enriched using the murine Pan T Cell Isolation Kit (Miltenyi Biotec), and TCD BM was prepared with CD90.2 Microbeads (Miltenyi Biotec). Mice were housed in sterilized micro-isolator cages and maintained on acidified water (pH < 3) for 3 weeks. Survival was monitored daily, and clinical GVHD scores were assessed weekly as described previously (41). Mice were euthanized when the clinical scores reached 6.5, in accordance with animal protocols approved by the Indiana University Institutional Review Board.
Human PBMCs ➔ NSG xenogeneic mouse model of GVHD and in vivo treatment with ALPN-101
For the study performed at the University of Minnesota, on study day −1 (SD–1), 70 female NSG mice (10 per group) were administered 10 mg of human gamma globulin subcutaneously in 100 μl, and test articles were administered intraperitoneally on the study days indicated in each figure. Pretreatment with human gamma globulin was included since treatment of NSG mice lacking endogenous B cells and IgG with human antibodies or Fc fusion proteins (including Fc1.1) can lead to “coating” of the human PBMC (in this case, the human T cells) with the drug, and subsequent engagement with FcγRI on both mouse and human effector cells can then deplete drug-bound target cells. Such an effect could lead to a misinterpretation of results; in other words, T cell depletion by this FcR-dependent mechanism might be interpreted as T cell inhibition. The preinjected soluble human IgG in the huPBMCs NSG model outcompetes the effectorless Fc on ICOSL-Fc for FcγRI binding and thus prevents erroneous FcR-mediated T cell depletion.
Human apheresis products from two healthy donors were collected on SD–1 and analyzed for expression of CD4, CD8, CD14, and CD19 by flow cytometry and then stored overnight at room temperature.
On SD0, mice were weighed and irradiated (1.8 Gy/180 rad) from an x-ray irradiator source. The apheresis units with values closest to 40% CD4+ were Ficoll-purified, and each mouse was injected intravenously with 1 × 107 human PBMCs in 200 μl of phosphate-buffered saline.
BWs and clinical scores were collected three times a week (Monday, Wednesday, and Friday), and blood was collected from mice on SD5 and SD15 to phenotype the circulating human CD4+ and CD8+ T cells by flow cytometry. Blood was collected for flow cytometry phenotyping as mice were euthanized throughout the study and on the last day of the study, and a portion of the blood was processed to serum for drug exposure analysis. The study was terminated on day 84. For disease activity scoring, mice were assessed for BW loss and a DAI score comprising the sum of scores for overall health and activity, skin and hair changes, and BW loss over the course of the study. Mice were euthanized by CO2 asphyxiation before study termination if they showed >20% BW loss from their starting weight or a combination of the following clinical signs: >10 to 20% weight loss from their starting weight, cold to touch, or lethargic, pale, hunched posture, and/or scruffy coat. The mean DAI scores were plotted for the time course of the experiment.
For studies conducted at the Indiana University, immunocompromised NSG mice received 300 or 350 cGy total body irradiation (137Cs source) on day −1. On day 0, mice were injected intravenously with 3.5 × 106 or 5 × 106 human PBMCs. Pretreatment with 10 mg of human gamma globulin was included in all experiments. Mice were injected intraperitoneally with 20 or 100 μg of ALPN-101 once per day or every other day, for a total of 3 or 11 doses, respectively. Mice were injected intraperitoneally with 20 μg of ALPN-101 on days −1 and +1 (two doses total) or 100 μg of ALPN-101 from days −1 to +14, every other day (11 doses total). For the studies described in Fig. 3, female NSG mice were administered 10 mg of human gamma globulin (Sigma-Aldrich) subcutaneously and then irradiated (100 cGy) from an x-ray irradiator source. On day 0, within 24 hours of irradiation, mice received 10 × 107 human PBMCs intravenously and were treated with Fc control, ALPN-101, or belatacept using the dose regimens described in the figure legend (20, 100, or 500 μg three times weekly × 4 weeks, or a single 100 μg dose on day 0).
Statistics
Phenotypic and functional data were compared using unpaired t test for two-group comparison and analysis of variance (ANOVA) test for three or more group comparison. Before analysis, the normality assumption was examined. All tests were two-sided at the significance level P ≤ 0.05. To account for the type I error inflation due to multiple comparisons, we applied the Bonferroni correction. A log-rank test (Mantel-Cox, Gehan-Breslow-Wilcoxon, or Mann-Whitney) was used for survival analysis in the various GVHD studies. The murine GVHD study analyses were performed using GraphPad Prism software version 8, and data are presented as mean ± SEM.
Supplementary Material
Fig. S1. pDCs and cDCs gating strategy.
Fig. S2. Frequencies of total DCs and cDCs in patient cohort.
Fig. S3. Frequencies of ICOSL+ cDCs in patient cohort.
Fig. S4. Frequencies of ICOSL+ pDCs in lower and upper GI-GVHD.
Fig. S5. Correlative and ROC analyses in patients with GVHD and GI-GVHD.
Fig. S6. Frequencies and absolute counts of murine CD11b−CD11c+CD103+B220+ splenic pDCs in a major mismatch model.
Fig. S7. Splenic Th17 cells coexpressing CXCR3 and RORγt in a major mismatch murine model.
Fig. S8. Frequencies of murine intestinal CD11b−CD11c+CD103+B220+ pDCs in a haploidentical allogeneic model.
Fig. S9. Frequencies of IFNγ/IL-17–producing T cells in the gut in a haploidentical allogeneic model.
Fig. S10. Cell population pretransplant, and model of transplant.
Fig. S11. Frequencies of human T cells and human pDCs in spleens of NSG mice receiving unstimulated or ICOSL+ pDCs.
Fig. S12. Schematic of the strategy used to generate the therapeutic lead Fc fusion protein designated ALPN-101.
Fig. S13. ALPN-101 potently suppresses human T cell responses in vitro.
Fig. S14. Human cytokine concentrations measured in terminal serum collected from mice in all treatment groups in the human PBMC-NSG GvHD study.
Fig. S15. Evaluation of the binding of anti-CD28 and anti-ICOS antagonist Fc fusion proteins to Jurkat cells expressing endogenous CD28 or transfected chimeric ICOS-CD28 molecules.
Fig. S16. Blockade of CD28- or ICOS-mediated costimulation by ALPN-101, anti-CD28, anti-ICOS, combination anti-CD28+ anti-ICOS, or belatacept.
Fig. S17. Dual blockade of CD28 and ICOS or CD28 alone prevents GVHD in the human PBMC-NSG model.
Fig. S18. Percent change in BW and flow analysis of T cells in blood from human PBMC-NSG mice treated with ALPN-101 with or without CsA.
Fig. S19. Frequency and absolute counts of human infiltrating splenic cells in the peri-transplant and prophylactic human PBMC-NSG study.
Fig. S20. Prophylactic blockade of ICOS/CD28 on donor pDCs using ALPN-101 in an aggressive xenogeneic GVHD model.
Table S1. Characteristics of the study patients.
Table S2. Causes of overall mortality in patients with GVHD symptoms (n = 117).
Table S3. Relative affinities of ALPN-101 to human CD28, ICOS, and CTLA-4 measured by BLI analysis.
Table S4. Summary of protocols used for the human PBMC-NSG GVHD studies.
Table S5. Final circulating T cell counts and statistical analysis for the GVHD survival, DAI, and BW loss data.
Table S6. Statistical analysis of terminal serum human cytokine concentrations in mice treated with repeat 100-μg doses of ALPN-101 or belatacept, or a single 100-μg dose of ALPN-101.
Table S7. Summary of statistical significance for survival between treatment groups in the human PBMC-NSG GVHD study.
Table S8. Additional correlation analyses of the flow cytometric data from the human PBMC-NSG GVHD study.
Table S9. Summary of statistical significance for survival between treatment groups in the human PBMC-NSG GVHD study.
Data file S1. Transcriptome analysis of sorted intestinal WT versus ICOSL−/− pDCs.
Data file S2. Transcriptome analysis of sorted intestinal WT versus ICOSL−/− CD4+Foxp3− T cells.
Acknowledgments:
We thank our colleagues at Alpine Immune Sciences, M. Kornacker, C. Navas, M. Rixon, S. MacNeil, J. Bhandari, J. Hoover, R. Sanderson, K. Kleist, F. Ahmed-Qadri, L. Hendrix, and J. Hillson for their contributions to the development of ALPN-101. We thank H. Konig (Indiana University, USA) and H. Hanenberg (University of Dusseldorf, Germany) for providing us with the human MOLM-14-EGFP cells. Editorial services were provided by N. R. Gough (BioSerendipity, LLC, Elkridge, MD).
Funding: This work was supported by the National Cancer Institute (R01 CA168814 to S.P.), the National Heart Lung and Blood Institute (R01 HL141432 to S.P., T32 HL007910 to D.A., and R01 11879 to B.R.B.), the National Institute of Allergy and Infectious Disease (R37 AI34495 to B.R.B.), and the Leukemia and Lymphoma Society (grant 1293-12 to S.P.). HuPBMC-NSG GVHD studies conducted at JAX were sponsored by Alpine Immune Sciences.
Footnotes
Competing interests: S.P. has a patent (US 20130115232A1 and WO 2013066369A3) on “Methods of detection of graft-versus-host disease” licensed to Viracor-IBT laboratories. B.R.B. reports consulting income from Kamon Pharmaceuticals Inc., Five Prime Therapeutics Inc., Regeneron Pharmaceuticals, Magenta Therapeutics, and BlueRock Therapeuetics and research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences Inc., Abbvie Inc., Leukemia and Lymphoma Society, Childrens’ Cancer Research Fund, and KidsFirst Fund and is a cofounder of Tmunity. S.R.D., K.E.L., L.E., R.W., S.D.L., S.M., J.Y., E.R., M.S., K.H., C.J.G., M.F.W., R.M.S., K.M.S., and S.L.P. are current or former employees and/or shareholders of Alpine Immune Sciences.
Data and materials availability: ALPN-101 and Fc control proteins are available through a material transfer agreement with AIS; direct inquiries to S.R.D. All data associated with this paper are present in the paper and/or in the Supplementary Materials.
REFERENCES AND NOTES
- 1.Zeiser R, Blazar BR, Acute graft-versus-host disease—Biologic process, prevention, and therapy. N. Engl. J. Med 377, 2167–2179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin PJ, McDonald GB, Sanders JE, Anasetti C, Appelbaum FR, Deeg HJ, Nash RA, Petersdorf EW, Hansen JA, Storb R, Increasingly frequent diagnosis of acute gastrointestinal graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant 10, 320–327 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, Zhang Q, Wong CH, Wang H, Chin A, Gomez A, Harris AC, Levine JE, Choi SW, Couriel D, Reddy P, Ferrara JL, Paczesny S, ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N. Engl. J. Med 369, 529–539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald GB, Tabellini L, Storer BE, Lawler RL, Martin PJ, Hansen JA, Plasma biomarkers of acute GVHD and nonrelapse mortality: Predictive value of measurements before GVHD onset and treatment. Blood 126, 113–120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartwell MJ, Ozbek U, Holler E, Renteria AS, Major-Monfried H, Reddy P, Aziz M, Hogan WJ, Ayuk F, Efebera YA, Hexner EO, Bunworasate U, Qayed M, Ordemann R, Wolfl M, Mielke S, Pawarode A, Chen YB, Devine S, Harris AC, Jagasia M, Kitko CL, Litzow MR, Kroger N, Locatelli F, Morales G, Nakamura R, Reshef R, Rosler W, Weber D, Wudhikarn K, Yanik GA, Levine JE, Ferrara JL, An early-biomarker algorithm predicts lethal graft-versus-host disease and survival. JCI Insight 2, e89798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, Liu L, Gomez A, Zhang J, Ramadan A, Zhang Q, Choi SW, Zhang P, Greenson JK, Liu C, Jiang D, Virts E, Kelich SL, Chu HW, Flynn R, Blazar BR, Hanenberg H, Hanash S, Paczesny S, Proteomics analysis reveals a Th17-prone cell population in presymptomatic graft-versus-host disease. JCI Insight 1, e86660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, Liu L, Bakoyannis G, Jiang D, Chu HW, Lobera M, Yang J, Wilkes DS, Du J, Gartlan K, Hill GR, MacDonald KP, Espada EL, Blanco P, Serody JS, Koreth J, Cutler CS, Antin JH, Soiffer RJ, Ritz J, Paczesny S, Blazar BR, An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight 2, e92111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crepeau RL, Ford ML, Challenges and opportunities in targeting the CD28/CTLA-4 pathway in transplantation and autoimmunity. Expert Opin. Biol. Ther 17, 1001–1012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortes-Cerisuelo M, Laurie SJ, Mathews DV, Winterberg PD, Larsen CP, Adams AB, Ford ML, Increased pretransplant frequency of CD28+CD4+TEM predicts belatacept-resistant rejection in human renal transplant recipients. Am. J. Transplant 17, 2350–2362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banchereau J, Steinman RM, Dendritic cells and the control of immunity. Nature 392, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Laouar Y, Welte T, Fu XY, Flavell RA, STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 19, 903–912 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Reizis B, Plasmacytoid dendritic cells: Development, regulation, and function. Immunity 50, 37–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, Schindler C, Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol 184, 2638–2645 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koyama M, Hashimoto D, Aoyama K, Matsuoka K, Karube K, Niiro H, Harada M, Tanimoto M, Akashi K, Teshima T, Plasmacytoid dendritic cells prime alloreactive T cells to mediate graft-versus-host disease as antigen-presenting cells. Blood 113, 2088–2095 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Banovic T, Markey KA, Kuns RD, Olver SD, Raffelt NC, Don AL, Degli-Esposti MA, Engwerda CR, MacDonald KP, Hill GR, Graft-versus-host disease prevents the maturation of plasmacytoid dendritic cells. J. Immunol 182, 912–920 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Markey KA, MacDonald KP, Hill GR, Recipient plasmacytoid DCs are not required to prime allogeneic T-cell responses after BMT. Blood 113, 6038–6039 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Blazar BR, McKenna HJ, Panoskaltsis-Mortari A, Taylor PA, Flt3 ligand (FL) treatment of murine donors does not modify graft-versus-host disease (GVHD) but FL treatmentof recipients post-bone marrow transplantation accelerates GVHD lethality. Biol. Blood Marrow Transplant 7, 197–207 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Covassin L, Laning J, Abdi R, Langevin DL, Phillips NE, Shultz LD, Brehm MA, Human peripheral blood CD4 T cell-engrafted non-obese diabetic-scid IL2rγnull H2-Ab1 tm1Gru Tg (human leucocyte antigen D-related 4) mice: A mouse model of human allogeneic graft-versus-host disease. Clin. Exp. Immunol 166, 269–280 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patton J, Vuyyuru R, Siglin A, Root M, Manser T, Evaluation of the efficiency of human immune system reconstitution in NSG mice and NSG mice containing a human HLA.A2 transgene using hematopoietic stem cells purified from different sources. J. Immunol. Methods 422, 13–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehx G, Somja J, Warnatz HJ, Ritacco C, Hannon M, Delens L, Fransolet G, Delvenne P, Muller J, Beguin Y, Lehrach H, Belle L, Humblet-Baron S, Baron F, Xenogeneic graft-versus-host disease in humanized NSG and NSG-HLA-A2/HHD mice. Front. Immunol 9, 1943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levin SD, Evans LS, Bort S, Rickel E, Lewis KE, Wu RP, Hoover J, MacNeil S, La D, Wolfson MF, Rixon MW, Dillon SR, Kornacker MG, Swanson R, Peng SL, Novel immunomodulatory proteins generated via directed evolution of variant IgSF domains. Front. Immunol 10, 3086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramadan AM, Daguindau E, Rech JC, Chinnaswamy K, Zhang J, Hura GL, Griesenauer B, Bolten Z, Robida A, Larsen M, Stuckey JA, Yang CY, Paczesny S, From proteomics to discovery of first-in-class ST2 inhibitors active in vivo. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Ramadan AM, Griesenauer B, Li W, Turner MJ, Liu C, Kapur R, Hanenberg H, Blazar BR, Tawara I, Paczesny S, ST2 blockade reduces sST2-producing T cells while maintaining protective mST2-expressing T cells during graft-versus-host disease. Sci. Transl. Med 7, 308ra160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, Leong W, Grumet FC, Blume KG, Stockerl-Goldstein KE, Levy R, Shizuru JA, Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell transplant recipients. Blood 99, 1442–1448 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG, Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 285, 412–415 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, Lineburg KE, Cheong M, Robb RJ, Markey KA, Varelias A, Malissen B, Hammerling GJ, Clouston AD, Engwerda CR, Bhat P, Macdonald KP, Hill GR, Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat. Med 18, 135–142 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD, Donor APCs are required for maximal GVHD but not for GVL. Nat. Med 10, 987–992 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Waller EK, Logan BR, Harris WA, Devine SM, Porter DL, Mineishi S, McCarty JM, Gonzalez CE, Spitzer TR, Krijanovski OI, Linenberger ML, Woolfrey A, Howard A, Wu J, Confer DL, Anasetti C, Improved survival after transplantation of more donor plasmacytoid dendritic or naïve T cells from unrelated-donor marrow grafts: Results from BMTCTN 0201. J. Clin. Oncol 32, 2365–2372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonial S, Akhtari M, Kaufman J, Torre C, Lechowicz MJ, Flowers C, Sinha R, Khoury HJ, Langston AA, Waller EK, Mobilization of hematopoietic progenitors from normal donors using the combination of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor results in fewer plasmacytoid dendritic cells in the graft and enhanced donor T cell engraftment with Th1 polarization: Results from a randomized clinical trial. Biol. Blood Marrow Transplant 19, 460–467 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Ramos MI, Perez SG, Aarrass S, Helder B, Broekstra P, Gerlag DM, Reedquist KA, Tak PP, Lebre MC, FMS-related tyrosine kinase 3 ligand (Flt3L)/CD135 axis in rheumatoid arthritis. Arthritis Res. Ther 15, R209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu L, A Flt3L encounter: mTOR signaling in dendritic cells. Immunity 33, 580–582 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Ferrara JL, Cytokine dysregulation as a mechanism of graft versus host disease. Curr. Opin. Immunol 5, 794–799 (1993). [DOI] [PubMed] [Google Scholar]
- 33.Despoix N, Walzer T, Jouve N, Blot-Chabaud M, Bardin N, Paul P, Lyonnet L, Vivier E, Dignat-George F, Vely F, Mouse CD146/MCAM is a marker of natural killer cell maturation. Eur. J. Immunol 38, 2855–2864 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Li J, Semple K, Suh W-K, Liu C, Chen F, Blazar BR, Yu X-Z, Roles of CD28, CTLA4, and inducible costimulator in acute graft-versus-host disease in mice. Biol. Blood Marrow Transplant 17, 962–969 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betjes MG, Clinical consequences of circulating CD28-negative T cells for solid organ transplantation. Transpl. Int 29, 274–284 (2016). [DOI] [PubMed] [Google Scholar]
- 36.de Graav GN, Hesselink DA, Dieterich M, Kraaijeveld R, Weimar W, Baan CC, Down-regulation of surface CD28 under belatacept treatment: An escape mechanism for antigen-reactive T-Cells. PLOS ONE 11, e0148604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaitsu M, Issa F, Hester J, Vanhove B, Wood KJ, Selective blockade of CD28 on human T cells facilitates regulation of alloimmune responses. JCI Insight 2, e89381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor PA, Panoskaltsis-Mortari A, Freeman GJ, Sharpe AH, Noelle RJ, Rudensky AY, Mak TW, Serody JS, Blazar BR, Targeting of inducible costimulator (ICOS) expressed on alloreactive T cells down-regulates graft-versus-host disease (GVHD) and facilitates engraftment of allogeneic bone marrow (BM). Blood 105, 3372–3380 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Koura DT, Horan JT, Langston AA, Qayed M, Mehta A, Khoury HJ, Harvey RD, Suessmuth Y, Couture C, Carr J, Grizzle A, Johnson HR, Cheeseman JA, Conger JA, Robertson J, Stempora L, Johnson BE, Garrett A, Kirk AD, Larsen CP, Waller EK, Kean LS, In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: A first-in-disease trial. Biol. Blood Marrow Transplant 19, 1638–1649 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Watkins BK, Tkachev V, Furlan SN, Hunt DJ, Betz K, Yu A, Brown M, Poirier N, Zheng HB, Taraseviciute A, Colonna L, Mary C, Blancho G, Soulillou JP, Panoskaltsis-Mortari A, Sharma P, Garcia A, Strobert E, Hamby K, Garrett A, Deane T, Blazar BR, Vanhove B, Kean LS, CD28 blockade controls T cell activation to prevent graft-versus-host disease in primates. J. Clin. Invest 128, 3991–4007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, Ferrara JL, An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 88, 3230–3239 (1996). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. pDCs and cDCs gating strategy.
Fig. S2. Frequencies of total DCs and cDCs in patient cohort.
Fig. S3. Frequencies of ICOSL+ cDCs in patient cohort.
Fig. S4. Frequencies of ICOSL+ pDCs in lower and upper GI-GVHD.
Fig. S5. Correlative and ROC analyses in patients with GVHD and GI-GVHD.
Fig. S6. Frequencies and absolute counts of murine CD11b−CD11c+CD103+B220+ splenic pDCs in a major mismatch model.
Fig. S7. Splenic Th17 cells coexpressing CXCR3 and RORγt in a major mismatch murine model.
Fig. S8. Frequencies of murine intestinal CD11b−CD11c+CD103+B220+ pDCs in a haploidentical allogeneic model.
Fig. S9. Frequencies of IFNγ/IL-17–producing T cells in the gut in a haploidentical allogeneic model.
Fig. S10. Cell population pretransplant, and model of transplant.
Fig. S11. Frequencies of human T cells and human pDCs in spleens of NSG mice receiving unstimulated or ICOSL+ pDCs.
Fig. S12. Schematic of the strategy used to generate the therapeutic lead Fc fusion protein designated ALPN-101.
Fig. S13. ALPN-101 potently suppresses human T cell responses in vitro.
Fig. S14. Human cytokine concentrations measured in terminal serum collected from mice in all treatment groups in the human PBMC-NSG GvHD study.
Fig. S15. Evaluation of the binding of anti-CD28 and anti-ICOS antagonist Fc fusion proteins to Jurkat cells expressing endogenous CD28 or transfected chimeric ICOS-CD28 molecules.
Fig. S16. Blockade of CD28- or ICOS-mediated costimulation by ALPN-101, anti-CD28, anti-ICOS, combination anti-CD28+ anti-ICOS, or belatacept.
Fig. S17. Dual blockade of CD28 and ICOS or CD28 alone prevents GVHD in the human PBMC-NSG model.
Fig. S18. Percent change in BW and flow analysis of T cells in blood from human PBMC-NSG mice treated with ALPN-101 with or without CsA.
Fig. S19. Frequency and absolute counts of human infiltrating splenic cells in the peri-transplant and prophylactic human PBMC-NSG study.
Fig. S20. Prophylactic blockade of ICOS/CD28 on donor pDCs using ALPN-101 in an aggressive xenogeneic GVHD model.
Table S1. Characteristics of the study patients.
Table S2. Causes of overall mortality in patients with GVHD symptoms (n = 117).
Table S3. Relative affinities of ALPN-101 to human CD28, ICOS, and CTLA-4 measured by BLI analysis.
Table S4. Summary of protocols used for the human PBMC-NSG GVHD studies.
Table S5. Final circulating T cell counts and statistical analysis for the GVHD survival, DAI, and BW loss data.
Table S6. Statistical analysis of terminal serum human cytokine concentrations in mice treated with repeat 100-μg doses of ALPN-101 or belatacept, or a single 100-μg dose of ALPN-101.
Table S7. Summary of statistical significance for survival between treatment groups in the human PBMC-NSG GVHD study.
Table S8. Additional correlation analyses of the flow cytometric data from the human PBMC-NSG GVHD study.
Table S9. Summary of statistical significance for survival between treatment groups in the human PBMC-NSG GVHD study.
Data file S1. Transcriptome analysis of sorted intestinal WT versus ICOSL−/− pDCs.
Data file S2. Transcriptome analysis of sorted intestinal WT versus ICOSL−/− CD4+Foxp3− T cells.