

Abstract
In a conjugated polymer based single-particle heterojunction, stochastic fluctuations of the photogenerated hole population lead to spontaneous fluorescence switching. We found that 405 nm irradiation can induce charge recombination and activate the single-particle emission. Based on these phenomena, we developed a novel class of semiconducting polymer dots that can operate in two superresolution imaging modes. The spontaneous switching mode offers efficient imaging of large areas, with <10 nm localization precision, while the photoactivation/deactivation mode offers slower imaging, with further improved localization precision (~1 nm), showing advantages in resolving small structures that require high spatial resolution. Superresolution imaging of microtubules and clathrin-coated pits was demonstrated, under both modes. The excellent localization precision and versatile imaging options provided by these nanoparticles offer clear advantages for imaging of various biological systems.
Keywords: Fluorescent Probes, Photoactivation, Photoswitching, Polymers, Superresolution Imaging
Graphical Abstract

By controlling the population of photogenerated holes, we demonstrated dual-mode superresolution imaging with semiconducting polymer dots. The localization precision achieved is 3–8 times better than the typical resolution obtained by localization of dyes and fluorescent proteins, which allows the hollow structure of microtubules to be resolved.
Introduction
Superresolution imaging methods are powerful tools for studying biological structures and processes beyond the diffraction limit.[1] The performance of single particle localization based superresolution methods depends critically on the photophysical properties of the fluorescent probe used. In order to resolve one fluorophore at a time, the fluorescent probe needs to exhibit either photoactivation behavior or stochastic photoswitching.[1e, 2] These requirements limit the choice of fluorescent probes for superresolution imaging, and in certain wavelength ranges the performance of available fluorescent probes is less than optimal. Therefore, there is considerable interest in the development of improved fluorescent probes and photoactivation/photoswitching mechanisms for superresolution microscopy.
Semiconducting polymer based fluorescent probes exhibit high brightness and good photostability.[3] Their applications, however, are mostly limited to conventional microscopy and non-localization based superresolution microscopy (reported spatial resolution: 70–150 nm) due to lack of a photoactivation or stochastic switching mechanism.[4] Semiconducting polymers exhibit spontaneous blinking through photo-generation and elimination of holes, which are known to be fluorescence quenchers.[5] However, hole generation in neat semiconducting polymers typically involves photoinduced electron transfer to molecular oxygen or defects.[6] Such processes are not well controlled, which makes it difficult to optimize the optical properties for superresolution imaging.
In this work, we developed a novel approach to control charge carrier generation and recombination dynamics in semiconducting polymers, which forms the basis for a new class of ultrabright, photoactivatable single-nanoparticle photoswitches for superresolution microscopy. Semiconducting polymers developed for use in light-emitting devices were mixed with an electron-accepting polymer (poly[(N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl)-alt-5,5′-(2,2′-bithiophene)] (PNDI-2T) or its derivative) to form nanoparticles. In these nanoscale heterojunctions, charge carriers were efficiently generated through photoinduced electron transfer. The large number of holes accumulated in the electron-donating polymer phase resulted in nearly complete quenching of single-particle emission. However, stochastic fluctuation of the hole population due to recombination led to occasional fluorescence bursts.[7] We found that the single-particle emission can be effectively activated by 405 nm excitation, likely through reduction of the hole population in the nanoparticles caused by a photoinduced recombination process. The discrete switching and light-induced activation phenomena were exploited for superresolution microscopy. Four kinds of semiconducting polymer dots (Pdots) of different emission colors were prepared and evaluated for their performance in superresolution imaging. We demonstrated that, under different imaging settings, the Pdots operate in two imaging modes that suit different application scenarios. In the spontaneous switching mode, 405 nm excitation was only used when necessary, at low power, to adjust blinking duty cycle, the Pdots cycle through “on” and “off” states spontaneously and offer efficient superresolution imaging of large areas/structures, with <10 nm localization precision. In the photoactivation/deactivation mode, periodic 405 nm laser pulses were used to randomly activate the Pdots, the Pdots cycle through “on” and “off” states slower, but further improve the expected localization precision to as high as ~1 nm, showing advantages in resolving small structures that require high spatial resolution. In either mode, the Pdots were able to resolve microtubule (MT) networks and the hollow structure of clathrin-coated pits (CCPs). Two-color superresolution imaging was also demonstrated. Overall, the optimal localization precision achieved is 3–8 times better than the typical resolution obtained by localization of dyes and photoactivatable fluorescent proteins.[2, 8] The excellent brightness and versatile imaging options provided by these nanoparticles offer clear advantages for imaging of various biological systems.
Results and Discussion
Pdot Preparation and Characterization
The Pdots were prepared using a nanoprecipitation method described previously.[3a, 9] We studied four kinds of semiconducting polymers that are used in fluorescent nanoparticles, polymer light-emitting devices and photovoltaic devices: poly[(9,9-dihexyl-2,7-(2-cyanodivinylene)-fluorenylenyl-2,7-diyl)] (CN-PDHFV), poly[(9,9-dioctyl-2,7-divinylenefluorenylene)-alt-co-{2-methoxy5-(2-ethylhexyloxy)-1,4-phenylene}] (PFPV), poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadiazole)] (F8BT), and poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-(1-cyanovinylene-1,4-phenylene)] (CN-PPV) (Figure 1a). Each polymer was blended with the electron-accepting polymer PNDI-2T or its derivative, poly[(N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl)-alt-5,5′-(3,3′-difluoro-2,2′-bithiophene)] (PNDI-2F) (Figure 1a). The choice of electron-accepting polymer (PNDI-2T or PNDI-2F) was based on the energy levels of the light-emitting polymer to ensure efficient charge generation. Each light-emitting polymer was co-precipitated with the corresponding electron-accepting polymer as well as with poly(styrene-co-maleic anhydride) (PSMA) to form single-particle type II heterojunctions.[10] PSMA hydrolyzes during the co-precipitation process, providing handles for the downstream bioconjugation.[11] The resulting Pdot size was typically ~15 nm based on dynamic light scattering (DLS) (Figure 1b) and transmission electron microscopy (TEM) measurements (Figure S1).
Figure 1.
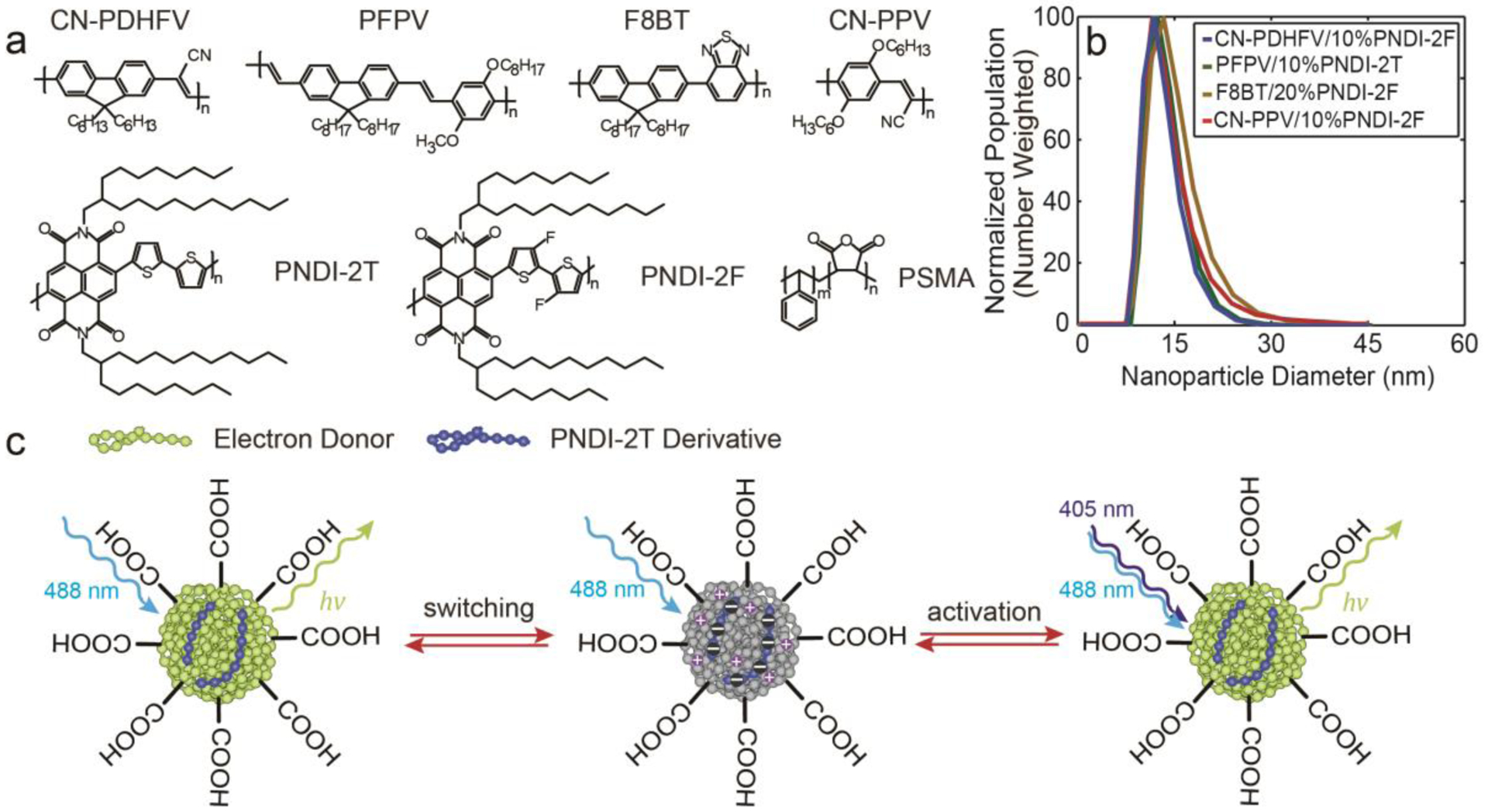
a) Chemical structures of CN-PDHFV, PFPV, F8BT, CN-PPV, PNDI-2T, PNDI-2F, and PSMA. b) Number-weighted particle size distributions of 10% PNDI-2F-doped CN-PDHFV Pdots, 10% PNDI-2T-doped PFPV Pdots, 20% PNDI-2F-doped F8BT Pdots, and 10% PNDI-2F-doped CN-PPV Pdots, determined using DLS. The corresponding particle sizes were 14.5±5.1 nm, 13.4±4.6 nm, 17.2±6.5 nm, 15.9±5.4 nm, respectively. c) Illustration of the photoswitching and photoactivation mechanisms of the Pdots.
Photophysical Properties of the Pdots
To characterize the photophysical properties of the Pdots, single-particle imaging was performed with the Pdots immobilized on glass coverslips (Figure 2a–h). The Pdots were excited close to their absorption maximum (typical excitation power density, ~2 kW/cm2) and imaged in oxygen scavenging glucose oxidase (GLOX) buffer for 10 min, at a 100 Hz frame rate. In contrast to undoped Pdots, Pdots that contained PNDI-2T or PNDI-2F exhibited pronounced photoblinking. The photophysical properties of the Pdots were found to be highly sensitive to the electron acceptor doping concentration. When the electron acceptor content is too low, the quencher population is not sufficient to completely quench the single-particle emission, thus the fluorescence intensity of the Pdots fluctuates between multiple levels without turning “off”. When the electron acceptor content is too high, the frequency of “on” events becomes too low for imaging. Therefore, for each light-emitting polymer, we tested a range of electron acceptor doping levels and chose a level that offered robust “on” and “off” switching with a suitable frequency. The duration of “on” events typically ranged from milliseconds to several seconds, whereas “off” events typically lasted tens of seconds (Figure 2i). It should be noted that the timescales of charge separation and geminate recombination at the electron donor/acceptor interface should be well below our experimental temporal resolution.[12] The slow blinking dynamics that we observed is likely due to accumulation and elimination of holes occurring at charge carrier traps in the host polymer. Thus, we hypothesize that the “on” and “off” transitions reflect fluctuations in the population of trapped holes in the Pdots. To qualitatively visualize this effect, we simulated stochastic hole polaron population fluctuation using a polaron generation rate determined from the single-step “on” to “off” transitions (Figure S2–4) and a recombination rate estimated from previously reported hole polaron lifetimes in semiconducting polymers (Figure S5; details of modeling and analysis are provided in the supporting information).[5c, 13] Based on previously estimated hole polaron quenching efficiency (~10%) in Pdots of similar sizes,[13–14] we assumed that roughly 10 polarons can completely quench the single-particle emission. As shown in Figure 2j, k, under the simulated experimental conditions, the hole polaron population is typically more than enough to completely quench the single-particle fluorescence, and only low probability large fluctuations in the quencher population lead to rare fluorescence bursts.
Figure 2.
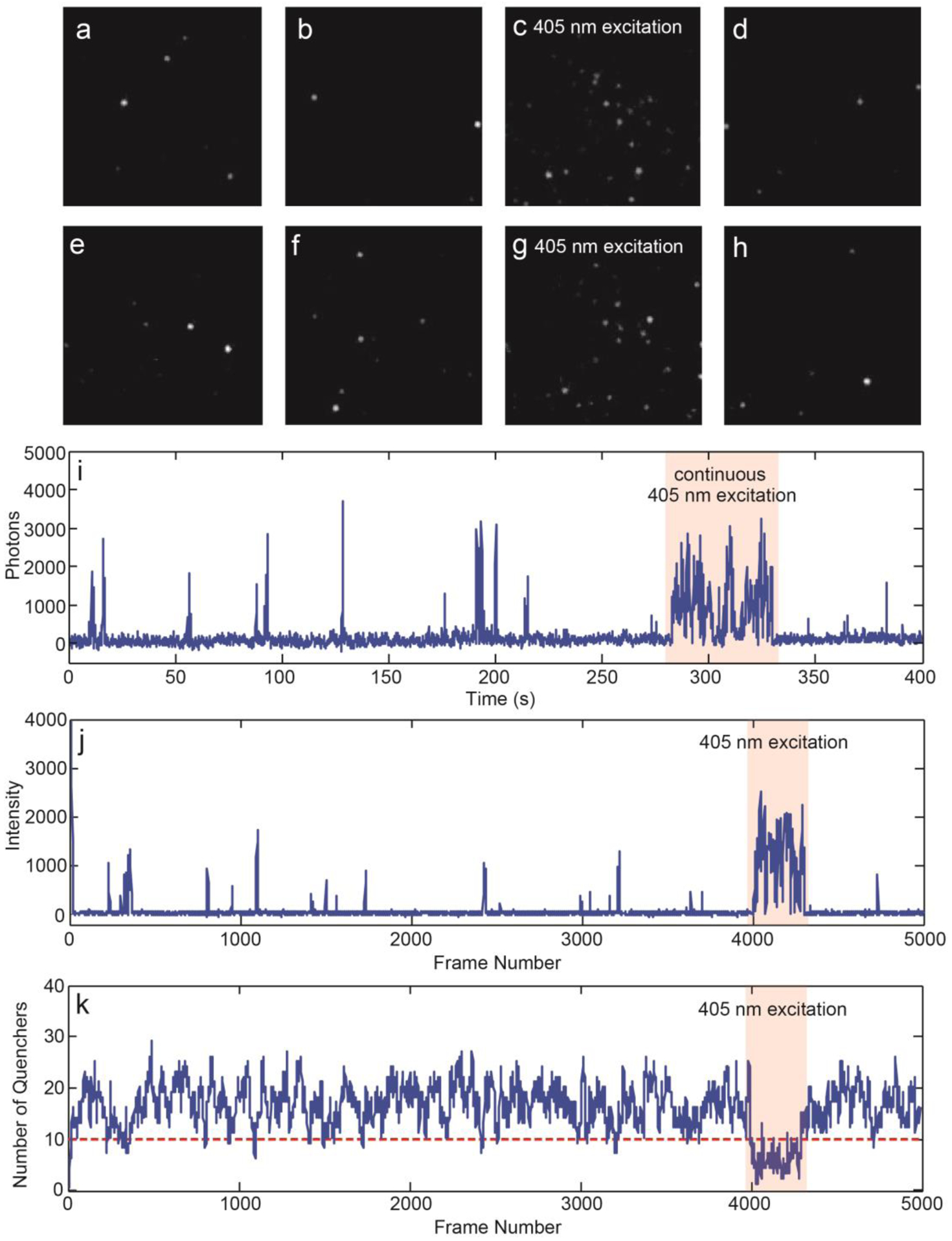
a-h) Fluorescence microscopy image sequence, showing photoblinking and photoactivation behavior of 10% PNDI-2F-doped CN-PPV Pdots. i) Singe-particle fluorescence intensity trajectory of 10% PNDI-2F-doped CN-PPV Pdots. Photoactivation behavior is highlighted by the red band. j) Simulated fluorescence intensity trajectory of 10% PNDI-2F-doped CN-PPV Pdots. k) Corresponding quencher population fluctuation trajectory used to simulate the fluorescence intensity trajectory shown in panel j.
In addition to spontaneous photoblinking, we found that the single-particle emission could be activated by 405 nm excitation (Figure 2a–h). For each activation cycle, a random set of Pdots were activated. The activation effect can last for seconds after turning off 405 nm excitation, after which the Pdots revert back to their normal duty cycle. It is likely that 405 nm excitation reduces the hole polaron population in the Pdots and increases the probability of switching “on” events (Figure 2j, k). It should be noted that most of the light-emitting polymers studied here have weak absorption at 405 nm, whereas PNDI-2T and PNDI-2F exhibit a two-peak absorption spectrum with peaks at 700 nm and 390 nm, corresponding to transitions from S0 to S1 and to a higher delocalized state, respectively.[15] In contrast to the pronounced photoactivation phenomenon observed under 405 nm excitation, exciting PNDI-2T or its derivative to the S1 state did not result in observable photoactivation of the Pdots. Considering the energy level alignment of the system (Figure 3a–d), one possibility is that PNDI-2T and PNDI-2F in the higher excited state transfer electrons to the lowest unoccupied molecular orbital (LUMO) of the light-emitting polymers’ radical cations, which results in elimination of holes in the light-emitting polymer phase. This process may be facilitated by the delocalized nature of the higher excited state, as indicated by ultrafast pump-push studies.[16] It is also possible that the higher excited state of PNDI-2T or PNDI-2F participates in an Auger recombination process to detrapped electron in the PNDI-2T/PNDI-2F phase.[17] Escaped electrons could recombine with holes at the interface and reduce the hole population. Overall, the photoactivation mechanism is likely complex with multiple possible origins, and requires further ultrafast and single-molecule investigations to elucidate in more detail.
Figure 3.
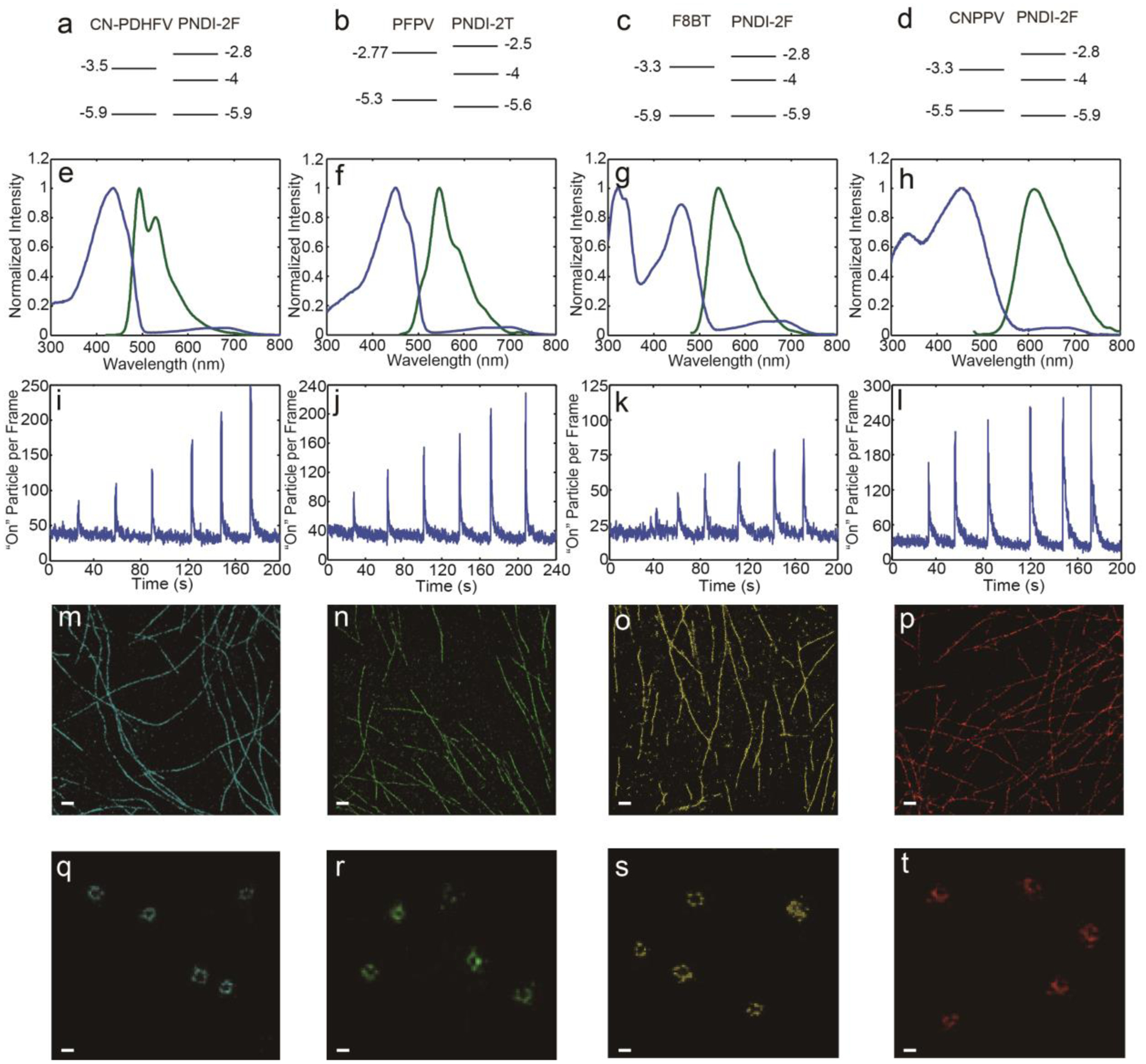
a-d) Energy level alignment of a) 10% PNDI-2F-doped CN-PDHFV Pdots, b) 10% PNDI-2T-doped PFPV Pdots, c) 20% PNDI-2F-doped F8BT Pdots, and d) 10% PNDI-2F-doped CN-PPV Pdots, plotted according to previously reported HOMO and LUMO energies of CN-PDHFV,[18] PFPV,[19] F8BT,[20] CN-PPV,[21] PNDI-2T,[22] and PNDI-2F.[23] e-h) Absorption (blue) and fluorescence spectra (green) of e) 10% PNDI-2F-doped CN-PDHFV Pdots, f) 10% PNDI-2T-doped PFPV Pdots, g) 20% PNDI-2F-doped F8BT Pdots, and h) 10% PNDI-2F-doped CN-PPV Pdots. i-l) Switched “on” particle per frame trajectories of i) 10% PNDI-2F-doped CN-PDHFV Pdots, j) 10% PNDI-2T-doped PFPV Pdots, k) 20% PNDI-2F-doped F8BT Pdots, and l) 10% PNDI-2F-doped CN-PPV Pdots, acquired after duty cycle induction. The six peaks from left to right correspond to 405 nm laser activation at 15, 30, 60, 120, 240, and 480 W/cm2, respectively. m-p) Reconstructed superresolution images of MTs obtained using m) 10% PNDI-2F-doped CN-PDHFV Pdots, n) 10% PNDI-2T-doped PFPV Pdots, o) 20% PNDI-2F-doped F8BT Pdots, and p) 10% PNDI-2F-doped CN-PPV Pdots (scale bars, 1 μm), under spontaneous switching mode. q-t) Reconstructed superresolution images of CCPs obtained using q) 10% PNDI-2F-doped CN-PDHFV Pdots, r) 10% PNDI-2T-doped PFPV Pdots, s) 20% PNDI-2F-doped F8BT Pdots, and t) 10% PNDI-2F-doped CN-PPV Pdots (scale bars, 100 nm), under activation/deactivation mode.
Above, we discussed some general properties of the PNDI-2T or PNDI-2F doped Pdots. The performance of the PNDI-2T- and PNDI-2F-doped Pdots varied depending on the composition, as discussed below. For each light-emitting polymer, we tested a range of electron acceptor polymer doping levels and chose a level that offered optimum photophysical properties. Based on these results, we chose four kinds of Pdots for further investigation: CN-PDHFV doped with 10% PNDI-2F, PFPV doped with 10% PNDI-2T, F8BT doped with 20% PNDI-2F, and CN-PPV doped with 10% PNDI-2F. The performance of these Pdots was first characterized in the spontaneous switching mode. When the Pdots were first exposed to excitation, most were switched “on” at the beginning. The fluorescence intensity then rapidly decayed for hundreds of milliseconds to a few seconds, and “on”/“off” blinking behavior started to occur. The “on” event frequency (duty cycle) also decreased for up to tens of seconds until reaching a steady state. The initial fluorescence intensity decay and duty cycle induction likely reflect the growth of the quencher population from zero to a steady-state value (Figure S5; details of modeling and analysis are provided in the supporting information). To determine the duty cycle induction time of the Pdots, we analyzed the single-particle fluorescence intensity trajectories with a sliding time window of 100 s to determine when the duty cycle stabilized. The induction times and duty cycles determined under 2 kW/cm2 excitation power density are summarized in Table 1. The steady-state duty cycles are in the same range as those of typical dyes used in stochastic optical reconstruction microscopy.[2] Typically, a long induction time was observed together with a high steady-state blinking duty cycle, both indicating less efficient charge generation in the Pdots. Of the Pdots tested, F8BT doped with 20% PNDI-2F exhibited the longest induction time and the highest duty cycle. 10% PNDI-2T-doped PFPV Pdots exhibited the lowest steady-state duty cycle and equilibrated quickly upon exposure to excitation, indicating very efficient charge generation and transport to traps throughout the Pdots. 10% PNDI-2F-doped CN-PDHFV and 10% PNDI-2F-doped CN-PPV Pdots showed intermediate performance, with an induction time of tens of seconds and a duty cycle of 1–2×10−3. To measure photostability, we estimated the fraction of surviving Pdots after imaging for 600 s (Table 1, Figure S6; procedures provided in the supporting information). 10% PNDI-2F-doped CN-PDHFV and 20% PNDI-2F-doped F8BT Pdots both yielded ~80% survival fraction after 600 s imaging, indicating good photostability and promising performance for superresolution microscopy. In addition to duty cycle and photostability, another important parameter of a superresolution fluorescence probe is the photon number per switching cycle, which dictates localization precision. Of the four Pdots tested, 20% PNDI-2F-doped F8BT Pdots yielded the highest number of photons per switching event (Table 1). All of the Pdots exhibited good brightness with typically a few thousand photons detected per “on” event (Table 1), yielding <10 nm theoretical and <15 nm experimental localization uncertainty (Figure S7, Table S1, localization analysis provided in the supporting information).
Table 1.
Photophysical properties of the Pdots
| 10% PNDI-2F-doped CN-PDHFV Pdots | 10% PNDI-2T-doped PFPV Pdots | 20% PNDI-2F-doped F8BT Pdots | 10% PNDI-2F-doped CN-PPV Pdots | |
|---|---|---|---|---|
| Duty cycle induction time (s) | 28±16 | 14±5 | 108±72 | 42±28 |
| Equilibrium duty cycle | 1.4±0.6×10−3 | 0.9±0.4×10−3 | 3.2±2.2×10−3 | 1.9±1.1×10−3 |
| Photon number per cycle in switching mode | 4.2±3.7×103 | 3.2±2.9×103 | 6.2±6.4×103 | 2.8±2.5×103 |
| Photon number per cycle in activation mode | 1.1±0.7×104 | 8.2±5.3×103 | 1.6±1.3×104 | 7.8±4.7×103 |
| Survival fraction after 600 s | 85% | 65% | 78% | 55% |
In the activation/deactivation mode, the Pdots were placed under a constant excitation that is close to their absorption maximum (typical power density, ~4 kW/cm2), in GLOX buffer containing 1% beta-mercaptoethanol (BME). This condition offers further improved activation/deactivation reversibility (Figure S8) and reduced spontaneous blinking.[7] To test the photoactivation response, the Pdots were activated under six different 405 nm intensities ranging from 15 to 480 W/cm2 (Figure 3i–l). For each intensity, the Pdots were exposed to a 405 nm laser for 0.5 s. For each type of Pdots, a clear photoactivation response was observed that an increased number of Pdots were switched “on” per frame. Depending on the Pdots, it took up to 10 s for the Pdot to return to the equilibrium. The number of detected photon per activation event was dictated by the activation laser intensity. Stronger activation laser pulse leads to more charge carrier recombined, thus results in stronger Pdots emission and longer “on” state duration. However, activation laser pulse that is too strong results in many Pdots activated in a diffraction limited spot, which complicates the analysis. In imaging of biological samples, the intensity of the periodic activation laser pulses was adjusted to ensure appropriate “on” Pdots density, while the activation laser frequency was adjusted so that for each cycle, all the Pdots return to the equilibrium before the next activation pulse arrive. Typically, the activation/deactivation mode takes 2–3 times longer imaging time to acquire the same amount of localization points as the spontaneous switching mode. The “on” state photon number in table 1 was determined at the typical activation laser power for each Pdot, which are several fold higher than those obtained under the spontaneous switching mode (Figure S9), yielding 1.3–2.5 nm theoretical and 3.2–5.6 nm experimental localization uncertainty (Table S1, localization analysis provided in the supporting information). If imaging efficiency is a concern, higher activation laser power can be used to accelerate data acquisition in the activation/deactivation mode. The acquired data requires additional processing to reduce clustering artifacts (multiple switched “on” Pdots in a diffraction limited spot). Instead of directly performing localization analysis with raw images, a frame-by-frame subtraction of images needs to be performed for each activation cycle. Since Pdots switching “off” is a stochastic process, frame-by-frame subtraction separates out Pdots that are simultaneously activated in a diffraction limited spot. Localization analysis can then be performed with the subtracted images. This method has been previously used to increase duty cycle tolerance and extend the choice of fluorophores for superresolution microscopy.[1c] An alternative method to reduce clustering artifacts is to check point spread function widths in each image and reject the ones that are above a certain threshold, which are potentially from multiple Pdots.
Superresolution Imaging of MTs and CCPs
We used MTs and CCPs as model systems to evaluate the performance of the four Pdots in superresolution imaging. The Pdots were conjugated to streptavidin or to a secondary antibody via a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide-catalyzed reaction.[10b, 11] BS-C-1 cells labeled with Pdots were excited close to the Pdot absorption maximum, with a typical excitation power density of 2–4 kW/cm2, depending on the imagine mode. The experimental frame rate was set to closely match the “on” duration of the Pdots. GLOX buffer was used for the spontaneous switching mode, while GLOX buffer containing 1% BME was used for the activation/deactivation mode. For the spontaneous switching mode, the 405 nm laser was only used when necessary, at low power, to adjust the blinking duty cycle and ensure imaging efficiency, while for the activation/deactivation mode, periodic 405 nm laser pulses were used to randomly activate Pdots. The Pdots exhibited a good capability for resolving 2D structures of MTs, in both the spontaneous switching (Figure 3m–p) and activation/deactivation modes (Figure S10). However, the activation/deactivation mode requires longer imaging time (20 mins) to acquire the same quality image as the spontaneous switching mode (5–10 min). To resolve the structure of CCPs, we adapted a 3D astigmatism scheme described previously.[1b] Basically, a cylindrical lens with long focal length was introduced into the detection light path. As a result, the imaging focal plane was slightly different along the two axes. The axial position was indicated by the shape of the single-particle point spread function; the calibration curve (Figure S11) was determined by imaging Pdots immobilized on a coverslip. During imaging, a commercial autofocus system was engaged to minimize axial focus drift. In this case, the Pdots performs better in the activation/deactivation mode, providing clearly resolved hollow structure of CCPs (Figure 3q–t). Hollow structures were also observed in the images acquired under the spontaneous switching mode (Figure S10). However, due to the lower localization precision, the images were typically not as clear and in some cases, the fraction of resolvable hollows was lower. In addition to localization precision, the quality of superresolution images is also affected by labeling density, which is related to the size of the labels. However, it should be noted that superresolution studies typically involve immunostaining using Immunoglobulin G (IgG) antibodies, which have a comparable size (14.5 nm between the two arms) as the Pdots. For immunostained MTs and CCPs, the inner layer consists of IgG antibodies bond to target proteins, while the outer layer consists of Pdots linked to antibodies (Figure S12, additional modeling and discussion is provided in the supporting information). Considering the sizes of IgGs and Pdots, as well as the structure of immunostained MTs and CCPs, we concluded that, in our studies, Pdots did not result in significantly lower labeling density as compared to dyes (Figure S12, Figure S13). In the rare situation that requires extremely high labeling density, nanobodies are typically used instead of IgG antibodies. In such case, Pdot size can be conveniently reduced using the cross-flow filtration method that we previously reported to suit the need of the application.[5c]
Here, we provided two examples of 3D projection images of MTs and CCPs, which were acquired with 10% PNDI-2F-doped CN-PDHFV Pdots (Figure 4a, b), under spontaneous switching mode. Different colors in the images indicate different axial positions. Examination of closely packed MT filaments revealed sub-diffraction limit resolution. The two white rectangles in Figure 4a highlight MTs that are close together along Z axis and X, Y plane, respectively. The corresponding axial and lateral intersections were plotted in Figure 4c, d. In both cases, MTs separated by ~100 nm can be clearly differentiated (the difference in distribution widths of MTs in Figure 4c, d is due to the two-fold difference in the lateral and axial localization precision of the astigmatism method, as shown in Figure S7). Under the activation/deactivation mode, the lateral resolution of 10% PNDI-2F-doped CN-PDHFV Pdots can be further improved. To quantitatively compare the resolution of the two superresolution imaging modes and confocal imaging, we provided zoomed in images of single MT filaments resolved using the three methods (Figure 4 e, f, g, additional images were provided in the supporting information, Figure S14). Analysis of the MT cross-sections indicates that the hollow MT structure was clearly resolved by the activation/deactivation mode and was partially resolved by the spontaneous switching mode (Figure 4 h). In contrast, the cross-section profile of the MT in the confocal image was broad and featureless, suggesting that confocal imaging was unable to resolve the fine structure of MTs. Analysis of individual peaks in the cross-section plot indicates that the lateral resolutions achieved under activation/deactivation, spontaneous switching and confocal imaging were 4.3, 10.3, and 300 nm, respectively. Both superresolution imaging modes yielded a peak separation of 45 nm, consistent with the sizes of MTs, IgGs and Pdots.
Figure 4.
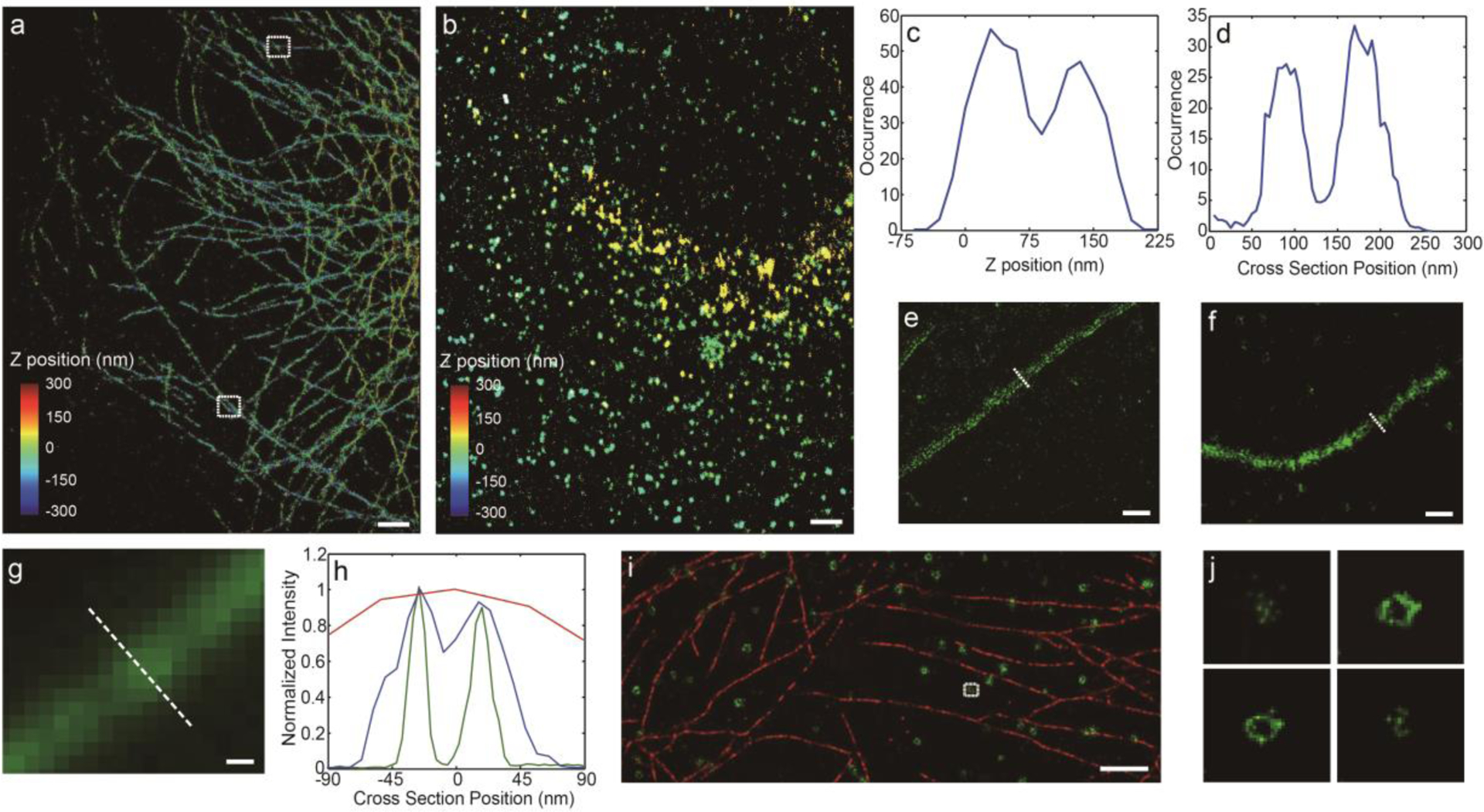
a), b) 3D projection images of a) MTs and b) CCPs, obtained using 10% PNDI-2F-doped CN-PDHFV Pdots (scale bar, 5 μm), under spontaneous switching mode. c) Z intersection plot of MTs, corresponding to the upper rectangle in panel a. d) Lateral intersection plot of MTs, corresponding to the lower rectangle in panel a. e), f) Lateral slices of 3D superresolution image of MTs labeled with 10% PNDI-2F-doped CN-PDHFV Pdots, acquired under e) activation/deactivation and f) spontaneous switching mode. Scale bar, 100 nm. g) Confocal image of MTs, scale bar 100 nm. h) Lateral intersection plots of MTs, corresponding to the white dashed line in panel e (green), f (blue) and g (red), showing clearly resolved (green), partially resolve (blue) and completely unresolved hollow structure (red). Analysis revealed that the lateral resolutions achieved under activation/deactivation, spontaneous switching and confocal imaging were 4.3, 10.3, and 300 nm, respectively. Both superresolution imaging modes yielded a peak separation of 45 nm, consistent with the sizes of MTs, IgGs and Pdots. i) Two-color superresolution images of MTs (red) and CCPs (green). 10% PNDI-2F-doped CN-PPV Pdots were used to label MTs, and 10% PNDI-2F-doped CN-PDHFV Pdots were used to label CCPs. Scale bar, 5 μm. j) A series of lateral slices of the CCP highlighted in panel i.
Based on the emission spectra, 10% PNDI-2F-doped CN-PDHFV and 10% PNDI-2F-doped CN-PPV Pdots were chosen to demonstrate two-color imaging. 10% PNDI-2F-doped CN-PPV Pdots were used to label MTs, and 10% PNDI-2F-doped CN-PDHFV Pdots were used to label CCPs. With appropriate emission filters, these two Pdots showed minimal cross-talk in imaging. One lateral slice of the two-color image is plotted in Figure 4i, showing clearly resolved MT and ring-shaped CCP structures. For the highlighted CCP, we plotted lateral slices at four different axial positions (Figure 4j). At the top and bottom of the CCP, we observed a small cluster of localized points; in the middle, a ring-shaped structure can be observed, consistent with the hollow spherical structure of CCPs.[1b]
Conclusion
In this work, we described a new class of semiconducting polymer-based photoswitches. Semiconducting polymers that are commonly used in fluorescence imaging were co-precipitated with electron-accepting polymers to form nanoparticles. Charge carriers were efficiently generated in these nanoscale heterojunctions through photoinduced electron transfer. The single-particle emission was almost completely quenched by the large number of accumulated hole polarons. However, stochastic fluctuation of the hole population led to occasional bursts of fluorescence. We found that 405 nm excitation effectively reduced the charge carrier population in Pdots, providing a facile method for activating single-particle emission. We designed four kinds of Pdots based on these phenomena and characterized their performance in superresolution imaging. The Pdots can operate in two superresolution imaging modes that suit different application scenarios. The spontaneous switching mode offers efficient, superresolution imaging of large areas, with <10 nm localization precision, while the photoactivation/deactivation mode offers slower imaging, with further improved localization precision (~1 nm), showing clear advantages in resolving small structures that requires high spatial resolution. MT networks as well as the hollow structure of CCPs were clearly resolved using the Pdots, under both modes. Overall, the optimal localization precision that we achieved is 3–8 times better than the typical resolution obtained by localization of dyes and photoactivatable fluorescent proteins. The strategy described here should serve as a general method for designing semiconducting polymer-based superresolution probes, providing a basis for a new class of fluorescent nanoparticles for use in superresolution microscopy.
Supplementary Material
Acknowledgments
This study was supported by NSF grant CHE-1412694 and NIH grant R01MH113333.
Footnotes
Conflicts of interest
The authors declare no competing financial interest.
References
- [1].a) Rust MJ, Bates M, Zhuang X, Nat. Methods 2006, 3, 793–796; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang B, Wang W, Bates M, Zhuang X, Science 2008, 319, 810–813; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Burnette DT, Sengupta P, Dai Y, Lippincott-Schwartz J, Kachar B, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 21081–21086; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang W, Shen H, Shuang B, Hoener BS, Tauzin LJ, Moringo NA, Kelly KF, Landes CF, J. Phys. Chem. Lett 2016, 7, 4524–4529; [DOI] [PubMed] [Google Scholar]; e) Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF, Science 2006, 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- [2].Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X, Nat. Methods 2011, 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Wu C, Bull B, Szymanski C, Christensen K, McNeill J, ACS Nano 2008, 2, 2415–2423; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Traina CA, Bakus RC, Bazan GC, J. Am. Chem. Soc 2011, 133, 12600–12607; [DOI] [PubMed] [Google Scholar]; c) Jiang Y, McNeill J, Chem. Rev 2017, 117, 838–859; [DOI] [PubMed] [Google Scholar]; d) Chen H, Zhang J, Chang K, Men X, Fang X, Zhou L, Li D, Gao D, Yin S, Zhang X, Biomaterials 2017, 144, 42–52; [DOI] [PubMed] [Google Scholar]; e) Wu C, Chiu DT, Angew. Chem. Int. Ed 2013, 52, 3086–3109; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yu J, Rong Y, Kuo C-T, Zhou X-H, Chiu DT, Anal. Chem 2017, 89, 42–56; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Nese A, Lebedeva NV, Sherwood G, Averick S, Li Y, Gao H, Peteanu L, Sheiko SS, Matyjaszewski K, Macromolecules 2011, 44, 5905–5910. [Google Scholar]
- [4].a) Chen X, Liu Z, Li R, Shan C, Zeng Z, Xue B, Yuan W, Mo C, Xi P, Wu C, ACS Nano 2017, 11, 8084–8091; [DOI] [PubMed] [Google Scholar]; b) Kuo C-T, Thompson AM, Gallina ME, Ye F, Johnson ES, Sun W, Zhao M, Yu J, Wu I-C, Fujimoto B, Nat. Commun 2016, 7, 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu Y, Ruan H, Zhao R, Dong Z, Li W, Tang X, Yuan J, Fang X, Adv. Opt. Mater 2018, 6, 1800333; [Google Scholar]; d) Zhang X, Chamberlayne CF, Kurimoto A, Frank NL, Harbron EJ, Chem. Commun 2016, 52, 4144–4147. [DOI] [PubMed] [Google Scholar]
- [5].a) Vanden Bout DA, Yip W-T, Hu D, Fu D-K, Swager TM, Barbara PF, Science 1997, 277, 1074–1077; [Google Scholar]; b) Gesquiere AJ, Park S-J, Barbara PF, J. Am. Chem. Soc 2005, 127, 9556–9560; [DOI] [PubMed] [Google Scholar]; c) Jiang Y, McNeill J, Nat. Commun 2018, 9, 4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Yan M, Rothberg L, Papadimitrakopoulos F, Galvin M, Miller T, Phys. Rev. Lett 1994, 73, 744; [DOI] [PubMed] [Google Scholar]; b) Yu J, Hu D, Barbara PF, Science 2000, 289, 1327–1330. [DOI] [PubMed] [Google Scholar]
- [7].Jiang Y, Novoa M, Nongnual T, Powell R, Bruce TF, McNeill JD, Nano Lett 2017, 17, 3896–3901. [DOI] [PubMed] [Google Scholar]
- [8].McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL, Nat. Methods 2009, 6, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu C, Szymanski C, McNeill J, Langmuir 2006, 22, 2956–2960. [DOI] [PubMed] [Google Scholar]
- [10].a) Yu J, Wu C, Zhang X, Ye F, Gallina ME, Rong Y, Wu IC, Sun W, Chan YH, Chiu DT, Adv. Mater 2012, 24, 3498–3504; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu C, Hansen SJ, Hou Q, Yu J, Zeigler M, Jin Y, Burnham DR, McNeill JD, Olson JM, Chiu DT, Angew. Chem. Int. Ed 2011, 50, 3430–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu C, Schneider T, Zeigler M, Yu J, Schiro PG, Burnham DR, McNeill JD, Chiu DT, J. Am. Chem. Soc 2010, 132, 15410–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Soon YW, Clarke TM, Zhang W, Agostinelli T, Kirkpatrick J, Dyer-Smith C, McCulloch I, Nelson J, Durrant JR, Chem. Sci 2011, 2, 1111–1120; [Google Scholar]; b Clarke TM, Durrant JR, Chem. Rev 2010, 110, 6736–6767. [DOI] [PubMed] [Google Scholar]
- [13].Jiang Y, Nongnual T, Groff L, McNeill J, J. Phys. Chem . C 2018, 122, 1376–1383. [Google Scholar]
- [14].Yu J, Wu C, Tian Z, McNeill J, Nano Lett. 2012, 12, 1300–1306. [DOI] [PubMed] [Google Scholar]
- [15].a) Steyrleuthner R, Schubert M, Howard I, nzer B, Schilling K, Chen Z, Saalfrank P, Laquai F, Facchetti A, Neher D, J. Am. Chem. Soc 2012, 134, 18303–18317; [DOI] [PubMed] [Google Scholar]; b) Caironi M, Bird M, Fazzi D, Chen Z, Di Pietro R, Newman C, Facchetti A, Sirringhaus H, Adv. Funct. Mater 2011, 21, 3371–3381. [Google Scholar]
- [16].a) Bakulin AA, Rao A, Pavelyev VG, van Loosdrecht PH, Pshenichnikov MS, Niedzialek D, Cornil J, Beljonne D, Friend RH, Science 2012, 335, 1340–1344; [DOI] [PubMed] [Google Scholar]; b) Bakulin AA, Silva C, Vella E, J. Phys. Chem. Lett 2016, 7, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Street R, Schoendorf M, Roy A, Lee J, Phys. Rev. B 2010, 81, 205307. [Google Scholar]
- [18].Aoki Y, Shakutsui M, Fujita K, Thin Solid Films 2009, 518, 493–496. [Google Scholar]
- [19].Jin S-H, Kang S-Y, Kim M-Y, Chan YU, Kim JY, Lee K, Gal Y-S, Macromolecules 2003, 36, 3841–3847. [Google Scholar]
- [20].Zaumseil J, Donley CL, Kim JS, Friend RH, Sirringhaus H, Adv. Mater 2006, 18, 2708–2712. [Google Scholar]
- [21].Li Y, Cao Y, Gao J, Wang D, Yu G, Heeger AJ, Synth. Met 1999, 99, 243–248. [Google Scholar]
- [22].Lee W, Lee C, Yu H, Kim DJ, Wang C, Woo HY, Oh JH, Kim BJ, Adv. Funct. Mater 2016, 26, 1543–1553. [Google Scholar]
- [23].Uddin MA, Kim Y, Younts R, Lee W, Gautam B, Choi J, Wang C, Gundogdu K, Kim BJ, Woo HY, Macromolecules 2016, 49, 6374–6383. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.