

Abstract
Bacterial communities are governed by a wide variety of social interactions, some of which are antagonistic with potential significance for bacterial warfare. Several antagonistic mechanisms, such as killing via the type VI secretion system (T6SS), require killer cells to directly contact target cells. The T6SS is hypothesized to be a highly potent weapon, capable of facilitating the invasion and defence of bacterial populations. However, we find that the efficacy of contact killing is severely limited by the material consequences of cell death. Through experiments with Vibrio cholerae strains that kill via the T6SS, we show that dead cell debris quickly accumulates at the interface that forms between competing strains, preventing physical contact and thus preventing killing. While previous experiments have shown that T6SS killing can reduce a population of target cells by as much as 106-fold, we find that, as a result of the formation of dead cell debris barriers, the impact of contact killing depends sensitively on the initial concentration of killer cells. Killer cells are incapable of invading or eliminating competitors on a community level. Instead, bacterial warfare itself can facilitate coexistence between nominally antagonistic strains. While a variety of defensive strategies against microbial warfare exist, the material consequences of cell death provide target cells with their first line of defence.
Keywords: bacteria, emergence, antagonism
1. Introduction
Bacteria commonly inhabit biofilms in the form of crowded, surface-attached microbial consortia embedded within a viscous matrix of polymers. Interactions between different bacterial strains and species govern the spatial organization and composition of biofilms [1–3], and ultimately affect the proliferation and survival of individual strains. These interactions can turn deadly. Bacteria have evolved many mechanisms to kill each other within biofilms [4,5], many of them requiring direct contact between cells [4,6–9]. One such contact killing mechanism is the broadly prevalent type VI secretion system (T6SS) in Gram-negative bacteria [10]. A significant amount of work has produced a detailed picture of the T6SS. Details are emerging of the T6SS structure, toxins and regulation [10–20]. However, the importance of this lethal activity in natural communities remains unclear [21,22]. Experiments have primarily focused on the outcome of competitions between T6SS-proficient ‘killers’ and target strains that lack T6SS activity, but the dynamics of T6SS killing are much less studied [23] (though dynamic simulations have made a number of successful predictions [24–28]). Understanding the impact of the T6SS requires experimental observation of contact killing in microbial communities as a function of time and isolated from other factors. This is a crucial step in assessing the ecological role of contact killing over short and long time scales.
T6SS-mediated killing is widely considered a potent weapon. In biofilms grown from a mixture of T6SS-proficient bacteria and target strains on planar agar pads, killer cells decreased the abundance of target cells by as much as 106-fold within 3 h [15,23,29–31]. Based on these competition assays, the T6SS is hypothesized to play important roles in inter- and intra-strain competition; for example, facilitating invasion of colonized space, elimination of competitors and defence against invaders and cheaters in biofilms [21,28,32–34]. However, in these competition assays T6SS-mediated killing is rarely able to completely eliminate all susceptible target cells, even when killer cells start at a numerical advantage (a 10 : 1 number ratio of killer : target cells is often used) [23,24,30,31,35]. Further, while the killing rate is typically very high shortly after inoculating competing strains on agar pads, killing nearly halts a few hours later, despite the presence of target cells [23]. This dramatic decrease in killing occurs even when the killer strain expresses a constitutively active T6SS [23]. While some studies report defence mechanisms that mitigate or counteract T6SS attacks [36–41], it is difficult to isolate alterations in T6SS activity over time as developing biofilms become increasingly heterogeneous [42,43] and constantly change [44–48]: biomass increases, nutrient and oxygen concentrations drop, excreted waste products accumulate and cellular behaviour changes owing to signalling from secreted public goods. As a result, a detailed picture of how T6SS killing proceeds within biofilms and how T6SS-mediated killing rates change over time remains elusive.
Here, we present the spatio-temporal dynamics of T6SS-mediated killing. We used the T6SS-proficient killer strain Vibrio cholerae C6706 and mutants of it, which secrete lethal effectors that cause cell death [30,35,37,49–51]. Through microscopy experiments, we show that while T6SS-mediated killing is effective on first contact between competing strains, surprisingly, killing nearly ceases after a few hours because of the accumulation of dead cell debris. Contact killing experiments typically focus on living cells as they measure the number of surviving target cells after a certain time of inoculation [15,29–31,39,52–55]. This approach was fundamental in discovering the deadly effect of T6SS [7], the characteristic spatial structure that emerges from contact killing [2,24,26,28], and microbial defence strategies against T6SS attacks [36–39]. Here, we focus on the role of dead cells and dead cell debris. We confirm that dead cell debris accumulates at the interface between competing strains and eventually prevents contact, thus halting killing. Paradoxically, contact killing may thus play a protective role in biofilms, facilitating the formation and coexistence of separate clonal domains [56,57].
2. Results
To study how T6SS killing proceeds within biofilms, we optically recorded the spatio-temporal dynamics of biofilms comprising two engineered strains of V. cholerae, a model organism for studying the T6SS [7]. These strains are isogenic, and only differ in their T6SS toxins and immunity modules, their ability to express T6SS and their fluorescent proteins [24]. To isolate the effects of killing from other effects related to changes in biofilm height (which can impact cellular behaviour [43]), we grew biofilms in confinement between a lysogeny broth (LB) agar pad and a glass coverslip (figure 1a), which limited vertical growth to less than 6 μm. (See electronic supplementary material for details on strains and sample preparation.)
Figure 1.
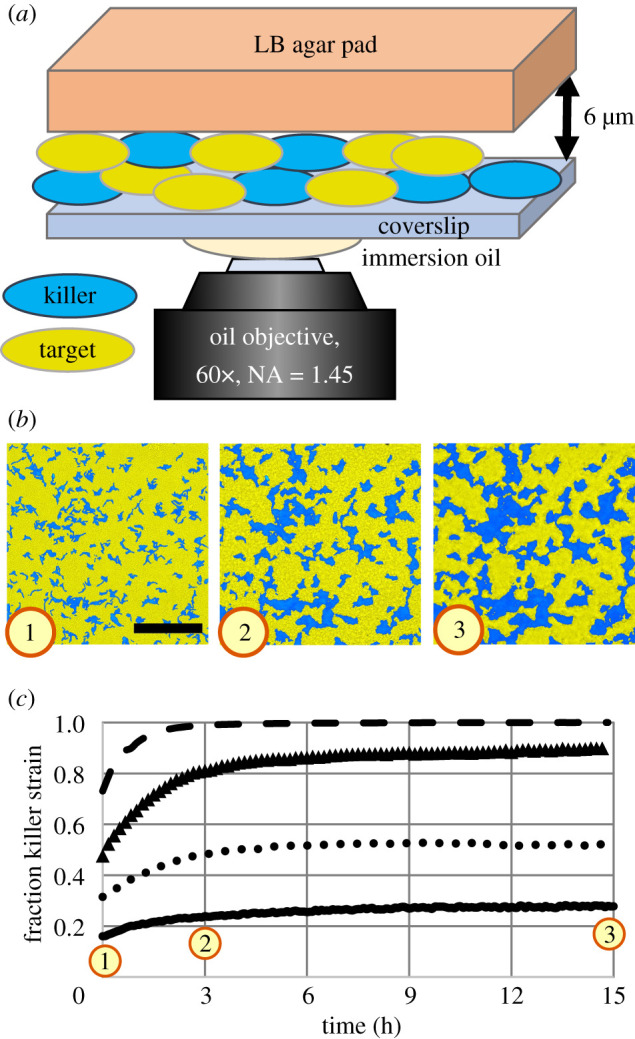
Contact killing slows substantially over time. (a) Cartoon depiction of biofilm samples for imaging. Confinement between an agar pad and a glass coverslip leads to a maximum biofilm thickness of approximately 6 μm. (b) Fluorescence images of a unidirectional competition between killer cells (blue) and target cells (yellow) at 0 h, 3 h and 15 h (left to right, respectively). The initial relative abundances were 0.09 killer cells and 0.91 target cells. T6SS-mediated contact killing leads to the formation of clonal domains, which increase in size over time. Scale bar: 50 μm. (c) Relative abundance of the killer cells over time for four different initial conditions (killer cell relative abundance during inoculation: 0.5 (dashed line), 0.33 (triangles), 0.17 (dotted line) and 0.09 (solid line)) measured from time-lapse images as depicted in (b). In all cases, the relative abundance of killer cells initially increased quickly, followed by a substantially slower increase after 3 h. (See also electronic supplementary material, video 1.)
2.1. Spatio-temporal dynamics
As a first step, using confocal time-lapse microscopy we investigated the temporal dynamics of unidirectional killing: the killer was a T6SS-proficient (T6SS+) V. cholerae strain, while the target was a susceptible, T6SS-defective (T6SS−) V. cholerae strain. We observed the formation of clonal domains (figure 1b) upon reproduction and killing. This phenomenon is reminiscent of domain formation observed in populations of mutual killer strains (i.e. both strains are T6SS+) [24], even though in our experiments one strain—the target strain—was engineered to be defective at T6SS killing. While we observed that domains of the killer strain expanded quickly at early times, domain growth later slowed substantially (figure 1b). We quantified the temporal dynamics of mixed populations of killer and target strains (figure 1c) by measuring the relative abundance of the fluorescent killer strain in the microscopy images over 15 h, for four different initial ratios of killer and target cells (see electronic supplementary material for further details on image analysis). Note that the mixing ratios do not reflect the relative abundance of killer cells at 0 h. Biofilms were grown from relatively low cell concentrations (OD600 = 1). However, we began measuring the relative abundance of the fluorescent strain only after a dense cell layer has formed, which we define to be when time = 0 h, after which the vast majority of killing takes place. Killer populations increased their relative abundance from 12% to 42%. In contrast, we performed control experiments with two non-killer strains. When killing was absent, the population of the fluorescent strain changed by less than 3% over 15 h for various mixing ratios (electronic supplementary material, figure S2). Therefore, demographic changes in biofilms with T6SS-active strains can mainly be attributed to killing. In all cases of unidirectional killing, the killer strain initially increased its population rapidly and reached approximately of its final size after approximately 3 h. Killing dramatically slowed afterwards. We observed similar temporal dynamics in experiments with mutually killing V. cholerae strains (electronic supplementary material, figure S4a,b and video 1). Again, a transition from rapid killing to almost no killing occurred after approximately 3 h. These observations are consistent with previously reported observations of small but long-lived target populations [23]. Why does T6SS-mediated killing stop after just a few hours?
2.2. Cell debris barrier
Surprisingly, we found that the boundary between competing strains was visible in images recorded with bright-field microscopy (figure 2a). Dark outlines become visible in bright-field images (figure 2a, top image); these dark outlines align with the interface between domains of killer and target cells in fluorescence images (figure 2a, bottom image). Time-lapse images showed that these dark outlines form at early times in biofilms with killing, but they are absent at all times in non-killer biofilms, i.e. those with two isogenic T6SS− strains (electronic supplementary material, figure S3). Small clonal domains still emerged in non-killer biofilms as non-motile, divided cells typically remain close after reproduction (figure 2b, bottom image), but the interface between these clonal domains did not appear dark in bright-field images (figure 2b, top image). The ability to visualize the interface between competing strains using bright-field microscopy is not expected since the two isogenic strains do not differ in their material properties—including index of refraction—so they appear identical when imaged with bright-field microscopy. The presence of dark lines in biofilms with T6SS killing suggests that there occurred a change in material properties (e.g. index of refraction) at the border between strains. Based on this observation, and the data presented above, we hypothesized that dead cell debris accumulates as cells are killed. Such cell debris may eventually prevent competing cells from contacting. Similar observations have been made previously when studying T6SS-mediated interactions with non-lethal effectors [58–60].
Figure 2.

Characterization of dead cell debris. (a) Representative bright-field image alone (top) and overlaid with fluorescence channel (bottom) of biofilms exhibiting unidirectional killing, recorded after 7 h of growth. Killer cells express superfolder green fluorescent protein (sfGFP) (cyan) while target cells are unlabelled. The entire field of view is densely packed with bacteria. (b) Same as (a) but the killer strain is engineered to be T6SS-deficient; no T6SS killing occurs and dark outlines are absent. (c,d) Merged bright-field and fluorescence images of unidirectional killing at start (0 h) (c) and after 7 h (d). Again, the killer expresses sfGFP (visible as cyan) while the target is unlabelled (grey). The DNA of compromised cells, i.e. dead cell debris, is labelled red via PI. Scale bar (a–d): 30 μm. (e) Intensity profile of different microscope images across a clonal patch (integrated vertically over the orange box in (d)) in the fluorescence images and bright-field image (inverted grey scale). (f) Normalized mean intensity of PI signal (red curve) and inverted bright-field signal (black curve) within a distance r from the interface between competing strains in (d). (g) Killing event, where a fluorescent killer cell kills a non-fluorescent target cell, which subsequently turns red. The dead cell debris is relocated by neighbouring cells that exert forces upon growth. Scale bar: 4 μm. (h) Counting the number of discrete PI-labelled cells over time demonstrates that PI-labelled dead cell debris persists long after cell death. Data were recorded from individual target cells densely surrounded by killer cells (initial target to killer number ratio of 1 : 50).
To test our hypothesis, we visualized dead cell debris in growing biofilms with propidium iodide (PI). PI binds to the DNA of cells with a compromised membrane and exhibits high red fluorescence. While stained dead cells appeared throughout the biofilm during the earliest stages of growth when clonal domains are small (figure 2c; see also electronic supplementary material, video 2), at later times the dead cell stain was clearly localized at the interface between large clonal domains (figure 2d). The PI signal is well aligned with both the interface between strains and the dark outlines seen with bright-field microscopy (figure 2e). The PI signal exhibits a peak at the same position (distance of 6.2 and 31.3 μm) where the cell fluorescence declines to about 30% of its maximum value. From a Gaussian fit we found that both peaks in PI signal and bright-field signal differ in position by less than 0.3 μm and differ in width (i.e. standard deviation of the peak) by less than 0.1 μm. This sub-micron alignment of signals suggests that dark outlines observed via bright-field microscopy correspond to a substantial amount of cell debris at the interfaces between patches.
To quantify the localization of dead cell debris at interfaces throughout the biofilm, we measured the mean intensity of PI signal as a function of distance from the interface between strains (figure 2f; see electronic supplementary material for more details on the image analysis). The intensity of the PI signal decays with distance from the strain interface, and reaches half its maximum value at a distance of 1.4 μm. We applied the same image analysis to bright-field images and characterized the dark outlines at the strain interfaces. The dark outlines lead to a similar decaying curve, reaching half its maximum value at a distance of about 1.8 μm (figure 2f) or higher, depending on the chosen threshold value during image analysis (electronic supplementary material, figure S1c). Both curves confirm that dead cell debris is highly localized at the interface between strains. Crucially, the estimated dead cell debris layer thickness is larger than the length of a V. cholerae cell.
To account for the observed slow rate of killing after 3 h, the dead cell debris that separates competing strains must also be stable over long periods of time. We observed that debris from one individual dead cell remained clearly visible for at least 60 min, even as it was relocated via forces exerted by neighbouring cells as those reproduce and die (figure 2g). However, the emergence of more dead cells in close proximity inhibits tracking for longer times.
To quantify the persistence of dead cell material over long times, we mixed 98% T6SS+ killer cells and 2% T6SS− target cells and inoculated at high density (OD600 = 10), so experiments began with close-packed cellular monolayers. Target cells were very far from each other, allowing us to isolate and track individual stained dead cells over long times (approx. 10 h). All 117 individual target cells died within 1.2 h of inoculation (figure 2h), and were tracked by PI labelling afterwards. The number of dead cells with detectable PI signal slowly decreased over time. The decrease in the number of PI-labelled dead cells may be due to a local loss in the presence of dead cell debris, e.g. the material may degrade and diffuse away. Note that the PI-labelled area per dead cell also decreased over time (electronic supplementary material, figure S5), which may be caused by degradation or compaction of dead cell debris. However, despite the observed decrease of dead cell debris, over 80% of dead cells displayed a clear PI signal after 4 h, and the majority of dead cells (over 50%) still displayed a clear PI signal after 10 h. Thus, a substantial amount of dead cell debris persisted over several hours.
We can derive a simple estimate for the time that it takes a dead cell debris barrier to form in our experiments. A single layer of dead cells is sufficient to halt killing. The time it takes a single layer of dead cells to form depends on the kill rate, which we estimate from experiments in which individual target cells are surrounded by killer cells (figure 2h). Target cells died after τ = 0.43 ± 0.02 h on average (N = 117 individual target cells). In a random, close-packing of non-spherical cells, each cell is expected to contact, on average, between z = 6 (cell monolayer [61]) and z = 10 (three-dimensional [62]) other cells. Assuming that killer cells fire the T6SS apparatus in random directions [11], a contact between killer and target cells leads to a killing event between z · τ = 2.58 ± 0.12 h and 4.30 ± 0.20 h on average. This time scale agrees with our experimental finding in figure 1c.
Accumulation of dead cell debris is also present in competitions between non-isogenic strains. To test the accumulation of debris between non-isogenic strains, we competed the previously used V. cholerae killer (T6SS+) against other killer strains (four other environmental isolates of V. cholerae [29]) and a non-killer strain (Vibrio harveyi) (electronic supplementary material, figure S6). Moreover, we competed a killing-deficient (T6SS−) variant of the V. cholerae strain against other T6SS+ killer strains (four other environmental isolates of V. cholerae and Enterobacter cloacae [63,64]) to test whether debris accumulation can be independent of the toxins used by different strains (electronic supplementary material, figure S7). In every strain combination, we observed the signatures of killing inhibition due to dead cell debris, i.e. localized dead cell stain (PI) that aligns with both the interface between strains as well as dark outlines in bright-field microscopy.
The evidence presented in figures 1 and 2 suggests that accumulated dead cell debris prevents contact between cells and thus prevents contact killing. However, these data cannot rule out counter-hypotheses such as a change in T6SS gene expression, nutrient density or oxygen concentration. To directly test if the presence of dead cell debris hinders killing, we mechanically disturbed the structural organization of the biofilms and thus broke down dead cell debris barriers, without otherwise altering biofilm conditions (see sketch in figure 3a). First, we studied a co-culture of killer and target V. cholerae strains. We inoculated the strains at a low initial concentration (OD600 = 1) between an agar plate and a glass coverslip, and began the measurement after a dense layer of cells formed. Killer cells initially expanded their population; by 3 h, the expansion of killer cells halted (figure 3b). After 4.5 h we sheared the biofilm by rotating the coverslip with respect to the agar pad in small circular motions (diameter ∼2 mm), until both strains and the dead cell debris were well mixed. After the perturbation, we observed an immediate increase in the fraction of killer cells, indicating that killing resumed (figure 3b). However, the killer strain took over space almost completely and the remaining target cell domains were too small for us to observe if dead cell debris barriers formed again to separate killer and target cells.
Figure 3.

Mechanical perturbation of dead cell debris barriers between domains of competing strains. (a) Sketch of mechanical perturbation experiments. The agar pad was rotated against the glass coverslip, mixing both strains and the dead cell debris within the biofilm. (b) For unidirectional killing (V. cholerae killer against target strain), the fraction of killer cells saturated after an initial increase. After mechanical perturbation at 4.5 h, the fraction of killers increased again as killing resumed, almost eliminating target cells. (See also electronic supplementary material, video 3.) (c–h) Time-lapse fluorescence images of mechanical perturbation of a mutual killer biofilm show that phase separation occurred repeatedly: (c) after a cellular monolayer has formed, (d) before and (e) after the first mechanical perturbation at 5 h, (f) before and (g) after the second perturbation at 19.5 h, and (h) the final biofilm structure. In the fluorescence images, one killer strain expresses sfGFP (cyan) while the other killer strain is unlabelled (appears black). In every image, the field of view is densely packed with cells. Dead cell debris was labelled with PI (red). Scale bar (c–h): 50 μm. (i) The characteristic length, L, of domains in mutual killer biofilms increases upon coarsening with time, and abruptly decreases after both mechanical perturbation events. However, L increases after each perturbation, indicating that killing resumed (filled circles). In contrast, in unperturbed biofilms L plateaus (empty circles).
Thus, we performed a new perturbation experiment with two ‘mutual’ killer V. cholerae strains, i.e. each strain was T6SS+ and able to kill the other strain. At 5 h, well after population changes had dramatically slowed (figure 3d), we sheared the biofilm (figure 3e), thoroughly mixing the two strains and the dead cell debris. After mixing, we observed that large clonal domains again formed over time, indicating that killing had resumed. Further, we observed that these domains became separated by dead cell debris and eventually killing again ceased (figure 3f). After shearing and mixing the biofilm a second time (at 19.5 h; figure 3g), we again observed the growth of clonal domains that eventually became separated by dead cell debris (figure 3h). These findings demonstrate that T6SS killing was prevented by dead cell debris barrier formation, and not by other factors such as nutrient depletion or changes in cell behaviour or cell density.
We quantified the growth, and mechanical destruction, of clonal domains by measuring the characteristic length of domains, L, of the fluorescent killer strain (figure 3i, filled circles) (see electronic supplementary material for details). L grows rapidly during the first ∼3 h, and much more slowly after that time. Upon the first mixing event, L immediately drops to the size of about three cells (figure 3e,g). After that, L increases again, demonstrating that killing had resumed. We obtained a qualitatively similar trend when mixing the biofilm a second time, as indicated by a sudden decrease in L, followed by an increase. As a control, we measured L for a biofilm that was not mechanically perturbed. The characteristic domain length that emerges after approximately 3 h in the undisturbed biofilm increases by less than over the next 37 h (empty circles in figure 3h); in other words, the characteristic length of domains remains nearly constant after initial domain formation has occurred.
2.3. Limited invasion via contact killing
The above results show that the accumulation of dead cell debris can limit the utility of T6SS-mediated killing within biofilms. Contact killing initially eliminates opponent cells, structuring the biofilm population—but only until dead cell debris accumulates and killing nearly ceases. These observations suggest that the T6SS may have limited ability to facilitate biofilm invasion and that completely taking over a biofilm from a small number of T6SS+ cells would be unlikely. To test the ability of T6SS-facilitated invasion, we examined the behaviour of single killer cells in dense environments.
We mixed 1% fluorescent killer cells with 99% non-fluorescent, susceptible target cells, which were otherwise isogenic to the killers. We inoculated and confined an initially dense monolayer of cells (OD600 = 10) on LB agar pads such that single killer cells were completely surrounded by target cells. We also performed control experiments in which the 1% fluorescent cells were defective killer cells (T6SS−). We found that after 24 h of growth the final killer population was only approximately 1.5× larger than the final fluorescent defective killer control population (figure 4b,c). In particular, clonal domains of killer cells had a mean size of 18.9 μm2 (standard deviation of 26.7 μm2); defective killer control cells formed clonal domains with a mean size of 10.6 μm2 (standard deviation of 14.9 μm2). Thus, while killer cells expanded their population more than killing-deficient cells, they were incapable of invading the existing biofilm and eliminating their competitors.
Figure 4.
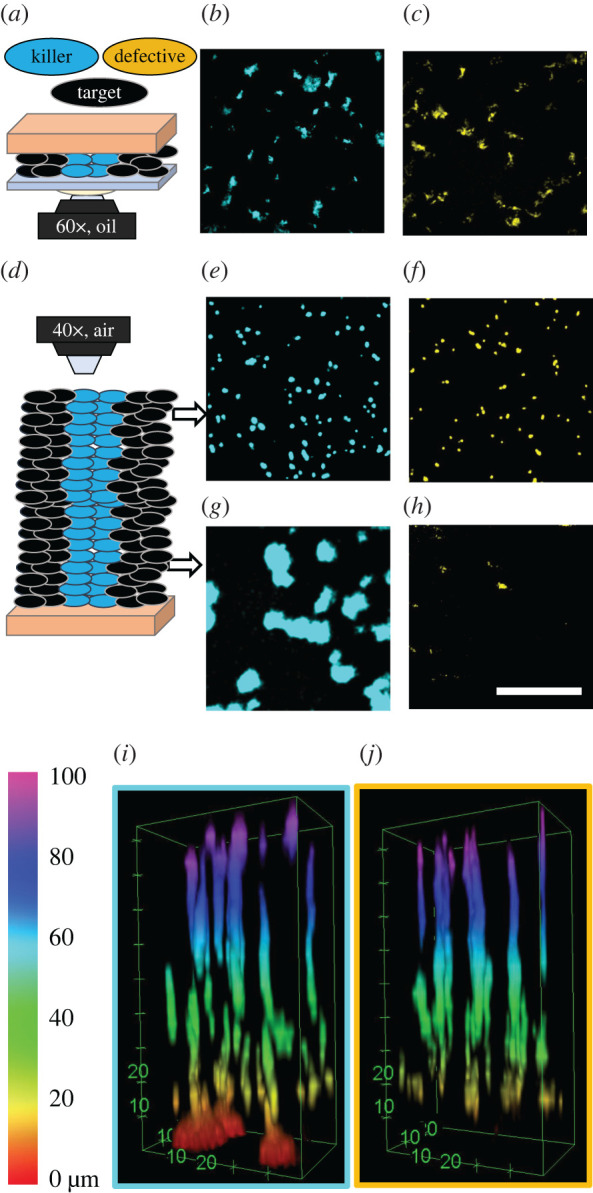
Invasion of dense biofilms by individual cells imaged with confocal microscopy after 24 h of biofilm growth. Biofilms grew from a dense cell monolayer containing 1% fluorescent cells, which were either T6SS+ killer cells (marked blue) or T6SS− defective killer cells (marked yellow). The remaining 99% of the population were non-fluorescent target cells, appearing black. In a confined geometry (schematic in (a)), killer cells (b) expanded their population only approximately 1.50 times more than defective killer cells (c). In unconfined biofilms (schematic in (d)) the final abundance of fluorescent cells depended on the distance above the agar surface. At the top of the biofilm, killer cells (e) only expanded their population 1.16 times more than defective killer cells (f). At the bottom, the killer population (g) was 275 times larger than the defective killer population (h), which was almost absent. Scale bar b,c,e–h: 50 μm. (i,j) Three-dimensional stack of unconfined biofilms showing that killer (i) and defective killer (j) cells performed different only within the bottom ∼10 μm. The colour bar indicates the distance from the agar pad.
Up to this point, all presented experiments were performed in confinement, i.e. biofilms were grown confined between an agar pad and a glass coverslip, to optimize the set-up for microscopy and exclude height-dependent differences [43,65]. While bacteria often inhabit confined geometries in natural settings [66], it is unclear if confinement itself impacts population dynamics. Thus, we next explored contact killing in unconfined environments, by growing biofilms without a coverslip limiting their height. For both mutual and unidirectional killing, we again observed that the (stronger) killer population rapidly increases for the first 3 h, at which point killing slows substantially (electronic supplementary material, figure S4c,d) and a layer of dead cell debris separates competing strains.
We also repeated the experiments on expansion of active and defective single killer cells in dense environments but without confinement. For both killer and defective killer experiments, biofilms reach a height of 92 μm ± 2 μm after 24 h of growth. Similar to the results under confinement, the relative abundances of killer and non-killer control populations differed only slightly (figure 4i,j): killer strains outperformed non-killer strains by a factor of only 1.64 throughout the whole biofilm. However, we observed that killer and defective killer strains exhibit markedly different behaviour near the agar surface. At the top of the unconfined biofilm, the killer population was only 1.16 times greater than the defective killer population (figure 4e,f). The mean sizes of clonal domains were 8.0 μm2 (standard deviation 6.0 μm2) and 7.4 μm2 (standard deviation 4.9 μm2) for killer cells and defective killer cells, respectively. At the bottom of the biofilm (next to the agar surface), some killer cells expanded into large clonal domains (figure 4g); the mean diameter of clonal domains of killer cells within 4 μm of the agar surface was 115.4 μm2 (standard deviation 146.7 μm2). In contrast, the defective killer cells were nearly eliminated at the bottom (figure 4h). (Importantly, the remaining space in figure 4e–h is occupied by a non-killer strain lacking fluorescent proteins.) In the representative biofilm stacks shown in figure 4i,j, within 4 μm of the agar surface, the final population of killer cells was 270 times greater than the population of defective killer cells. Thus, although killer cells were only slightly better at invading unconfined biofilms than defective killer cells, killer cells were able to capture territory at the inoculation surface more efficiently.
It is likely that physical effects observed in studies of lateral range expansions [67–70] also play a role in the upward growth of unconfined biofilms. Previous studies found that proliferating cells in crowded environments interact mechanically [71–73], pushing cells towards the expanding cell front. In analogy, during vertical biofilm growth, the few labelled, non-killer cells may be mechanically pushed off the agar surface by neighbouring cells that proliferate. These pushing forces may be diminished near dead cells, which do not reproduce, helping killer cells surrounded by dead cells to remain near the agar surface. In a related vein, it has been shown that rod-shaped bacteria at a solid–liquid interface undergo a mechanically driven transition from planar to vertical orientation during biofilm growth [73–75]. Such a transition probably also occurs in biofilms grown on agar pads. Such cellular reorientations could impact the number and extent of cell–cell contacts, thus altering the kill rate, and may also damage the debris interface. In fact, our time-lapse analysis of unconfined biofilms (electronic supplementary material, figure S4d) shows that the relative abundances of killer cells at the top and the bottom of the biofilm were established during early stages of biofilm growth—when the structural transition probably happens—and remains unchanged afterwards. In a related vein, it was recently reported that cells near the substrate and cells above the substrate behave differently [76], which is consistent with our observations of killer cells at and above the agar surface. However, understanding the roles of mechanical pushing and cellular reorganization in detail would require single-cell resolution experiments, which is beyond the scope of this work.
2.4. Phase separation dynamics
Finally, we analysed the ‘coarsening’ behaviour of clonal domains during the mechanical shearing experiments (figure 3c–i). Previous works predicted that the observed phase separation is part of the broad model A, or model A’, universality class [24,28]. As a result, the characteristic domain length, L, is predicted to scale as with time t. The mean structure factor, SL, scales as SL ∼ t [24]. Here, we confirm that killing-mediated phase separation follows these dynamics, despite the accumulation of a dead cell debris barrier (figure 5). Following the analysis in [24], we calculated the Fourier-transformed structure factor, S(q), which provides a measure for the frequency of structure sizes with wavenumber q. We determined the characteristic wavenumber, qm, as the mean wavenumber q weighted by S(q). From this, we obtained the characteristic domain length, L, which is inversely proportional to qm, and the mean structure factor SL, which is the height of S(q) at qm (see supplementary methods in the electronic supplementary material for details). SL exhibits linear behaviour each time phase separation occurs (figure 5a). However, all three curves exhibit a kink, where the slope decreases by a factor of 31, 9 or 4, respectively. This indicates a change in the speed of domain coarsening, which is probably caused by the establishment of debris barriers. We further found that L approximately follows a square root scaling with time (figure 5b) at early times after both perturbation events. However, this scaling analysis depends on when we set time t = 0, which is experimentally ambiguous and moreover changes as the speed of coarsening decreases. This ambiguity with respect to time can be avoided by plotting SL versus L. As predicted, it follows the time-independent, universal scaling of SL ∼ L2 for all data in the mechanical shearing experiment (figure 5c).
Figure 5.

Structural analysis of repeated phase separation during the perturbation experiment in figure 3c–i. Time dependence of (a) the mean structure factor SL and (b) the characteristic domain length L. (c) Time-independent structural analysis, SL versus L, for each recurrence of coarsening (with same colour code as in (a)). Dashed lines indicate the slope that corresponds to model A coarsening.
3. Discussion
Microbial antagonism is common in biofilms [4], and many mechanisms exist that kill on contact. Contact killing has previously been proposed to virtually eliminate susceptible competitor cells of similar numbers within a few hours [9,29,31,77,78] (unless target cells reproduce sufficiently quickly [26]). Surprisingly, we found that killing-induced changes in the target population dramatically slow after approximately 3 h, independent of the initial or final relative abundances of killer cells, and independent of the final amount of contact between competing strains (which varies by a factor of 25 across the different cases explored in figure 1c). In fact, even when the T6SS-proficient strain had captured over 99% of the population, small domains of target cells, only a few microns in size, persisted for hours (dashed curve in figure 1c).
In this work, we focused on dead cells, finding that the accumulation of dead cell debris is responsible for the observed dramatic decrease in contact killing. By mechanically shearing biofilms, we directly show that killing resumes once dead cell debris barriers are destroyed. Shearing only modifies the positions of cells and dead cell debris, excluding many other hypotheses (such as changes in T6SS gene expression, target cell susceptibility or metabolic activity). We further demonstrate that despite the formation of dead cell barriers the killing-mediated phase separation of competing strains still exhibits dynamics of the model A universality class, which has been predicted previously [24,28].
At first sight, findings presented here might appear to disagree with conclusions drawn from T6SS competition assays, in which T6SS killing decreases the abundance of target cells by several orders of magnitude [7,13,15,29,31]. However, killing competition assays often start with a majority of killer cells over target cells, and the final abundance of target cells has a nonlinear relationship with the initial abundance of killer cells. Decreasing the initial abundance of killer cells leads to enhanced survival of the target cells [23]. This nonlinear phenomenon agrees with our microscopy results; we found that, for the same pair of strains, target inhibition can be as high as 99.97% (three orders of magnitude) or as low as 79.47% (less than one order of magnitude) as the fraction of killer cells during inoculation was varied from 0.50 to 0.09, respectively. In fact, despite the excess of killer cells in competition assays, the target strain is rarely, if ever, completely eliminated [7,13,15,29,31]. Previous time-lapse competition assays found that the number of surviving target cells remains remarkably constant after a quick decline in the first few hours [23], consistent with the temporal dynamics we observed via microscopy. Therefore, our observations are in agreement with and explain results from previously reported competition assays. Combined, these results demonstrate that the killing efficiency must be studied and discussed in context, with the initial abundances considered. For example, traditional competition assays show that, for some strains, a large number of killer cells can kill a small number of target cells effectively, but they do not show how a small number of killer cells would perform against a large number of target cells.
Even though the efficacy of killing may be limited by the accumulation of dead cell debris, the utility remains quite high. First, it initially facilitates the formation of clonal domains. Depending on the diffusion length of secreted goods [79], this initial domain formation may be sufficient to favour intra-strain cooperation [24,80,81] or reciprocal benefits between different strains [2,82]. Second, killing can prevent social cheaters from emerging in a population. Such ’policing’ effects have been observed in cases where social behaviour is linked with T6SS regulation [83]. Finally, we found that contact-killing strains are able to capture and maintain territory near the surface on which they were inoculated; killing-deficient cells have a much lower probability of remaining near the surface. Occupying nutrient-rich territories may have long-term benefits, beyond the scope of our experiments [84].
The physical inhibition of killing observed here is comparable to other phenomena as seen in contact-dependent growth inhibition experiments [85]. Such a passive defence is starkest in scenarios where the target strain cannot fight back, but the accumulation of dead cell debris eventually prevents killing and protects the target strain from elimination. Such physical protection stands in stark contrast to active, species-dependent defence mechanisms that are controlled genetically [36,38,40,41]. Physical barriers represent an emergent first line of defence that do not require active sensing or control and are species independent. Other defence mechanisms, e.g. immunity proteins acquired through horizontal gene transfer [86], may help protect small numbers of target cells and become relevant if cells touch directly, before the debris barrier forms or after the debris barrier is broken down.
In this vein, it is important to note that the efficacy of T6SS killing, and thus potentially the role of debris accumulation, can vary among different combinations of strains [87]. In fact, previous measurements demonstrated that the killing ability of V. cholerae strains in traditional competition assays can vary by one to seven orders of magnitude [29]. These observations suggest that T6SS killing may be highly effective in some but not all scenarios. The results presented here suggest that the efficacy of T6SS killing also depends on the stability of the cell debris barrier and the rate at which cells can overcome the barrier. The barrier may be broken down through the predation and consumption of dead cells [88], by secreted enzymes (such as lipases or DNases), by shear flow [89] or by other mechanical perturbations, among other potential mechanisms. The rate at which dead cell debris breaks down may also be impacted by the chemical environment and the action of the delivered toxins [87,90], which are known to exhibit a wide range of effects from growth inhibition to lysis [20,21,35,91,92]. Further, while we observed similar characteristics of cell debris accumulation across different co-culture competitions containing V. cholerae, E. cloacae and V. harveyi, the material and physical characteristics of dead cell debris may vary across different combinations of competing strains or species, impacting how long accumulated cell debris prevents contact killing. For example, if competing strains grow at different rates, reproduction may allow the faster growing strain to push through the barrier. Further, motility may enable cells to penetrate barriers as well.
Yet, while several mechanisms may make the debris barrier less stable, dead cell debris is always an obstacle as dead bacteria do not instantaneously disappear. The relative ability or inability of dead cell debris to inhibit contact killing is not a question of if this effect is present, but instead depends on the time scale on which dead cell debris accumulates and how long debris barriers persist. In other words, while the steric hindrance by dead cell debris is likely to be quite general, the impact of debris barriers is specific to the experimental or ecological details [87].
The accumulation of dead cell debris and barrier formation may hold wide-ranging consequences in a variety of contexts. We observe that dead cell debris facilitates the coexistence of antagonistic strains, including allowing killer cells to coexist with non-killer strains that cannot fight back. This concept may apply to other modes of microbial killing [4], such as killing mechanisms that act over longer distances via diffusible deadly bio-molecules [93], phages [94] and bacteriocins [95]. These killing mechanisms will typically only be effective within some diffusion length. Killing may thus be hindered if a dead cell debris barrier longer than the diffusion length forms. Moreover, dead cells locally alter the chemical and material composition, promoting biofilm dispersal [96], providing a source of nutrients [97] or protecting against antibiotics [98,99]. Finally, the phenomena we observe are reminiscent of territorial resource competition seen in a variety of ecosystems at various scales [100]. Previous models have suggested that such effects play important roles in maintaining diversity [101,102]. Specifically, the barrier formation we observe is similar to gap formation that occurs during competition between plants [103,104].
In conclusion, it is striking that contact killing indirectly facilitates the coexistence of antagonistic strains [105]. These results suggest that the T6SS, and perhaps other contact killing mechanisms, may not always prompt a microbial ‘arms race’. Instead, T6SS-mediated killing may also stabilize diverse communities and the increase in dead cell biomass may facilitate bacterial interaction and survival against external attacks. Further, these results align with recent works which question the ecological purpose of bacterial production of antibiotics [84,106]. Nevertheless, the fact that contact killing facilitates coexistence suggests that the impact of the T6SS in bacterial consortia is complex, and the T6SS is more than simply a potent weapon.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data accessibility
All experimental datasets are available at https://osf.io/vhmdn.
Authors' contributions
G.S. and P.J.Y. designed the study. C.C. and S.L.N. engineered mutant strains and prepared liquid cultures. G.S. carried out experiments and analysed the data. G.S. and P.J.Y. wrote the manuscript. All authors gave feedback during project development, reviewed the manuscript and gave final approval for publication. P. J. Y. and B. K. H. supervised the project.
Competing interests
We declare we have no competing interests.
Funding
G.S. is grateful for funding from the German National Academy of Sciences Leopoldina (LDPS 2017-03). B.K.H. acknowledges funding from the Gordon and Betty Moore Foundation (grant no. 6790.13), the School of Biological Sciences Abell Fellowship and the College of Sciences Cullen Peck Scholars Award. P.J.Y. acknowledges funding from the Coulter Foundation and the Georgia CTSA. P.J.Y. and B.K.H. acknowledge funding from the NSF Biomaterials (BMAT-2003721).
References
- 1.Liu W, Jacquiod S, Brejnrod A, Russel J, Burmølle M, Sørensen SJ. 2019. Deciphering links between bacterial interactions and spatial organization in multispecies biofilms. ISME J. 13, 3054–3066. ( 10.1038/s41396-019-0494-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadell CD, Drescher K, Foster KR. 2016. Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol. 14, 589–600. ( 10.1038/nrmicro.2016.84) [DOI] [PubMed] [Google Scholar]
- 3.Yanni D, Márquez-Zacarías P, Yunker PJ, Ratcliff WC. 2019. Drivers of spatial structure in social microbial communities. Curr. Biol. 29, R545–R550. ( 10.1016/j.cub.2019.03.068) [DOI] [PubMed] [Google Scholar]
- 4.Granato ET, Meiller-Legrand TA, Foster KR. 2019. The evolution and ecology of bacterial warfare. Curr. Biol. 29, R521–R537. ( 10.1016/j.cub.2019.04.024) [DOI] [PubMed] [Google Scholar]
- 5.García-Bayona L, Comstock LE. 2018. Bacterial antagonism in host-associated microbial communities. Science 361, eaat2456 ( 10.1126/science.aat2456) [DOI] [PubMed] [Google Scholar]
- 6.Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309, 1245–1248. ( 10.1126/science.1115109) [DOI] [PubMed] [Google Scholar]
- 7.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl Acad. Sci. USA 103, 1528–1533. ( 10.1073/pnas.0510322103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nudleman E, Wall D, Kaiser D. 2005. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science 309, 125–127. ( 10.1126/science.1112440) [DOI] [PubMed] [Google Scholar]
- 9.Souza DP. et al. 2015. Bacterial killing via a type IV secretion system. Nat. Commun. 6, 6453 ( 10.1038/ncomms7453) [DOI] [PubMed] [Google Scholar]
- 10.Bingle LEH, Bailey CM, Pallen MJ. 2008. Type VI secretion: a beginner’s guide. Curr. Opin. Microbiol. 11, 3–8. ( 10.1016/j.mib.2008.01.006) [DOI] [PubMed] [Google Scholar]
- 11.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. 2012. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483, 182–186. ( 10.1038/nature10846) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L, Lezan E, Schmidt A, Basler M. 2019. Abundance of bacterial type VI secretion system components measured by targeted proteomics. Nat. Commun. 10, 2584 ( 10.1038/s41467-019-10466-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cianfanelli FR, Monlezun L, Coulthurst SJ. 2016. Aim, load, fire: the type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 24, 51–62. ( 10.1016/j.tim.2015.10.005) [DOI] [PubMed] [Google Scholar]
- 14.Ho BT, Dong TG, Mekalanos JJ. 2014. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe 15, 9–21. ( 10.1016/j.chom.2013.11.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watve SS, Thomas J, Hammer BK. 2015. CytR is a global positive regulator of competence, type VI secretion, and chitinases in Vibrio cholerae. PLoS ONE 10, 0138834 ( 10.1371/journal.pone.0138834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoued A, Brunet YR, Durand E, Aschtgen M-S, Logger L, Douzi B, Journet L, Cambillau C, Cascales E. 2014. Architecture and assembly of the type VI secretion system. Biochim. Biophys. Acta 1843, 1664–1673. ( 10.1016/j.bbamcr.2014.03.018) [DOI] [PubMed] [Google Scholar]
- 17.Filloux A, Hachani A, Bleves S. 2008. The bacterial type VI secretion machine: yet another player for protein transport across membranes. Microbiology 154, 1570–1583. ( 10.1099/mic.0.2008/016840-0) [DOI] [PubMed] [Google Scholar]
- 18.Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ. 2009. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl Acad. Sci. USA 106, 4154–4159. ( 10.1073/pnas.0813360106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joshi A, Kostiuk B, Rogers A, Teschler J, Pukatzki S, Yildiz FH. 2017. Rules of engagement: the type VI secretion system in Vibrio cholerae. Trends Microbiol. 25, 267–279. ( 10.1016/j.tim.2016.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crisan CV, Hammer BK. 2020. The Vibrio cholerae type VI secretion system: toxins, regulators and consequences. Environ. Microbiol. 22, 4112–4122. ( 10.1111/1462-2920.14976) [DOI] [PubMed] [Google Scholar]
- 21.Russell AB, Peterson SB, Mougous JD. 2014. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 12, 137–148. ( 10.1038/nrmicro3185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz S, Hood RD, Mougous JD. 2010. What is type VI secretion doing in all those bugs?. Trends Microbiol. 18, 531–537. ( 10.1016/j.tim.2010.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. 2010. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc. Natl Acad. Sci. USA 107, 19 520–19 524. ( 10.1073/pnas.1012931107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNally L, Bernardy E, Thomas J, Kalziqi A, Pentz J, Brown SP, Hammer BK, Yunker PJ, Ratcliff WC. 2017. Killing by type VI secretion drives genetic phase separation and correlates with increased cooperation. Nat. Commun. 8, 14371 ( 10.1038/ncomms14371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallespir Lowery N, Ursell T. 2019. Structured environments fundamentally alter dynamics and stability of ecological communities. Proc. Natl Acad. Sci. USA 116, 379–388. ( 10.1073/pnas.1811887116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borenstein DB, Ringel P, Basler M, Wingreen NS. 2015. Established microbial colonies can survive type VI secretion assault. PLoS Comput. Biol. 11, e1004520 ( 10.1371/journal.pcbi.1004520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalziqi A, Ng SL, Yanni D, Steinbach G, Hammer BK, Yunker PJ. 2019 Viscosity independent diffusion mediated by death and reproduction in biofilms. (http://arxiv.org/abs/1901.01350. )
- 28.Lavrentovich MO, Nelson DR. 2019. Nucleation of antagonistic organisms and cellular competitions on curved, inflating substrates. Phys. Rev. E 100, 042406 ( 10.1103/PhysRevE.100.042406) [DOI] [PubMed] [Google Scholar]
- 29.Bernardy EE, Turnsek MA, Wilson SK, Tarr CL, Hammer BK. 2016. Diversity of clinical and environmental isolates of Vibrio cholerae in natural transformation and contact-dependent bacterial killing indicative of type VI secretion system activity. Appl. Environ. Microbiol. 82, 2833–2842. ( 10.1128/AEM.00351-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crisan CV. et al 2019. Analysis of Vibrio cholerae genomes identifies new type VI secretion system gene clusters. Genome Biol. 20, 163 ( 10.1186/s13059-019-1765-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng J, Ho B, Mekalanos JJ. 2011. Genetic analysis of anti-amoebae and anti-bacterial activities of the type VI secretion system in Vibrio cholerae. PLoS ONE 6, e23876 ( 10.1371/journal.pone.0023876) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vacheron J, Péchy-Tarr M, Brochet S, Heiman CM, Stojiljkovic M, Maurhofer M, Keel C. 2019. T6SS contributes to gut microbiome invasion and killing of an herbivorous pest insect by plant-beneficial Pseudomonas protegens. ISME J. 13, 1318–1329. ( 10.1038/s41396-019-0353-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verster AJ, Ross BD, Radey MC, Bao Y, Goodman AL, Mougous JD, Borenstein E. 2017. The landscape of type VI secretion across human gut microbiomes reveals its role in community composition. Cell Host Microbe 22, 411–419.e4. ( 10.1016/j.chom.2017.08.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coulthurst SJ. 2013. The type VI secretion system—a widespread and versatile cell targeting system. Res. Microbiol. 164, 640–654. ( 10.1016/j.resmic.2013.03.017) [DOI] [PubMed] [Google Scholar]
- 35.Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD. 2013. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512. ( 10.1038/nature12074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hersch SJ. et al 2020. Envelope stress responses defend against type six secretion system attacks independently of immunity proteins. Nat. Microbiol. 5, 706–714. ( 10.1038/s41564-020-0672-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyata ST, Unterweger D, Rudko SP, Pukatzki S. 2013. Dual expression profile of type VI secretion system immunity genes protects pandemic Vibrio cholerae. PLoS Pathog. 9, e1003752 ( 10.1371/journal.ppat.1003752) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toska J, Ho BT, Mekalanos JJ. 2018. Exopolysaccharide protects Vibrio cholerae from exogenous attacks by the type 6 secretion system. Proc. Natl Acad. Sci. USA 115, 7997–8002. ( 10.1073/pnas.1808469115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basler M, Ho B, Mekalanos J. 2013. Tit-for-tat: type VI secretion system counterattack during bacterial cell-cell interactions. Cell 152, 884–894. ( 10.1016/j.cell.2013.01.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong TG, Dong S, Catalano C, Moore R, Liang X, Mekalanos JJ. 2015. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl Acad. Sci. USA 112, 2181–2186. ( 10.1073/pnas.1425007112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lories B, Roberfroid S, Dieltjens L, De Coster D, Foster KR, Steenackers HP. 2020. Biofilm bacteria use stress responses to detect and respond to competitors. Curr. Biol. 30, 1231–1244; e4. ( 10.1016/j.cub.2020.01.065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair HAS, Periasamy S, Yang L, Kjelleberg S, Rice SA. 2017. Real time, spatial, and temporal mapping of the distribution of c-di-GMP during biofilm development. J. Biol. Chem. 292, 477–487. ( 10.1074/jbc.M116.746743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Wang G, Hao M. 2015. Modeling of the Bacillus subtilis bacterial biofilm growing on an agar substrate. Comput. Math. Methods Med. 2015, 581829 ( 10.1155/2015/581829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wentland EJ, Stewart PS, Huang C-T, McFeters GA. 1996. Spatial variations in growth rate within Klebsiella pneumoniae colonies and biofilm. Biotechnol. Prog. 12, 316–321. ( 10.1021/bp9600243) [DOI] [PubMed] [Google Scholar]
- 45.Xu KD, Stewart PS, Xia F, Huang C-T, McFeters GA. 1998. Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl. Environ. Microbiol. 64, 4035–4039. ( 10.1128/AEM.64.10.4035-4039.1998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anwar H, Strap JL, Costerton JW. 1992. Establishment of aging biofilms: possible mechanism of bacterial resistance to antimicrobial therapy. Antimicrob. Agents Chemother. 36, 1347–1351. ( 10.1128/AAC.36.7.1347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartolini M. et al. 2018. Regulation of biofilm aging and dispersal in Bacillus subtilis by the alternative sigma factor SigB. J. Bacteriol. 201, e00473 ( 10.1128/JB.00473-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartolini M. et al. 2019. Regulation of biofilm aging and dispersal in Bacillus subtilis by the alternative sigma factor SigB. J. Bacteriol. 201, e00473 ( 10.1128/JB.00473-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brooks TM, Unterweger D, Bachmann V, Kostiuk B, Pukatzki S. 2013. Lytic activity of the Vibrio cholerae type VI secretion toxin VgrG-3 is inhibited by the antitoxin TsaB. J. Biol. Chem. 288, 7618–7625. ( 10.1074/jbc.M112.436725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altindis E, Dong T, Catalano C, Mekalanos J. 2015. Secretome analysis of Vibrio cholerae type VI secretion system reveals a new effector-immunity pair. mBio 6, e00075 ( 10.1128/mBio.00075-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong TG, Ho BT, Yoder-Himes DR, Mekalanos JJ. 2013. Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl Acad. Sci. USA 110, 2623–2628. ( 10.1073/pnas.1222783110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hood RD. et al. 2010. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37. ( 10.1016/j.chom.2009.12.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hachani A, Lossi NS, Filloux A. 2013. A visual assay to monitor T6SS-mediated bacterial competition. J. Vis. Exp. 73, e50103 ( 10.3791/50103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Church SR, Lux T, Baker-Austin C, Buddington SP, Michell SL. 2016. Vibrio vulnificus type 6 secretion system 1 contains anti-bacterial properties. PLoS ONE 11, e0165500 ( 10.1371/journal.pone.0165500) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishikawa T, Sabharwal D, Bröms J, Milton DL, Sjöstedt A, Uhlin BE, Wai SN. 2012. Pathoadaptive conditional regulation of the type VI secretion system in Vibrio cholerae O1 strains. Infect. Immun. 80, 575–584. ( 10.1128/IAI.05510-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Czárán TL, Hoekstra RF, Pagie L. 2002. Chemical warfare between microbes promotes biodiversity. Proc. Natl Acad. Sci. USA 99, 786–790. ( 10.1073/pnas.012399899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Czaran TL, Hoekstra RF. 2003. Killer-sensitive coexistence in metapopulations of micro-organisms. Proc. R. Soc. Lond. B 270, 1373–1378. ( 10.1098/rspb.2003.2338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong M, Liang X, Smart M, Tang L, Moore R, Ingalls B, Dong TG. 2016. Microbial herd protection mediated by antagonistic interaction in polymicrobial communities. Appl. Environ. Microbiol. 82, 6881–6888. ( 10.1128/AEM.02210-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zepeda-Rivera MA, Saak CC, Gibbs KA. 2018. A proposed chaperone of the bacterial type VI secretion system functions to constrain a self-identity protein. J. Bacteriol. 200, e00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wenren LM, Sullivan NL, Cardarelli L, Septer AN, Gibbs KA. 2013. Two independent pathways for self-recognition in Proteus mirabilis are linked by type VI-dependent export. mBio 4, 265 ( 10.1128/mBio.00374-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delaney G, Weaire D, Hutzler S, Murphy S. 2005. Random packing of elliptical disks. Philos. Mag. Lett. 85, 89–96. ( 10.1080/09500830500080763) [DOI] [Google Scholar]
- 62.Donev A, Cisse I, Sachs D, Variano EA, Stillinger FH, Connelly R, Torquato S, Chaikin PM. 2004. Improving the density of jammed disordered packings using ellipsoids. Science 303, 990–993. ( 10.1126/science.1093010) [DOI] [PubMed] [Google Scholar]
- 63.Stephens WZ, Burns AR, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan BJM. 2016. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 10, 644–654. ( 10.1038/ismej.2015.140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlomann BH, Wiles TJ, Wall ES, Guillemin K, Parthasarathy R. 2019. Sublethal antibiotics collapse gut bacterial populations by enhancing aggregation and expulsion. Proc. Natl Acad. Sci. USA 116, 21 392–21 400. ( 10.1073/pnas.1907567116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Renslow RS, Majors PD, McLean JS, Fredrickson JK, Ahmed B, Beyenal H. 2010. In situ effective diffusion coefficient profiles in live biofilms using pulsed-field gradient nuclear magnetic resonance. Biotechnol. Bioeng. 106, 928–937. ( 10.1002/bit.22755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bar-On YM, Phillips R, Milo R. 2018. The biomass distribution on Earth. Proc. Natl Acad. Sci. USA 115, 6506–6511. ( 10.1073/pnas.1711842115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schreck CF, Fusco D, Karita Y, Martis S, Kayser J, Duvernoy M-C, Hallatschek O. 2019. Impact of crowding on the diversity of expanding populations. bioRxiv 743534 ( 10.1101/743534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giometto A, Nelson DR, Murray AW. 2018. Physical interactions reduce the power of natural selection in growing yeast colonies. Proc. Natl Acad. Sci. USA 115, 11 448–11 453. ( 10.1073/pnas.1809587115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hallatschek O, Hersen P, Ramanathan S, Nelson DR. 2007. Genetic drift at expanding frontiers promotes gene segregation. Proc. Natl Acad. Sci. USA 104, 19 926–19 930. ( 10.1073/pnas.0710150104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farrell FD, Gralka M, Hallatschek O, Waclaw B. 2017. Mechanical interactions in bacterial colonies and the surfing probability of beneficial mutations. J. R. Soc. Interface 14, 20170073 ( 10.1098/rsif.2017.0073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalziqi A, Yanni D, Thomas J, Ng SL, Vivek S, Hammer BK, Yunker PJ. 2018. Immotile active matter: activity from death and reproduction. Phys. Rev. Lett. 120, 018101 ( 10.1103/PhysRevLett.120.018101) [DOI] [PubMed] [Google Scholar]
- 72.Copenhagen K, Alert R, Wingreen NS, Shaevitz JW. 2020 Topological defects induce layer formation in Myxococcus xanthus colonies. (http://arxiv.org/abs/2001.03804. )
- 73.Yan J, Sharo AG, Stone HA, Wingreen NS, Bassler BL. 2016. Vibrio cholerae biofilm growth program and architecture revealed by single-cell live imaging. Proc. Natl Acad. Sci. USA 113, E5337–E5343. ( 10.1073/pnas.1611494113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beroz F, Yan J, Meir Y, Sabass B, Stone HA, Bassler BL, Wingreen NS. 2018. Verticalization of bacterial biofilms. Nat. Phys. 14, 954–960. ( 10.1038/s41567-018-0170-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drescher K, Dunkel J, Nadell CD, van Teeffelen S, Grnja I, Wingreen NS, Stone HA, Bassler BL. 2016. Architectural transitions in Vibrio cholerae biofilms at single-cell resolution. Proc. Natl Acad. Sci. USA 113, E2066–E2072. ( 10.1073/pnas.1601702113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qin B, Fei C, Bridges AA, Mashruwala AA, Stone HA, Wingreen NS, Bassler BL. 2020. Cell position fates and collective fountain flow in bacterial biofilms revealed by light-sheet microscopy. Science 369, 71–77. ( 10.1126/science.abb8501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coulthurst S. 2019. The type VI secretion system: a versatile bacterial weapon. Microbiology 165, 503–515. ( 10.1099/mic.0.000789) [DOI] [PubMed] [Google Scholar]
- 78.Chen C, Yang XB, Shen XH. 2019. Confirmed and potential roles of bacterial T6SSs in the intestinal ecosystem. Front. Microbiol. 10, 1484 ( 10.3389/fmicb.2019.01484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Co AD, Vliet Sv, Kiviet DJ, Schlegel S, Ackermann M. 2020. Short-range interactions govern the dynamics and functions of microbial communities. Nat. Ecol. Evol. 4, 366–375. ( 10.1038/s41559-019-1080-2) [DOI] [PubMed] [Google Scholar]
- 80.Julou T, Mora T, Guillon L, Croquette V, Schalk IJ, Bensimon D, Desprat N. 2013. Cell-cell contacts confine public goods diffusion inside Pseudomonas aeruginosa clonal microcolonies. Proc. Natl Acad. Sci. USA 110, 12 577–12 582. ( 10.1073/pnas.1301428110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dobay A, Bagheri HC, Messina A, Kümmerli R, Rankin DJ. 2014. Interaction effects of cell diffusion, cell density and public goods properties on the evolution of cooperation in digital microbes. J. Evol. Biol. 27, 1869–1877. ( 10.1111/jeb.12437) [DOI] [PubMed] [Google Scholar]
- 82.Momeni B, Waite AJ, Shou W. 2013. Spatial self-organization favors heterotypic cooperation over cheating. Elife 2, e00960 ( 10.7554/eLife.00960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Majerczyk C, Schneider E, Greenberg EP. 2016. Quorum sensing control of type VI secretion factors restricts the proliferation of quorum-sensing mutants. Elife 5, e14712 ( 10.7554/eLife.14712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25. ( 10.1038/nrmicro2259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bottery MJ, Passaris I, Dytham C, Wood AJ, van der Woude MW. 2019. Spatial organization of expanding bacterial colonies is affected by contact-dependent growth inhibition. Curr. Biol. 29, 3622–3634; e5. ( 10.1016/j.cub.2019.08.074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas J, Watve SS, Ratcliff WC, Hammer BK. 2017. Horizontal gene transfer of functional type VI killing genes by natural transformation. mBio 8, e00654 ( 10.1128/mBio.00654-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith WPJ, Vettiger A, Winter J, Ryser T, Comstock LE, Basler M, Foster KR. 2020. The evolution of the type VI secretion system as a disintegration weapon. PLoS Biol. 18, e3000720 ( 10.1371/journal.pbio.3000720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pérez J, Moraleda-Muñoz A, Marcos-Torres FJ, Muñoz-Dorado J. 2016. Bacterial predation: 75 years and counting! Environ. Microbiol. 18, 766–779. ( 10.1111/1462-2920.13171) [DOI] [PubMed] [Google Scholar]
- 89.Thomen P, Robert J, Monmeyran A, Bitbol A-F, Douarche C, Henry N. 2017. Bacterial biofilm under flow: first a physical struggle to stay, then a matter of breathing. PLoS ONE 12, e0175197 ( 10.1371/journal.pone.0175197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Unterweger D, Miyata ST, Bachmann V, Brooks TM, Mullins T, Kostiuk B, Provenzano D, Pukatzki S. 2014. The Vibrio cholerae type VI secretion system employs diverse effector modules for intraspecific competition. Nat. Commun. 5, 3549 ( 10.1038/ncomms4549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Durand E, Cambillau C, Cascales E, Journet L. 2014. VgrG, Tae, Tle, and beyond: the versatile arsenal of type VI secretion effectors. Trends Microbiol. 22, 498–507. ( 10.1016/j.tim.2014.06.004) [DOI] [PubMed] [Google Scholar]
- 92.Yang X, Long M, Shen X. 2018. Effector–immunity pairs provide the T6SS nanomachine its offensive and defensive capabilities. Molecules 23, 1009 ( 10.3390/molecules23051009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8, 423–435. ( 10.1038/nrmicro2333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kortright KE, Chan BK, Koff JL, Turner PE. 2019. Phage therapy: a renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe 25, 219–232. ( 10.1016/j.chom.2019.01.014) [DOI] [PubMed] [Google Scholar]
- 95.Scholl D. 2017. Phage tail-like bacteriocins. Annu. Rev. Virol. 4, 453–467. ( 10.1146/annurev-virology-101416-041632) [DOI] [PubMed] [Google Scholar]
- 96.Mai-Prochnow A, Evans F, Dalisay-Saludes D, Stelzer S, Egan S, James S, Webb JS, Kjelleberg S. 2004. Biofilm development and cell death in the marine bacterium Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 70, 3232–3238. ( 10.1128/AEM.70.6.3232-3238.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schink SJ, Biselli E, Ammar C, Gerland U. 2019. Death rate of E. coli during starvation is set by maintenance cost and biomass recycling. Cell Syst. 9, 64–73; e3. ( 10.1016/j.cels.2019.06.003) [DOI] [PubMed] [Google Scholar]
- 98.Podlesek Z, Butala M, Šakanović A, Žgur Bertok D. 2016. Antibiotic induced bacterial lysis provides a reservoir of persisters. Antonie Van Leeuwenhoek 109, 523–528. ( 10.1007/s10482-016-0657-x) [DOI] [PubMed] [Google Scholar]
- 99.Auschill TM, Arweiler NB, Netuschil L, Brecx M, Reich E, Sculean A. 2001. Spatial distribution of vital and dead microorganisms in dental biofilms. Arch. Oral Biol. 46, 471–476. ( 10.1016/S0003-9969(00)00136-9) [DOI] [PubMed] [Google Scholar]
- 100.Wu J, Loucks OL. 1995. From balance of nature to hierarchical patch dynamics: a paradigm shift in ecology. Q. Rev. Biol. 70, 439–466. ( 10.1086/419172) [DOI] [Google Scholar]
- 101.Weiner BG, Posfai A, Wingreen NS. 2019. Spatial ecology of territorial populations. Proc. Natl Acad. Sci. USA 116, 17 874–17 879. ( 10.1073/pnas.1911570116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amarasekare P. 2003. Competitive coexistence in spatially structured environments: a synthesis. Ecol. Lett. 6, 1109–1122. ( 10.1046/j.1461-0248.2003.00530.x) [DOI] [Google Scholar]
- 103.Liao J, Bogaert J, Nijs I. 2015. Species interactions determine the spatial mortality patterns emerging in plant communities after extreme events. Sci. Rep. 5, 11229 ( 10.1038/srep11229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brokaw NVL. 1985. Chapter 4—Treefalls, regrowth, and community structure in tropical forests. In The ecology of natural disturbance and patch dynamics (eds STA Pickett, PS White), pp. 53–69. San Diego, CA: Academic Press.
- 105.Ratzke C, Barrere J, Gore J. 2020. Strength of species interactions determines biodiversity and stability in microbial communities. Nat. Ecol. Evol. 4, 376–383. ( 10.1038/s41559-020-1099-4) [DOI] [PubMed] [Google Scholar]
- 106.Pishchany G, Kolter R. 2020. On the possible ecological roles of antimicrobials. Mol. Microbiol. 113, 580–587. ( 10.1111/mmi.14471) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental datasets are available at https://osf.io/vhmdn.