

Abstract
The retinal pigment epithelium (RPE) is a particularly vulnerable tissue to age-dependent degeneration. Over the lifespan, the RPE develops an expanded endo-lysosomal compartment to maintain the high efficiency of phagocytosis and degradation of photoreceptor outer segments (POS) necessary for photoreceptor survival. As the assembly and activation of the mechanistic target of rapamycin complex 1 (mTORC1) occur on the lysosome surface, increased lysosome mass with aging leads to higher mTORC1 activity. The functional consequences of hyperactive mTORC1 in the RPE are unclear. In the current study, we used integrated high resolution metabolomic and genomic approaches to examine mice with RPE-specific deletion of the tuberous sclerosis 1 (Tsc1) gene which encodes an upstream suppressor of mTORC1. Our data show that RPE cells with constitutively high mTORC1 activity were reprogramed to be hyperactive in glucose and lipid metabolism. Lipolysis was suppressed, mitochondrial carnitine shuttle was inhibited, while genes involved in fatty acid (FA) biosynthesis were upregulated. The metabolic changes occurred prior to structural changes of RPE and retinal degeneration. These findings have revealed cellular events and intrinsic mechanisms that contribute to lipid accumulation in the RPE cells during aging and age-related degeneration.
Keywords: mTOR, aging, metabolism, AMD, lipid
Introduction:
Mechanistic target of rapamycin (mTOR) is the core component of the multi-subunit complexes mTORC1 and mTORC2. The serine/threonine kinase activity of mTOR controls cell growth, metabolism, and survival (1–3). A rich body of evidence links mTORC1 signaling to mammalian aging. Suppressing mTORC1, either by genetic manipulations or by the administration of its pharmacological inhibitor rapamycin, can extend the lifespan of C. elegans, Drosophila, and mice (4–9). Deregulated mTORC1 activity has been reported in different types of tissues and cells in aged mice (9, 10). While the beneficial effects of mTOR reduction in longevity are well accepted, the roles of the mTOR pathway in age-related pathologies are less clear. Overactivation of mTORC1 is implicated in the pathogenesis of neurodegenerative diseases (11, 12), and mTORC1 inhibition has shown promising therapeutic effects in experimental models of neurodegeneration (13). In clinical trials, allosteric or catalytic inhibitors of mTOR enhanced immune function in the elderly (14, 15). However, mTORC1 inhibition has also been linked to increased incidence of some age-related diseases, such as cataracts (8, 16), and may aggravate metabolic syndromes in diabetes (17). The varying clinical outcomes of long-term mTOR inhibition indicate distinct roles of mTOR in the (patho)physiology of the specific tissues and cells that are involved in each degenerative disease (8, 17).
The eye is a highly specialized organ and is composed of many post-mitotic, terminally differentiated cells/tissues that are particularly susceptible to age-related degeneration. Among them, RPE has a unique dependence on mTORC1. The RPE is a monolayer tissue lying between the neural retina and choriocapillaris. It provides essential support to the metabolic functions of the retina (18). Degeneration of the RPE is a primary lesion in AMD, the leading cause of irreversible blindness in the elderly (19). mTORC1 is vital to RPE physiology. In response to phagocytosis of POS, mTORC1 in the RPE displays rhythmic activation and deactivation and likely directs the cellular degradation machinery to the turnover of engulfed POS (20, 21). Active mTORC1 inhibits autophagy, which competes with phagocytosis for key protein components such as LC3, Vps34, Atg5, and Atg7 (22). With aging, RPE cells display abnormally high mTORC1 activity, which is at least partially due to the increased lysosome docking of mTORC1 components (10, 20). Abnormal mTORC1 activation in the RPE has also been reported in animal models of RPE and retinal degeneration that were generated through impairing different cellular pathways, including mitochondrial biogenesis, protein folding, and lipid metabolism (23–25). While rapamycin can attenuate progressive RPE and retinal degeneration in mice with deficiency of mitochondrial transcription factor A in the RPE (23), clinical trials of rapamycin had conflicting results in patients with advanced AMD (26, 27). The causative link between high mTORC1 activity and RPE degeneration, and especially the underlying mechanisms, remain elusive.
In the current study, we explored the roles of hyperactive mTORC1 signaling in RPE degeneration and defined the underlying mechanisms using a mouse model of bestrophin-Cre-driven knockout of Tsc1 and subsequent mTORC1 activation in the RPE. These conditional knockout mice developed RPE and photoreceptor degeneration, with clinical and pathological features of AMD. A combined high-resolution metabolomics (HRM) and transcriptome analyses revealed metabolic reprogramming towards lipogenesis at the cost of increased glycolysis via the mTORC1-SREBP1 axis in RPE tissue with constitutive mTORC1 activation. Together, our data suggest that abnormal mTORC1 activation in the aged RPE could be a driving force in the lipid accumulation process that is closely linked to the pathogenesis of AMD.
MATERIALS AND METHODS
Antibodies and chemicals
The antibodies against SREBP1 was purchased from Novus Biologicals, LLC (Centennial, CO, USA). The antibodies against HSC70 and Lamin A/C were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). All other primary antibodies were from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies were obtained from Life Technologies (Grand Island, NY, USA) or LI-COR Biosciences (Lincoln, NE, USA). Alexa Fluor® 647 Phalloidin and 4′,6-Diamidino-2-Phenylindole (DAPI) were purchased from Life Technologies. Oil red O was obtained from MilliporeSigma (St. Louis, MO, USA).
Mice
Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch and the University of Oklahoma Health Science Center (OUHSC). RPE-specific Tsc1 knockout mice (RPE_TSC1 KO) were generated by mating TSC1tm1Djk/J (Stock 005680) and C57BL/6-Tg(BEST1-cre)1Jdun/J (Stock 017557) mice from the Jackson Laboratory (Bar Harbor, ME, USA). The animals were housed under cyclic 12-hour light/dark conditions and fed ad libitum. Genotyping was performed either following the protocols recommended by the Jackson laboratory or by Transnetyx, Inc. (Cordova, TN, USA). Rd8 mutation in the Crb1 gene (28) was routinely monitored and was not detected in the colonies. Euthanasia was performed by exposing mice to carbon dioxide delivered at 2 liters/min through a three-stage gas regulator. All procedures were conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and the NIH Guide for the Care and Use of Laboratory Animals. Both male and female mice were used for the experiments.
In vivo examination of mouse fundus
Animals were anesthetized by intraperitoneal injection of a mixture of ketamine (50–75 mg/kg) and dexmedetomidine (1–5 mg/kg). The pupil was dilated with Tropicamide Ophthalmic Solution (Bausch & Lomb, Rochester, NY, USA), and covered with Gonak™ hypromellose ophthalmic demulcent solution (Akorn, Lake Forest, IL, USA). Fundus photographs were taken with a Micron-IV camera (Phoenix Research Laboratories, Pleasanton, CA) with cornea-contacting lens (29). Spectral-domain optical coherence tomography (OCT) scan was performed on a Bioptigen Envisu Imaging System (Leica Microsystems Inc, Buffalo Grove, IL, USA) with a mouse retina lens.
Electroretinography (ERG)
Scotopic and photopic ERG responses were recorded with a Diagnosys Espion E2 ERG system (Diagnosys, LLC, Lowell, MA, USA), which is provided by the OHUSC Vision Research Core (30). Mice were dark-adapted overnight, anesthetized with ketamine/dexmedetomidine, and their pupils were dilated with Tropicamide ophthalmic drops. The ground needle electrode and the reference electrode were placed in the tail and subdermally between the eyes of the animal, respectively. After applying one small drop of Gonak™ on each eye, gold wire electrodes were placed on the cornea. Scotopic ERG responses were evoked by flashing light with intensity of 0.0002, 0.002, 0.02, 2, 20, 200, 400 cd s/m2. A- and b-wave amplitudes were recorded in the Espion software. The animal was then light-adapted for 10 minutes with a background light intensity of 20 cd s/m2 (31). Photopic ERGs were recorded with pulse intensity of 5, 10 and 20 cd s/m2.
Histopathology and immunofluorescence staining
Paraffin sections of eyes were prepared as previously described (32), using 4% paraformaldehyde-fixed tissues. For each mouse eye, sagittal sections of 5-μm thickness were cut from the cornea to the optic nerve and stained with hematoxylin and eosin (H&E). To count the number of photoreceptor nuclei in the outer nuclear layer (ONL), serial sections were cut along the horizontal meridian. Ten sections from the perioptic nerve area that spanned a distance of ~150 μm were picked for quantification of the number of ONL nuclei (29). For immunofluorescence staining, sagittal cryosections (8 μm thickness) prepared from the cornea to the optic nerve or whole-mount tissues of RPE/choroid were used (18). After blocking in phosphate buffered saline (PBS) with 10% fetal bovine serum (FBS) (v/v) and 0.5% Triton X-100 (v/v), the tissues were incubated with primary antibodies followed by Alexa Fluor–conjugated secondary antibodies (Thermo Fisher Scientific). Nuclei were counterstained with DAPI. For RPE flat-mounts, the F-actin dye phalloidin (Life Technologies) was used to delineate the RPE boundaries. Images were acquired with a Carl Zeiss AxioVision microscope equipped with ApoTome (Zeiss, Jena, Germany) (29).
Western blot analysis
RPE/choroid tissues were prepared as described previously (21). Tissues were lysed in buffer containing CelLytic M Cell Lysis Reagent (Sigma-Aldrich) and 2× Laemmli Sample Buffer (Bio-Rad) at a 1:1 ratio, supplemented with 10 mM sodium glycerophosphate, 10 mM sodium pyrophosphate, 1 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktails. After sonication, samples were resolved on SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were probed with specific antibodies, and signals were detected by an Odyssey Infrared Imaging System (LI-COR).
Electron Microscopy (EM)
After euthanasia, mice were perfused with 2.5% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.4) through the left ventricle (32). Eyes were enucleated and fixed in the same buffer for 12 h at room temperature, followed by post-fixation, dehydration, and embedding in epoxy resin (33). Semi-thin sections (1 μm) through the optic nerve were prepared, stained with toluidine blue and examined by light microscopy. Ultrathin sections (0.1 μm) of selected areas were then prepared and stained with uranyl acetate and lead citrate for EM (CM-12 TEM; Philips).
Liquid Chromatography (LC)-Ultra high-Resolution Mass Spectrometry (MS)-based HRM
RPE/choroid tissues were harvested 30 minutes before lights on as previously described (21), snap-frozen with liquid nitrogen, and sent to the Clinical Biomarkers Laboratory of Emory University. Mice at the age of 1.5 ~ 3 months were used. Frozen tissue samples were processed for LC-MS analyses as described (34). Briefly, 50 μL of sample aliquots were extracted with 100 μL acetonitrile containing 2.5 μL internal standard of a mixture of 14 stable isotope standards. Proteins were precipitated and pelleted by incubation at 4 °C for 30 min followed by centrifugation at 21,000 g for 10 min at 4°C. Metabolic extracts in the supernatants were loaded via an autosampler onto a LC-Fourier transform MS system (Accela-LTQ Velos Orbitrap, ThermoFisher). Electrospray ionization was performed in positive ion mode. Triplicated samples were analyzed for mass-to-charge ratios (m/z) range from 85–2000. Identities of acylcarnitines and phosphatidylcholines were based upon accurate mass match (within 5 ppm) and MS/MS product ion spectra (Level 2 by criteria of Schymanski et al) (35). Other lipids from mummichog pathway enrichment analysis were based on Identity Level 5 (35).
Data Processing and Extraction
Data collection occurred continuously throughout 10 min of chromatographic separation. Raw files were converted to .cdf files using Xcalibur file converter from ThermoFisher. Data extraction was performed using apLCMS (36) and xMSanalyzer (37) softwares, which annotate m/z ratio, retention time and ion intensity. The m/z features with a median CV of 50% or less and a minimum mean Pearson correlation coefficient of 0.7 between technical replicates, were retained for data analyses (38).
RNA sequencing (RNA-seq) and bioinformatics analyses
Mouse eyes were enucleated from euthanized 6-week old RPE_Tsc1 KO and flox littermates 3 h after lights on. After removal of the anterior segment and the neural retina, RPE/choroid tissues were harvested and RNA was prepared using RNeasy kit (Qiagen, Germantown, MD). Two eyes from the same animal were used for one sample. RNA-seq was performed by BGI (Cambridge, MA).
For data analyses, we applied DESeq2 package in R Studio to perform differential gene expression analysis (39). Pre-ranked Gene Set Enrichment Analysis (GSEA) (40) was used to identify sets of genes overrepresented in significant pathways differentiating 6 week-old RPE_Tsc1 KO from flox littermates using enrichment score statistics. The GSEA analysis tool (version 2.0) was downloaded from the website (http://www.broadinstitute.org/gsea/index.jsp) at the Broad Institute. Hallmark gene sets from Molecular Signatures Database (MSigDB) were used to compute overlaps in the current analysis. Sets containing 15–500 genes were analyzed.
Quantitative RT-PCR analyses of RNA isolated from RPE/choroid tissues
Total RNA was prepared from RPE/choroid tissues as described previously (29). Mice at the age of 5 ~ 8 weeks were used. cDNA was synthesized using M-MLV Reverse Transcriptase and oligo(dT)15 primer (Promega, Madison, WI, USA). Taqman probe-based real-time PCR assays were designed with the Universal Library Probe approach (29). Primers used for gene-specific PCR amplifications were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Primer sequences used are as the followings: Acacb (5′-TGAATCTCACGCGCCTACTA-3′;5′-GCCTCTCTTCACCAGATGGA-3′), Acsl6 (5′-CAAAGGTGCAATGCTCACC-3′; 5′-GGGGCCCACTGACTCTCT-3′), Actin (5′-CTAAGGCCAACCGTGAAAAG-3′; 5′-ACCAGAGGCATACAGGGACA-3′), Aldh2 (5′-GAGCAGACACCGCTCACC-3′; 5′-TCCGGGAACGATATTGACC-3′), Aldoc (5′-CGTAGGCATCAAGGTTGACA-3′ ; GAGCACAGCGTTCCAAGAG-3′), Bdh1 (5′-TCTCCGAAGAGCCAAAGGT-3′; 5′-CAAACTTGGTGATGCAGTATGG-3′), Elovl1 (5′-CCCCTCTGGTGACAGAAAGT-3′; 5′-CCATCCTGGCTAAGGACTCA-3′), Gapdh (5′-AAG AGG GAT GCT GCC CTT AC-3′; 5′-CCA TTT TGT CTA CGG GAC GA-3′), Hif1 (5′-CATGATGGCTCCCTTTTTCA-3′; 5′-GTCACCTGGTTGCTGCAATA-3′), Hk1 (5′-TGTGAGATTGGACTCATCGTG-3′; 5′-CCACCATCTCCACGTTTTTC-3′), Hmgcs2 (5′-TGAACGAGTGGATGAGATGC-3′; 5′-GGGGAATTGTTGCATGGAT-3′), Ldhb (5′-CAG AGA AGC TTG GCA TTC ATC-3′; 5′-AGA CTC CTG CCA CAT TCA CC −3′), Oxct1 (5′-GAAGCTGATGCGGATCTCA-3′; 5′-GAATGACTCATCGCTGGAGAA-3′), Pfkm (5′-GGGGAAGGGCATCTTTGA-3′; 5′-GTCAAAGGGAGTTGGGCTTC-3′), Pfkp (5′-GAGGGACCCCATCTGCAT-3′; 5′-GTAGCTTCCAGCAAGGCAAT-3′), Pgk1 (5′-CCA AGG CTT TGG AGA GTC C −3′; 5′-GAT CAG CTG GAT CTT GTC TGC −3′), Scd1 (5′-GCTCTACACCTGCCTCTTCG-3′; 5′-GCCGTGCCTTGTAAGTTCTG-3′), Slc2a8 (5′-ACA TCT CGG AAA TCG CCT AC-3′; 5′-CCA GCG CCA CTC TAG GAC −3′), Slc25a1 (5′-GCCCTATGGAGACCATCAAG-3′; 5′-TTCCCTTTAGCCCTTGTTCC-3′), and Srebf1 (5′-TCAAGCAGGAGAACCTGACC-3′; 5′-TCATGCCCTCCATAGACACA-3′).
Measurement of RPE glucose metabolism
Eye-cups were prepared from euthanized 6 week-old RPE_Tsc1 KO and flox littermates, with retina removed. To measure glucose uptake, the posterior eye-cup was incubated with 2-deoxy-glucose (2-DG) diluted in PBS (1 mM) for 10 minutes, and the uptake of 2-DG was determined using a luciferase-based Glucose Uptake-Glo™ Assay kit (Promega, Madison, WI, USA) following the manufacturer’s instructions. To measure the lactate release, the eye-cup was incubated in PBS, with no additional glucose, for up to 45 minutes. The medium was collected at 15, 30, and 45 minutes and measured for the level of lactate using a Lacate-Glo™ Assay kit (Promega) following the manufacturer’s instructions.
Primary mouse RPE cultures were used for metabolic assays to measure the rates of oxygen consumption and extracellular acidification through glycolysis. To isolate the RPE, posterior eye-cups were digested with 2% dispase-0.05% Trypsin for 45 min at 37 °C, and the RPE were gently brushed off from the choroid with a pipette tip (41). Cells were washed, seeded in 96-well microplates (Agilent, Santa Clara, CA, USA), and cultured in Minimum Essential Medium Eagle, Alpha Modification, supplemented with 10% FBS, N1 supplements (MilliporeSigma), nonessential amino acid, 1 mM sodium pyruvate, and 2 mM glutamine. Cells reached confluence after 5 ~ 6 days in culture. Glycolysis was measured using the Glycolysis Stress Test kit on a Seahorse XF Analyzers (Agilent), following the manufacturer′s instructions. The data collection and analysis were performed using the Wave software (Agilent).
ATP measurement
RPE/choroid tissues were prepared from 6 ~ 8 week-old RPE_Tsc1 KO and flox littermates. ATP was extracted with 5% trichloroacetic acid, diluted in PBS and measured using an ENLITEN® ATP assay system (Promega) following the manufacturer’s instructions.
Statistics
Statistical analyses were performed with GraphPad Prism software. Normality of the data distribution was evaluated using the Kolmogorov-Smirnov test. Between-group differences were assessed by Student’s t-test or Mann-Whitney test, with the level of significance presented as p values. For multiple group comparisons, Two-Way analysis of variance (ANOVA) was used, followed by Tukey’s or Sidak’s Multiple Comparisons Test.
Results:
Constitutive RPE mTORC1 activation led to RPE degeneration
To achieve mTORC1 hyper-activation in the RPE, we generated RPE_Tsc1 KO lines by crossing mice containing the loxp-flanked Tsc1 alleles (42) with mice expressing Cre recombinase driven by the bestrophin 1 promoter (43). The depletion of Tsc1 in the RPE was confirmed by Western blot and quantitative RT-PCR analyses (Fig. 1A and Fig. S1A). Cre protein expression was confined to the RPE tissue and was not detected in the neural retina (Fig. 1B and Fig. S1B). About 80% of the RPE of the conditional knockout line expressed Cre (Fig. 1C). Relieving mTORC1 from upstream inhibition by the TSC1/2 complex led to increased mTORC1 activation in the RPE as assessed by the phosphorylation status of its surrogate substrate protein ribosome S6 (Fig. 1A and Fig. S1B).
Fig. 1. Phenotypic Characterization of RPE-Specific Tsc1 KO Mice.

(A) Western blot analysis of RPE tissue from 4 ~ 6 weeks old RPE_Tsc1 KO and Tsc1 flox littermate. Actin was used as the loading control. The data are representative of three different experiments. (B) Immunostaining of Cre recombinase on cryosections prepared from the posterior eye of RPE Tsc1_KO at the age of 4 ~ 6 weeks. Red: Cre; Blue: Nuclei. The immunostaining was repeated with cryosections prepared from three different animals. (C) Immunostaining of flat-mounted RPE/choroid tissue with an antibody specific for Cre. Green: Cre; Red: F-actin; Blue: Nuclei. The data are representative of three different experiments. Animals at the age of 6 ~ 8 weeks were used. (D) OCT scan of mice at 8 ~ 11 months. RPE_Tsc1 KO displayed hyper-reflective spots (arrowheads) resembling subretinal drusenoid deposits (SDD). (E-J) H&E-stained histology sections of the posterior eyes from Tsc1 flox littermates and Tsc1 RPE-specific knockouts at 8 ~ 11 months. The RPE_Tsc1 KO displayed outer retina pathology, including subretinal cell infiltration (blue arrows in Fig. 1F and I), OS disorganization (white arrows in Fig. 1G), RPE cyst and lysis (asterisk in Fig. 1F), hypopigmentation (Fig. 1J black arrows), hyperpigmentation (Fig. 1J white arrows), hypertrophy (blue arrowheads in Fig. 1G) and atrophy of the RPE (black arrowheads in Fig. 1G), anuclear material accumulation on the apical face of the RPE (Fig. 1H), and choroid thinning (Fig. 1E, G and H). (K) Quantification of the numbers of rows of nuclei in the outer nuclei layer (n=5 ~ 6 per genotype). Values are presented as mean ± SEM, * indicates a statistically significant difference between different genotypes (*p < 0.05, ** p < 0.01, two-way ANOVA followed by Sidak’s multiple comparison tests). Scale bar: 50 μm (B, H-J); 20 μm (C); 100 μm (E-G). ELM: external limiting membrane; IS/OS: inner segments/outer segments; RPE: retinal pigment epithelium; GCL: ganglion cell layer; IPL: inner plexiform layer; INL: inner nuclei layer; OPL: outer plexiform layer; ONL: outer nuclei layer; CH: choroid.
On fundoscopic examination, RPE_Tsc1 KO mice showed RPE pigment changes and retinal white or yellowish spots at the age of 8 ~ 10 months (44) (Fig. S2). Tsc1-floxed littermates and mice expressing Best-Cre had normal fundi comparable to those of wild-type mice at 10 months (Fig. S2). On OCT scan, hyperreflective spots, corresponding to subretinal drusenoid deposits (SDD; pseudodrusen) (44, 45), were detected between the RPE and inner and outer segments junction of the photoreceptor cells in the eyes of RPE_Tsc1 KO mice (Fig. 1D) at 8 ~ 10 months. SDD was not seen in age-matched Tsc1-floxed littermates (Fig. 1D).
Formaldehyde-fixed, paraffin-embedded histology sections were used to examine RPE and retinal pathology. Control Tsc1-floxed mice had normal retina and RPE (Fig. 1E). At the same age range of 8 ~ 11 months, RPE of KO animals showed various signs of degeneration, including the formation of RPE cysts, hypo- and hyper-pigmentation, hypertrophy, and atrophy (Fig. 1, F - J). Subretinal cell infiltration was frequently detected in the RPE_Tsc1 KO mice (Fig. 1 F and I). The nuclei of the infiltrated cells in the photoreceptor inner segment layer were often without pigments, while cells in the outer segments, particularly those close to the RPE, were enwrapped by pigments resembling monocyte-derived cells. Infiltration of subretinal microglia is commonly observed in retinal diseases, including AMD (46, 47). When we stained cryosections of the posterior eyes and flat-mounted RPE tissues with an antibody specific for IBA1, there were IBA1-positive cells in the OS and the apical side of the RPE, supporting microglia/macrophage infiltration in the eyes from RPE_Tsc1 KO mice (Fig. S3).
When the numbers of the ONL nuclei were counted, moderate but significant thinning was found in the retina of RPE_Tsc1 KO mice at the age of 8 ~ 11 months, as compared to the retina of age-matched flox littermates in both females and males (Fig. 1K). The loss of photoreceptor cells was not caused by potential Cre toxicity (48), as the ONL thickness of bestrophin-Cre mice was comparable to that of wild-type mice at the age examined (Fig. S4).
Early retinal functional changes in RPE-TSC1 KO mice
To quantitatively assess RPE pathology, we measured the mean area of RPE cells from RPE_Tsc1 KO mice and Tsc1-floxed littermates. An increase in RPE cell size is routinely used as an indicator of RPE pathology (49). We stained flat-mounted RPE/choroid tissues from control and knockout groups of mice at different ages (1.5 ~ 2 months, 4 ~ 5 months, and 8 ~ 11 months) with F-actin dye phalloidin, which delineates the boundaries of RPE cells, and quantified the mean area of RPE cells at the middle-peripheral region (21). In the Tsc1-floxed littermates at all age groups, the majority of the RPE cells were polygonal with a well-defined honeycomb shape (Fig. 2A). At the age of 1.5 to 2 months, the RPE of the KO mice had a comparable mean area to that of the flox littermates (Fig. 2A and B), while some cells had irregular morphology resembling mixed shaped RPE cells as described by Curcio and colleagues (49). With increased age, the RPE from the KO animals had more mixed and round shaped cells, and some cells had multi-nuclei instead of mono-/bi- nuclei of healthy RPE (50, 51). Accompanied by the irregularity of the morphology, there was an age-dependent increase in the average size of the RPE. The significant increase in the mean area was identified at the age of 4 ~ 5 months, and the change was more prominent when analyzed at 8 ~ 11 months (Fig. 2B), suggesting focal loss of TSC1-deficient RPE and filling in of the gaps by neighboring RPE cells. The number of RPE cells in the mid-peripheral area decreased in KO mice after 4 ~ 5 months (Fig. 2C).
Fig. 2. RPE Degeneration and Retinal Dysfunction in RPE Tsc1_KO Mice.
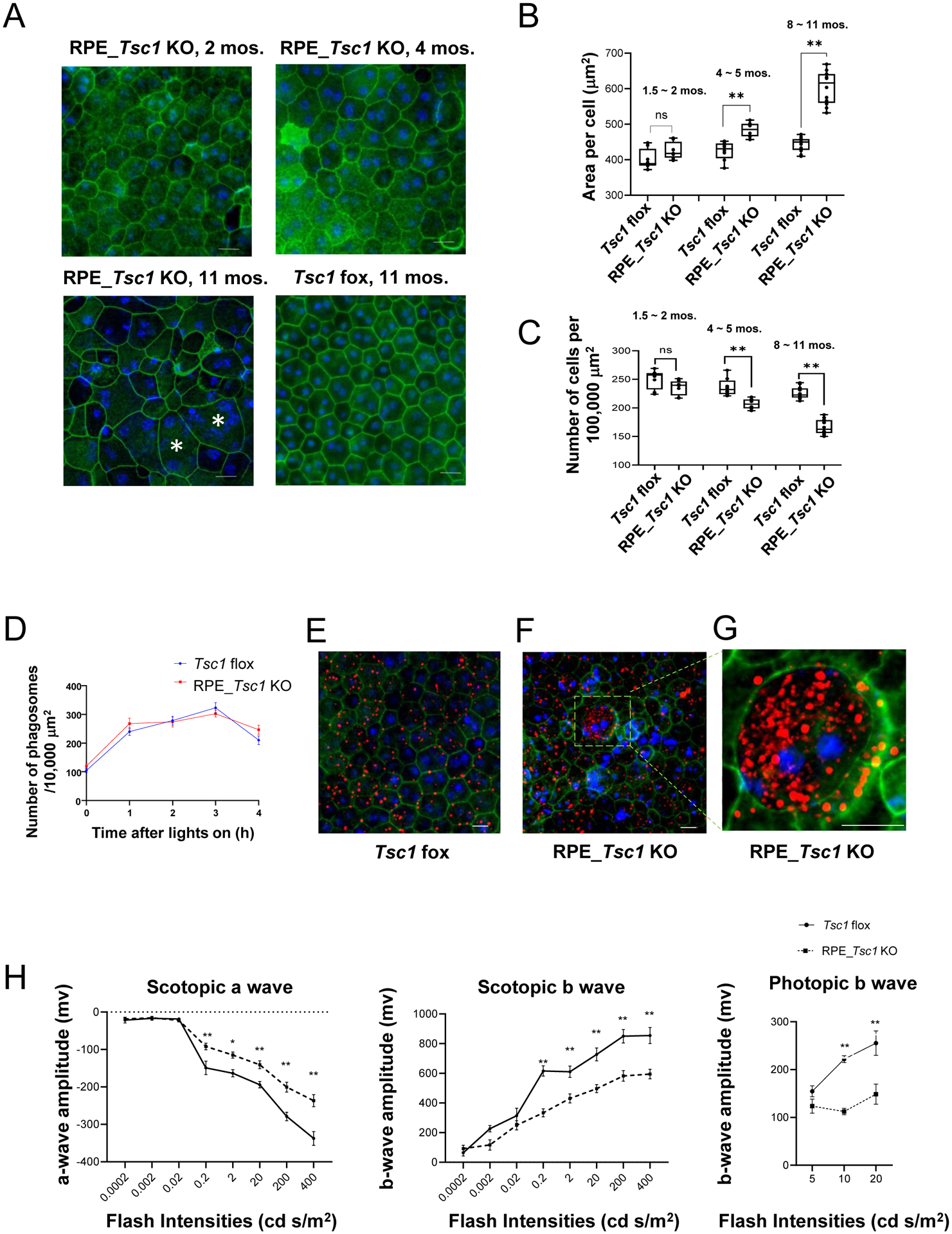
(A) Representative images of flat-mounted RPE/choroid tissues stained with F-actin dye phalloidin (Green) and nuclei dye DAPI (Blue) at the indicated ages, * indicated cells with more than two nuclei (Scale bar: 20 μm). (B) Quantitation of the mean area and (C) average density of RPE cells in the mid-periphery regions data are presented as mean ± SEM, N=8 per genotype. * indicates a statistically significant difference between two groups (**p < 0.01, two-way ANOVA followed by Sidak’s multiple comparisons test). (D) Quantification of the rhodopsin-positive structures on immunostained RPE/choroid whole mounts at different time points after lights on (N=6 ~ 8 per group). No statistical difference was observed between the two groups (two-way ANOVA followed by Sidak’s multiple comparisons test). (E) Representative image of rhodopsin-staining of flat-mounted RPE tissue from Tsc1 flox mice. (F) Cell with abnormal accumulation of rhodopsin-positive structure on flat-mounted RPE with Tsc1 KO. (G) is the enlarged image of the box area in (F). (H) ERG response of scotopic a- and b- wave, and photopic b-wave amplitudes of RPE_Tsc1 KO and flox littermates at the age of 4~ 5 months. Values are presented as mean ± SEM, N=8 per genotype * indicates a statistically significant difference between different genotypes (* p <0.05, ** p < 0.01, two-way ANOVA followed by Sidak’s multiple comparisons test). Scale bar: 20 μm.
A primary physiological function of the RPE is to engulf and degrade the shed POS. We and others have previously shown that RPE mTORC1 can be activated by POS (21, 52), indicating a link between mTORC1 signaling and phagocytosis. We therefore performed quantitative measurements of POS uptake and degradation in control and RPE_Tsc1 KO mice following the morning burst of POS shedding, using animals at 2- to 3- months of age. RPE_Tsc1 KO mice maintained the rhythmic phagocytotic activities with a peak at 3 h after lights on in the morning (Fig. 2D and Fig. S5). Compared to the Tsc1-floxed littermates, there was a trend of increase in the remaining POS in the RPE from KO mice at 4 h after lights on (Fig. 2D). However, the difference did not reach statistical significance. In addition, on flat-mounted RPE tissue from RPE_Tsc1 KO mice, we identified cells that had accumulated rhodopsin-positive structures (Fig. 2G). Such cells were detected in over 80% of the flat-mounted RPE tissues from KO animals, although their percentage was low and only accounted for less than 1% of the entire RPE layer. In the Tsc1-floxed littermates, RPE cells with similar aberrant structures were rare. We only observed one such cell from 40 flat mounts from control mice.
To measure retinal functional changes, we recorded ERG responses in animals at the age of 4 ~ 5 months, when mild RPE pathology were developed in the RPE_Tsc1 KO mice. Mice with Tsc1 deletion in the RPE had decreases in the amplitudes of scotopic a- and b-waves, and photopic b-wave (Fig. 2H), suggesting the loss of neuronal function. Collectively, these data demonstrate that hyperactive mTORC1 in the RPE caused RPE degeneration, followed by photoreceptor dysfunction and degeneration.
Metabolic reprogramming in TSC1-deficient RPE
On semi-thin sections of the posterior eyes from the RPE_Tsc1 KO mice, consistent with the findings from H&E-stained histological sections and immunostaining of the cryosections of the outer retina, we detected subretinal cell infiltration (Fig. 3B) and abnormal amorphous structures between the OS and the RPE (Fig. 3C and D). A prominent feature we observed in the TSC1-deficient RPE is the abnormal accumulation of lipids at relatively young age (4 ~ 5 months). In the RPE, there were structures reminiscent of lipid droplets (LDs) that tended to be clustered. Multiple LDs, instead of single ones, were often observed in the same RPE cell on semi-thin sections (Fig. 3D). At the ultra-structure level, various signs of RPE pathology were identified in the RPE_Tsc1 KO mice in addition to the LDs (Fig. 3E–N). On the apical face, there were areas with subretinal cell (Fig. 3G) or membrane whorl-like (Fig. 3I and 3K) structures. Abnormal intracellular structures, such as lipofuscin and undigested POS, were detected in the Tsc1-KO RPE (Fig. 3L). Their basal infoldings had depositions of electron-dense materials, although the cisternae were still present (Fig. 3N). No differences in the thicknesses of the Bruch’s membrane were detected between control and Tsc1- KO RPE at 4 ~ 5 months.
Fig. 3.

Lipid accumulation in the RPE of Tsc1 KO mice. (A-D) Toluidine blue-stained semi-thin sections of the posterior eyes from Tsc1 flox and RPE_Tsc1 KO at 4 ~ 5 months. The latter had cell infiltration (B) and accumulation of amorphous materials (C) on the apical face of the RPE, and accumulation of LD (D). (E to N) Transmission electron microscopy images of mice at 4 ~ 5 M. RPE-specific Tsc1 KO showed subretinal cell infiltration (G), membrane whorls on the apical faces of the RPE (blue arrows in I and K), LD with abnormal size (I) or abundance (J), preidroplet mitochondria (L, *), lipofuscin (LF) (L), undigested POS structure (L, green arrows), and mild disorganized basal infoldings (N). RPE hypo- (I) and hyper-pigmentation (K) were also detected. (O) and (P) Oil Red O staining of neural lipids on cryosections of posterior eyes prepared from RPE_Tsc1 KO animals and the Tsc1 flox counterparts. Scale bar: 10 μm (A-G), 2 μm (H-K), 1 μm (M and N), 400 nm (L), and 50 μm (O and P).
We validated the accumulation of lipids in the TSC1-deficient RPE by Oil Red O staining of the cryosections of posterior eyes (Fig. 3J). Oil Red has specificity towards neutral lipids such as triglyceride and esterified cholesterol (53). POS has neutral lipid compositions and, therefore, can serve as a positive control for the staining. In control mice (Fig. 3O), there was no staining in the RPE, although OS was stained. In the Tsc1 KO tissue, Oil Red O staining was detected on the basal side of the RPE and in the Bruch′s membrane (Fig. 3P).
The abnormal accumulation of lipid droplets indicates metabolic changes in the affected RPE. Given the importance of lipid metabolism in the pathogenesis of AMD (19, 54) and the roles of mTORC1-mediated signaling as a central regulator of metabolism (17), we performed LC/MS-based HRM analyses to examine changes in RPE metabolome and metabolic pathways associated with TSC1 deficiency. Mice at 1.5 ~ 3 months of age were used to detect early changes directly caused by hyperactivated mTORC1. HRM analyses identified 7524 metabolic features (termed “metabolites” hereafter for simplicity purpose) (Supplemental Table 1) in the mouse RPE. Principal-component analyses (Fig. 4A) revealed that the metabolomes of the control and Tsc1 KO RPE had profound differences. There were 452 metabolites whose abundances were significantly different between the two groups (p<0.05, 204 higher and 248 lower in T1 KO than in the flox littermates, Fig. S6A and B). In order to protect against type 2 errors and for optimal capture of all differentially expressed features, no cut-off threshold was set for fold change during initial data mining. Upon further examination, 80 metabolites were found to have more than 2-fold differences between the two groups.
Fig. 4. LC/MS-based measurements of high-resolution RPE metabolome.
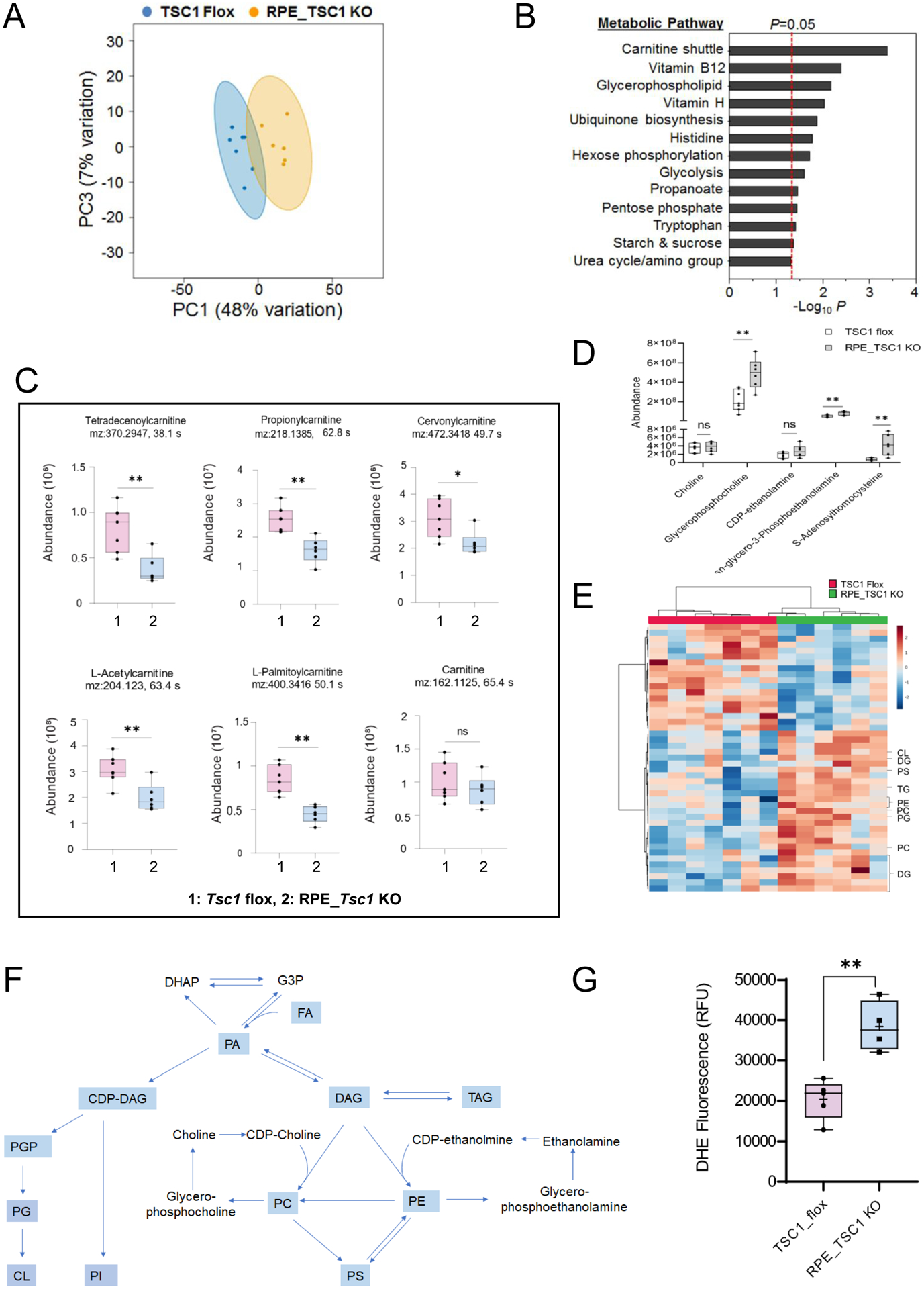
Baseline metabolic changes associated with Tsc1 deletion in the RPE were measured in tissues collected before morning burst of phagocytosis. (A) Pairwise principal component (PC) score plot of HRM data from 7 Tsc1 flox (blue) and 6 Tsc1 KO (yellow) RPE/choroid samples. (B) Metabolic pathways significantly affected by constitutively active mTORC1 signaling in the RPE/Choroid, as determined by mummichog pathway enrichment analysis. (C) and (D) Abundances of acylcarnitines and intermediates of glycerophospholipid metabolic pathways in the RPE/choroid tissues from Tsc1 flox and RPE_Tsc1 KO (* p < 0.05, ** p < 0.01, 2 tailed student t-tests). (E) Heatmap showing glycerophospholipids, DG and TG that are significantly different between flox and TSC1-deficient RPE/choroid. A total of 18 lipid species decreased and 27 increased in KO (P< 0.05). (F) Schematic diagram of pathways of glycerophospholipid biosynthesis in mammalian cells. Metabolites containing FA moieties are in light blue. (G) The level of reactive oxygen species in RPE/CH tissue as measured by dihydroethidium (DHE) fluorescence. 5 and 4 animals were used for the flox and KO group, respectively. (** p < 0.01, two-tailed student t-test). Abbreviations: DHAP: dihydroxyacetone phosphate; G3P: glycerol-3-phosphate; PA: phosphatidic acid; PGP: phosphatidylglycerol phosphate; PG: phosphatidylglycerol; CL: cardiolipin; PI: Phosphatidylinositol; PC: phosphatidylcholine; PE: phosphatidylethanolamine; PS: phosphatidylserine.
Next we performed pathway enrichment analysis using mummichog software (55), on 140 metabolites that are associated with major metabolic pathways and showed significant differences between control and Tsc1 KO RPE. As shown in Fig. 4B, four of the top five metabolic pathways were closely related to FA metabolism, and the most affected one in the Tsc1 KO RPE tissue was carnitine shuttle. Carnitine shuttle mediates the transport of activated long-chain FAs (acyl-CoA) into the mitochondrial matrix in the form of acylcarnitine conjugates and is required for β-oxidation of long-chain FAs. Many species of acylcarnitines had decreased abundance in Tsc1 KO RPE, including cervonyl carnitine, palmitoylcarnitine, tetradecenoylcarnitine, and octadecenylcarnitine (Fig. 4C), indicating the mitochondrial import of long-chain FAs was inhibited by high mTORC1 activity. There was no significant change in the level of carnitine (Fig. 4C), suggesting the decreased acyl-carnitine intermediates were not caused by limited availability of carnitine.
In addition to the carnitine shuttle, TSC1-deficient RPE had altered glycerophospholipid metabolism (p < 0.05, Fig. 4B). When examined the metabolites involved in that pathway, the intermediates such as glycerophosphocholine and glycerophosphoethanolamine were more abundant in the RPE with higher mTORC1 activity (Fig. 4 D). Furthermore, a variety of phospholipids species, including phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylserine (PS), cardiolipin (CL), as well as diacylglycerol (DAG) and triacylglycerol (TAG), had significantly higher amounts in Tsc1-KO RPE compared to that in the Tsc1 flox control (Fig. 4 E and F). Increased accumulation of neutral lipids and phospholipids, along with the decreased mitochondrial import of long-chain FAs via the carnitine shuttle, suggest that the high mTORC1 activity inhibited the lipid turnover and utilization in the RPE. The beneficial effects of Omega-3 long-chain polyunsaturated FAs in preventing AMD had been postulated and supported by clinical studies (56, 57). However, in TSC1-deficient RPE, we did not find significant changes in the metabolites of that pathway (Table S2). Lipid peroxidation is a major source of oxidative stress; consistently, the TSC1-deficient RPE had higher level of reactive oxygen species as measured by dihydroethidium (DHE) fluorescence (Fig. 4G).
RNA-seq analyses using RPE/choroid tissue from either RPE-Tsc1 KO or floxed littermates further supported metabolic reprogramming in the TSC1-deficient RPE. Among the 16,108 genes analyzed, 475 (2.9%) had over 2-fold changes in their expression levels with an adjusted p-value < 0.001. 256 genes were significantly upregulated and 219 genes were downregulated (Fig. S7A). GSEA of the genes that were significantly upregulated in the TSC1-deficient RPE (p < 0.05, FDR q < 0.2) confirmed the upregulation of mTORC1 pathway and FA metabolism (p < 0.05) (Fig. S5B). Reactome analysis also supported that the depletion of Tsc1 had a general impact on the profiles of genes involved in lipid metabolism (p = 0.009), with altered expression of 100 genes out of a total of 746 genes and 189 reactions out of 812 total reactions analyzed. When we examined individual aspects of FA metabolism (biosynthesis/elongation/oxidation/ketone body metabolism), we found that genes mediating major steps of FA biosynthesis and elongation were upregulated in Tsc1-KO tissue (Fig. 5A and B). Genes encoding key proteins that participate in both ketogenesis (Hmgcs2) and ketolysis (Bdh1 and Oxct1) also showed upregulation at mRNA level in the KO tissue. The aberrant mTORC1 did not upregulate expression of genes involved in the FA oxidation (FAO) except for Cpt1 and Cpt2, which encode transporter protein of the carnitine shuttle. Quantitative RT-PCR measurements confirmed the upregulation of mRNAs involved in FA biosynthesis and ketone body metabolism in the RPE tissue with hyperactive mTORC1 (Fig. 5C).
Fig. 5. Aberrant fatty acid biosynthesis and utilization in the Tsc1-deficient RPE.
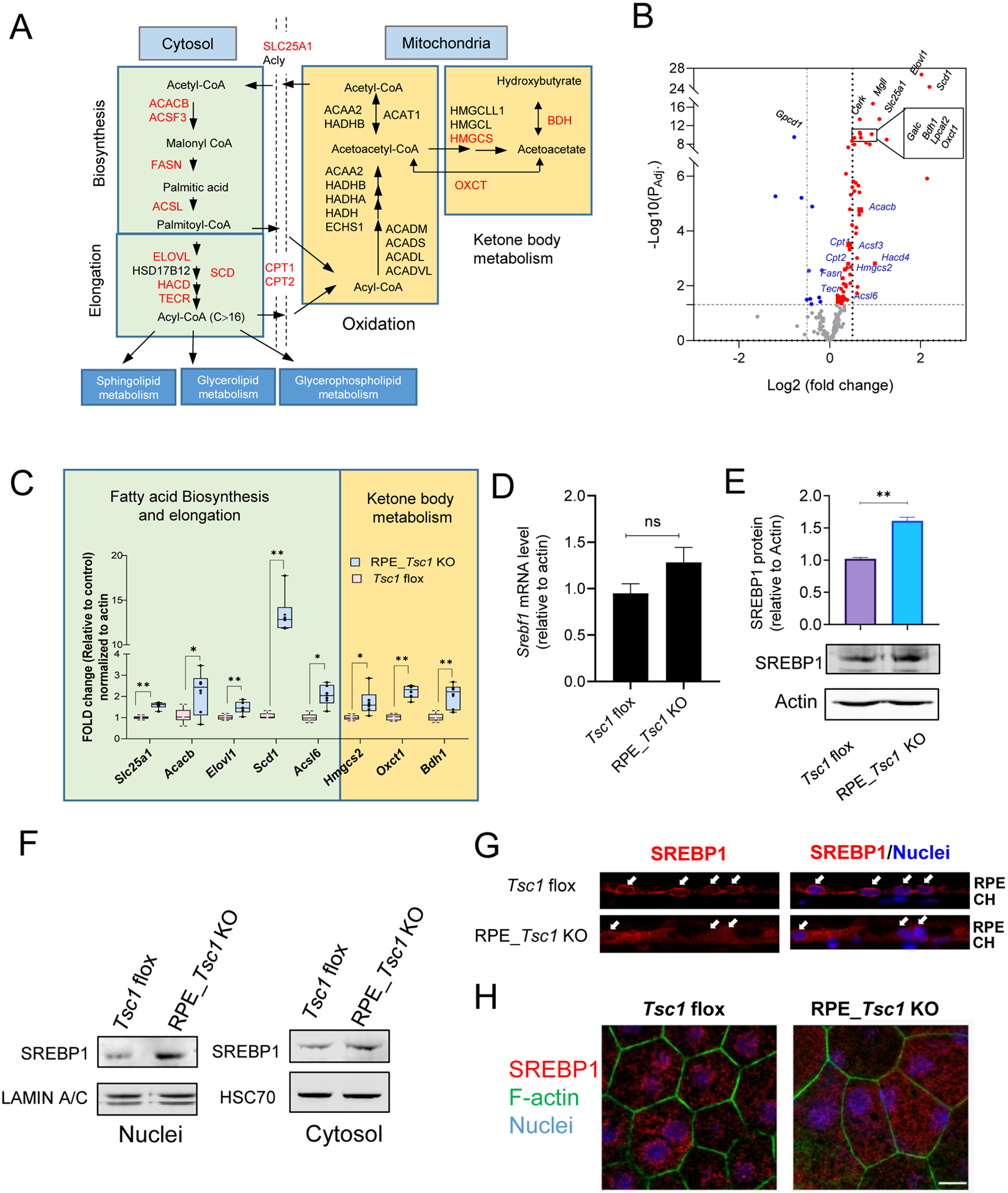
(A) Schematic illustration of FA metabolic pathways. Genes showed upregulation by RNA-seq analysis in FA biosynthesis/elongation/degradation were highlighted in Red. (B) Volcano plot of differential gene expression between control and TSC1-deficient RPE from RNA-seq. The genes analyzed are involved in FA metabolic pathways including FA biosynthesis, elongation, degradation, glycerophospholipid metabolism, glycerolipid metabolism, and sphingolipid metabolism as illustrated in (A). The horizontal dotted line indicates adjusted p value of 0.05. The vertical dotted lines correspond to two-fold change. Blue indicates decreased expression, and red indicates increased expression in the Tsc1 KO RPE. The top 10 genes with most significant changes are labeled in black. (C) Quantitative RT-PCR of genes involved in fatty acid biosynthesis and utilization, after normalization to the expression level of β-actin. (* p < 0.05, ** p < 0.01, two-tailed Student’s t-test, 6 biological repeats for each group). (D) and (E) Comparison mRNA and protein level of SREBP1 between the RPE/choroid tissues isolated from control and RPE_Tsc1 KO mice. 7 and 3 biological replicates were used for each assay, respectively. (F) Subcellular distribution of SREBP1 in RPE tissue/choroid by western blot. (G) and (H) Immunostaining of SREBP1 on cryosection or flat mounted RPE/choroid tissue. Three biological repeats were used for each assay. Scale bar: 20 μm.
In lipogenic tissues like the liver and adipocytes, mTOR controls de novo synthesis of triglycerides and other lipid molecules from carbohydrates by regulating the activity of the sterol regulatory element-binding protein SREBP1 (58). In the TSC1-deficient RPE, many of the upregulated lipogenic genes, including Fasn, Acacb, Elovl1, and Scd1 (Fig. 5B and C), are established targets of SREBP1 (59–61). The mRNA level of Srebf1 (the gene encoding SREBP1) in RPE/choroid was not different between Tsc1 KO RPE and Tsc1-floxed controls (Fig. 5D). However, its protein level was increased in the Tsc1 KO RPE as compared to that from the control littermates (Fig. 5E), supporting post-transcriptional regulation (58). When examined for its subcellular localization, immunostaining of SREBP1 protein on the cryosections of posterior eye (Fig. 5G) and flat-mounted RPE/choroid tissue (Fig. 5H) both showed an increased presence of SREBP1 in nuclei of RPE with Tsc1 deletion. Subcellular fractionation and immunoblotting of cytosolic and nuclear proteins of the RPE tissue further confirmed the nuclear translocation of SREBP1 after Tsc1 KO (Fig. 5F). Thus, constitutively high mTORC1 in the RPE activated SREBP1 and upregulated its downstream lipogenic genes. Collectively, data from integrated metabolome and transcriptome analyses defined a previously unrecognized role of mTORC1 in RPE lipid metabolism.
Lipid biosynthesis relies on glycolysis to provide precursor metabolites such as acetyl-CoA (Fig. 5A). Consistent with the increased lipogenesis, we found an upregulation of glycolysis in the Tsc1-KO RPE. Pathway enrichment analysis by GSEA revealed that differentially expressed genes in the transcriptome of Tsc1-KO RPE were over represented in glycolysis pathway (Fig. 6A). Genes encoding glycolytic enzymes, especially those participating in the committing steps of the pathway, had higher expression levels in the knockout tissue (Fig. 6 A and B). The upregulation of selected glycolysis genes from RNA-seq was further validated by quantitative RT-PCR (Fig 6 C).
Fig. 6. Increased glycolysis in Tsc1-deficient RPE.
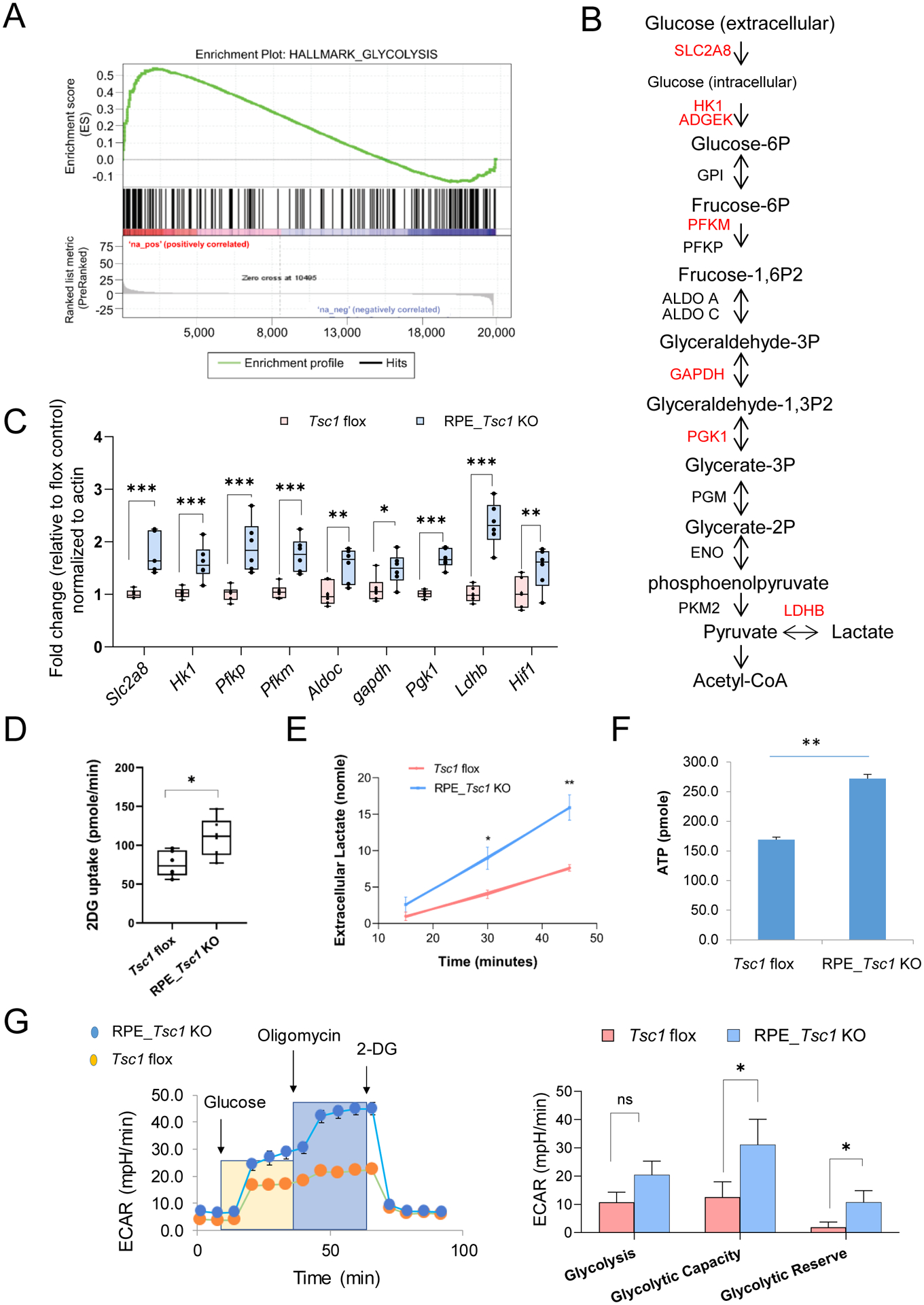
(A) GSEA-based enrichment plot of glycolysis pathway significant at FDR q-value < 0.05. (B) Schematic illustration of glycolysis. Genes upregulated in TSC1-deficient RPE were highlighted in red. (C) Quantitative RT-PCR of genes involved in glycolytic pathway. 6 biological replicates were used for each genotype. (* p < 0.05, ** p < 0.01, two-tailed Student’s t-test, 6 biological repeats for each group). (D) and (E) Rates of glucose uptake and lactate secretion from RPE/Choroid tissue prepared from RPE-Tsc1 KO or flox littermates. 6 biological replicates were used for each genotype. (* p < 0.05, ** p < 0.01, two-tailed Student’s t-test). (F) Measurement of ATP levels in RPE/choroid tissues (* p < 0.05, two-tailed student t’s test, N=8). (G) Metabolic assay of measuring glycolysis capacity in primary mouse RPE culture established from RPE_Tsc1 KO mice and Tsc1 flox controls. N=4 (* p < 0.05, two-tailed Student’s t-test).
In addition to gene expression, we measured glucose uptake and lactate secretion using isolated RPE tissue with ex vivo organ culture. The RPE/choroid explants from RPE_Tsc1 KO mice had increased glucose uptake and higher rate of lactate release into the conditioned medium, as compared to the RPE from Tsc1-floxed littermates (Fig. 6 D and E). Extracellular medium acidification rates were measured in primary RPE isolated from either the RPE_Tsc1 KO mice or Tsc1-floxed controls (Fig. 6F). The metabolic assay showed that TSC1-deficient RPE had higher glycolytic capacity and reserve compared to the littermate RPE (Fig. 6F). The metabolic switch between fatty acid utilization and glycolysis did not interfere with RPE energy production. The cellular ATP level was higher in Tsc1_KO RPE tissue (Fig. 6G). A similar observation was reported from the iPSC-derived RPE of AMD patients that also show lipid droplet accumulation (62).
Discussion:
Fine-tuned mTORC1 signaling is critical for cells/tissues to perform orchestrated functions to achieve homeostasis and survival, and aberrant mTORC1 activity is implicated in aging and the pathogenesis of age-related diseases. Here, using a murine model with tissue-specific deletion of Tsc1, we reported that hyperactive mTORC1 led to progressive RPE degeneration, followed by the functional loss and structural changes of the retina. Similar pathology findings, including thinning of the outer retina and decreased ERG responses, were reported in a recently published study by Huang and colleagues using the similar RPE-specific Tsc1 knockout mice (63). Together with data from many other studies (23, 25, 64), the results suggest that abnormally high mTORC1 activity is likely to be a common molecular mechanism that is elicited by converging chronic stress pathways leading to age-related RPE degeneration.
In the RPE_Tsc1 KO model, the loss of RPE cells became significant at 4 ~ 5 months (Fig. 2B). Molecular and biochemical changes, as shown on RPE ultrastructure (Fig. 3), metabolome (Fig. 4) and gene expression (Fig. 5 and 6), occurred at ~ 2 months and before the structural damages detected at light microscopy level. Iba1-positive cells were detected in the subretinal space of RPE_Tsc1 KO mice (Fig. S3), suggesting chronic inflammation is a contributing factor. ONL thinning and loss of ERG responses were detected with a similar time course as reported by Huang et al. (63), linking high mTORC1 activity of the RPE to retinal dysfunction. Noticeably in both studies, RPE basal infoldings had only moderate changes with deposition of electron-dense materials (Fig. 3N), suggesting that at least at relatively young age abnormally high mTORC1 does not disrupt the RPE transport functions at the basal side. Maintaining an optimal level of mTORC1 activity is also essential for the health of photoreceptor neurons, although mTORC1 is often considered as a survival pathway in cone cells (65). A recent study by Cheng et al. reported that the deletion of Tsc1 in photoreceptor cells caused retinal and RPE degeneration, accumulation of lipids and lipoproteins and retinal vascular lesions in mice at an advanced age (66).
AMD is a common disease involving the degeneration of the outer retina, RPE and choroid (19). Other than the genetic predispositions, RPE cells have intrinsic properties rendering them more vulnerable to age-dependent degeneration. Life-long phagocytosis and processing of the internalized POS stress the cellular degradation pathways of the RPE (67). With aging, the phagolysosome system becomes less efficient (10). Undigested POS, damaged mitochondria, intermediate structures of autophagy, and lipofuscin accumulate in aged RPE (68). In response to the degradative stress, aged RPE upregulates lysosome biogenesis (10). The increased mass of lysosomes provides additional docking sites for mTORC1 assembly (69). Therefore, aged RPE cells display increased mTORC1 activity and are slower in recovery for mTORC1 to return to the baseline level (10). These previous observations, together with the results presented here, suggest that constitutive RPE mTORC1 activation likely initiates a vicious cycle among aging, mTORC1 activity, and degeneration of the RPE and the retina. Deregulated lipid metabolism is a key pathological event downstream of mTORC1.
Human histopathological evidence indicates potential roles of deregulated lipid metabolism in the pathogenesis of AMD, and genetic studies further corroborate the involvement (19, 70–72). However, epidemiologic data do not consistently support the association of the serum lipid levels and AMD, indicating the hallmark change of extracellular lipid deposition on both apical and basolateral sides of the RPE in AMD patients may result from a local effect instead of systemic dyslipidemia (73). Previous studies demonstrate that RPE produces lipoproteins (74, 75). Tsc1 KO RPE had neutral lipid accumulation (Fig. 3P) and SDD formation (Fig. 1D), two of the pathological features of AMD. Data from metabolomic and genomic studies suggest that decreased lipolysis and increased lipogenesis both contributed to lipid accumulation.
On HRM data, diacylglycerol, triacylglycerol, phosphatidic acid, and a variety of phospholipid species were increased in the TSC1-deficient RPE (Fig. 2D–2F). In adipocytes, mTORC1 inhibits the breakdown of neutral lipid and release of fatty acid from lipid droplets, likely by regulating the activities of hormone-sensitive lipases (76, 77). Because of the phagocytosis activity, RPE has an unusual load of OS membranes that provide a rich source of phospholipids and neutral lipids. Under normal physiological conditions, lipid contents of POS are efficiently processed and recycled by the RPE (78, 79). With aging and high mTORC1 activity, however, the breakdown, turnover or trafficking processes can be impaired, which will result in the accumulation of lipids and lipid metabolites. In our study we measured the RPE metabolome before the morning burst of phagocytosis. The accumulation of the intermediate lipid metabolites indicated baseline levels in the Tsc1 KO RPE, rather than the response to phagocytosis. Future studies can be performed to examine the kinetics of POS-induced metabolomes changes in the RPE.
RPE cells are lipogenic (46) (80) and mTORC1 has well-established roles in lipid biosynthesis. GSEA of RNA-seq data revealed an enrichment of upregulated genes in FA biosynthesis (Fig. 5A and 5B). SREBP is a crucial transcription factor controlling the expression of these genes, and nuclear translocation of SREBP was detected in Tsc1 KO RPE (Fig. 5 F–G). Furthermore, FA utilization via mitochondrial β-oxidation was likely to be suppressed by high mTORC1 activity. Acylcarnitine metabolites were lower in Tsc1 KO RPE (Fig. 4C), while the genes encoding proteins in FAO were not changed (Fig. 5A). In cells with accumulated lipids, mitochondria are often found to be bound to lipid droplets (81). Similar structures are observed on EM images of Tsc1 KO RPE (Fig. 3I). The peridroplet mitochondria have distinct metabolic profiles and are less efficient in utilizing fatty acids as energy substrates (81). How mTORC1 controls mitochondria structure and function in the RPE can be explored in future studies.
In the outer retina, the photoreceptors, RPE and choroid rely on the specialized ecosystem for their energy production and utilization, and metabolic changes have emerged as a driving force in retinal diseases (18, 82, 83). Healthy RPE use different energy fuels (amino acids, fatty acids, and lactate) besides glucose for oxidative phosphorylation (18, 84–87). The efficient energy metabolism in the RPE ensures adequate delivery of glucose from the choroidal supply to the retina and maintains the function and integrity of neighboring photoreceptor neurons. Shifting of metabolic status to glycolysis is closely related to the loss of epithelial features of the RPE, and forced conversion of the metabolic pathway in the RPE led to retinal degeneration in murine models (23, 83). Data from our metabolomic and genomic analyses demonstrate that hyperactive mTORC1 activity leads to metabolic reprogramming to a more lipogenic state at the cost of increased glycolysis in the RPE (Fig. 6). The increased utilization of glucose by RPE aerobic glycolysis could disrupt the ecosystem between the RPE and photoreceptor neurons. The limited supply of energy substrates likely caused the loss of photoreceptor ERG responses and a moderate thinning of the ONL layer, which occurred secondary to RPE changes in the conditional knockout mice. The decrease in all three ERG waves suggests that the impairment may result from multiple cell types including rod, cone and other interneurons in the inner retina.
Metabolites of the FA β-oxidation are known to inhibit glucose utilization (88, 89). The inhibitory effects of mTORC1 on carnitine shuttle (Fig. 4) likely contributed to the increased glycolysis. An intriguing finding from RNA-seq data is the upregulation of mRNAs encoding enzymes in ketone body production and ketolysis (Fig. 5C). While we did not find upregulation of genes of FAO, there was an increase in the mRNA of ketogenic enzyme Hmgcs2 in Tsc1-KO RPE. One possible explanation is that under our experimental conditions, RPE ketone body synthesis utilized acetyl-CoA produced from sources other than fatty acids, such as ketogenic amino acids (90). In fact, results from metabolomic studies supported the possibility. The metabolic pathway related to Tryptophan was among the top pathways altered in Tsc1-KO RPE, and Tryptophan is one of the seven ketogenic amino acids (Fig. 4B).
Despite the apparent role of mTORC1 in RPE aging and age-related degeneration, clinical trials of rapamycin in patients with advanced AMD were not successful (27, 91). A possible explanation could be that the retina and RPE have distinct metabolic pathways, and mTORC1 signal is a survival factor of photoreceptor cells (65). Accordingly, rapamycin treatment caused retinal toxicity in AMD patients. Meanwhile, mTORC1 is on the top of the signaling network and controls multiple avenues of cellular processes. Disrupting mTORC1 complex by rapamycin will lead to almost complete inhibition of its downstream events without distinction. Thereby, further dissecting downstream pathways in a tissue-specific context is warranted to address the therapeutic effects of mTORC1 reduction in age-related diseases.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01EY026999 (YC), R01EY 028773 (JC), R01EY025218 (TGW), R01-026545 (TGW), NIEHS grants R01ES023485 (DPJ and YMG) and NIHS10 OD018006 (DPJ), BrightFoucs Foundation grant M2017186 (YC), and Welch Foundation Q0035 (TGW). The authors will like to acknowledge the Oklahoma University Health Science Center (OUHSC) Vision Research Facilities for the Live Animal Imaging Core, Genotyping Core and Cellular Imaging Core services. The cores are supported by NIH/NEI grant P30EY027125 to Dr. Michelle C. Callegan and an unrestricted grant from Research to Prevent Blindness to the Dean McGee Eye Institute. We also will like to thank the Molecular Biology and Cytometry Research Shared Resource at OUHSC which provided the Metabolic Analysis of Live Cells service.
Nonstandard Abbreviations:
- AMD
age-related macular degeneration
- DAPI
4′,6-diamidino-2-phenylindole
- EM
electron microscopy
- ERG
Electroretinography
- FA
fatty acid
- FBS
fetal bovine serum
- GSEA
Gene Set Enrichment Analysis
- H&E
hematoxylin and eosin
- HRM
High-resolution metabolomics
- LD
lipid droplet
- mTOR
mechanistic target of rapamycin
- mTORC1
mechanistic target of rapamycin complex 1
- m/z
mass-to-charge ratio
- OCT
optical coherence tomography
- PBS
phosphate buffered saline
- POS
photoreceptor outer segment
- RNA-seq
RNA sequencing
- RPE
retinal pigment epithelium
- SDD
subretinal drusenoid deposits
- SREBP1
Sterol regulatory element-binding protein 1
- TSC1
tuberous sclerosis complex 1
Reference
- 1.Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yip CK, Murata K, Walz T, Sabatini DM, and Kang SA (2010) Structure of the human mTOR complex I and its implications for rapamycin inhibition. Molecular cell 38, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laplante M, and Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, and Withers DJ (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, and Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, and Muller F (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. [DOI] [PubMed] [Google Scholar]
- 7.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, and Partridge L (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, Fergusson MM, Rovira II, Allen M, Springer DA, Lago CU, Zhang S, DuBois W, Ward T, deCabo R, Gavrilova O, Mock B, and Finkel T (2013) Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep 4, 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Liu Y, Liu Y, and Zheng P (2009) mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu B, Xu P, Zhao Z, Cai J, Sternberg P, and Chen Y (2014) Subcellular distribution and activity of mechanistic target of rapamycin in aged retinal pigment epithelium. Investigative ophthalmology & visual science 55, 8638–8650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zoncu R, Efeyan A, and Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caccamo A, Majumder S, Richardson A, Strong R, and Oddo S (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285, 13107–13120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bove J, Martinez-Vicente M, and Vila M (2011) Fighting neurodegeneration with rapamycin: mechanistic insights. Nature reviews. Neuroscience 12, 437–452 [DOI] [PubMed] [Google Scholar]
- 14.Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, and Klickstein LB (2018) TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med 10, eaaq1564. [DOI] [PubMed] [Google Scholar]
- 15.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, and Klickstein LB (2014) mTOR inhibition improves immune function in the elderly. Sci Transl Med 6, 268ra179. [DOI] [PubMed] [Google Scholar]
- 16.Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, and Miller RA (2012) Rapamycin slows aging in mice. Aging Cell 11, 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saxton RA, and Sabatini DM (2017) mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, Linton JD, Farnsworth CC, Sloat SR, Rountree A, Sweet IR, Lindsay KJ, Parker ED, Brockerhoff SE, Sadilek M, Chao JR, and Hurley JB (2017) Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller JW (2013) Age-related macular degeneration revisited--piecing the puzzle: the LXIX Edward Jackson memorial lecture. American journal of ophthalmology 155, 1–35 e13 [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Wang J, Cai J, and Sternberg P (2010) Altered mTOR signaling in senescent retinal pigment epithelium. Investigative ophthalmology & visual science 51, 5314–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu B, Egbejimi A, Dharmat R, Xu P, Zhao Z, Long B, Miao H, Chen R, Wensel TG, Cai J, and Chen Y (2018) Phagocytosed photoreceptor outer segments activate mTORC1 in the retinal pigment epithelium. Sci Signal 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, and Ferguson TA (2013) Noncanonical autophagy promotes the visual cycle. Cell 154, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C, Yasumura D, Li X, Matthes M, Lloyd M, Nielsen G, Ahern K, Snyder M, Bok D, Dunaief JL, LaVail MM, and Vollrath D (2011) mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. The Journal of clinical investigation 121, 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zigler JS Jr., Zhang C, Grebe R, Sehrawat G, Hackler L Jr., Adhya S, Hose S, McLeod DS, Bhutto I, Barbour W, Parthasarathy G, Zack DJ, Sergeev Y, Lutty GA, Handa JT, and Sinha D (2011) Mutation in the betaA3/A1-crystallin gene impairs phagosome degradation in the retinal pigmented epithelium of the rat. Journal of cell science 124, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaur G, Tan LX, Rathnasamy G, La Cunza N, Germer CJ, Toops KA, Fernandes M, Blenkinsop TA, and Lakkaraju A (2018) Aberrant early endosome biogenesis mediates complement activation in the retinal pigment epithelium in models of macular degeneration. Proc Natl Acad Sci U S A 115, 9014–9019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nussenblatt RB, Byrnes G, Sen HN, Yeh S, Faia L, Meyerle C, Wroblewski K, Li Z, Liu B, Chew E, Sherry PR, Friedman P, Gill F, and Ferris F 3rd. (2010) A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina 30, 1579–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petrou PA, Cunningham D, Shimel K, Harrington M, Hammel K, Cukras CA, Ferris FL, Chew EY, and Wong WT (2014) Intravitreal sirolimus for the treatment of geographic atrophy: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci 56, 330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, and Caspi RR (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investigative ophthalmology & visual science 53, 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Liang Y, Liu Y, Xu P, Flamme-Wiese MJ, Sun D, Sun J, Mullins RF, Chen Y, and Cai J (2017) Choroidal gammadelta T cells in protection against retinal pigment epithelium and retinal injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 31, 4903–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin Y, Moiseyev G, Petrukhin K, Cioffi CL, Muthuraman P, Takahashi Y, and Ma JX (2018) A novel RPE65 inhibitor CU239 suppresses visual cycle and prevents retinal degeneration. Biochim Biophys Acta Mol Basis Dis 1864, 2420–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benchorin G, Calton MA, Beaulieu MO, and Vollrath D (2017) Assessment of Murine Retinal Function by Electroretinography. Bio Protoc 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, and Cai J (2011) Age-related retinopathy in NRF2-deficient mice. PloS one 6, e19456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilliam JC, Chang JT, Sandoval IM, Zhang Y, Li T, Pittler SJ, Chiu W, and Wensel TG (2012) Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 151, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Go YM, Kim CW, Walker DI, Kang DW, Kumar S, Orr M, Uppal K, Quyyumi AA, Jo H, and Jones DP (2015) Disturbed flow induces systemic changes in metabolites in mouse plasma: a metabolomics study using ApoE(−)/(−) mice with partial carotid ligation. Am J Physiol Regul Integr Comp Physiol 308, R62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, and Hollender J (2014) Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol 48, 2097–2098 [DOI] [PubMed] [Google Scholar]
- 36.Yu T, Park Y, Li S, and Jones DP (2013) Hybrid feature detection and information accumulation using high-resolution LC-MS metabolomics data. J Proteome Res 12, 1419–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T, and Jones DP (2013) xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinformatics 14, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Go YM, Walker DI, Liang Y, Uppal K, Soltow QA, Tran V, Strobel F, Quyyumi AA, Ziegler TR, Pennell KD, Miller GW, and Jones DP (2015) Reference Standardization for Mass Spectrometry and High-resolution Metabolomics Applications to Exposome Research. Toxicol Sci 148, 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, and Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Shen D, Wang VM, Yu CR, Wang RX, Tuo J, and Chan CC (2012) Enhanced apoptosis in retinal pigment epithelium under inflammatory stimuli and oxidative stress. Apoptosis 17, 1144–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, and Onda H (2002) A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and upregulation of p70S6 kinase activity in Tsc1 null cells. Human molecular genetics 11, 525–534 [DOI] [PubMed] [Google Scholar]
- 43.Iacovelli J, Zhao C, Wolkow N, Veldman P, Gollomp K, Ojha P, Lukinova N, King A, Feiner L, Esumi N, Zack DJ, Pierce EA, Vollrath D, and Dunaief JL (2011) Generation of Cre transgenic mice with postnatal RPE-specific ocular expression. Investigative ophthalmology & visual science 52, 1378–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zweifel SA, Spaide RF, Curcio CA, Malek G, and Imamura Y (2010) Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology 117, 303–312 e301 [DOI] [PubMed] [Google Scholar]
- 45.Chen L, Messinger JD, Zhang Y, Spaide RF, Freund KB, and Curcio CA (2019) SUBRETINAL DRUSENOID DEPOSIT IN AGE-RELATED MACULAR DEGENERATION: Histologic Insights Into Initiation, Progression to Atrophy, and Imaging. Retina [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choudhary M, Ismail EN, Yao PL, Tayyari F, Radu RA, Nusinowitz S, Boulton ME, Apte RS, Ruberti JW, Handa JT, Tontonoz P, and Malek G (2020) LXRs regulate features of age-related macular degeneration and may be a potential therapeutic target. JCI Insight 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, and Sennlaub F (2007) CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. Journal of Clinical Investigation 117, 2920–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He L, Marioutina M, Dunaief JL, and Marneros AG (2014) Age- and gene-dosage-dependent cre-induced abnormalities in the retinal pigment epithelium. The American journal of pathology 184, 1660–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gambril JA, Sloan KR, Swain TA, Huisingh C, Zarubina AV, Messinger JD, Ach T, and Curcio CA (2019) Quantifying Retinal Pigment Epithelium Dysmorphia and Loss of Histologic Autofluorescence in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 60, 2481–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodenstein L, and Sidman RL (1987) Growth and development of the mouse retinal pigment epithelium. II. Cell patterning in experimental chimaeras and mosaics. Dev Biol 121, 205–219 [DOI] [PubMed] [Google Scholar]
- 51.Chen M, Rajapakse D, Fraczek M, Luo C, Forrester JV, and Xu H (2016) Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell 15, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muniz-Feliciano L, Doggett TA, Zhou Z, and Ferguson TA (2017) RUBCN/rubicon and EGFR regulate lysosomal degradative processes in the retinal pigment epithelium (RPE) of the eye. Autophagy 13, 2072–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rudolf M, and Curcio CA (2009) Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem 57, 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Handa JT, Cano M, Wang L, Datta S, and Liu T (2017) Lipids, oxidized lipids, oxidation-specific epitopes, and Age-related Macular Degeneration. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 430–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, and Pulendran B (2013) Predicting network activity from high throughput metabolomics. PLoS Comput Biol 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.SanGiovanni JP, Chew EY, Agron E, Clemons TE, Ferris FL 3rd, Gensler G, Lindblad AS, Milton RC, Seddon JM, Klein R, Sperduto RD, and Age-Related Eye Disease Study Research, G. (2008) The relationship of dietary omega-3 long-chain polyunsaturated fatty acid intake with incident age-related macular degeneration: AREDS report no. 23. Arch Ophthalmol 126, 1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Souied EH, Delcourt C, Querques G, Bassols A, Merle B, Zourdani A, Smith T, Benlian P, and Nutritional AMDTSG (2013) Oral docosahexaenoic acid in the prevention of exudative age-related macular degeneration: the Nutritional AMD Treatment 2 study. Ophthalmology 120, 1619–1631 [DOI] [PubMed] [Google Scholar]
- 58.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, and Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell 39, 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Z, Wang Y, Feng X, Bao C, He Q, Bao L, Hao H, and Wang Z (2016) Rapamycin Inhibits Expression of Elongation of Very-long-chain Fatty Acids 1 and Synthesis of Docosahexaenoic Acid in Bovine Mammary Epithelial Cells. Asian-Australas J Anim Sci 29, 1646–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Im SS, Hammond LE, Yousef L, Nugas-Selby C, Shin DJ, Seo YK, Fong LG, Young SG, and Osborne TF (2009) Sterol Regulatory Element Binding Protein 1a Regulates Hepatic Fatty Acid Partitioning by Activating Acetyl Coenzyme A Carboxylase 2 (vol 29, pg 4864, 2009). Mol Cell Biol 29, 5974–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, Wakelam M, Harris AL, and Schulze A (2013) Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Golestaneh N, Chu Y, Cheng SK, Cao H, Poliakov E, and Berinstein DM (2016) Repressed SIRT1/PGC-1alpha pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J Transl Med 14, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J, Gu S, Chen M, Zhang SJ, Jiang Z, Chen X, Jiang C, Liu G, Radu RA, Sun X, Vollrath D, Du J, Yan B, and Zhao C (2019) Abnormal mTORC1 signaling leads to retinal pigment epithelium degeneration. Theranostics 9, 1170–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shang P, Valapala M, Grebe R, Hose S, Ghosh S, Bhutto IA, Handa JT, Lutty GA, Lu L, Wan J, Qian J, Sergeev Y, Puertollano R, Zigler JS Jr., Xu GT, and Sinha D (2017) The amino acid transporter SLC36A4 regulates the amino acid pool in retinal pigmented epithelial cells and mediates the mechanistic target of rapamycin, complex 1 signaling. Aging cell 16, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Punzo C, Kornacker K, and Cepko CL (2009) Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci 12, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng SY, Cipi J, Ma S, Hafler BP, Kanadia RN, Brush RS, Agbaga MP, and Punzo C (2020) Altered photoreceptor metabolism in mouse causes late stage age-related macular degeneration-like pathologies. Proc Natl Acad Sci U S A 117, 13094–13104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strauss O (2005) The retinal pigment epithelium in visual function. Physiol Rev 85, 845–881 [DOI] [PubMed] [Google Scholar]
- 68.Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W Jr., Ding J, Bowes Rickman C, and Boulton M (2014) Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10, 1989–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YT, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GH, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Said S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanche H, Deleuze JF, Igo RP Jr., Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA Jr., Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NT, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CC, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JR, Allikmets R, Wang JJ, Schaumberg DA, Klein BE, Hagstrom SA, Chowers I, Lotery AJ, Leveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BH, Abecasis GR, and Heid IM (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48, 134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spaide RF, Ooto S, and Curcio CA (2018) Subretinal drusenoid deposits AKA pseudodrusen. Surv Ophthalmol 63, 782–815 [DOI] [PubMed] [Google Scholar]
- 72.Ratnapriya R, Sosina OA, Starostik MR, Kwicklis M, Kapphahn RJ, Fritsche LG, Walton A, Arvanitis M, Gieser L, Pietraszkiewicz A, Montezuma SR, Chew EY, Battle A, Abecasis GR, Ferrington DA, Chatterjee N, and Swaroop A (2019) Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet 51, 606–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klein R, Myers CE, Buitendijk GH, Rochtchina E, Gao X, de Jong PT, Sivakumaran TA, Burlutsky G, McKean-Cowdin R, Hofman A, Iyengar SK, Lee KE, Stricker BH, Vingerling JR, Mitchell P, Klein BE, Klaver CC, and Wang JJ (2014) Lipids, lipid genes, and incident age-related macular degeneration: the three continent age-related macular degeneration consortium. Am J Ophthalmol 158, 513–524 e513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Landowski M, Kelly U, Klingeborn M, Groelle M, Ding JD, Grigsby D, and Bowes Rickman C (2019) Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc Natl Acad Sci U S A 116, 3703–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang L, Li CM, Rudolf M, Belyaeva OV, Chung BH, Messinger JD, Kedishvili NY, and Curcio CA (2009) Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest Ophthalmol Vis Sci 50, 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ricoult SJ, and Manning BD (2013) The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO reports 14, 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, and Madeo F (2012) FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell metabolism 15, 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen H, and Anderson RE (1993) Metabolism in frog retinal pigment epithelium of docosahexaenoic and arachidonic acids derived from rod outer segment membranes. Exp Eye Res 57, 369–377 [DOI] [PubMed] [Google Scholar]
- 79.Gordon WC, Rodriguez de Turco EB, and Bazan NG (1992) Retinal pigment epithelial cells play a central role in the conservation of docosahexaenoic acid by photoreceptor cells after shedding and phagocytosis. Curr Eye Res 11, 73–83 [DOI] [PubMed] [Google Scholar]
- 80.Storti F, Klee K, Todorova V, Steiner R, Othman A, van der Velde-Visser S, Samardzija M, Meneau I, Barben M, Karademir D, Pauzuolyte V, Boye SL, Blaser F, Ullmer C, Dunaief JL, Hornemann T, Rohrer L, den Hollander A, von Eckardstein A, Fingerle J, Maugeais C, and Grimm C (2019) Impaired ABCA1/ABCG1-mediated lipid efflux in the mouse retinal pigment epithelium (RPE) leads to retinal degeneration. eLife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Benador IY, Veliova M, Liesa M, and Shirihai OS (2019) Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell metabolism 29, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swarup A, Samuels IS, Bell BA, Han JYS, Du J, Massenzio E, Abel ED, Boesze-Battaglia K, Peachey NS, and Philp NJ (2019) Modulating GLUT1 expression in retinal pigment epithelium decreases glucose levels in the retina: impact on photoreceptors and Muller glial cells. Am J Physiol Cell Physiol 316, C121–C133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kurihara T, Westenskow PD, Gantner ML, Usui Y, Schultz A, Bravo S, Aguilar E, Wittgrove C, Friedlander M, Paris LP, Chew E, Siuzdak G, and Friedlander M (2016) Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adijanto J, and Philp NJ (2014) Cultured primary human fetal retinal pigment epithelium (hfRPE) as a model for evaluating RPE metabolism. Exp Eye Res 126, 77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adijanto J, Du J, Moffat C, Seifert EL, Hurle JB, and Philp NJ (2014) The retinal pigment epithelium utilizes fatty acids for ketogenesis. The Journal of biological chemistry 289, 20570–20582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Du J, Yanagida A, Knight K, Engel AL, Vo AH, Jankowski C, Sadilek M, Tran VT, Manson MA, Ramakrishnan A, Hurley JB, and Chao JR (2016) Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc Natl Acad Sci U S A 113, 14710–14715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tyni T, Johnson M, Eaton S, Pourfarzam M, Andrews R, and Turnbull DM (2002) Mitochondrial fatty acid beta-oxidation in the retinal pigment epithelium. Pediatr Res 52, 595–600 [DOI] [PubMed] [Google Scholar]
- 88.Randle PJ, Sugden PH, Kerbey AL, Radcliffe PM, and Hutson NJ (1978) Regulation of pyruvate oxidation and the conservation of glucose. Biochem Soc Symp, 47–67 [PubMed] [Google Scholar]
- 89.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, Covington JD, Bajpeyi S, Ravussin E, Kraus W, Koves TR, and Mynatt RL (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell metabolism 15, 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Puchalska P, and Crawford PA (2017) Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab 25, 262–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong WT, Dresner S, Forooghian F, Glaser T, Doss L, Zhou M, Cunningham D, Shimel K, Harrington M, Hammel K, Cukras CA, Ferris FL, and Chew EY (2013) Treatment of geographic atrophy with subconjunctival sirolimus: results of a phase I/II clinical trial. Investigative ophthalmology & visual science 54, 2941–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.