

Abstract
Aim: Capecitabine is a prodrug that is metabolized to its active form, 5-fluorouracil (5-FU), in three enzymatic steps. This prospective pharmacokinetic study evaluated cytidine deaminase (CDA) activity, the second drug-metabolizing enzyme that generates 5’-deoxy-5-fluorouridine (5’-DFUR) from 5’-deoxy-5-fluorocytidine (5’-DFCR), as well as creatinine clearance (CLcr). Patients and Methods: Patients with colorectal cancer who received capecitabine plus oxaliplatin were selected. Pharmacokinetics of capecitabine and its metabolites, and CDA activity in plasma were analyzed. Results: Eighteen patients were examined. The area under the plasma concentration-time curve (AUC) of 5’-DFUR showed a significant inverse correlation with CLcr (p=0.003). The metabolic ratio, i.e. the ratios of the AUC of 5’-DFUR plus that of 5-FU to the AUC of 5’-DFCR, significantly increased when CLcr decreased (p=0.001) but did not depend on plasma CDA activity. Conclusion: Metabolism of 5’-DFCR to form 5’-DFUR increased as CLcr decreased but the mechanism remains unknown.
Keywords: 5’-DFCR, 5’-DFUR, capecitabine, cytidine deaminase, creatinine clearance
Capecitabine is an oral fluoropyrimidine that is designed to be finally metabolized to an active metabolite, 5-fluorouracil (5-FU). After oral administration, capecitabine is rapidly absorbed through the intestine and is metabolized to 5’-deoxy-5-fluorocytidine (5’-DFCR) by carboxylesterase in the liver. Subsequently, 5’-DFCR is converted to 5’-deoxy-5-fluorouridine (5’-DFUR) by cytidine deaminase (CDA) in the liver and tumor tissues. It is finally converted to 5-FU by thymidine phosphorylase, which is predominantly expressed in tumor tissues (1,2).
A previous pharmacokinetic study demonstrated that the area under the plasma concentration–time curve (AUC) of 5’-DFUR (AUC5’-DFUR) in patients with renal impairment was significantly higher than that in patients with normal renal function, resulting in a higher incidence of capecitabine-induced adverse events in patients with renal impairment (3). Therefore, reduction of the starting dose of capecitabine is recommended according to measures of the patient’s renal function, such as creatinine clearance (CLcr) (3). It is believed that increased systemic exposure to 5’-DFUR in patients with impaired renal function is likely to be caused by delayed renal excretion of the metabolite. However, according to the pharmacokinetic parameters of capecitabine and its metabolites, the maximum concentration (Cmax) of 5’-DFUR was higher in patients with renal impairment than that in patients with normal renal function but the elimination half-life (t1/2) was similar in both patient groups (3), suggesting that 5’-DFUR might accumulate due to the increased production of 5’-DFUR rather than by its delayed renal excretion. Therefore, we hypothesized that systemic exposure to 5’-DFUR is associated with the metabolic activity of CDA, which primarily generates 5’-DFUR from 5’-DFCR, rather than the delayed renal excretion of 5’-DFUR.
This study carefully evaluated the effects of CDA activity, as well as CLcr, on systemic exposure to 5’-DFCR and 5’-DFUR in patients with colorectal cancer who received treatment with capecitabine plus oxaliplatin (Cape-OX).
Patients and Methods
Chemicals. Capecitabine, 5-FU, and 5’-DFUR were obtained from FUJIFILM Wako Pure Chemical Corporation (Tokyo, Japan), 5’-DFCR was from Tokyo Chemical Industry (Tokyo, Japan), and 2’-deoxy-2’,2’-difluorouridine (dFdU) was from Toronto Research Chemicals (Toronto, Canada). All chemicals and solvents were of the highest grade commercially available.
Study design. The primary objective of this study was to prospectively investigate the pharmacokinetic profiles of capecitabine and its metabolites and to evaluate the role of CDA activity, as well as CLcr, on the systemic exposure to 5’-DFCR and 5’-DFUR. The study was conducted in accordance with the principles of the Declaration of Helsinki, and the study protocol (UMIN000019244) was approved by the Institutional Review Board and Ethics Committee. All patients provided written informed consent before the enrollment in the study.
Patients. Eligible patients were 20 years or older, had histologically confirmed colonic or rectal cancer, and received Cape-OX as postoperative adjuvant chemotherapy. Patients also had an Eastern Cooperative Oncology Group performance status of 0-2, and were required to maintain adequate organ functions, including hematological function (absolute leukocyte count, ≥3.0×103/mm3; hemoglobin, ≥9.0 g/dl; platelets, ≥10×104/mm3) and renal function (CLcr of ≥30 ml/min). CLcr was calculated using the Cockcroft and Gault equation: CLcr (ml/min)=[140–age (years)]×weight (kg)/
[(72×serum creatinine (mg/dl)] (4). If patients were female, the calculated values were multiplied by 0.85.
Treatment. Cape-OX chemotherapy consisted of oral capecitabine twice daily within 30 min after breakfast and dinner for 2 weeks of a 3-week cycle plus intravenous oxaliplatin given over the course of 120 min at a dose of 130 mg/m2 on day 1 of each cycle for eight cycles. Because no pharmacokinetic drug-drug interaction between capecitabine and oxaliplatin has been reported (5), oxaliplatin was administered on day 1. The first dose of capecitabine was given on the morning of day 1, and the last dose was given on the evening of day 14. The actual dose of capecitabine administered was determined according to the patient’s body surface area (BSA): 2,400 mg/day when BSA was less than 1.36 m2; 3,000 mg/day when BSA was 1.36 m2 or higher but less than 1.66 m2; 3,600 mg/day when BSA was 1.66 m2 or higher but less than 1.96 m2; and 4,200 mg/day when BSA was 1.96 m2 or higher.
The dose of capecitabine was adjusted according to the patient’s renal function (3). For patients with moderately renal impairment (CLcr of 30 to 50 ml/min), the starting dose was reduced to 75% of the standard dose, and for those with mildly impaired renal function (CLcr of 50 to 80 ml/min), the starting dose was as same as that for patients with normal renal function (CLcr of more than 80 ml/min).
Patients generally received dexamethasone and a serotonin receptor 5-Hydroxytryptamine-3 antagonist as antiemetic prophylaxis. Other supportive care in individual patients was left at the discretion of the treating physicians.
Blood sampling for pharmacokinetic analysis. Blood samples for pharmacokinetic analysis were obtained from the patients before taking capecitabine and then at 30, 60, 120, 180, 240, 300, 360, and 480 min on day 1 of the first cycle. At each time point, 5 ml of blood was collected in EDTA tubes containing 50 μl of a 10 mg/ml solution of tetrahydrouridine (Calbiochem-Novabiochem, La Jolla, CA, USA), a cytidine deaminase inhibitor, and the sample was centrifuged immediately. The plasma was stored at –80˚C until analysis.
Sample acquisition of CDA activity. A blood sample for CDA activity analysis was obtained from the patients before taking capecitabine on day 1 of the first cycle. Five milliliters of blood were collected into a heparinized tube, and the sample was centrifuged immediately. The plasma was stored at –80˚C until analysis.
Measurement of capecitabine, 5’-DFCR, 5’-DFUR, and 5-FU concentrations. Plasma concentrations of capecitabine, 5’-DFCR, 5’-DFUR, and 5-FU were measured using a reverse-phase high-performance liquid chromatography (HPLC) as described previously (6).
Pharmacokinetic parameters. The plasma concentration–time data of 5’-DFCR, 5’-DFUR, and 5-FU were analyzed by a standard non-compartmental method using WinNonlin®, version 6.4 software (Pharsight, Mountain View, CA, USA) as described previously (6). The AUC was calculated from zero to the last measurable concentration. Since the Cmax and AUC of capecitabine and its metabolites increase linearly with administered dose (7), the parameters of Cmax and AUC were adjusted by the actual capecitabine doses. The metabolic activity of conversion from 5’-DFCR to 5’-DFUR was assessed by the metabolic ratio, defined as the AUC of 5’-DFUR plus 5-FU (AUC5’-DFUR+5-FU) to the AUC of 5’-DFCR (AUC5’-DFCR).
CDA activity in plasma. The AUC of gemcitabine, which is known to be detoxified by the liver CDA, is significantly correlated with plasma CDA activity (8). Therefore, this study used CDA activity in peripheral blood as a surrogate for hepatic CDA activity.
The time-dependent formation of 5’-DFUR was assessed with a 5’-DFCR concentration of 50 μM in 150 μl of plasma. Linearity was obtained up to 10 min of incubation time. The reaction was stopped by mixing the plasma sample with 450 μl of acetonitrile containing 2.2 μM dFdU (internal standard) in a vortex mixer. The mixture was then centrifuged at 16,000×g for 10 min at 4˚C. The supernatant was transferred to another tube and evaporated to dryness at 45˚C in an SPD1010 SpeedVac™ System. The residue was reconstituted in 200 μl of 10 mM sodium phosphate buffer (pH 4.8), and a 100-μl portion was injected into an HPLC system. The concentration of 5’-DFUR was determined as described above.
Statistical analyses. Spearman’s rank correlation test was performed to evaluate the correlation of CLcr and CDA activity with pharmacokinetic parameters and the correlation between CLcr and CDA activity.
Results
Patient characteristics. A total of 18 patients were enrolled between November 2015 and November 2018 (Table I). The median serum creatinine level was 0.71 mg/dl (range=0.54-1.06 mg/dl), and the median CLcr was 72.0 ml/min (range=49.0-99.7 ml/min). The dose of capecitabine was 2,400 mg/day in two patients (BSA=1.24 and 1.61 m2; the starting dose of capecitabine was reduced from 3,000 mg/day to 2,400 mg/day because CLcr was 49 ml/min in the patient with a BSA of 1.61 m2), 3,000 mg/day in 12 patients (median BSA=1.55 m2; range=1.40-1.65 m2), and 3,600 mg/day in 4 patients (median BSA=1.69 m2; range=1.66-1.80 m2).
Table I. Baseline patient characteristics (N=18).

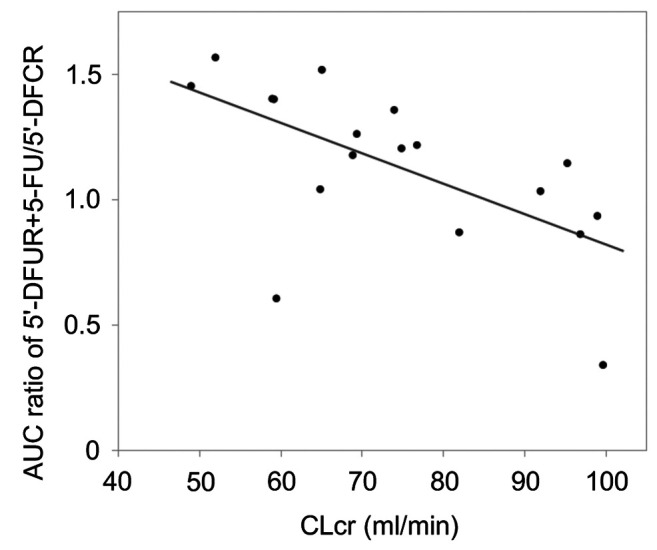
Pharmacokinetic analyses. Blood samples from 18 patients were all obtained at the scheduled time for appropriate pharmacokinetic analyses (Table II). The AUC5’-DFUR was significantly inversely correlated with CLcr (Figure 1A, r=–0.657, p=0.003). The Cmax ratio of 5’-DFUR/5’-DFCR also significantly inversely correlated with CLcr (Figure 1B, r=–0.554, p=0.017). In contrast, there was no significant correlation between the t1/2 of 5’-DFUR and CLcr (Figure 1C, r=–0.003, p=0.990). Interestingly, the metabolic ratio (AUC5’-DFUR+5-FU/AUC5’-DFCR) was significantly correlated with the patient’s CLcr (Figure 2, r=–0.702, p=0.001).
Table II. Pharmacokinetic parameters of capecitabine and its metabolites. Data are the mean±standard deviation.

5’-DFCR: 5’-Deoxy-5-fluorocytidine; 5’-DFUR: 5’-deoxy-5-fluorouridine; 5-FU: 5-fluorouracil; Cmax: maximum plasma concentration (doseadjusted); tmax: time of maximum concentration; AUC: area under the plasma concentration–time curve (dose-adjusted); t1/2: elimination half-life. an=10, bn=6.
Figure 1. Correlation between pharmacokinetic parameters of capecitabine metabolites and creatinine clearance (CLcr). A: The area under the plasma concentration–time curve (AUC)/dose of 5’-deoxy-5-fluorouridine (5’-DFUR) and CLcr were significantly inversely correlated (r=– 0.657, p=0.003). B: The maximum concentration (Cmax) ratio of 5’-DFUR/5’-deoxy-5-fluorocytidine (5’-DFCR) and CLcr were significantly inversely correlated (r=–0.554, p=0.017). C: The elimination half-life (t1/2) of 5’-DFUR and CLcr were not correlated (r=–0.003, p=0.990).

Figure 2. Correlation between the metabolic activity of the conversion from 5’-deoxy-5-fluorocytidine (5’-DFCR) to 5’-deoxy-5-fluorouridine (5’-DFUR) and creatinine clearance (CLcr). The area under the plasma concentration–time curve (AUC) ratio of 5’-DFUR+5-fluorouracil (5- FU)/5’-DFCR and CLcr were significantly inversely correlated (r=– 0.702, p=0.001).
The median plasma CDA activity was 53.7 pmol (range=30.4-197.5 pmol). The distribution of CDA activity in 17 patients ranged from 30.4 to 76.6 pmol but one patient showed CDA activity of 197.5 pmol, which was higher than the upper limit of the 99% confidence interval. Contrary to the initial expectation, the metabolic ratio (AUC5’-DFUR+5-FU/AUC5’-DFCR) did not depend on plasma CDA activity (Figure 3, r=–0.258, p=0.919). The plasma CDA activity was not significantly correlated with CLcr (Figure 4, r=0.137, p=0.587).
Figure 3. Correlation between the metabolic activity of the conversion from 5’-deoxy-5-fluorocytidine (5’-DFCR) to 5’-deoxy-5-fluorouridine (5’-DFUR) and cytidine deaminase (CDA) activity. CDA activity in plasma is expressed as the formation of 5’-DFUR (pmol) during a 10- min incubation of 150 μl plasma containing 50 μM 5’-DFCR at 37˚C. The area under the plasma concentration–time curve (AUC) ratio of 5’- DFUR+5-fluorouracil (5-FU)/5’-DFCR and plasma CDA activity showed no significant correlation (r=–0.258, p=0.919).
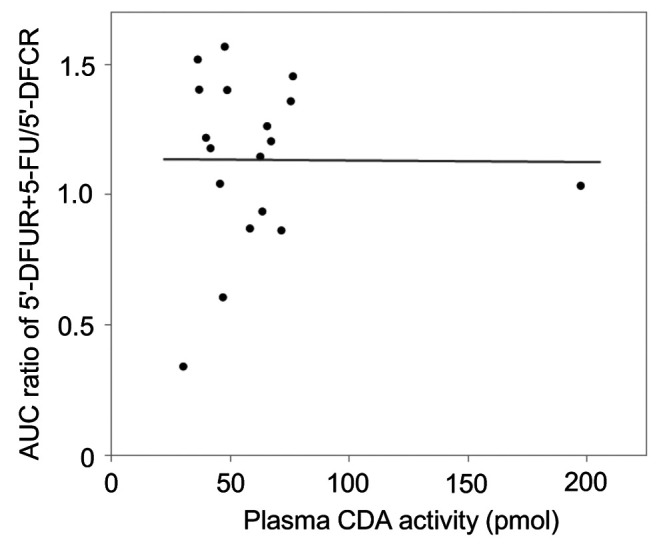
Figure 4. Correlation between plasma cytidine deaminase (CDA) activity and creatinine clearance (CLcr). CDA activity in plasma is expressed as the formation of 5’-deoxy-5-fluorouridine (5’-DFUR) (pmol) during a 10-min incubation of 150 μl plasma containing 50 μM 5’-deoxy-5-fluorocytidine (5’-DFCR) at 37˚C. Plasma CDA activity and CLcr showed no significant correlation (r=0.137, p=0.587).

Discussion
This study reconfirmed the capecitabine pharmacokinetics finding that AUC5’-DFUR is negatively correlated with CLcr, but did not support our initial hypothesis that AUC5’-DFUR would be raised by increased CDA enzyme activity, rather than by delayed excretion of the metabolite. Interestingly, the metabolic activation of 5’-DFCR to generate 5’-DFUR correlated inversely with CLcr, while the t1/2 of 5’-DFUR did not, although the underlying mechanism for this remains unknown.
Because 5’-DFUR is known to be excreted via the kidney (9), it is natural that AUC5’-DFUR in patients with impaired renal function is higher than that in patients with normal renal function. In fact, the inverse correlation between the AUC5’-DFUR and CLcr in this study is consistent with the findings of a pivotal study that assessed capecitabine pharmacokinetics in patients with impaired renal function (3). According to that study, the AUCs of capecitabine, 5’-DFCR, and 5-FU did not differ among the groups of patients with different renal function, whereas the AUC5’-DFUR in patients with moderately and severely impaired renal function was higher than that in patients with normal and mildly impaired renal function. The increased AUC5’-DFUR is generally explained by the delayed renal excretion of this metabolite in patients with impaired renal function. However, the t1/2 of 5’-DFUR was not extended, and the Cmax of 5’-DFUR was elevated by 91% in patients with impaired renal function, which implies that the accumulation of 5’-DFUR appears to be caused by unknown mechanisms in capecitabine metabolism rather than by delayed renal excretion.
According to our study, the metabolic ratio (AUC5’-DFUR+5-FU/AUC5’-DFCR) inversely correlated with CLcr. Therefore, we investigated whether or not CDA activity is involved in a mechanism of enhanced conversion from 5’-DFCR to 5’-DFUR by measuring plasma CDA activity. Unfortunately, however, the CDA activity was not associated with the metabolic ratio and there was also no correlation between CDA activity and CLcr. Therefore, an unknown mechanism other than CDA-mediated 5’-DFCR metabolism might exist to explain our findings. A similar phenomenon was reported for gemcitabine, where the metabolic activity converting gemcitabine to an inactive metabolite dFdU catalyzed by CDA increased as the renal function decreased (10), hence a common mechanism may exist.
Our study had several limitations as follows: (i) There was only one patient with moderately impaired renal function (CLcr=49 ml/min) in this study. Although the pharmacokinetic parameters of this patient were consistent with those of patients with mildly impaired renal function, it may be difficult to conclude that the findings of our study might be extrapolated to patients with more severely impaired renal function. On the contrary, the inverse correlation between the AUC5’-DFUR and renal function in patients with normal or preserved renal function would provide important insight into accurately understanding capecitabine metabolism. (ii) This study did not measure the plasma concentration of the final metabolite of 5-FU, α-fluoro-β-alanine (FBAL). A previous study found that the AUC of FBAL was higher in patients with moderately and severely impaired renal function, as was the AUC5’-DFUR. Unlike 5’-DFUR, the t1/2 of FBAL was apparently extended as renal function declined, which also implies that the increased AUCs of 5’-DFUR and FBAL in patients with impaired renal function have different pharmacokinetic backgrounds (3). (iii) Hepatic CDA activity was not directly measured in this practical study and was estimated based on plasma CDA activity. However, in a previous pharmacokinetic study, the CDA-mediated metabolism of gemcitabine was demonstrated to be correlated with the plasma CDA activity (8). Therefore, we believe that plasma CDA activity is a reliable surrogate marker for hepatic CDA activity. (iv) One patient had an extremely high plasma CDA activity as compared with the other patients. As one possible explanation of this, the patient might have a variant allele of the CDA gene which might enhance the catalytic activity. However, we did not analyze genetic polymorphisms of the CDA gene, and genetic analysis was beyond the purpose of the present study.
In conclusion, there was an interesting correlation between CLcr and the metabolic activation of generating 5’-DFUR from 5’-DFCR in patients who received capecitabine treatment but the underlying mechanism remains unknown.
Conflicts of Interest
YA reports receiving research funds and personal fees from Chugai, and research funds from Yakult Honsha. KF, NM, TS, OM, TK, NH, GN, and TI have nothing to disclose.
Authors’ Contributions
TI, KF, and YA designed the research. TI, KF, NM, TS, OM, TK, NH, GN, and YA performed the research. TI, KF, NM, TS, and YA analyzed the data. TI wrote the article. All Authors read and approved the final article.
Acknowledgements
The Authors thank Ms. Yuka Murasaki for her technical assistance.
References
- 1.Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34(8):1274–1281. doi: 10.1016/s0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 2.Schüller J, Cassidy J, Dumont E, Roos B, Durston S, Banken L, Utoh M, Mori K, Weidekamm E, Reigner B. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol. 2000;45(4):291–297. doi: 10.1007/s002800050043. [DOI] [PubMed] [Google Scholar]
- 3.Poole C, Gardiner J, Twelves C, Johnston P, Harper P, Cassidy J, Monkhouse J, Banken L, Weidekamm E, Reigner B. Effect of renal impairment on the pharmacokinetics and tolerability of capecitabine (Xeloda) in cancer patients. Cancer Chemother Pharmacol. 2002;49(3):225–234. doi: 10.1007/s00280-001-0408-0. [DOI] [PubMed] [Google Scholar]
- 4.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 5.Farkouh A, Ettlinger D, Schueller J, Georgopoulos A, Scheithauer W, Czejka M. A rapid and simple HPLC assay for quantification of capecitabine for drug monitoring purposes. Anticancer Res. 2010;30(12):5207–5211. [PubMed] [Google Scholar]
- 6.Sekido M, Fujita KI, Kubota Y, Ishida H, Takahashi T, Ohkuma R, Tsunoda T, Ishikawa F, Shibanuma M, Sasaki Y. Rabeprazole intake does not affect systemic exposure to capecitabine and its metabolites, 5’-deoxy-5-fluorocytidine, 5’-deoxy-5-fluorouridine, and 5-fluorouracil. Cancer Chemother Pharmacol. 2019;83(6):1127–1135. doi: 10.1007/s00280-019-03837-y. [DOI] [PubMed] [Google Scholar]
- 7.Budman DR, Meropol NJ, Reigner B, Creaven PJ, Lichtman SM, Berghorn E, Behr J, Gordon RJ, Osterwalder B, Griffin T. Preliminary studies of a novel oral fluoropyrimidine carbamate: Capecitabine. J Clin Oncol. 1998;16(5):1795–1802. doi: 10.1200/jco.1998.16.5.1795. [DOI] [PubMed] [Google Scholar]
- 8.Sugiyama E, Kaniwa N, Kim SR, Kikura-Hanajiri R, Hasegawa R, Maekawa K, Saito Y, Ozawa S, Sawada J, Kamatani N, Furuse J, Ishii H, Yoshida T, Ueno H, Okusaka T, Saijo N. Pharmacokinetics of gemcitabine in Japanese cancer patients: The impact of a cytidine deaminase polymorphism. J Clin Oncol. 2007;25(1):32–42. doi: 10.1200/jco.2006.06.7405. [DOI] [PubMed] [Google Scholar]
- 9.de Bruijn EA, van Oosterom AT, Tjaden UR, Reeuwijk HJ, Pinedo HM. Pharmacology of 5’-deoxy-5-fluorouridine in patients with resistant ovarian cancer. Cancer Res. 1985;45(11 Pt 2):5931–5935. [PubMed] [Google Scholar]
- 10.Serdjebi C, Gattacceca F, Seitz JF, Fein F, Gagnière J, François E, Abakar-Mahamat A, Deplanque G, Rachid M, Lacarelle B, Ciccolini J, Dahan L. Population pharmacokinetics of gemcitabine and dFdU in pancreatic cancer patients using an optimal design, sparse sampling approach. Ther Drug Monit. 2017;39(3):290–296. doi: 10.1097/ftd.0000000000000399. [DOI] [PubMed] [Google Scholar]