

Abstract
Calcific uremic arteriolopathy (CUA) is a rare, life-threatening disease, typically affecting patients with end-stage renal disease. It is characterized by widespread vascular calcification, endothelial fibrosis and end-organ ischemia. The mortality rate is high with infection and sepsis being the most common causes of death. Common therapies include restoration of calcium and phosphorous homeostasis, wound care and pain control. Although soft tissue calcification is a known complication in children with advanced renal disease, the incidence of CUA in pediatrics remains unknown. Additionally, current literature regarding its management in pediatric patients is lacking. We report the case of a 17-year-old African–American male patient with end-stage renal disease secondary to Wegener’s granulomatosis who developed CUA after 3 years on peritoneal dialysis. Treatment with sodium thiosulfate (STS) and hyperbaric oxygen (HBO) therapy alone was ineffective, forcing the patient to undergo bilateral below the-knee-amputation (BKA) 5 months after presentation. It was not until peritoneal dialysis had been changed to daily hemodialysis, while continuing STS and HBO therapy, that the patient demonstrated complete resolution of CUA on repeat bone scan. Based on these findings, and the extremely high mortality rate associated with this disease, CUA management requires early and aggressive intervention with multi-faceted therapy, including prompt conversion from peritoneal dialysis to hemodialysis, STS infusions and hyperbaric oxygen therapy.
Keywords: Calciphylaxis, Sodium thiosulfate, Hyperbaric oxygen, Wegener’s granulomatosis
Introduction
Calcific uremic arteriolopathy (CUA) is a life-threatening syndrome of vascular calcification, intimal fibrosis and small-vessel thrombosis. It is a rapidly progressive condition, resulting in painful necrosis of the skin and subcutaneous tissues. These areas can become ulcerated and infected, which often necessitate limb amputation [1, 2]. Once considered a very rare condition, CUA now has reported prevalence rates as high as 4.1% in adults with end-stage-renal disease (ESRD). Increased awareness, along with the extensive use of calcium-containing phosphate binders and vitamin D analogues, is likely responsible for the increased prevalence of this disease [3]. Treatment of CUA remains controversial, with many experimental approaches currently in practice, including sodium thiosulfate (STS) and hyperbaric oxygen (HBO) [1, 2]. Unfortunately, the prognosis of CUA remains poor, and mortality rates are high—greater than 80%. Death usually ensues after infection or sepsis, often within 6 months of presentation [2].
Although soft tissue calcification is a well-known complication in children with advanced renal disease [4], the incidence of CUA in pediatrics is unknown. In addition, very few guidelines are available in the current literature regarding its management in pediatric patients.
We report the case of a 17-year-old, peritoneal dialysis-dependent, male patient with CUA, who was initially treated with STS. Although STS successfully treated the patient’s pain, it did not halt the disease process. It was only with a more aggressive approach, including conversion from peritoneal dialysis to daily hemodialysis and HBO therapy, in addition to STS, that we were able to successfully control the clinical course of CUA.
Case report
A 17-year-old African–American patient with ESRD, who had been on peritoneal dialysis for 3 years, presented with erythema and swelling of bilateral first metatarsals. The patient’s past medical history was significant for ESRD secondary to Wegener’s granulomatosis, diagnosed by renal and nasal biopsies at age 14 years.
Hemodialysis was initiated at the time of diagnosis and then converted to peritoneal dialysis shortly thereafter. The patient’s clinical course was complicated by the following:
Right atrial thrombosis. This occurred 5 months after initiation of hemodialysis and was treated with warfarin anticoagulation for 2.5 years. Hypercoagulability was assessed at this time (including anti-cardiolipin antibody testing) and was unremarkable. After severe epistaxis requiring 3 separate blood transfusions, warfarin had been replaced with aspirin 4 months prior to the above presentation.
Hyperphosphatemia. Calcium and phosphorous homeostasis had been well controlled during the first year of dialysis with proper dietary restrictions and the use of calcium carbonate as a phosphate binder. Serum phosphorous levels began to increase progressively after 12–15 months of dialysis (Table 1). At this time, calcium carbonate dosing was increased, treatment with sevelamer hydrochloride was initiated and dietary restrictions were heavily emphasized.
Immunosuppression. The patient had been maintained on prednisone and cyclophosphamide for management of Wegener’s granulomatosis. After rising cytoplasmic anti-neutrophil cytoplasmic antibody (c-ANCA) titers and reports of joint pain, dosing of both medications had been increased 2 months prior to presentation with erythema and swelling of bilateral first metatarsals.
Table 1.
Average serum calcium, phosphorous, calcium × phosphate (Ca × PO4) product and intact parathyroid hormone (PTH) levels, based on monthly determinations obtained throughout 2005, 2006 and 2007
| Year | Ca (mg/dl) | PO4 (mg/dl) | Ca × PO4 | PTH (pg/ml) |
|---|---|---|---|---|
| 2005 | 9.3 ± 0.4 | 4.4 ± 0.6 | 40.5 ± 6.5 | 168 ± 52 |
| 2006 | 8.6 ± 0.7 | 5.9 ± 1.1 | 50.4 ± 10.8 | 559 ± 232 |
| 2007 | 9.4 ± 0.6 | 7.2 ± 1.6 | 68.1 ± 17.6 | 424 ± 203 |
| Average | 8.6 ± 2.1 | 5.7 ± 2.1 | 51.8 ± 20.3 | 367 ± 249 |
When the patient presented with erythema and swelling of the metatarsals, the initial lesion was diagnosed as cellulitis, but it did not improve after the administration of two courses of oral antibiotics. The patient subsequently developed severe pain in both legs as well as ischemic changes in his toes.
Laboratory investigations at this time revealed serum calcium 9.7 mg/dl, phosphorus 8.3 mg/dl (Ca × phosphate product 80.5) and parathyroid hormone level 321 pg/ml. A plain radiograph demonstrated vascular calcifications in both feet. A skin biopsy was not performed; however, the diagnosis of calciphylaxis was strongly supported by a technetium-99 bone scan, which demonstrated prominent vessel calcifications within the upper and lower extremities bilaterally, as well as areas of intensely increased activity in the soft tissues of the inferior buttock bilaterally (Fig. 1). At the time of the scan, the patient manifested evidence of ischemia only in both feet, with sparing of the upper extremities and buttocks.
Fig. 1.
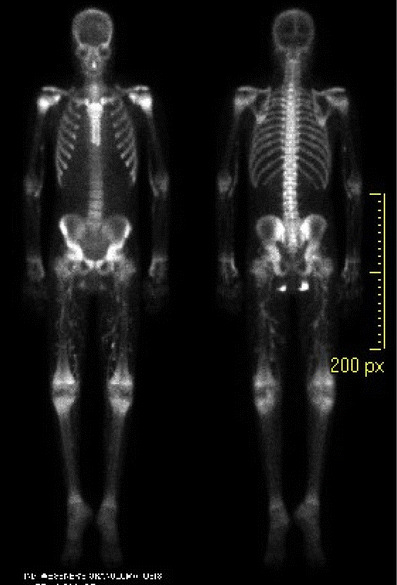
Technetium-99 bone scan demonstrating prominent vessels in bilateral lower extremities and areas of increased activity in the soft tissues of the inferior buttock bilaterally, both findings consistent with calcific uremic arteriolopathy
Once the diagnosis of CUA had been made, aggressive attempts to improve calcium–phosphorus homeostasis were implemented. This included dietary modification, discontinuation of calcium-containing phosphate binders and vitamin D analogues, a brief course of aluminum hydroxide (followed by sevelamer hydrochloride) and institution of a mid-day peritoneal exchange, in addition to ten overnight exchanges.
Therapy with STS was initiated 3 weeks after diagnosis, at a dose of 25 g/1.73 m2, with thrice-weekly administration, as previously recommended [1]. The patient’s pain was reduced rapidly with the initiation of STS infusions. After 7 treatments, his need for narcotic analgesia decreased dramatically. By the 15th infusion, the patient was pain free. Despite initiation of STS, necrosis of bilateral first metatarsals continued to spread proximally (Fig. 2). Evidence of poor perfusion to the patient’s hands also became apparent, as demonstrated by blue discoloration of the distal fingertips and delayed capillary refill. At this point, HBO therapy was implemented. The patient received treatment five times per week, each session lasting 90 min at 2.2 atm. After 15 infusions of STS and 4 weeks of HBO therapy, necrosis of the patient’s feet had become so advanced that he underwent bilateral below-the-knee amputation (BKA).
Fig. 2.
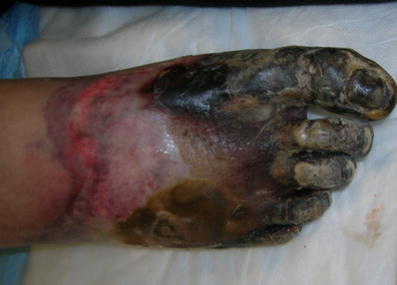
Advancement of necrosis, after 6 weeks of STS and 2 weeks of HBO therapy
Two months had passed from the initiation of STS therapy to the time of amputation. Average serum calcium and phosphorus levels during this time were 9.5 ± 0.4 mg/dl and 5.6 ± 0.9 mg/dl, respectively. In order to achieve better control of hyperphosphatemia, the patient was switched from peritoneal dialysis to hemodialysis, five times per week, immediately after amputation. His clinical course improved dramatically thereafter. While on hemodialysis, the patient continued to receive thrice weekly STS (for a total of 33 doses) and HBO therapy 3 to 5 times per week. Two months after the transition to hemodialysis, average serum phosphorus fell significantly to 3.3 ± 1.3 mg/dl (Fig. 3). At this time, ischemic changes of the hands had completely resolved, and a repeat bone scan showed no abnormal tracer activity suggestive of vascular calcification in any location, including the previously affected areas (upper extremities and buttocks).
Fig. 3.
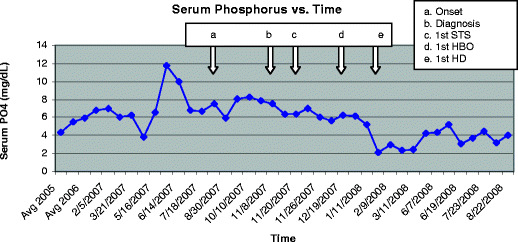
Serum phosphorus over time. a The patient presented with bilateral first metatarsal erythema and swelling. b Diagnosis of CUA confirmed via bone scan. c First infusion of STS. d First HBO treatment. e Conversion from peritoneal dialysis to daily hemodialysis (HD) and bilateral BKA
The patient continued to receive hemodialysis 5 times per week (without STS or HBO therapy) until he underwent successful deceased donor renal transplantation, approximately 7 months after the last bone scan. He has shown no clinical evidence of recurrence thus far. With aggressive physical therapy and steadfast perseverance, the patient is now able to ambulate well, with the assistance of prosthetic limbs.
Discussion
This case is an example of severe CUA necessitating bilateral BKA in a pediatric patient with ESRD. Only with aggressive management using multiple modalities, including conversion to daily hemodialysis, STS infusions and HBO therapy, were we able to reverse the process of calciphylaxis and salvage his upper extremities.
The exact pathogenesis of CUA is not fully understood. Chronic inflammation, disordered calcium−phosphorus metabolism (particularly hyperphosphatemia) and derangements in bone metabolism are likely culprits in the development of this disease [3]. It occurs most frequently in patients with ESRD; however, it has also been documented in patients with breast cancer, inflammatory bowel disease, alcohol-induced cirrhosis, primary hyperparathyroidism and chronic kidney disease [2, 5].
Wilmer and Magro suggest a 2 step mechanism of injury in CUA. Step 1 is the development of isolated vascular calcification, which begins with an imbalance between the positive and negative regulators of vascular calcification. Stimulatory agents of vascular calcification include calcium, phosphorus, vitamin D, bone morphogenic protein-2 (BMP-2), warfarin, reactive oxygen species, C-reactive protein, interleukin (IL)-6 and tumor necrosis factor alpha (TNF-α). Conversely, agents that inhibit vascular calcification include fetuin-A, matrix gamma carboxyglutamate (GLA) protein and IL-10 [5]. Step 2 reflects the consequences of vascular calcification, such as poor tissue perfusion, end-organ ischemia and development of subcutaneous and skin ischemia with ulceration [6].
Known risk factors for developing CUA include Caucasian race, female gender, longer than 1 year on dialysis, severe secondary hyperparathyroidism, adynamic osteodystrophy, hypoalbuminemia, obesity, type 2 diabetes mellitus (or metabolic syndrome), use of calcium-containing phosphate binders or vitamin D analogues, hypercoagulability (protein C and S deficiency), warfarin therapy, and the use of steroids or other immunosuppressive drugs (methotrexate) [2, 3, 6–9].
The association between warfarin therapy and CUA has been documented in a number of pediatric and adult patients [1, 10, 11]. This association is likely related to the fact that warfarin inhibits activation of the vitamin K-dependent calcification inhibitor, matrix GLA protein. Administration of warfarin to rats leads to extensive vascular calcification, presumably by this mechanism [12]. As noted above, our patient had received warfarin for 2 years prior to the diagnosis of CUA.
CUA has also been reported in several patients with vasculitis, both with and without chronic renal insufficiency. Lee et al. examined the 11 known cases of CUA with autoimmune disease, which included patients with systemic lupus erythematosus, rheumatoid arthritis, giant cell arteritis and sarcoidosis. The majority of these patients were female, presented with intractable lower extremity pain and ulceration and died after overwhelming sepsis [13]. The pathogenesis of CUA in the face of autoimmune disease (particularly without renal insufficiency) is also not clearly understood. Two studies have raised the possibility that steroids and other immunosuppressive drugs (methotrexate) are potential triggers [8, 9].
Our patient had several of the aforementioned risk factors, including 3 years of dialysis dependency, abnormal calcium/phosphorus metabolism, warfarin anticoagulation, and adynamic osteodystrophy, as well as an underlying vasculitis treated with prednisone and other immunosuppressive drugs.
Despite recent advances in the understanding of the pathophysiology of CUA, the most effective treatment remains unclear. Currently, management of CUA is largely empiric in nature and varies greatly from one center to another. Improved calcium and phosphorus metabolism is usually attempted through dietary changes, removal of calcium-based phosphate binders and vitamin D analogues, and increased dialysis. Common supportive care measures include aggressive pain control and wound care. Other experimental therapies, including parathyroidectomy (of most benefit to those individuals with extremely elevated parathyroid hormone levels), cinacalcet, bisphosphonates, low doses of plasminogen activator, STS and hyperbaric oxygen, have all shown some success in a variety of small studies or case reports [3, 5].
For our patient, conservative therapy provided little benefit. Parathyroidectomy and cinacalcet treatment were deferred, as his intact parathyroid hormone levels were relatively low (92–321 pg/ml), suggestive of adynamic bone. Although CUA most frequently occurs in parallel to secondary hyperparathyroidism, 7 other cases of CUA in patients with relatively low parathyroid hormone (PTH) values and low-turnover bone disease have been described [14]. While patients with adynamic bone have similar intestinal calcium absorption to those with hyperparathyroidism, the ability of bone to buffer calcium is diminished [15]. This phenomenon likely predisposes those with low bone turnover to soft tissue calcification, making parathyroidectomy a poor therapeutic option.
Despite maximizing conservative care for our patient, ischemia within the lower extremities continued to progress rapidly. Hence, more aggressive therapy with STS was initiated. STS is a highly soluble calcium chelator commonly used in the treatment of cyanide and cisplatin toxicity. Because of its antioxidant and calcium-chelating properties, STS has also been used to treat tumoral calcinosis and CUA. The literature on the efficacy of STS in CUA is limited to case reports (for a full review, see Hayden et al. [5]). All 13 of these patients (including 3 children) have shown a positive response to STS therapy, although these reports also document a variety of concomitant therapies.
STS therapy is usually well tolerated, but it can be associated with side effects that include nausea, vomiting, metabolic acidosis and hypocalcemia [5]. Our patient developed intermittent nausea and vomiting, both during and after STS infusions. This improved significantly with granisetron pre-medication. He also developed metabolic acidosis, which resolved with oral administration of sodium bicarbonate. Of note, the patient had one episode of ventricular tachycardia (five beats in total) with the first STS infusion, at which his time serum bicarbonate level was 13 mmol/L. This particular adverse side effect has not been reported previously. It is unclear whether this arrhythmia was due to the STS itself or to the STS-induced metabolic acidosis. It did not recur after correction of the metabolic acidosis. Hypocalcemia was not noted in our patient.
STS therapy for our patient resulted in a rapid and dramatic reduction in pain, but it did not prevent further progression of the disease. Thus, after 1 month of STS infusions, HBO therapy was initiated. HBO is often instituted after medical and/or surgical management has failed. It has been shown to be beneficial for adult dialysis patients with CUA [16–19]. This procedure enhances wound healing by improving oxygen delivery to the diseased hypoxic tissue, thereby stimulating fibroblast function, collagen formation and angiogenesis [20]. The process itself consists of the patient breathing 100% oxygen in a sealed chamber with an increased pressure, typically between 2 atm and 2.5 atm, for 60–90 minutes per session. Twenty-five to 40 consecutive sessions have been used in the treatment of CUA.
Despite success noted in previous case reports with only STS or HBO therapy, our patient’s disease continued to progress rapidly. Only 5 months after the initial presentation, ischemic necrosis had advanced to the point where bilateral BKA was necessary. Fortunately, with the subsequent transition made from peritoneal dialysis to daily hemodialysis, rapid clinical improvement ensued, as demonstrated by reperfusion in both hands and resolution of CUA on bone scan. Two of the 13 case reports of successful STS therapy in CUA referred to patients on peritoneal dialysis, but our case clearly demonstrates that the additional phosphate clearance from daily hemodialysis was life-saving [2, 21]. To date, there have been no other case reports that have demonstrated such a remarkable or rapid recovery from CUA after conversion from peritoneal dialysis to hemodialysis.
Although there are similarities to those in previous reports, this case of CUA is unique in several aspects. The patient’s young age, male gender, African–American race, underlying vasculitis (Wegener’s granulomatosis) and adynamic osteodystrophy are not typical features of CUA. In addition, while the aggressive nature of his disease was comparable to that of others with CUA, this patient’s resolution of symptoms and continued survival after initiation of a multi-faceted and aggressive approach is distinct from 80% of other CUA cases.
Given the destructive nature and high mortality rate of CUA, it seems prudent to employ a multi-faceted approach as early as possible. Thus, for a persistently hyperphosphatemic peritoneal dialysis patient with CUA, we would recommend early conversion to daily hemodialysis, alongside STS and HBO therapy. In our experience, we feel it is necessary to implement multiple therapies concurrently, in order to minimize the risk of amputation, life-long morbidity and death.
Acknowledgements
We thank Dr. Carlos Araya for his assistance in the care of this patient.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Araya CE, Fennell RS, Neiberger RE, Dharnidharka VR. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006;1:1161–1166. doi: 10.2215/CJN.01520506. [DOI] [PubMed] [Google Scholar]
- 2.Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104–1108. doi: 10.1053/j.ajkd.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Rogers NM, Teubner DJ, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150–157. doi: 10.1111/j.1525-139X.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- 4.Rogers NM, Teubner DJ, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150–157. doi: 10.1111/j.1525-139X.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- 5.Hayden MR, Goldsmith D, Sowers JR, Khanna R. Calciphylaxis: calcific uremic arteriolopathy and the emerging role of sodium thiosulfate. Int Urol Nephrol. 2008;40:443–451. doi: 10.1007/s11255-008-9373-4. [DOI] [PubMed] [Google Scholar]
- 6.Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172–186. doi: 10.1046/j.1525-139X.2002.00052.x. [DOI] [PubMed] [Google Scholar]
- 7.Meissner M, Gille J, Kaufmann R. Calciphylaxis: no therapeutic concepts for a poorly understood syndrome? J Dtsch Dermatol Ges. 2006;4:1037–1044. doi: 10.1111/j.1610-0387.2006.06127.x. [DOI] [PubMed] [Google Scholar]
- 8.Ozbalkan Z, Calguneri M, Onat AM, Ozturk MA. Development of calciphylaxis after long-term steroid and methotroxate use in a patient with rheumatoid arthritis. Intern Med. 2005;44:1178–1181. doi: 10.2169/internalmedicine.44.1178. [DOI] [PubMed] [Google Scholar]
- 9.Korkmaz C, Dündar E, Zubaroğlu I. Recognizing calcific uremic arteriolopathy in autoimmune disease: an emerging mimicker of vasculitis. Clin Rheumatol. 2002;21:66–69. doi: 10.1007/s100670200016. [DOI] [PubMed] [Google Scholar]
- 10.Asobie N, Wong E, Cook MG. Calciphylaxis in a diabetic patient provoked by warfarin therapy. Clin Exp Dermatol. 2008;33:342–344. doi: 10.1111/j.1365-2230.2007.02626.x. [DOI] [PubMed] [Google Scholar]
- 11.Coates T, Kirkland GS, Dymock RB, Murphy BF, Brealey JK, Mathew TH, Disney AP. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384–391. doi: 10.1053/ajkd.1998.v32.pm9740153. [DOI] [PubMed] [Google Scholar]
- 12.Howe AM, Webster WS. Warfarin exposure and calcification of the arterial system in the rat. Int J Exp Pathol. 2000;81:51–56. doi: 10.1046/j.1365-2613.2000.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JL, Naguwa SM, Cheema G, Gershwin ME. Recognizing calcific uremic arteriolopathy in autoimmune disease: an emerging mimicker of vasculitis. Autoimmun Rev. 2008;7:638–643. doi: 10.1016/j.autrev.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Mawad HW, Sawaya BP, Sarin R, Malluche HH. Calcific uremic arteriolopathy in association with low turnover uremic bone disease. Clin Nephrol. 1999;52:160–166. [PubMed] [Google Scholar]
- 15.Kurz P, Monier-Faugere MC, Bognar B, Werner E, Roth P, Vlachojannis J, Malluche HH. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. 1994;46:855–861. doi: 10.1038/ki.1994.342. [DOI] [PubMed] [Google Scholar]
- 16.Dean SM, Werman H. Calciphylaxis: a favorable outcome with hyperbaric oxygen. Vasc Med. 1998;3:115–120. doi: 10.1177/1358836X9800300205. [DOI] [PubMed] [Google Scholar]
- 17.Dwyer KM, Francis DM, Hill PA, Murphy BF. Calcific uraemic arteriolopathy: local treatment and hyperbaric oxygen therapy. Nephrol Dial Transplant. 2002;17:1148–1149. doi: 10.1093/ndt/17.6.1148. [DOI] [PubMed] [Google Scholar]
- 18.Podymow T, Wherrett C, Burns KD. Hyperbaric oxygen in the treatment of calciphylaxis: a case series. Nephrol Dial Transplant. 2001;16:2176–2180. doi: 10.1093/ndt/16.11.2176. [DOI] [PubMed] [Google Scholar]
- 19.Vassa N, Twardowski ZJ, Campbell J. Hyperbaric oxygen therapy in calciphylaxis-induced skin necrosis in a peritoneal dialysis patient. Am J Kidney Dis. 1994;23:878–881. doi: 10.1016/S0272-6386(12)80145-4. [DOI] [PubMed] [Google Scholar]
- 20.Leach RM, Rees PJ, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317:1140–1143. doi: 10.1136/bmj.317.7166.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mataic D, Bastani B. Intraperitoneal sodium thiosulfate for the treatment of calciphylaxis. Ren Fail. 2006;28:361–363. doi: 10.1080/08860220600583781. [DOI] [PubMed] [Google Scholar]