

Abstract
Objective
To evaluate short‐ and long‐term outcomes of African American (AA) participants of Scleroderma Lung Studies (SLS) I and II.
Methods
SLS I randomized 158 participants with systemic sclerosis‐interstitial lung disease (SSc‐ILD) to 1 year of oral cyclophosphamide (CYC) versus placebo. SLS II randomized 142 participants with SSc‐ILD to 1 year of oral CYC followed by 1 year of placebo versus 2 years of mycophenolate (MMF). Joint models compared the course of forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO) between AA and non‐AA, and Cox proportional hazard models assessed long‐term morbidity and mortality outcomes.
Results
In SLS I, there was no difference in the course of the FVC or DLCO between AA and non‐AA in either treatment arm. In SLS II, AA had an improved course of the FVC compared with non‐AA in the CYC arm; in the MMF arm, there was no difference in FVC course. There was no difference in DLCO course in either arm. Time to death and respiratory failure were similar for AA and non‐AA in SLS I. There was a trend for improved survival and time to respiratory failure in AA compared with non‐AA in SLS II. AA race was not independently associated with mortality in the SLS I or II in the Cox models.
Conclusion
Data from two randomized controlled trials demonstrated that AA patients with SSc‐ILD have similar morbidity and mortality outcomes compared with non‐AA patients. These findings contrast with the racial disparities described in prior observational studies and warrant further investigation.
Introduction
A number of observational studies have demonstrated that African American (AA) patients with systemic sclerosis (SSc) have a worse prognosis compared with non‐AA patients in the United States (1, 2, 3, 4). Not only is the incidence and prevalence of SSc higher among AAs (5, 6, 7), AA patients with SSc are more likely to have diffuse cutaneous sclerosis (1, 2, 3, 4), cardiac involvement (2), renal disease (2), as well as severe restrictive lung disease (1, 2, 4).
Single‐center observational cohort studies have demonstrated that mortality rates are higher in AA versus non‐AA patients with SSc, even when factors such as age and disease severity are considered (2, 4, 5). However, two studies have found that the association between AA race and mortality diminishes after adjustment for socioeconomic status (2, 4). The latter observation suggests that one should exert caution when interpreting the results of observational studies where bias (eg, systematic bias, selection bias, lead time bias) (8) and confounding factors may affect the relationship between race and health outcomes.
Using SSc cohorts from well‐controlled trials to examine outcomes among AA patients may further our understanding of racial disparities in SSc, as all patients in these studies received standard treatment, uniform follow‐up, and equal access to care. To this end, the purpose of the present study was to understand the severity and progression of interstitial lung disease (ILD) among AA patients who participated in two randomized controlled trials (RCTs) for SSc‐ILD (Scleroderma Lung Studies [SLS] I (9) and II (10)). A secondary goal was to compare long‐term morbidity and mortality outcomes between AA and non‐AA patients with SSc who participated in these trials.
PATIENTS ANDMETHODS
Study participants
All participants enrolled in SLS I (9) (NCT01762449; NCT00004563) and SLS II (10) (NCT00883129) were included in these analyses. Participating centers and investigators were similar for both trials. Eligibility criteria for both studies were also similar. Common inclusion criteria included age of 18 years or older, duration of disease that was 7 years or less from onset of the first non‐Raynaud’s symptom of SSc, forced vital capacity (FVC) 40% to 85% predicted (SLS I) or 40% to 80% predicted (SLS II), hemoglobin‐adjusted single‐breath diffusing capacity for carbon monoxide (DLCO) 40% or greater predicted (or 30%‐39% predicted if no evidence of clinically significant pulmonary hypertension), and evidence of any ground glass opacity (GGO) on high‐resolution computed tomography (HRCT). Key exclusion criteria included clinically significant pulmonary vascular disease and smoking within the previous 6 months. Please see the main SLS I (9) and II (10) manuscripts for a complete review of eligibility criteria for these trials. The Institutional Review Board of each site approved the studies, and only participants who provided informed consent were included in the present analyses.
SLS I and II study design
In SLS I, 158 participants were randomized to receive either oral cyclophosphamide (CYC) or matching placebo for 1 year, followed by an additional year of observation off‐treatment as previously published (9). In SLS II, 142 patients were randomized to receive either mycophenolate (MMF) for 2 years or oral CYC for 1 year followed by placebo for an additional year using a double‐dummy design to maintain the blinding as reported (10). The complete details of the SLS I and II study design have been previously published (9, 10).
SLS I and II assessment measurements
The FVC (primary SLS I and II end point) and DLCO (secondary SLS I and II end point) were measured every 3 months during the 24‐month study period for both SLS I and II (9, 10). HRCT thoracic imaging was obtained at baseline and at 24 months in SLS II and at baseline and at 12 months in SLS I. A computer‐aided design scoring system was used to quantify the extent of different patterns of ILD as previously described (11). Quantitative ILD (QILD) score was the sum of all abnormally classified scores, including scores for quantitative lung fibrosis (QLF, linear reticular markings with architectural distortion), GGO, and honeycomb changes (clustered air‐filled cysts with dense walls). Scores were calculated as percentage of total counted voxels for both the whole lung (WL), including both lungs, and for the zone (area‐equivalent upper, middle, or lower lung zone) of maximal involvement (ZM).
Long‐term morbidity and mortality outcomes
In SLS I, long‐term morbidity and mortality outcomes were assessed up to 12 years after the first patient was randomized. In SLS II, long‐term morbidity and mortality outcomes were assessed up to 8 years after the first patient was randomized. If survival status was unknown, survival time (in months) was censored at the date when the participant was last known to be alive. The methodology for the long‐term follow‐up studies of SLS I and II has been previously published (12). In SLS I and II, only two and zero patients, respectively, had no long‐term follow‐up information available on survival status. For this analysis, the morbidity outcome of interest was time to respiratory failure, which was defined as the need for supplemental oxygen and/or lung transplantation. In SLS I and II, only 2 and 11 patients, respectively, had no long‐term follow‐up information available on respiratory failure.
Statistical analysis
Baseline characteristics
Summary statistics were generated for baseline characteristics. A two‐sample t test or Wilcoxon rank‐sum test was used to compare continuous variables, and a χ2 test or Fisher exact test was used to compare categorical variables.
Primary outcome: progression of ILD
A joint model analysis was used to determine whether AA race was associated with progression of SSc‐ILD. The joint model (used also in the main SLS II analysis (10)) adjusts for nonignorable missing data due to treatment failure, death, and drop‐outs (13). The outcome for the primary outcome model was the course of FVC%‐predicted that was measured every 3 months from 3 to 24 months. The longitudinal model of the joint analysis included the following covariates: AA, baseline FVC %‐predicted, a linear spline time trend with knots at 12 months and 18 months for SLS I and at 12 months and 21 months for SLS II, and the interaction between AA and these time trends. The inflection points (knots) for the time trends were determined by examining the changes in the slopes of the FVC%‐predicted from 3 to 24 months as described in further detail in our manuscript on the joint model analysis (13). The same approach was employed for the DLCO%‐predicted analysis.
Linear regression models were created to evaluate the change in the QLF and QILD scores from baseline to 12 months (SLS I) or to 24 months (SLS II). The covariates for these models were baseline QLF/QILD scores, AA, and treatment arm.
Secondary outcomes: long‐term morbidity and mortality
The Kaplan‐Meier estimate was used to generate survival curves, and the log‐rank test was used to compare survival between groups. If survival status was unknown, survival time was censored at the date when the participant was last known to be alive. Cox proportional hazard models were created to evaluate time to death and time to respiratory failure. Based on our prior publication of long‐term outcomes in SLS I and II (12), the following covariates were included in these models: age, baseline modified Rodnan Skin Score (mRSS), and baseline FVC%‐predicted. The covariate of AA race was added to the models to assess the relationship between AA and survival/time to respiratory failure.
All tests were two‐sided. The joint analyses were performed using the R package JMbayes, and all other analyses were conducted in SAS v9.4 (The SAS Institute).
RESULTS
Participant characteristics
Baseline characteristics of SLS I and II participants appear in Table 1. Among the 158 SLS I participants, 26 (16.5%) were AA, and among the 142 SLS II participants, 33 (23.2%) were AA. In both studies, AA participants were younger than non‐AA participants. The total lung capacity (TLC)%‐predicted (SLS I and II) and the DLCO%‐predicted (SLS II only) were lower in AA compared with non‐AA participants; however, AAs have lower lung volumes than Caucasians, and the prediction equations used to calculate TLC do not adjust for race (14). There was no significant difference in the FVC%‐predicted between AA and non‐AA participants in SLS I; however, in SLS II, the FVC%‐predicted was slightly better in AA participants. In contrast to the prediction equations for the TLC, the prediction equation for the FVC%‐predicted does adjust for race (15). The average radiographic fibrosis and ILD scores were numerically higher (worse) in AA participants compared with non‐AA participants for both studies, although the difference was only statistically significant for the QILD‐ZM score in SLS II participants. Although not statistically significant, a greater percentage of AA participants of SLS I were Scl‐70 antibody positive, whereas a similar percentage of AA and non‐AA participants of SLS II were Scl‐70 antibody positive.
Table 1.
Baseline characteristics of AA and non‐AA SLS I and II participants
| Measure | SLS I | SLS II | ||
|---|---|---|---|---|
|
AA (N = 26) |
Non‐AA (N = 132) |
AA (N = 33) |
Non‐AA (N = 109) |
|
| Treatment, CYC | 12 (46.2%) | 67 (50.8%) | 19 (57.6%) | 54 (49.5%) |
| Age, years*, **, *** | 43.1 ± 12.8 | 49.5 ± 12.0 | 49.1 ± 9.2 | 53.2 ± 9.7 |
| Female | 19 (73.1%) | 92 (69.7%) | 26 (78.8%) | 79 (72.5%) |
| Education*** | ||||
| Eighth grade or less | 0 (0%) | 3 (2.3%) | 1 (3.0%) | 3 (2.8%) |
| Some high school | 5 (19.2%) | 4 (3.1%) | 0 (0%) | 3 (2.8%) |
| High school graduate | 8 (30.8%) | 28 (21.5%) | 7 (21.2%) | 24 (22.0%) |
| Trade school or some college | 2 (7.7%) | 9 (6.9%) | 10 (30.3%) | 27 (24.8%) |
| Received bachelor’s degree | 5 (19.2%) | 41 (31.5%) | 11 (33.3%) | 34 (31.2%) |
| Graduate or professional degree | 4 (15.4%) | 29 (22.3%) | 4 (12.1%) | 18 (16.5%) |
| Other | 2 (7.7%) | 16 (12.3%) | 0 | 0 |
| Diffuse | 19 (73.1%) | 75 (56.8%) | 23 (69.7%) | 60 (55.1%) |
| Disease duration, years | 2.8 ± 2.0 | 3.2 ± 2.1 | 2.6 ± 1.9 | 2.6 ± 1.7 |
| FVC, % predicted** | 65.8 ± 11.2 | 68.6 ± 12.3 | 69.4 ± 9.1 | 65.6 ± 9.0 |
| FEV1/FVC, % | 83.8 ± 7.3 | 82.6 ± 8.1 | 81.9 ± 6.1 | 82.8 ± 5.4 |
| TLC, % predicted*, ** | 63.3 ± 10.6 | 70.8 ± 13.2 | 58.8 ± 8.9 | 67.9 ± 10.9 |
| DLCO, % predicted*, *** | 38.7 ± 10.0 | 48.2 ± 12.9 | 55.0 ± 12.7 | 53.7 ± 12.7 |
| BDI (focal score; 0‐12) a | 6.2 ± 1.7 | 5.6 ± 1.9 | 7.0 ± 2.4 | 7.3 ± 2.1 |
| HAQ‐DI (score, 1‐3) b | 1.1 ± 0.7 | 0.8 ± 0.7 | 0.7 ± 0.6 | 0.7 ± 0.7 |
| mRSS (0‐51) | 16.4 ± 11.4 | 14.5 ± 10.9 | 15.9 ± 12.0 | 14.3 ± 10.0 |
| QLF‐WL % | 13.4 ± 13.6 | 9.7 ± 9.8 | 10.4 ± 7.5 | 8.0 ± 6.7 |
| QLF‐ZM % | 31.6 ± 27.2 | 25.7 ± 21.0 | 28.7 ± 21.4 | 21.0 ± 18.8 |
| QILD‐WL % | 40.9 ± 17.4 | 34.7 ± 16.8 | 33.4 ± 13.9 | 28.3 ± 13.9 |
| QILD‐ZM %** | 67.4 ± 18.1 | 56.6 ± 21.9 | 58.5 ± 18.6 | 49.0 ± 20.4 |
| Scl‐70 antibody | 8 (47.1%) | 28 (31.5%) | 14 (48.3%) | 47 (44.8%) |
| Anticentromere antibody | 0 (0%) | 3 (3.4%) | 0 (0%) | 3 (2.9%) |
| RNA polymerase III antibody | 2 (11.8%) | 15 (16.9%) | 4 (13.8%) | 14 (13.3%) |
Data reported are means ± SD or N (%).
Abbreviations: AA, African American; BDI, Baseline Dyspnea Index; CYC, cyclophosphamide; DLCO, single‐breath diffusing capacity for carbon monoxide; FEV1, forced expired volume in 1 second; FVC, forced vital capacity; HAQ‐DI, Health Assessment Questionnaire for Scleroderma‐Disability Index; HRCT, high‐resolution computed tomography; mRSS, modified Rodnan Skin Score; QILD‐WL %, quantitative extent of interstitial lung disease (fibrosis + ground glass opacity + honeycombing) in whole lung on HRCT; QILD‐ZM, quantitative extent of interstitial lung disease in the zone of maximal involvement on HRCT; QLF‐WL %, quantitative extent of lung fibrosis (reticulations) in whole lung on HRCT; QLF‐ZM %, quantitative extent of lung fibrosis in the zone of maximal involvement on HRCT; SLS, Scleroderma Lung Studies; TLC, total lung capacity.
High score denotes worse dyspnea.
High score denotes worse function.
p < 0.05 comparing AA vs. non‐AA in SLS I.
p < 0.05 comparing AA vs. non‐AA in SLS II.
p < 0.05 comparing AA in SLS I vs. AA in SLS II.
Alternate disease‐modifying medication usage during trial
In SLS I, one AA and one non‐AA participant in the placebo arm initiated treatment with CYC in the second year of the study. In SLS II, nine participants in the CYC arm initiated treatment with MMF in the second year of the study (two AA and seven non‐AA), whereas two participants in the MMF arm initiated treatment with rituximab in the second year of the study (both non‐AA).
Progression of SSc‐ILD: evolution of FVC%‐predicted
In both the CYC and placebo arms of SLS I, there was no significant difference in the course of the FVC%‐predicted between AA and non‐AA participants from 3 to 24 months (Figures 1 and 2; Supplementary Tables S1 and S2). In both SLS I study arms, it appeared that AA participants had a faster decline in the FVC%‐predicted from 18 to 24 months, but this was not statistically significant.
Figure 1.
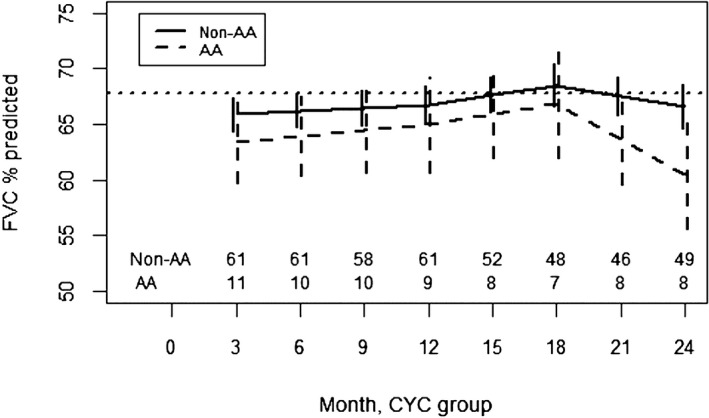
Course of the forced vital capacity (FVC)% from 3 to 24 months in African American (AA) and non‐AA participants assigned to cyclophosphamide (CYC) in Scleroderma Lung Studies I using a joint model analysis. Prespecified covariates for this model include the baseline FVC%‐predicted, AA race, time trends (3‐12 months, 12‐18 months, and 18‐24 months), and interactions between AA race and the time trends.
Figure 2.
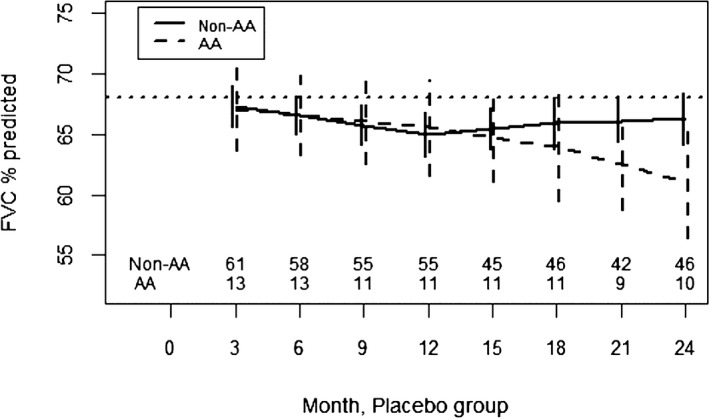
Course of the forced vital capacity (FVC)% from 3 to 24 months in African American (AA) and non‐AA participants assigned to placebo in Scleroderma Lung Studies I using a joint model analysis. Prespecified covariates for this model include the baseline FVC%‐predicted, AA race, time trends (3‐12 months, 12‐18 months, and 18‐24 months), and interactions between AA race and the time trends.
In the CYC arm of SLS II, AA participants had an improved response to CYC compared with non‐AA participants from 3 to 12 months (p = 0.008) (Figure 3; Supplementary Table S3). After CYC therapy concluded, AA participants subsequently experienced a faster decline in the FVC%‐predicted compared with non‐AA participants (p < 0.001). In the MMF arm of SLS II, there was no difference in the course of the FVC%‐predicted between AA and non‐AA participants from 3 to 24 months (Figure 4; Supplementary Table S4). Both groups experienced a similar improvement in the FVC%‐predicted over the course of the trial.
Figure 3.
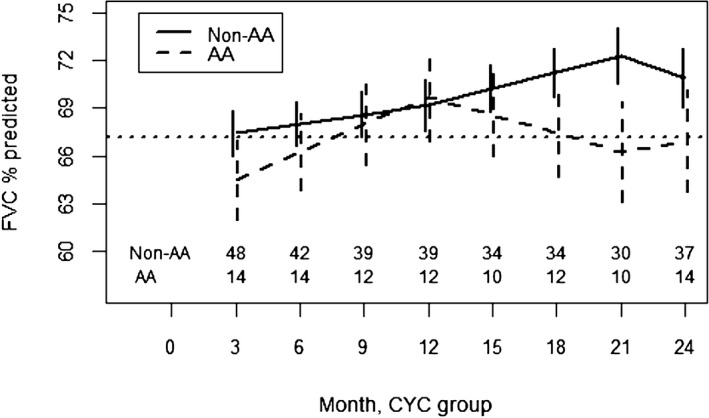
Course of the forced vital capacity (FVC)% from 3 to 24 months in African American (AA) and non‐AA participants assigned to cyclophosphamide (CYC) in Scleroderma Lung Studies II using a joint model analysis. Prespecified covariates for this model include the baseline FVC%‐predicted, AA race, time trends (3‐12 months, 12‐21 months, and 21‐24 months), and interactions between AA race and the time trends.
Figure 4.
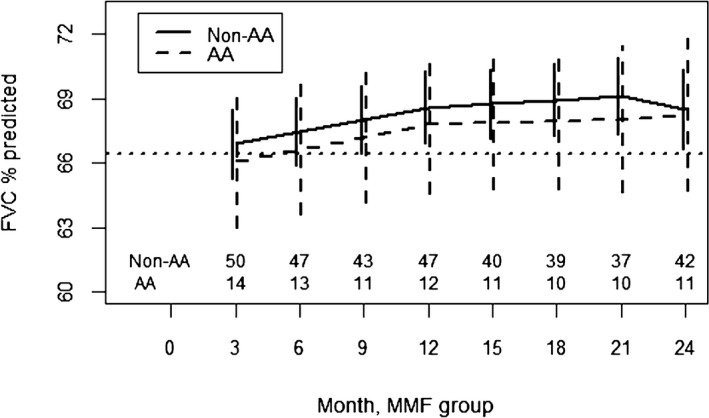
Course of the forced vital capacity (FVC)% from 3 to 24 months in African American (AA) and non‐AA participants assigned to mycophenolate (MMF) in Scleroderma Lung Studies II using a joint model analysis. Prespecified covariates for this model include the baseline FVC%‐predicted, AA race, time trends (3‐12 months, 12‐21 months, and 21‐24 months), and interactions between AA race and the time trends.
Progression of SSc‐ILD: evolution of DLCO%‐predicted
In the CYC arm of SLS I, there was no difference in the course of the DLCO%‐predicted between AA and non‐AA participants from 3 to 24 months (Supplementary Figure S1). In the placebo arm of SLS I, there was no difference in the course of the DLCO%‐predicted between AA and non‐AA participants from 3 to 24 months (Supplementary Figure S2).
In the CYC arm of SLS II, there was no difference in the course of the DLCO%‐predicted between AA and non‐AA participants from 3 to 24 months (Supplementary Figure S3). In the MMF arm of SLS II, there was also no difference in the course of the DLCO%‐predicted between AA and non‐AA participants from 3 to 24 months (Supplementary Figure S4).
Progression of ILD: change in radiographic fibrosis
In SLS I, there was no difference in the change in QLF‐WL/ZM or QILD‐WL/ZM from baseline to 12 months between AA and non‐AA participants (Supplementary Table S5). The linear regression models for the radiographic outcomes demonstrated a significant treatment effect favoring CYC as previously published (16).
In SLS II, there was no difference in the change in QLF‐WL/ZM or QILD‐WL/ZM from baseline to 24 months between AA and non‐AA participants (Supplementary Table S6). There was no difference in radiographic outcomes between participants randomized to MMF or CYC as previously reported (17).
Long‐term outcomes: time to death
During the 24‐month study period of SLS I, no AA participants died. Over the long‐term follow‐up period for SLS I, there was no significant difference in long‐term survival between AA and non‐AA participants (p value for log‐rank test = 0.54; Figure 5A). In the Cox model adjusting for baseline mRSS, FVC%‐predicted, and age, AA race was not independently associated with time to death (p = 0.29) (Supplementary Table S7).
Figure 5.
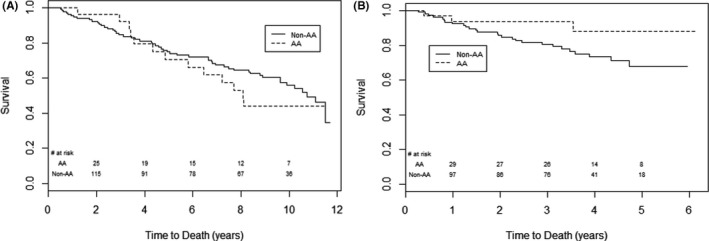
Time to death in African American (AA) versus non‐AA participants from the time of randomization in (A) Scleroderma Lung Studies (SLS) I and (B) SLS II. There was no significant difference in long‐term survival (p = 0.54 by log‐rank test; A) between AA and non‐AA participants of SLS I. There was a trend for improved survival in AA participants compared with non‐AA participants of SLS II (p = 0.07 by log‐rank test; B). The last known date they were known to be alive was used for the survival analysis.
During the 24‐month study period of SLS II, 2 AA participants (6.1%) died compared with 14 non‐AA participants (12.8%). In the long‐term follow‐up period, there was a trend for improved long‐term survival in AA compared with non‐AA participants in SLS II (p value for log‐rank test = 0.07; Figure 5B). However, in the Cox model adjusting for baseline mRSS, FVC%‐predicted, and age, AA race was not independently associated with time to death (p = 0.50) (Supplementary Table S8).
Long‐term outcomes: time to respiratory failure
In SLS I, there was no significant difference in time to respiratory failure (defined above as initiation of supplemental oxygen [N = 29] or lung transplantation [N = 3]) between AA and non‐AA participants (p value for log‐rank test = 0.37; Supplementary Figure S5). In the Cox model adjusting for baseline mRSS, FVC%‐predicted, and age, AA race was not independently associated with time to respiratory failure (p = 0.42; Supplementary Table S9).
In SLS II, there was a trend for improved (longer) time to respiratory failure (defined above as initiation of supplemental oxygen [N = 11] or lung transplantation [N = 1]) in AA compared with non‐AA participants (p value for log‐rank test = 0.10; Supplementary Figure S6). However, in the Cox model adjusting for baseline mRSS, FVC%‐predicted, and age, AA race was not independently associated with time to respiratory failure (p = 0.55; Supplementary Table S10).
DISCUSSION
This study is the first to demonstrate outcomes of AA patients with SSc who participated in two RCTs for SSc‐ILD: SLS I and II. AA participants from these two trials had similar efficacy outcomes as well as similar long‐term morbidity and mortality outcomes compared with non‐AA participants.
Consistent with prior observational studies (1, 2, 4), AA participants had evidence of more extensive ILD at baseline compared with non‐AA participants (based on radiographic measures but not on the degree of restriction on pulmonary function testings). Disease duration was similar between AA and non‐AA participants of SLS I and II, suggesting that AA participants likely had a faster rate of progression of their ILD. Although the baseline mRSS was similar between AA and non‐AA participants in these trials, a greater percentage (although not statistically significant) of the AA participants had diffuse cutaneous disease. Taken together, these findings support the observation that AA patients with SSc may exhibit a more severe phenotype of the disease state compared with non‐AA patients.
However, despite the disparities in disease severity appreciated at baseline, AA and non‐AA participants in both SLS I and II had similar rates of ILD progression during the 2‐year trials using various methods for defining disease progression (eg, FVC%‐predicted, DLCO%‐predicted, and changes in different patterns of radiographic fibrosis/ILD). These findings are in line with a prior analysis of data from three RCTs of patients with diffuse cutaneous SSc, which also found no racial differences in the course of the FVC%‐predicted among participants (18). The SENSCIS trial comparing nintedanib versus placebo for SSc‐ILD also demonstrated that there was no significant treatment‐by‐time‐by‐subgroup interaction for race, although AA represented only 6% of patients in this study (19). Interestingly, AA participants with SSc‐ILD randomized to CYC in SLS II actually experienced an improved course of FVC%‐predicted compared with non‐AA participants during the active treatment phase of the study, whereas in the MMF arm of SLS II, there was no difference in the course of the FVC%‐predicted between AA and non‐AA participants. One possible explanation for these observations is that AA may demonstrate a preferential treatment response to CYC. To properly test this hypothesis, one would have to design a trial in which AA participants with SSc‐ILD were randomized to treatment with CYC versus MMF.
During the placebo phase of SLS II‐CYC arm, AA participants had a faster decline in the FVC%‐predicted compared with non‐AA participants. This observation could also suggest that in addition to upfront aggressive therapy for SSc‐ILD, AA patients may need to be monitored more closely for ILD progression when therapy is stopped.
In contrast to prior observational studies (2, 4, 5), there was no difference in long‐term morbidity and mortality outcomes between AA and non‐AA participants in SLS I and II. Moreover, in SLS II, AA participants actually had a trend for improved long‐term survival and time to respiratory failure. However, after adjusting for baseline disease severity and age, AA race was not associated with any race‐related difference in long‐term morbidity or mortality. These findings suggest that there may be other factors mediating the relationship between AA race and health outcomes reported in prior observational studies, such as access to care. For instance, Moore and colleagues (4) demonstrated that household income was more strongly linked with survival than race. A number of other studies have demonstrated that the effect of race on health outcomes diminishes or disappears when socioeconomic position is considered (20). The present studies did not collect information regarding household income. Both studies did collect information about educational background, and there were no significant differences in the educational background of AA and non‐AA participants in either SLS I or SLS II; however, SLS II AA participants on average achieved a higher educational level than SLS I AA participants.
This study has certain limitations that deserve mention. First, the numbers of AA participants in SLS I and II were relatively small, and thus, our study may be underpowered to detect differential treatment effects. Furthermore, patients were not stratified by race at randomization. However, the overall findings were reassuringly similar for both study cohorts. Second, some may argue that AA patients wutg SSc who participate in clinical trials may possess unique attributes that distinguish them from AA patients with SSc who do not participate in clinical trials (eg, better access to care, higher socioeconomic status or education level), potentially compromising the generalizability of the findings. This is a reasonable concern; however, the same could be said for non‐AA patients who participate in clinical trials. A 10‐year longitudinal study of patients with rheumatoid arthritis demonstrated that the factors associated with patients’ continued participation in the study were: higher education, female sex, moderate to high levels of social support, employment, and fewer joint flares (21). Thus, the limited generalizability would apply to all racial groups in this trial and is a general criticism for clinical trials.
This study also has notable strengths. First, using two independent cohorts to study outcomes among AA participants minimizes the likelihood that our findings were due to chance alone. Second, this study assessed ILD progression using different methods for defining progression (physiologic, radiographic, long‐term morbidity and mortality) and measured these outcomes in a standardized manner and at multiple time points, which ultimately improves our confidence that we have captured the true evolution of ILD in these patients (as opposed to relying on two to three FVC data points measured at random intervals to understand disease progression). Third, because the patients in this study were from diverse geographic regions throughout the United States, our findings are likely translatable to most SSc patients in the United States who receive specialty care at academic centers of excellence for the management of SSc.
In summary, the present study demonstrated that when AA patients with SSc‐ILD are treated early and aggressively with upfront immunosuppressive therapy, they have similar rates of ILD progression and long‐term morbidity and mortality outcome, as non‐AA patients with SSc‐ILD. These findings should motivate us to help ensure that our AA patients with SSc have the same follow‐up and treatment opportunities as our non‐AA patients with SSc. Additional studies are needed to determine whether AA patients with SSc demonstrate preferential treatment responses to certain agents (eg, CYC vs. MMF) to ultimately help personalize the care of these patients and improve health outcomes.
AUTHOR CONTRIBUTIONS
All authors drafted the article or revised it critically for important intellectual content, and all authors gave final approval of the version of the article to be published.
Study conception and design
Volkmann, Roth, Khanna, Elashoff, Tashkin.
Acquisition of data
Volkmann, Roth, Clements, Furst, Khanna, Kim, Goldin, Tashkin.
Analysis and interpretation of data
Volkmann, Steen, Li, Assassi, Khanna, Elashoff, Tashkin.
Supporting information
Supplementary Material
Acknowledgments
We thank the patients, investigators, and coordinators who participated in the Scleroderma Lung Study I and II. Bristol Myers Squibb supplied cyclophosphamide for use in SLS I, and Hoffmann‐LaRoche supplied mycophenolate mofetil for use in SLS II.
Appendix 1.
The following persons and institutions participated in the Scleroderma Lung Study I: University of California at Los Angeles (UCLA), Los Angeles: P.J. Clements, D.P. Tashkin, R. Elashoff, J. Goldin, M. Roth, D. Furst, K. Bulpitt, D. Khanna, W.‐L.J. Chung, S. Viasco, M. Sterz, L. Woolcock, X. Yan, J. Ho, S. Vasunilashorn, I. da Costa; University of Medicine and Dentistry of New Jersey, New Brunswick: J.R. Seibold, D.J. Riley, J.K. Amorosa, V.M. Hsu, D.A. McCloskey, J.E. Wilson; University of Illinois Chicago, Chicago: J. Varga, D. Schraufnagel, A. Wilbur, M. Lopata, S. Arami, P. Cole‐Saffold; Boston University, Boston: R. Simms, A. Theodore, P. Clarke, J. Korn, K. Tobin, M. Nuite; Medical University of South Carolina, Charleston: R. Silver, M. Bolster, C. Strange, S. Schabel, E. Smith, J. Arnold, K. Caldwell, M. Bonner; Johns Hopkins School of Medicine, Baltimore: R. Wise, F. Wigley, B. White, L. Hummers, M. Bohlman, A. Polito, G. Leatherman, E. Forbes, M. Daniel; Georgetown University, Washington, D.C.: V. Steen, C. Read, C. Cooper, S. Wheaton, A. Carey, A. Ortiz; University of Texas at Houston, Houston: M. Mayes, E. Parsley, S. Oldham, T. Filemon, S. Jordan, M. Perry; University of California at San Francisco, San Francisco: K. Connolly, J. Golden, P. Wolters, R. Webb, J. Davis, C. Antolos, C. Maynetto; University of Alabama at Birmingham, Birmingham: B. Fessler, M. Olman, C. Sanders, L. Heck, T. Parkhill; University of Connecticut Health Center, Farmington: N. Rothfield, M. Metersky, R. Cobb, M. Aberles, F. Ingenito, E. Breen; Wayne State University, Detroit: M. Mayes, K. Mubarak, J.L. Granda, J. Silva, Z. Injic, R. Alexander; Virginia Mason Research Center, Seattle: D. Furst, S. Springmeyer, S. Kirkland, J. Molitor, R. Hinke, A. Mondt; Data Safety and Monitoring Board: Harvard Medical School, Boston — T. Thompson; Veterans Affairs Medical Center, Brown University, Providence, R.I. — S. Rounds; Cedars Sinai–UCLA, Los Angeles — M. Weinstein; Clinical Trials Surveys, Baltimore — B. Thompson; Mortality and Morbidity Review Committee: UCLA, Los Angeles — H. Paulus, S. Levy; Johns Hopkins University, Baltimore — D. Martin.
The following persons and institutions participated in the Scleroderma Lung Study II: University of Boston, Boston: A.C. Theodore, R.W. Simms, E. Kissin, F.Y. Cheong; Georgetown University, Washington, D.C.: V.D. Steen, C.A. Read Jr., C. Fridley, M. Zulmatashvili; Johns Hopkins University, Baltimore: R.A. Wise, F.M. Wigley, L. Hummers, G. Leatherman; Medical University of South Carolina, Charleston: R.M. Silver, C. Strange, F.N. Hant, J. Ham, K. Gibson, D. Rosson; University of California, Los Angeles (UCLA), Los Angeles: D.P. Tashkin, R.M. Elashoff, M.D. Roth, P.J. Clements, D. Furst, S. Kafaja, E. Kleerup, D. Elashoff, J. Goldin, E. Ariola, G. Marlis, J. Mason‐Berry, P. Saffold, M. Rodriguez, L. Guzman, J. Brook; University of California, San Francisco (UCSF), San Francisco: J. Golden, M.K. Connolly, A. Eller, D. Leong, M. Lalosh, J. Obata; University of Illinois, Chicago: S. Volkov, D. Schraufnagel, S. Arami, D. Franklin; Northwestern University, Chicago: J. Varga, J. Dematte, M. Hinchcliff, C. DeLuca, H. Donnelly, C. Marlin; University of Medicine and Dentistry of New Jersey, New Brunswick: D.J. Riley, V.M. Hsu, D.A. McCloskey; University of Michigan, Ann Arbor: K. Phillips, D. Khanna, F.J. Martinez, E. Schiopu, J. Konkle; University of Texas, Houston: M. Mayes, B. Patel, S. Assassi, F. Tan; National Jewish Health, Denver: A. Fischer, J. Swigris, R. Meehan, K. Brown, T. Warren, M. Morrison; University of Utah, Salt Lake City: M. B. Scholand, T. Frecht, P. Carey, M. Villegas; University of Minnesota, Minneapolis: J. Molitor, P. Carlson.
This work was supported in part by the Rheumatology Research Foundation (ERV); Scleroderma Foundation (ERV); and the following authors were supported by NIH National Heart, Lung, and Blood Institute: Dr. Tashkin (R01‐HL‐089758 and U01‐HL‐60587) and Dr. Elashoff (R01‐HL‐089901 and U01‐HL‐60606). Bristol Myers Squibb supplied cyclophosphamide for use in SLS I and Hoffmann‐LaRoche supplied mycophenolate mofetil for use in SLS II.
Dr. Volkmann reports receipt of consulting fees from Boehringer Ingelheim and Forbius, and grant support from Corbus and Forbius during the course of the study. Dr. Roth reports nonfinancial support from Hoffmann‐La Roche/Genentech during the course of the study. Dr. Khanna reports the following disclosures during the course of the study: grant support from NIH, Immune Tolerance Network, Bayer, BMS, Horizon, Pfizer; Consultant: Acceleron, Actelion, Amgen, Bayer, Blade Therapeutics, Boehringer Ingelheim, CSL Behring, Corbus, Cytori, Galapagos, Genentech/Roche, GSK, Horizon Merck, Mitsubishi Tanabe Pharma, Regeneron, Sanofi‐Aventis, and United Therapeutics; CME programs: Impact PH; Stocks: Eicos Sciences, Inc; Leadership/Equity position: medical lead, scleroderma development, CiviBioPharma/Eicos Sciences, Inc. No other disclosures relevant to this article were reported.
REFERENCES
- 1. Steen V, Domsic RT, Lucas M, Fertig N, Medsger TA Jr. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum 2012;64:2986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gelber AC, Manno RL, Shah AA, Woods A, Le EN, Boin F, et al. Race and association with disease manifestations and mortality in scleroderma: a 20‐year experience at the Johns Hopkins Scleroderma Center and review of the literature. Medicine (Baltimore) 2013;92:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morgan ND, Shah AA, Mayes MD, Domsic RT, Medsger TA Jr, Steen VD, et al. Clinical and serological features of systemic sclerosis in a multicenter African American cohort: analysis of the genome research in African American scleroderma patients clinical database. Medicine (Baltimore) 2017;96:e8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moore DF, Kramer E, Eltaraboulsi R, Steen VD. Increased morbidity and mortality of scleroderma in African Americans compared to non‐African Americans. Arthritis Care Res (Hoboken) 2019;71:1154–63. [DOI] [PubMed] [Google Scholar]
- 5. Laing TJ, Gillespie BW, Toth MB, Mayes MD, Gallavan RH Jr, Burns CJ, et al. Racial differences in scleroderma among women in Michigan. Arthritis Rheum 1997;40:734–42. [DOI] [PubMed] [Google Scholar]
- 6. Mayes MD, Lacey JV Jr, Beebe‐Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003;48:2246–55. [DOI] [PubMed] [Google Scholar]
- 7. Steen VD, Oddis CV, Conte CG, Janoski J, Casterline GZ, Medsger TA Jr. Incidence of systemic sclerosis in Allegheny County, Pennsylvania: a twenty‐year study of hospital‐diagnosed cases, 1963–1982. Arthritis Rheum 1997;40:441–5. [DOI] [PubMed] [Google Scholar]
- 8. Wang MT, Bolland MJ, Grey A. Reporting of limitations of observational research. JAMA Intern Med 2015;175:1571–2. [DOI] [PubMed] [Google Scholar]
- 9. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006;354:2655–66. [DOI] [PubMed] [Google Scholar]
- 10. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma‐related interstitial lung disease: Scleroderma Lung Study II (SLS‐II), a double‐blind, parallel group, randomised controlled trial. Lancet Respir Med 2016;4:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim HJ, Tashkin DP, Clements P, Li G, Brown MS, Elashoff R, et al. A computer‐aided diagnosis system for quantitative scoring of extent of lung fibrosis in scleroderma patients. Clin Exp Rheumatol 2010;28 Suppl 62:S26–35. [PMC free article] [PubMed] [Google Scholar]
- 12. Volkmann ER, Tashkin DP, Sim M, Li N, Goldmuntz E, Keyes‐Elstein L, et al. Short‐term progression of interstitial lung disease in systemic sclerosis predicts long‐term survival in two independent clinical trial cohorts. Ann Rheum Dis 2019;78:122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elashoff R, Li G, Li N. Joint modeling of longitudinal and time‐to‐event data In: A short course on survival analysis. Boca Raton, FL: CRC Press; 2016. [Google Scholar]
- 14. Crapo RO, Morris AH, Clayton PD, Nixon CR. Lung volumes in healthy nonsmoking adults. Bull Eur Physiopathol Respir 1982;18:419–25. [PubMed] [Google Scholar]
- 15. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999;159:179–87. [DOI] [PubMed] [Google Scholar]
- 16. Goldin J, Elashoff R, Kim HJ, Yan X, Lynch D, Strollo D, et al. Treatment of scleroderma‐interstitial lung disease with cyclophosphamide is associated with less progressive fibrosis on serial thoracic high‐resolution CT scan than placebo: findings from the scleroderma lung study. Chest 2009;136:1333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldin JG, Kim GH, Tseng CH, Volkmann E, Furst D, Clements P, et al. Longitudinal changes in quantitative lung disease on computed tomography after immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc 2018;15:1286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nashid M, Khanna PP, Furst DE, Clements PJ, Maranian P, Seibold J, et al. Gender and ethnicity differences in patients with diffuse systemic sclerosis—analysis from three large randomized clinical trials. Rheumatology (Oxford) 2011;50:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for systemic sclerosis‐associated interstitial lung disease. N Engl J Med 2019;380:2518–28. [DOI] [PubMed] [Google Scholar]
- 20. Williams DR. Race/ethnicity and socioeconomic status: measurement and methodological issues. Int J Health Serv 1996;26:483–505. [DOI] [PubMed] [Google Scholar]
- 21. Reisine S, Fifield J, Winkelman DK. Characteristics of rheumatoid arthritis patients: who participates in long‐term research and who drops out? [research support]. Arthritis Care Res 2000;13:3–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material