

Abstract
Patients with chronic lymphocytic leukemia (CLL) treated with Ibrutinib often present hemorrhagic complications. Platelets dysfunction is well documented by aggregometry and flow cytometry, but the mechanisms by which Ibrutinib treatment influences the platelets status is yet to be evaluated. The aim of this study is to identify platelet membrane parameters in chronic lymphocytic leukemia (CLL) that could be altered by Ibrutinib administration. In this paper we propose a set of fluorescence measurements of the following parameters: membrane fluidity, resting membrane potential, and reactive oxygen species production of platelets suspensions obtained from CLL patients treated or not with Ibrutinib as markers for platelets status in this pathological situation. Platelets from CLL patients treated with Ibrutinib have higher membrane fluidity, lower resting membrane potential and higher level of reactive oxygen species production compared to the untreated CLL patients. These patients are also presenting higher membrane fluidity and lower resting membrane potential compared to healthy volunteers.
Keywords: Ibrutinib, leukemia, platelets, membrane potential, ROS
Introduction
Patients with chronic lymphocytic leukemia (CLL) have usually associated a high risk of hemorrhagic complications which may be induced by anticoagulation or antiplatelet therapy (possibly associated with renal impairment, anemia, thrombocytopenia, or alcohol abuse) [1].
An increased activity of Bruton’s tyrosine kinase (BTK) has been found increased in B-cell malignancies such as CLL [2]. The BTK has a signaling role in platelet (PLT) activation via phospholipase Cγ2, by reducing the expression of the PLT membrane glycoprotein VI (GPVI). The PLT adhesion to collagen is realized through GPVI, and, once the adhesion occurred, a signaling cascade (involving Src kinases Fyn and Lyn, tyrosine kinase Syk, and Tec kinases BTK) is triggered, leading to the PLT activation [3,4].
Ibrutinib, a BTK inhibitor used in CLL treatment, was associated with unexpected serious bleedings [2,5]. Platelet dysfunction, as diagnosed by platelet aggregation tests, was identified as the main cause of bleeding in CLL patients treated with Ibrutinib [6-8]. Indeed, irreversible inhibition of BTK by Ibrutinib was associated with a decrease of PLT activation, resulting in a reduction of their aggregation capacity, as found in studies on PLTs from healthy persons or nonhuman primates [9].
The properties of platelet membrane modulate the activity of intrinsic proteins (like channels, pumps, signaling proteins, etc.) and expression of receptors [10,11].
Even though the pro-hemorrhagic effect of Ibrutinib is well documented [12], the way by which Ibrutinib influences the PLT membrane status is yet to be evaluated.
In this study we propose a set of biophysical parameters which can be relevant for the PLTs impairment in CLL patients treated with Ibrutinib. Fluorescence measurements of the following parameters: membrane fluidity (MF), resting membrane potential (RMP), and production of reactive oxygen species (ROS), were done on PLTs suspensions obtained from CLL patients treated or not with Ibrutinib. Our data may complement the information given by the standard techniques used to monitor the PLT activity (aggregometry and flow cytometry).
Materials and methods
Selection of patients
The study included 36 patients with CLL from the Hematology Department of Colentina Clinical Hospital and 16 healthy volunteers as controls.
Inclusion criteria were:
1) Male or female > 18 years of age.
2) Diagnosis of B-cell CLL, established according to International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria.
3) CLL patients treated with single-agent Ibrutinib must be first-line (patients ≥ 65 years old or with presence of 17p/TP53 mutation at FISH analyses) or relapsed/refractory (one or more prior therapies) CLL/SLL treatment settings. Patients must have accepted IWCLL criteria for initiation of therapy.
4) Patients in untreated group must be included in watch and wait group (never treated before enrolment).
5) Karnofsky performance score of ≥ 60.
6) Required baseline laboratory data (within 4 weeks prior to enrolment).
Required screening laboratory values:
•Platelets > 100×103/mL
•Hemoglobin > 10.0 g/dL
•Renal creatinine clearance > 40 ml/min
•Pregnancy-HCGc negative for women in fertile period
•CRP (reactive protein C) < 1.5 N
•Normal serum glucose, cholesterol, triglycerides and lipids
7) Signed informed consent indicates the acception of the subject regarding aspects of participation to the study.
Exclusion criteria were:
1) Known histological transformation from CLL to an aggressive lymphoma (Richter transformation).
2) History of a non-CLL active malignancy (solid tumor or myeloproliferative neoplasms, myelodysplastic syndromes) with ongoing treatment or less than 6 months after chemotherapy.
3) Prior or ongoing clinically significant illness, medical condition, surgical history, physical finding, or laboratory abnormality that could adversely affect the assessment of study results (dyslipidemia, diabetes mellitus, arterial hypertension stroke or myocardial infarction or arterial/vein thrombosis or pulmonary embolism).
4) Evidence of ongoing systemic bacterial, fungal, or viral infection at the time of enrolment in the study.
5) Ongoing alcohol or drug addiction.
All participants have given informed consent. The procedures used in the study were approved by the Hospital Ethics Committee. The diagnosis of CLL was established using the criteria proposed by the consensus guidelines of the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) [13]. CLL patients were divided into two subgroups based on the treatment received: treated with Ibrutinib (26 patients) and untreated (10 patients). The patients treated with Ibrutinib were receiving the drug for a maximum period of 18 months 420 mg daily, while untreated patients were on “watch and wait” status, never receiving any CLL medication. The patients were not presenting any hemorrhagic complications prior enrolment in the study. The control group had the median age of 51 years (min 29, max 74) and included 37.50% males.
The clinical and hematological characteristics of the patients are presented in Table 1.
Table 1.
Clinical and hematological characteristics of the patients recruited in the study
| Parameter | Classification | Subgroup of patients by treatment | p value | |
|---|---|---|---|---|
|
| ||||
| Untreated | Treated with Ibrutinib | |||
| Patients | % (number) | 27.8 (10) | 72.2 (26) | 0.01 |
| Age (years) | Median (min; max) | 66.5 (44; 82) | 69.0 (52; 91) | 0.91 |
| Sex | Male | 7 (19.44%) | 17 (47.22%) | 0.79 |
| Female | 3 (8.33%) | 9 (25%) | ||
| Stage (RAI Classification) | 1 | 19.44% | 2.77% | 0.003 |
| 2 | 8.33% | 47.22% | ||
| 3 | 2.77% | 8.33% | ||
| 4 | 0% | 11.1% | ||
| Cytogenetic mutations (TP53, 17p) | Present | 2.77% | 16.66% | 0.34 |
| No Present | 8.33% | 8.33% | ||
| Not determined | 16.66% | 47.22% | ||
| WBC count (×1000/uL) | Median (min; max) | 22.44 (5.35; 88.5) | 12.23 (3.3; 149.96) | 0.21 |
| Hemoglobin (g/dL) | Median (min; max) | 13.2 (10.5; 15) | 12.85 (6.3; 16.8) | 0.52 |
| Hematocrit (%) | Median (min; max) | 40.35 (33; 45,8) | 39.7 (21; 51) | 0.76 |
| Platelet Count (×1000/uL) | Median (min; max) | 174.5 (61; 273) | 164.00 (70; 337) | 0.75 |
| Lymphocytes count (×1000/uL) | Median (min; max) | 18.93 (1.1; 85) | 6.05 (0.83; 142.6) | 0.12 |
WBC = white blood cells. P values represent the significance of the differences between untreated and treated patients.
Preparation of PLTs suspensions
The protocol for the preparation of PLTs suspensions was designed to obtain non-activated platelets. The PLTs are very sensitive: they activate and aggregate easily under mechanical forces (e.g., fast pipetting, vigorous shaking or long time centrifugation), in presence of calcium and on contact with glass; therefore all these conditions have been avoided during the PLTs preparation. Blood sampling was performed in the morning (after overnight fasting). Blood was collected in sodium citrate tubes by venipuncture. Platelet rich plasma (PRP) was obtained by gentle centrifugation at 130× g for 10 minutes, at room temperature. PLTs were separated from PRP by gel-filtration chromatography on a Sepharose CL-2B column (Sigma, 2B300) in Tyrode buffer without calcium, with glucose (134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM Na2HPO4, 1 mM MgCl2, 10 mM HEPES, 5 mM D-glucose) [14]. This chromatographic separation of PLTs has the advantage to avoid mechanical activation of platelets which may occur when PLTs are pelleted by standard centrifugation. Moreover, 0.2 U/mL Apyrase (Apyrase grade I from potato, Sigma, A6132-5KU) were added to prevent PLTs aggregation during the manipulation. The optical density (OD) of PLTs suspension measured at 355 nm was adjusted to 0.8 ± 0.04 with Tyrode buffer. PLTs suspension was then split in equivalent samples for the following measurements: resting membrane potential (RMP), membrane fluidity and production of reactive oxygen species (ROS).
Membrane fluidity measurements
PLTs membrane fluidity was assessed by fluorescence anisotropy using N,N,N-Trimethyl-4-(6-phenyl-1,3,5-hexatrien-1-yl) phenylammonium p-toluenesulfonate (TMA-DPH) (Sigma, 43060) (10 mM stock solution in dimethyl formamide kept at -20°C). TMA-DPH is a rigid molecule with an electronic transition moment very well defined against the geometry of the molecule. The fluorophore molecules insert rapidly into the lipid bilayer (1 min) and remain trapped within the membrane for about 25 min [15]. When the sample is illuminated with polarized light, only those molecules that have the transition vector aligned parallel to the polarized excitation electric vector have the highest probability of excitation. The emitted light will be less polarized, depending on the movement possibilities of the fluorophore within the membrane during the fluorophore excited state lifetime. The degree of depolarization is quantified by a parameter called fluorescence anisotropy (r), defined as (Equation 1):
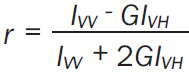 |
where G represents a correction factor (G = IHV/IHH) and I stands for the fluorescence intensities with excitation and emission polarizers oriented as specified by the indices (V for vertical, H for horizontal). The more fluid the lipid bilayer, the lower the anisotropy parameter r values.
TMA-DPH was added to the PLTs suspension (final concentration of 50 μM), incubated for 2 min at 37°C and fluorescence anisotropy (λ ex = 355 nm, λ em = 425 nm) was measured.
Resting membrane potential measurements
RMP was assessed by fluorescence, using the probe 3,3 Dipropylthiadicarbocyanine 5 (DiSC3-5) (Invitrogen, D306), 0.5 mM stock solution in dimethyl formamide, kept at -20°C [16]. The cationic dye DiSC3-5 is membrane permeant and distributes between cytoplasm and extracellular medium according to the electrochemical gradient: the more negative the interior, the higher the cytoplasmic concentration of the fluorophore. A specific feature of DISC3-5 is that its fluorescence quantum yield is much smaller inside the cell than outside. Thus, as the probe accumulates in the cell, the fluorescence intensity drops, reaching a plateau at a level characterizing the membrane potential. To transform the fluorescence intensity in units of mV, the membrane potential is clamped to the potassium electrochemical gradient by adding the specific potassium transporter Valinomycin. While K+ is leaking out of the cell, the membrane hyperpolarizes, more DiSC3-5 enters the cell and its fluorescence decreases to a new plateau. Then, as known concentrations of K+ are added outside the cell, the fluorescence intensities are reaching new plateau levels. The membrane potential is computed (for each plateau) using the Nernst equation (Equation 2):
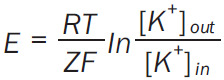 |
where E = PLT resting membrane potential (mV), R = universal gas constant (8.314 J.mol-1.K-1), T = absolute temperature (K), z = potassium ion relative electric charge (+1), F = Faraday constant (96485 C.mol-1), [K+]out = added (known) potassium concentration outside the cell, [K+]in = basal cytoplasmic concentration of potassium (135 mM). The computed membrane potential is represented as a function of the fluorescence intensities and fitted with a linear equation that allows the interpolation of fluorescence intensity corresponding to the first plateau.
DiSC3-5 at final concentration 2 μM was added to the PLTs suspension incubated at 37°C until reaching the fluorescence signal plateau (λ ex = 620 nm, λ em = 670 nm). Valinomycin (Sigma, V0627) (50 mM stock solution in ethanol kept at -20°C) was then added to the PLTs suspension (final concentration of 50 μM) and a new fluorescence plateau was obtained. Aliquots of 2 M KCl (Sigma) were added sequentially after each new plateau was obtained.
ROS production measurements
ROS levels were assessed by fluorescence using 2’,7’-Dichlorofluorescein diacetate (H2DCFDA) (Sigma Aldrich, 35845) (0.5 mM stock solution in methanol kept at -20°C). H2DCFDA is a membrane permeant, nonfluorescent compound which is deacetylated by cellular esterases after entering the cell, oxidized by ROS to a highly fluorescent compound (DCF). The fluorescence intensity is thus proportional to the amount of ROS produced by the cell.
H2DCFDA was added to the PLTs suspension (final concentration of 5 μM) and fluorescence intensity (λ ex = 488 nm, λ em = 523 nm) was measured continuously for 15 minutes (at 37°C). ROS production was assessed by normalizing the area under the fluorescence intensity curve to the OD of the sample.
All fluorescence measurements were done using a Horiba Jobin Yvon FluoroLog 3 spectrofluorometer (with two emission channels) s(USA).
Statistical analysis
Statistical analysis of data was performed using Mann-Whitney rank sum test (two variables) or Kruskal-Wallis test (more than two variables) for variables with abnormal distribution. The statistical significance was set for P = 0.05. For all statistical computations, FluorEssence™ 2.1 (developed by Horiba under OriginProTM) was used.
Results
Fluorescence anisotropy of PLTs membrane
The fluorescence anisotropy of PLTs membrane was found to be: median value for controls r = 0.096 (min 0.077, max 0.141), median value for untreated CLL patients r = 0.084 (min 0.069, max 0.141) while for patients treated with Ibrutinib, median r = 0.073 (min 0.040, max 0.121) (Figure 1). CLL patients treated with Ibrutinib have statistically higher fluidity of PLTs membrane (lower fluorescence anisotropy) as compared to CLL untreated patients (P = 0.010). There is a tendency (although not statistically significant) of an increased membrane fluidity in case of untreated CLL patients as compared to controls.
Figure 1.
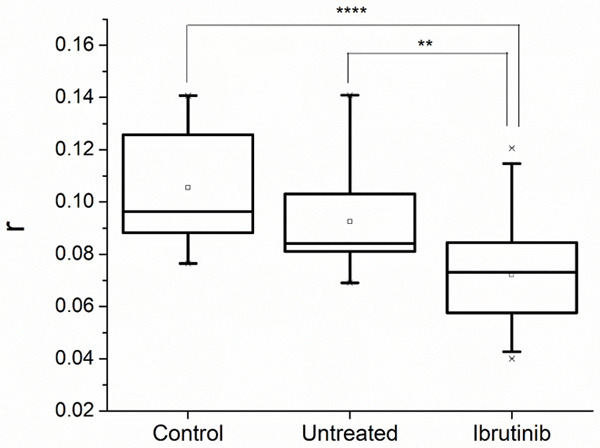
Fluorescence anisotropy parameter (r) measured in platelets suspensions from controls, and chronic lymphocytic leukemia patients, untreated and treated with Ibrutinib (Statistical significance ** = P < 0.01, **** = P < 0.0001).
Resting membrane potential of PLTs membrane
An example of an experimental curve and the corresponding RMP calibration using the Nernst equation for potassium clamped potential are presented in Figure 2.
Figure 2.
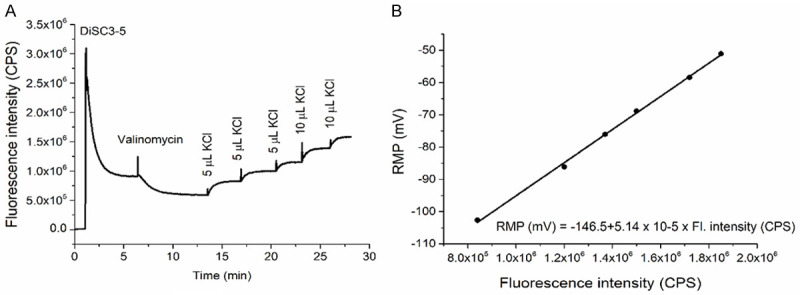
Measurement of resting membrane potential (RMP) in millivolts (mV). The experimental curve of the 3,3 Dipropylthiadicarbocyanine 5 (DiSC3-5) fluorescence intensity is shown in counts pers second (CPS) (A). The RMP computed on the base of Nernst equation is represented as a function of fluorescence intensity (B).
The median value of the RMP for controls was found to be -56 mV (min -37 mV, max -68 mV). The untreated CLL group presented a RMP median value of -58 mV (min -39 mV, max -72 mV), without statistical difference as compared to controls. The RMP for Ibrutinib treated CLL patients was -43 mV (min -21 mV, max -58 mV), with a statistically significant difference when compared to any of the previous two groups (P < 0.05) (Figure 3).
Figure 3.
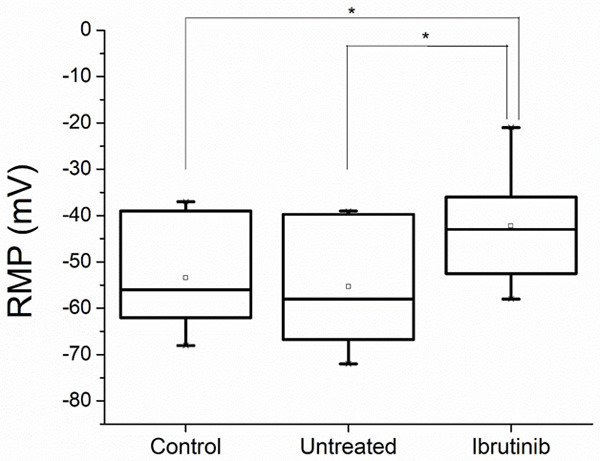
Resting membrane potential (RMP) in millivolts (mV) measured on platelets of controls and chronic lymphocytic leukemia patients untreated and treated with Ibrutinib (Statistical significance * = P < 0.05).
Evaluation of ROS production by PLTs
An example of an experimental curve of ROS production is presented in Figure 4.
Figure 4.
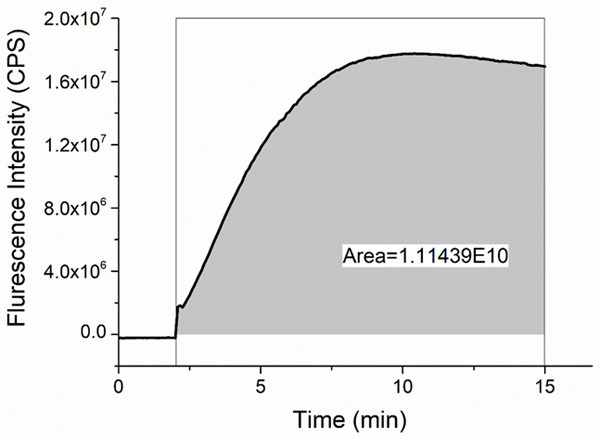
Measurement of reactive oxygen species production. Experimental curve of the Dichlorofluorescein fluorescence intensity (in counts per second - CPS) is presented and the area under the curve is computed.
Higher levels of ROS production were observed for PLTs from patients treated with Ibrutinib: median value 6.12×1010 (min 4.17×1010, max 2.07×1011) compared to untreated patients: median value 4.65×1010 (min 1.08×107, max 5.85×1010), with a statistically significant difference between these groups (P = 0.014). In case of untreated CLL patients, there is a slightly lower ROS production as compared to controls (median value 5.52×1010, min 3.94×1010, max 1.58×1011) (Figure 5).
Figure 5.
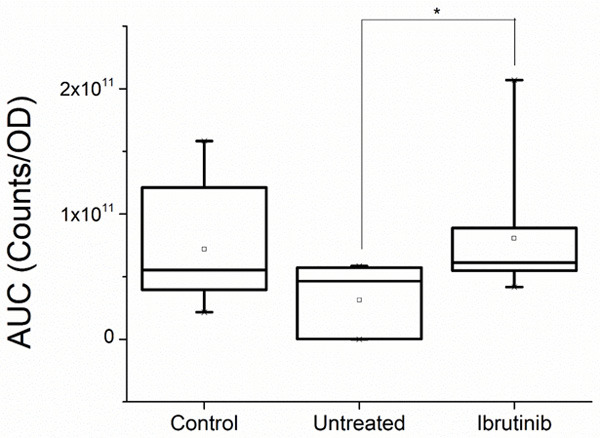
Reactive oxygen species (ROS) production measured on platelets of controls and chronic lymphocytic leukemia patients untreated and treated with Ibrutinib. The ROS production is proportional to the area under the curve (AUC) measured in counts of fluorescence per optical density (OD) of the sample, as shown in Figure 4 (Statistical significance * = P < 0.05).
Discussion
Membrane fluidity as seen by fluorescence anisotropy is a membrane parameter which quantifies the wobbling movements of the lipid tails within the membrane bilayer. Alteration of these movements was shown to be correlated to the expression and activity of various PLTs membrane receptors [16-19].
There are few reports regarding the complex correlation of the membrane fluidity and the receptor expression on lymphocytes in CLL patients, having an impact on the defective transmembrane signaling [20-22]. To our knowledge, no reports are published regarding the relationship between platelet membrane fluidity and PLTs functions in CLL.
Kozubski et al. reported that Alzheimer patients present platelets with significantly increased membrane fluidity, when measured by Electron Spin Resonance technique. In the same time the PLTs reactivity was significantly decreased, since a reduced expression of GPIb in the resting and activated platelets was observed [17].
Vinik et al. found a decreased membrane fluidity in PLTs of diabetic patients which is associated with enhanced GP receptor binding of agonists [23].
In our study, we found that Ibrutinib treatment determined a statistically significant increase in the membrane fluidity of PLTs compared to untreated patients (Figure 3). Compared to controls, the untreated patients presented a slight increase of membrane fluidity, although not statistically significant.
Even though we cannot propose a molecular mechanism, we may speculate that the membrane fluidity is modulating the expression of PLTs membrane receptors.
Moreover, it is known that Ibrutinib is modifying the expression of receptors GPVI and GPIbα [2,12]. In case of our patients treated with Ibrutinib, the increased membrane fluidity may down regulate the expression of receptors, as reported by above mentioned articles.
According to Friedhoff and Sonenberg [16], the membrane potential of normal human platelets was estimated to be -52 to -60 mV. In our study the median value of RMP of controls was -56 mV in good agreement with the data reported in literature. In case of CLL patients we obtained a median value of RMPs -58 mV (min -39 mV, max -72 mV) without statistical difference compared to controls. The administration of Ibrutinib to these CLL patients was found to be associated to a depolarization of the PLTs membrane: RMP median value of -43 mV (min -21 mV, max -58 mV) with a statistically significant difference when compared to controls and untreated patients (P < 0.05).
According to Smith et al., there are two types of potassium channels in the PLTs membranes: calcium-gated and voltage-gated channels. In the early stages of platelet activation, the PLTs potential is reported to hyperpolarize to approximately -80 mV, but later, it may depolarize due to an opening of various channels [24].
We do not know if the Ibrutinib is the cause of the PLTs depolarization found by our study in the treated patients, but we may speculate that it interferes with the intracellular calcium flux. In vitro studies on Jurkat cells, have shown that the pre-treatment with Ibrutinib diminishes the intracellular calcium flux in response to T cell receptor stimulation [25]. In a study using platelets from patients under Ibrutinib treatment, Bye and collaborators reported that Ibrutinib inhibits the Ca2+ elevation and platelets aggregation evoked by collagen-related peptide (CRP)-XL and collagen [4]. Moreover, it is also known that under Ibrutinib, the patients are presenting non active PLTs [12]. This status may be attributed to the shading process induced by Ibrutinib on various membrane receptors. For instance, Ibrutinib produces a time- and dose-dependent shedding of GPIb-IX complex and GPIIbIIIa, but not of GPVI and GPV [26]. Bye and collaborators [27] confirm the effect of Ibrutinib on the integrin, but they are also evidencing an inhibition effect of Ibrutinib on GPVI signaling, resulting in formation of unstable thrombi. They are attributing the inhibition effect of Ibrutinib to the clot retraction and signaling evoked by platelet adhesion to immobilized fibrinogen.
The production of ROS was found to be higher in CLL patients treated with Ibrutinib when compared to untreated ones (P = 0.014). All CLL patients treated with Ibrutinib included in this study were in refractory status (second or third line of therapy) or newly diagnosed (first line with associated high cytogenetic risk-TP53 or 17p mutations present). The difference in ROS productions between treated and untreated patients could be explained by these characteristics of the Ibrutinib group. Untreated patients were belonging to a low risk CLL group (no TP53 or 17p mutations or IGHV unmutated). Similar results were recently reported by D’Arena and collaborators [28]. Their CLL patients with advanced stages of disease or characterized by markers of disease aggressiveness were presenting an increased ROS production and decreased antioxidant defense mechanisms.
Oxidative stress was also considered as a prognostic factor of unfavorable disease evolution in some other reports in which a high ROS production was found in CLL patients [29]. Moreover, the mitochondrial metabolism and ROS production was shown to be elevated in CLL [30]. High level of ROS production is due to the mitochondrial respiratory chain alterations of cells in CLL: defective oxidative phosphorylation or an enhanced mitochondrial biogenesis. Another mechanism of increased ROS production is related to the modified activity of membrane-bound NADPH-oxidase (NOX) complex [31]. Presence of high levels of ROS induces genetic instability, development of drug resistance and less harness of cell signaling [31]. In case of pathological conditions like those of heart failure patients, Mondal and collaborators proved that high level of ROS production is associated with platelet apoptosis and depolarized platelets and induces bleeding complication [32]. Moreover, the integrin α2bβ3, GPIbα and GPVI shedding and platelet dysfunction have been associated with high ROS production by [33,34].
In conclusion, platelets from CLL patients treated with Ibrutinib have higher membrane fluidity, lower resting membrane potential and higher level of ROS production compared to the untreated CLL patients. These patients are also presenting higher membrane fluidity and lower resting membrane potential compared to healthy volunteers. Ongoing experiments are intended to correlate these preliminary data with information about platelets activity as resulting from standard of care techniques in the attempt to find mechanistic explanation for our observations.
Acknowledgements
This work was partially supported by UEFISCDI grant PED141/2017. We thank Professor Eugenia Kovacs for critical reviewing of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Georgantopoulos P, Yang H, Norris LB, Bennett CL. Major hemorrhage in chronic lymphocytic leukemia patients in the US veterans health administration system in the pre-ibrutinib era: incidence and risk factors. Cancer Med. 2019;8:2233–2240. doi: 10.1002/cam4.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shatzel JJ, Olson SR, Tao DL, McCarty OJT, Danilov AV, DeLoughery TG. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J Thromb Haemost. 2017;15:835–847. doi: 10.1111/jth.13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luu S, Gardiner EE, Andrews RK. Bone marrow defects and platelet function: a focus on MDS and CLL. Cancers (Basel) 2018;10:147. doi: 10.3390/cancers10050147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost. 2016;14:918–930. doi: 10.1111/jth.13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mock J, Kunk PR, Palkimas S, Sen JM, Devitt M, Horton B, Portell CA, Williams ME, Maitland H. Risk of major bleeding with Ibrutinib. Clin Lymphoma Myeloma Leuk. 2018;18:755–761. doi: 10.1016/j.clml.2018.07.287. [DOI] [PubMed] [Google Scholar]
- 6.Seiter K, Stiefel MF, Barrientos J, Shaikh A, Ahmed N, Baskind P, Liu D. Successful treatment of ibrutinib-associated central nervous system hemorrhage with platelet transfusion support. Stem Cell Investig. 2016;3:27. doi: 10.21037/sci.2016.06.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberelli MA, Innocenti I, Sica S, Laurenti L, De Candia E. PO-54 - clinical and laboratory characterization of platelet dysfunction caused by ibrutinib treatment in patients with chronic lymphocytic leukemia. Thromb Res. 2016;140(Suppl 1):S196. doi: 10.1016/S0049-3848(16)30187-6. [DOI] [PubMed] [Google Scholar]
- 8.Bye AP, Unsworth AJ, Desborough MJ, Hildyard CAT, Appleby N, Bruce D, Kriek N, Nock SH, Sage T, Hughes CE, Gibbins JM. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1:2610–2623. doi: 10.1182/bloodadvances.2017011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rigg RA, Aslan JE, Healy LD, Wallisch M, Thierheimer ML, Loren CP, Pang J, Hinds MT, Gruber A, McCarty OJ. Oral administration of Bruton’s tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol. 2016;310:C373–80. doi: 10.1152/ajpcell.00325.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golovastov VV, Krasovskaya LA, Anissimova EV, Shchukin VN, Badyakina AO, Nesmeyanova MA. Unbalanced phospholipid composition of escherichia coli membranes affects PPHO promoter activity. Mol Biol. 2005;39:260–268. [PubMed] [Google Scholar]
- 11.Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 12.Aguilar C. Ibrutinib-related bleeding: pathogenesis, clinical implications and management. Blood Coagul Fibrinolysis. 2018;29:481–487. doi: 10.1097/MBC.0000000000000749. [DOI] [PubMed] [Google Scholar]
- 13.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating M, Montserrat E, Chiorazzi N, Stilgenbauer S, Rai KR, Byrd JC, Eichhorst B, O’Brien S, Robak T, Seymour JF, Kipps TJ. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–2760. doi: 10.1182/blood-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- 14.Aurbach K, Spindler M, Haining EJ, Bender M, Pleines I. Blood collection, platelet isolation and measurement of platelet count and size in mice-a practical guide. Platelets. 2019;30:698–707. doi: 10.1080/09537104.2018.1528345. [DOI] [PubMed] [Google Scholar]
- 15.Shinitzky M, Barenholz Y. Fluidity parameters of lipid regions determined by fluorescence polarization. Biochim Biophys Acta. 1978;515:367–394. doi: 10.1016/0304-4157(78)90010-2. [DOI] [PubMed] [Google Scholar]
- 16.Friedhoff LT, Sonenberg M. The membrane potential of human platelets. Blood. 1983;61:180–185. [PubMed] [Google Scholar]
- 17.Kozubski W, Swiderek M, Kloszewska I, Gwozdzinski K, Watala C. Blood platelet membrane fluidity and the exposition of membrane protein receptors in Alzheimer disease (AD) patients--preliminary study. Alzheimer Dis Assoc Disord. 2002;16:52–54. doi: 10.1097/00002093-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Watala C, Golański J, Walkowiak B, Baj Z, Pietrucha T, Tchórzewski H, Bodalski J, Ciemiewski CS. Does reduced membrane lipid fluidity underlie the altered thrombin-induced expression of integrin αIIbβ3 and PADGEM-140 in membranes of platelets from diabetic juveniles? Platelets. 1996;7:173–180. doi: 10.3109/09537109609023577. [DOI] [PubMed] [Google Scholar]
- 19.Watala C, Golański J, Boncler MA, Pietrucha T, Gwoździński K. Membrane lipid fluidity of blood platelets: a common denominator that underlies the opposing actions of various agents that affect platelet activation in whole blood. Platelets. 1998;9:315–327. doi: 10.1080/09537109876564. [DOI] [PubMed] [Google Scholar]
- 20.Daefler S, Krueger GR. Lack of dynamic lipid changes after binding of interleukin 2 in chronic lymphatic leukemia lymphocytes indicates defective transmembrane signalling. Anticancer Res. 1989;9:743–748. [PubMed] [Google Scholar]
- 21.Daefler S, Krueger GR. Expression of proliferation and differentiation antigens in response to modulation of membrane fluidity in chronic lymphocytic leukemia lymphocytes. Anticancer Res. 1989;9:501–506. [PubMed] [Google Scholar]
- 22.Daefler S, Krueger GR, Mödder B, Deliconstantinos G. Cell membrane fluidity in chronic lymphocytic leukemia (CLL) lymphocytes and its relation to membrane receptor expression. J Exp Pathol. 1987;3:147–154. [PubMed] [Google Scholar]
- 23.Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care. 2001;24:1476–1485. doi: 10.2337/diacare.24.8.1476. [DOI] [PubMed] [Google Scholar]
- 24.Mahaut-Smith MP. The unique contribution of ion channels to platelet and megakaryocyte function. J Thromb Haemost. 2012;10:1722–1732. doi: 10.1111/j.1538-7836.2012.04837.x. [DOI] [PubMed] [Google Scholar]
- 25.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, Stefanovski MR, Chappell DL, Frissora FW, Smith LL, Smucker KA, Flynn JM, Jones JA, Andritsos LA, Maddocks K, Lehman AM, Furman R, Sharman J, Mishra A, Caligiuri MA, Satoskar AR, Buggy JJ, Muthusamy N, Johnson AJ, Byrd JC. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobie G, Kuriri FA, Omar MMA, Alanazi F, Gazwani AM, Tang CPS, Sze DM, Handunnetti SM, Tam C, Jackson DE. Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin αIIbβ3 in mice and humans. Blood Adv. 2019;3:4298–4311. doi: 10.1182/bloodadvances.2019000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib inhibits platelet integrin αIIbβ3 outside-in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol. 2015;35:2326–2335. doi: 10.1161/ATVBAHA.115.306130. [DOI] [PubMed] [Google Scholar]
- 28.D’Arena G, Vitale C, Perbellini O, Coscia M, La Rocca F, Ruggieri V, Visco C, Di Minno NMD, Innocenti I, Pizza V, Deaglio S, Di Minno G, Giudice A, Calapai G, Musto P, Laurenti L, Iorio EL. Prognostic relevance of oxidative stress measurement in chronic lymphocytic leukaemia. Eur J Haematol. 2017;99:306–314. doi: 10.1111/ejh.12918. [DOI] [PubMed] [Google Scholar]
- 29.D’Arena G, Seneca E, Migliaccio I, De Feo V, Giudice A, La Rocca F, Capunzo M, Calapai G, Festa A, Caraglia M, Musto P, Iorio EL, Ruggieri V. Oxidative stress in chronic lymphocytic leukemia: still a matter of debate. Leuk Lymphoma. 2019;60:867–875. doi: 10.1080/10428194.2018.1509317. [DOI] [PubMed] [Google Scholar]
- 30.Roy Chowdhury S, Banerji V. Targeting mitochondrial bioenergetics as a therapeutic strategy for chronic lymphocytic leukemia. Oxid Med Cell Longev. 2018;2018:2426712. doi: 10.1155/2018/2426712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123:2663–2672. doi: 10.1182/blood-2013-10-532200. [DOI] [PubMed] [Google Scholar]
- 32.Mondal NK, Sorensen EN, Hiivala NJ, Feller ED, Pham SM, Griffith BP, Wu ZJ. Intraplatelet reactive oxygen species, mitochondrial damage and platelet apoptosis augment non-surgical bleeding in heart failure patients supported by continuous-flow left ventricular assist device. Platelets. 2015;26:536–544. doi: 10.3109/09537104.2014.948840. [DOI] [PubMed] [Google Scholar]
- 33.Mondal NK, Chen Z, Trivedi JR, Sorensen EN, Pham SM, Slaughter MS, Griffith BP, Wu ZJ. Oxidative stress induced modulation of platelet integrin α2bβ3 expression and shedding may predict the risk of major bleeding in heart failure patients supported by continuous flow left ventricular assist devices. Thromb Res. 2017;158:140–148. doi: 10.1016/j.thromres.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mondal NK, Chen Z, Trivedi JR, Sorensen EN, Pham SM, Slaughter MS, Griffith BP, Wu ZJ. Association of oxidative stress and platelet receptor glycoprotein GPIbα and GPVI shedding during nonsurgical bleeding in heart failure patients with continuous-flow left ventricular assist device support. ASAIO J. 2018;64:462–471. doi: 10.1097/MAT.0000000000000680. [DOI] [PMC free article] [PubMed] [Google Scholar]