

Abstract
Nanophthalmos and posterior microphthalmos are ocular abnormalities in which both eyes are abnormally small, and typically associated with extreme hyperopia. We recruited 40 individuals from 13 kindreds with nanophthalmos or posterior microphthalmos, with 12 probands subjected to exome sequencing. Nine probands (69.2%) were assigned a genetic diagnosis, with variants in MYRF, TMEM98, MFRP, and PRSS56. Two of four PRSS56 families harboured the previously described c.1066dupC variant implicated in over half of all reported PRSS56 kindreds, with different surrounding haplotypes in each family suggesting a mutational hotspot. Individuals with a genetic diagnosis had shorter mean axial lengths and higher hyperopia than those without, with recessive forms associated with the most extreme phenotypes. These findings detail the genetic architecture of nanophthalmos and posterior microphthalmos in a cohort of predominantly European ancestry, their relative clinical phenotypes, and highlight the shared genetic architecture of rare and common disorders of refractive error.
Keywords: microphthalmia, nanophthalmos, posterior microphthalmos, axial length, PRSS56, MFRP, TMEM98, MYRF
Introduction
Clear vision requires precise regulation of ocular growth, such that the axial length of the eye matches the optical focal plane created by the cornea and lens. A discrepancy between the two results in refractive error, which is the leading cause of visual impairment and the second leading cause of blindness worldwide.1
Important insight into the regulation of ocular axial length has come from the study of microphthalmia. Nanophthalmos and posterior microphthalmos are subtypes of microphthalmia without other ocular malformations, associated with symmetrically reduced axial length and high hyperopia. Posterior microphthalmos is characterised by a shorter posterior segment of the eye, with the anterior segment typically of normal length.2 Conversely, nanophthalmos is characterised by shorter posterior and anterior segments, with a predisposition to primary angle-closure glaucoma.2 Posterior microphthalmos and nanophthalmos can be allelic3, suggesting both conditions may represent a continuum of the same phenotypic spectrum.4 Most cases are explained by biallelic variants in MFRP3,5 and PRSS566–8, with rare examples of monoallelic variants in TMEM989,10 and MYRF11. PRSS56 and TMEM98 variants have since been implicated in multiple independent genome-wide association studies of myopia.12
Here we assembled a cohort of 13 nanophthalmos or posterior microphthalmos kindreds, describe their underlying genetic cause, and detail their relative clinical phenotypes.
Material and Methods
Subjects
Subjects referred with a diagnosis of nanophthalmos or posterior microphthalmos were included if presenting with bilateral and symmetrically small eyes, with axial lengths <20mm in both eyes (a threshold previously shown to account for individuals <3SD from the population mean, and proposed as a definition for nanophthalmos).13 Of the 13 families, 4 were sporadic, 3 suggestive of autosomal dominant inheritance, and 6 suggestive of autosomal recessive inheritance. Not all subjects had anterior chamber depth and/or fundus imaging, thus a distinction between nanophthalmos and posterior microphthalmos was not routinely made. Exclusion criteria included anterior segment coloboma, or multiple non-ophthalmic syndromic features. Ethics approval was granted by the Southern Adelaide Clinical Human Research Ethics Committee.
Sequencing
Exome sequencing was performed as previously described.14 Briefly, genomic DNA was extracted from blood or saliva, subjected to Agilent SureSelect exome capture, and paired-end libraries sequenced on an Illumina HiSeq. Alignment and variant calling were performed according to GATK best practices, with variant annotation using VEP. RNAseq data was generated as previously described.15
Results
Forty individuals diagnosed with nanophthalmos or posterior microphthalmos were recruited from 13 families, with 69.2% of self-reported European ancestry (Figure 1A, Supplementary Table 1). Exome sequencing was performed on one or more members of each family, with the exception of NNO03, where a causative variant was identified by capillary sequencing. When including two previously reported pedigrees10,16, a suspected genetic cause was identified in 9/13 kindreds (69.2%), in genes including MYRF (1), TMEM98 (1), MFRP (3), and PRSS56 (4) (Figure 1B–E). All 11 unique variants were rare or absent in gnomAD, and classified as pathogenic or likely pathogenic by ACMG criteria in 10/11 cases, and recessive variants confirmed to be in trans by segregation testing (Supplementary Table 2). Of the four families without a candidate genetic variant(s), two were sporadic, one suggestive of recessive inheritance, and another suggestive of dominant inheritance: none were found to be segregating rare coding variants in MYRF, TMEM98, MFRP, or PRSS56.
Figure 1: Genetic characterisation of a nanophthalmos and posterior microphthalmos cohort.
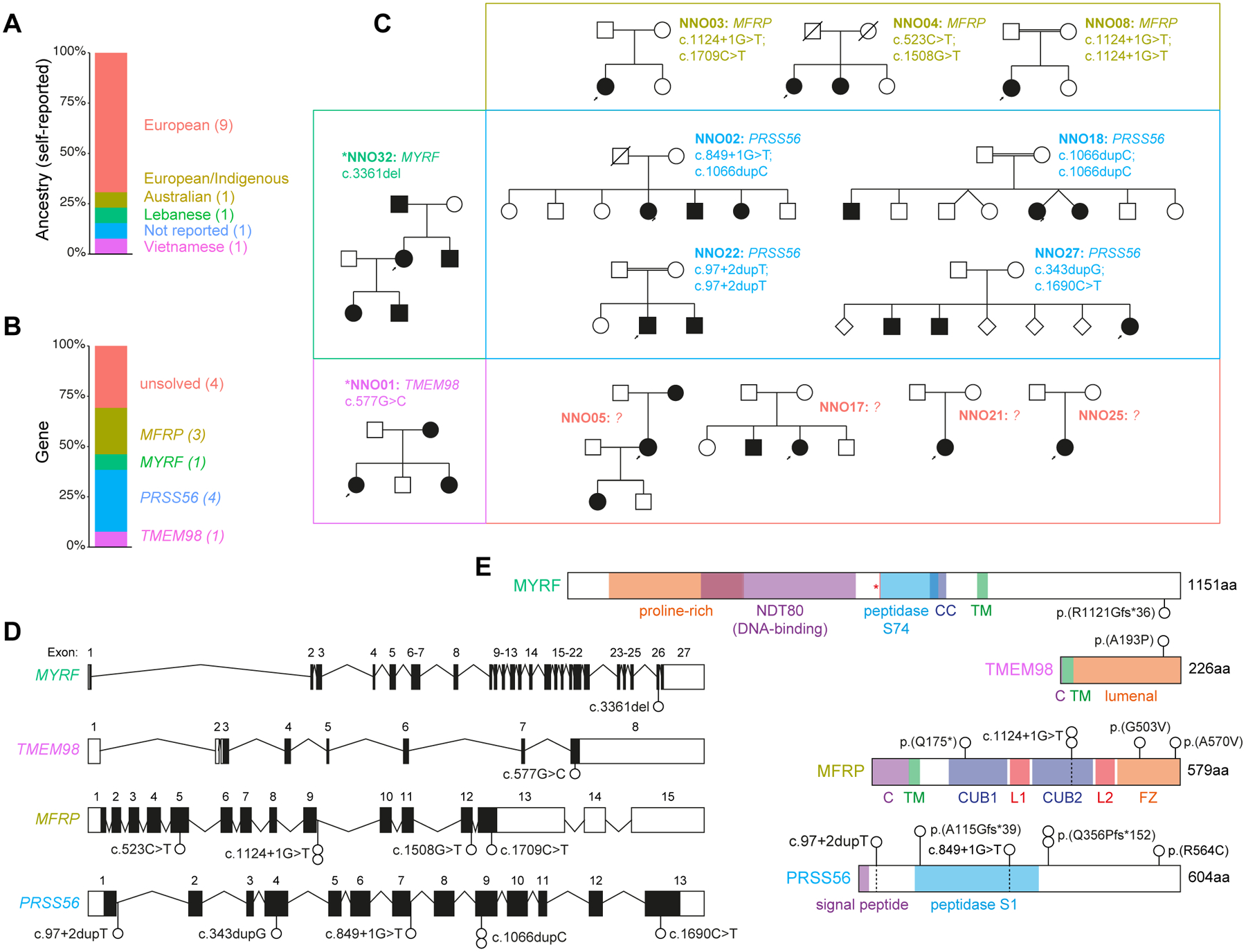
(A) Self-reported ancestry of the 13 probands.
(B) Proportion of probands with suspected pathogenic variants in the indicated genes.
(C) Pedigrees grouped by affected gene. Asterisks indicate previously reported pedigrees.
(D) Schematic of loci and variants identified.
(E) Protein schematics with variants identified. C, cytoplasmic domain; TM, transmembrane domain; CUB, Complement C1r/C1s, Uegf, Bmp1 domain; L, LDL-receptor class A domain; FZ, frizzled domain; CC, coiled-coil domain.
Dominant pathogenic frameshift and missense variants were identified in MYRF (p.(Arg1121GlyfsTer36)) and TMEM98 (p.(Ala193Pro)) respectively, both of which were previously reported by our group.10,16 Recessive pathogenic variants were more common, identified across seven families in MFRP and PRSS56. We identified four predicted pathogenic variants in MFRP across three probands, including two with no previous disease association (p.(Ala570Val) and p.(Gly503Val)). All four probands with predicted pathogenic variants in PRSS56 had rare compound heterozygous or homozygous variants, including previously unreported essential splice (c.849+1G>T and c.97+2dupT), frameshift (p.(Ala115GlyfsTer39)), and missense variants (p.(Arg564Cys)), as well as two instances of the previously reported c.1066dupC frameshift variant. The PRSS56 c.1066dupC variant (also reported as c.1059_1066insC) was initially reported in multiple Tunisian families with posterior microphthalmos sharing a common ancestor ~1,850 years ago (Figure 2A).6,8,17 Another nine cases explained by the c.1066dupC variant were subsequently reported in a Saudi Arabian cohort of microphthalmia and nanophthalmos/posterior microphthalmos, accounting for 69% (9/13) of all cases with PRSS56 variants in this report4, as well as in Egyptian and Lebanese patients.18
Figure 2: Origins of PRSS56 c.1066dupC in a mutational hotspot.
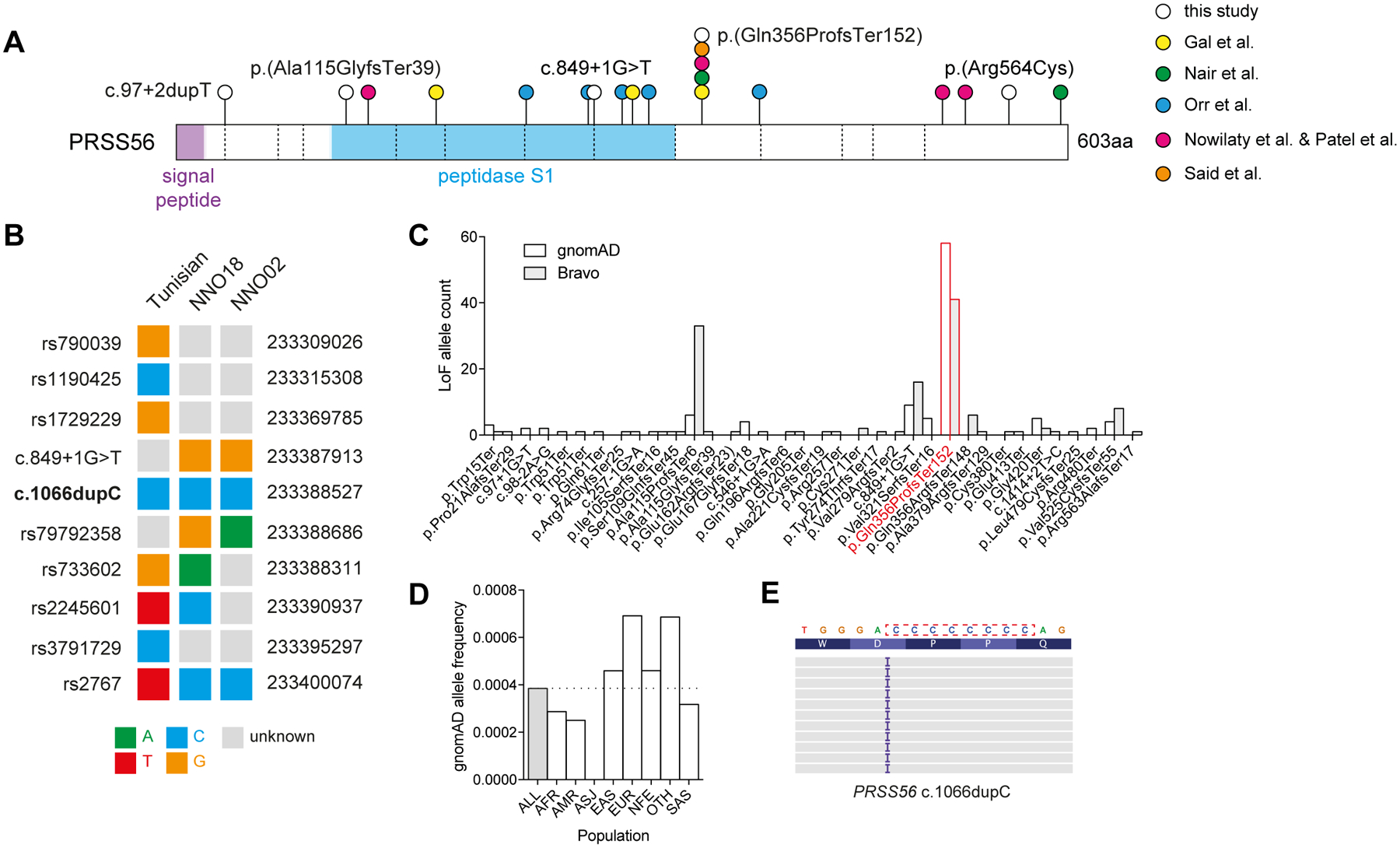
(A) PRSS56 protein schematic with variants identified in this study (white circles), or previous studies (coloured circles). Dashed vertical lines indicate exon boundaries. (B) Haplotype structure surrounding the PRSS56 c.1066dupC variant in a previously reported Tunisian founder, and two unrelated probands. (C) Frequency of PRSS56 loss-of-function variants (nonsense, essential splice, frameshift) reported in gnomAD r2.0.2 and Bravo (TOPMed Freeze5) collections. The c.1066dupC variant is highlighted in red. (D) Frequency of c.1066dupC across multiple ancestries within gnomAD. (E) IGV representation of the left-shifted c.1066dupC variant within a cytosine mononucleotide repeat.
To determine the origin of the c.1066dupC frameshift variant in our Australian kindreds, we compared surrounding haplotypes to the previously described Tunisian founder haplotype (Figure 2B).17 Three phased variants were genotyped in both the NNO18 proband and the Tunisian founder, yet the genotypes in each were divergent, suggesting that the two alleles had arisen independently. Similarly, the NNO02 haplotype was divergent from the Tunisian founder and NNO18 proband, suggesting that all three frameshift variants arose independently on different haplotypes.
c.1066dupC was the most frequent loss-of-function PRSS56 variant in the gnomAD database (Figure 2C), representing over half (58/112) of all reported PRSS56 loss-of-function variants. c.1066dupC variants were distributed across seven superpopulations at low frequencies, consistent with their independent emergence (Figure 2D). PRSS56 c.1066 was also noted to be multiallelic, with insertions and deletions at the same position (Figure 2C), both of which altered the length of an eight-nucleotide cytosine mononucleotide repeat (Figure 2E).
Individuals with a single-gene diagnosis had reduced ocular axial length, with the majority also highly hyperopic (Figure 3A–B, Supplementary Table 3). Mean axial length was shorter in all groups with a genetic diagnosis compared to those without (adjusted P<0.029), with the PRSS56 subset also having shorter mean axial length than the dominant TMEM98 (adjusted P=0.00003) and MYRF (adjusted P=0.00004) subsets. Mean refractive error was also higher in PRSS56 and MFRP groups compared to individuals without a genetic diagnosis (adjusted P<0.006), and when compared to the MYRF group (adjusted P<0.021). Chorioretinal folds, and/or choroidal effusions or retinal detachments were documented in 2/4 PRSS56 pedigrees and 1/3 MFRP pedigrees, with extra-ocular features (in the form of dextrocardia or congenital diaphragmatic hernia) only observed in 3/5 members of the MYRF pedigree (Supplementary Table 1). Within tissues of cadaveric human eyes, expression of the dominant genes MYRF and TMEM98 were more similar to one another, and higher, than the recessive genes MFRP and PRSS56 (Figure 3C), consistent with the known regulation of TMEM98 expression by MYRF.11 Also notable was the relatively high expression of PRSS56 in retina, where it is known to be derived from late retinal progenitor cells or Müller glia to promote elongation of ocular axial length both before and after eye opening.19 Less is known about the tissue-specific roles of MFRP, MYRF, and TMEM98, however.
Figure 3: Clinical and transcriptional phenotypes of dominant and recessive forms of nanophthalmos.
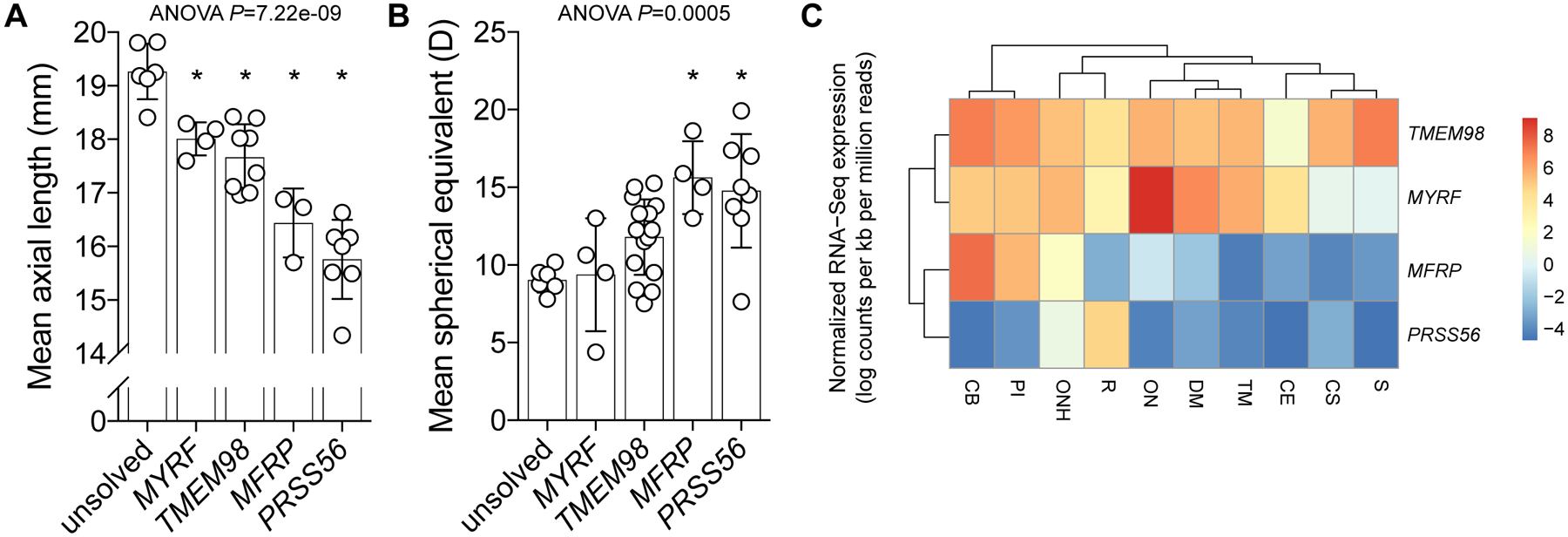
Mean ocular axial length (A) and spherical equivalent (B) of affected individuals stratified by genetic diagnosis. For both phenotypes, group means were significantly different by one-way ANOVA (P<0.0005), with asterisks (*) indicating a significant difference from the unsolved cohort (Tukey multiple comparison testing, P<0.03). (C) Relative mean expression dendrogram (normalised log counts per kb per million mapped reads) of genes in dissected human adult cadaveric eye tissue. S, sclera; CS, corneal stroma; CE, corneal epithelium; TM, trabecular meshwork; DM, Descemet’s membrane; ON, optic nerve; ONH, optic nerve head; PI, peripheral iris; CB, ciliary body.
Discussion
Here we present an Australian cohort of nanophthalmos or posterior microphthalmos cases in which 9/13 families were explained by variants in one of four genes. Mean axial length was shorter, and hyperopia higher, in individuals with a genetic diagnosis compared to those without. Recessive forms (MFRP and PRSS56) were associated with a more severe phenotype than dominant forms (TMEM98 and MYRF): recessive cases had a mean axial length range of 14.345–16.88mm, with dominant cases having a non-overlapping range of 16.96–18.42mm. Affected individuals without a genetic diagnosis were milder still, with a mean axial length range of 18.41–19.815mm, suggesting that these may be due to hypomorphic variants in the four known genes which were not detectable by exome sequencing (such as deep intronic or regulatory variants), or variants in genes yet to be associated with this phenotype, or alternatively may represent the smallest extreme of the polygenic distribution for axial length.12
PRSS56 was the most commonly implicated gene in this cohort (4/13, 30.8%) and other cohorts of posterior microphthalmos and nanophthalmos (27.3%−61.5%).4,18,20 Including the four described here, at least 27 reported pedigrees have PRSS56-associated posterior microphthalmos or nanophthalmos4,6–8,17,18,20, with at least 15 (55.6%) sharing the c.1066dupC variant.8,17 Two families here harbour at least one c.1066dupC variant, on haplotypes distinct from each other and from a previously reported Tunisian pedigree, suggesting the existence of a mutational hotspot.
While loss-of-function PRSS56 variants are associated with reduced axial length and hyperopia, gain-of-function variants might therefore be expected to cause increased axial length and myopia. Consistent with this, a p.(Ala30Thr) variant of PRSS56 has been identified in genome-wide association studies of myopia, suggesting it may increase axial length through a gain-of-function mechanism.12 PRSS56 itself is a soluble serine protease, potentially sensitive to protease inhibitors or monoclonal antibodies, and therefore a candidate target for treatment of refractive error.
In summary, we reveal considerable overlap between genes associated with rare and common refractive error phenotypes, a mutational hotspot responsible for the most frequent single variant associated with nanophthalmos or posterior microphthalmos worldwide, and a clinical distinction between recessive and dominant forms of the condition.
Supplementary Material
Acknowledgments:
Supported by the National Health and Medical Research Council (Project Grant APP1107098), the Ophthalmic Research Institute of Australia, The Rebecca L Cooper Medical Research Foundation, and the Flinders Foundation. DGM was supported in part by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900, and by the National Human Genome Research Institute grant R01 HG009141.
Footnotes
Conflict of Interest Statement: The authors report no conflicts of interest.
Data Availability Statement: Variants have been submitted to the ClinVar database.
References
- 1.Bourne RRA, Stevens GA, White RA, et al. Causes of vision loss worldwide, 1990–2010: a systematic analysis. Lancet Glob Health. 2013;1(6):e339–e349. [DOI] [PubMed] [Google Scholar]
- 2.Khan AO. Posterior microphthalmos versus nanophthalmos. Ophthalmic Genet. 2008;29(4):189. [DOI] [PubMed] [Google Scholar]
- 3.Aldahmesh MA, Nowilaty SR, Alzahrani F, et al. Posterior microphthalmos as a genetically heterogeneous condition that can be allelic to nanophthalmos. Arch Ophthalmol. 2011;129(6):805–807. [DOI] [PubMed] [Google Scholar]
- 4.Nowilaty SR, Khan AO, Aldahmesh MA, Tabbara KF, Al-Amri A, Alkuraya FS. Biometric and molecular characterization of clinically diagnosed posterior microphthalmos. Am J Ophthalmol. 2013;155(2):361–372.e7. [DOI] [PubMed] [Google Scholar]
- 5.Sundin OH, Leppert GS, Silva ED, et al. Extreme hyperopia is the result of null mutations in MFRP, which encodes a Frizzled-related protein. Proc Natl Acad Sci U S A. 2005;102(27):9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gal A, Rau I, El Matri L, et al. Autosomal-recessive posterior microphthalmos is caused by mutations in PRSS56, a gene encoding a trypsin-like serine protease. Am J Hum Genet. 2011;88(3):382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr A, Dubé M-P, Zenteno JC, et al. Mutations in a novel serine protease PRSS56 in families with nanophthalmos. Mol Vis. 2011;17:1850–1861. [PMC free article] [PubMed] [Google Scholar]
- 8.Nair KS, Hmani-Aifa M, Ali Z, et al. Alteration of the serine protease PRSS56 causes angle-closure glaucoma in mice and posterior microphthalmia in humans and mice. Nat Genet. 2011;43(6):579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khorram D, Choi M, Roos BR, et al. Novel TMEM98 mutations in pedigrees with autosomal dominant nanophthalmos. Mol Vis. 2015;21:1017–1023. [PMC free article] [PubMed] [Google Scholar]
- 10.Awadalla MS, Burdon KP, Souzeau E, et al. Mutation inTMEM98in a Large White Kindred With Autosomal Dominant Nanophthalmos Linked to 17p12-q12. JAMA Ophthalmol. 2014;132(8):970. [DOI] [PubMed] [Google Scholar]
- 11.Garnai SJ, Brinkmeier ML, Emery B, et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. 2019;15(5):e1008130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tedja MS, Wojciechowski R, Hysi PG, et al. Genome-wide association meta-analysis highlights light-induced signaling as a driver for refractive error. Nat Genet. 2018;50(6):834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day AC, Khawaja AP, Peto T, et al. The small eye phenotype in the EPIC-Norfolk eye study: prevalence and visual impairment in microphthalmos and nanophthalmos. BMJ Open. 2013;3(7):e003280. doi: 10.1136/bmjopen-2013-003280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siggs OM, Souzeau E, Pasutto F, et al. Prevalence of FOXC1 Variants in Individuals With a Suspected Diagnosis of Primary Congenital Glaucoma. JAMA Ophthalmol. January 2019. doi: 10.1001/jamaophthalmol.2018.5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siggs OM, Souzeau E, Taranath DA, et al. Congenital glaucoma with anterior segment dysgenesis in individuals with biallelic CPAMD8 variants. doi: 10.1101/297077 [DOI] [Google Scholar]
- 16.Siggs OM, Souzeau E, Breen J, et al. Autosomal dominant nanophthalmos and high hyperopia associated with a C-terminal frameshift variant in. Mol Vis. 2019;25:527–534. [PMC free article] [PubMed] [Google Scholar]
- 17.Said MB, Chouchène E, Salem SB, et al. Posterior microphthalmia and nanophthalmia in Tunisia caused by a founder c.1059_1066insC mutation of the PRSS56 gene. Gene. 2013;528(2):288–294. [DOI] [PubMed] [Google Scholar]
- 18.Patel N, Khan AO, Alsahli S, et al. Genetic investigation of 93 families with microphthalmia or posterior microphthalmos. Clin Genet. 2018;93(6):1210–1222. [DOI] [PubMed] [Google Scholar]
- 19.Paylakhi S, Labelle-Dumais C, Tolman NG, et al. Müller glia-derived PRSS56 is required to sustain ocular axial growth and prevent refractive error. PLoS Genet. 2018;14(3):e1007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo C, Zhao Z, Chen D, et al. Detection of Clinically Relevant Genetic Variants in Chinese Patients With Nanophthalmos by Trio-Based Whole-Genome Sequencing Study. Invest Ophthalmol Vis Sci. 2019;60(8):2904–2913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.