

Abstract
The mucopolysaccharidoses (MPS) are a family of lysosomal storage disorders characterized by deficient activity of enzymes that degrade glycosaminoglycans (GAGs). Abnormal development of the vertebrae and long bones is a hallmark of skeletal disease in several MPS subtypes; however, the underlying cellular mechanisms remain poorly understood. The objective of this study was to conduct an ultrastructural examination of how lysosomal storage differentially affects major skeletal cell types in MPS I and VII using naturally occurring canine disease models. We showed that both bone and cartilage cells from MPS I and VII dog vertebrae exhibit significantly elevated storage from early in postnatal life, with storage generally greater in MPS VII than MPS I. Storage was most striking for vertebral osteocytes, occupying more than forty percent of cell area. Secondary to storage, dilation of the rough endoplasmic reticulum (ER), a marker of ER stress, was observed most markedly in MPS I epiphyseal chondrocytes. Significantly elevated immunostaining of light chain 3B (LC3B) in MPS VII epiphyseal chondrocytes suggested impaired autophagy, while significantly elevated apoptotic cell death in both MPS I and VII chondrocytes was also evident. The results of this study provide insights into how lysosomal storage differentially effects major skeletal cell types in MPS I and VII, and suggests a potential relationship between storage, ER stress, autophagy, and cell death in the pathogenesis of MPS skeletal defects.
Keywords: bone, canine, cartilage, electron microscopy, endoplasmic reticulum stress, lysosomal storage, mucopolysaccharidosis
The mucopolysaccharidoses (MPS) are genetic, lysosomal storage disorders characterized by abnormal accumulation of glycosaminoglycans. Skeletal abnormalities are prevalent in several MPS subtypes. This study provides insights into how lysosomal storage differentially effects major skeletal cell types in MPS I and VII vertebrae at the onset of postnatal growth, and suggests a potential relationship between storage, ER stress, autophagy and cell death in the pathogenesis of MPS skeletal defects.
1. INTRODUCTION
The mucopolysaccharidoses are a family of inherited lysosomal storage disorders caused by mutations in genes encoding enzymes responsible for the step‐wise degradation of glycosaminoglycans (GAGs) (Neufeld and Muenzer, 2001). Enzyme deficiencies lead to incomplete degradation and abnormal accumulation of GAGs in cells and tissues resulting in multi‐organ manifestations. Progressive skeletal abnormalities, including growth retardation, kyphoscoliosis and joint dysplasia, are hallmarks of most subtypes including MPS I (Hurler Syndrome) and MPS VII (Sly Syndrome). MPS I is characterized by deficient α‐L‐iduronidase (IDUA) activity, which results in incomplete degradation and storage of dermatan and heparan sulfate GAGs. MPS VII is characterized by deficient β‐glucuronidase (GUSB) activity, resulting in incomplete degradation and storage of dermatan, heparan, and chondroitin sulfate GAGs (Neufeld and Muenzer, 2001).
In previous work using naturally occurring canine models of these diseases, we showed that both MPS I and VII dogs exhibit failures of endochondral ossification during postnatal growth. These failures include delayed cartilage‐to‐bone conversion in the vertebral and long bone epiphyses (Smith et al., 2010; Chiaro et al., 2013; Peck et al., 2015; Peck et al., 2019), and low bone volume and mineral density in the vertebral metaphyses (Chiaro et al., 2013). Vertebrae and long bones are formed through the process of endochondral ossification, in which a condensation of mesenchymal cells forms a cartilaginous template for future ossification (Mackie et al., 2008). These cells differentiate into chondrocytes, which proliferate and then mature through prehypertrophic, hypertrophic, and terminal differentiation stages, before undergoing programmed death to make way for bone‐forming osteoblasts (Mackie et al., 2008). Chondrocytes may also transdifferentiate into osteoblasts (Aghajanian and Mohan, 2018). This process of endochondral bone formation occurs in primary and then secondary ossification centers, and also at the growth plates facilitating longitudinal bone growth (Mackie et al., 2008). Bone remodeling (formation and resorption) continues in primary and secondary ossification centers and is mediated by bone‐resorbing osteoclasts and bone‐forming osteoblasts (Mackie et al., 2008).
Osteoclasts originate from the bone marrow and are differentiated from macrophages or myeloid precursors upon binding of essential ligands including receptor activation of NF‐кB ligand (RANKL) and macrophage colony‐stimulating factor (M‐CSF) by RANK and M‐CSF receptor (c‐FMs), respectively (Väänänen and Laitala‐Leinonen, 2008). Mononuclear osteoclast precursors attach to the bone surface and are induced to undergo cell–cell fusion by signals from the bone matrix. Differentiated, multinuclear osteoclasts re‐organize their ruffled border to dissolve bone mineral and degrade organic bone matrix, which also requires abundant mitochondria to provide energy (Miyamoto, 2011). Bone‐forming osteoblasts are endoplasmic reticulum (ER)‐rich cells derived from mesenchymal progenitor cells and act adjacent to each other to produce bone matrix (Dudley and Spiro, 1961). The rate of bone remodeling (formation versus resorption) is regulated by osteocytes, which are long‐lived cells derived from osteoblasts that are entombed within the mineralized bone matrix (Nakashima and Takayanagi, 2011). Prior studies have reported lysosomal storage in several major skeletal cell types (chondrocytes, osteoclasts, osteoblasts, and osteocytes) of human MPS patients and animal models (Silveri et al., 1991; Beck et al., 1992; Auclair et al., 2006; Yasuda et al., 2013; Kuehn et al., 2015). To our knowledge, no prior studies have undertaken a quantitative, comparative analysis of lysosomal storage across each of these cell types.
Lysosomes function to degrade or recycle cellular waste and foreign materials, and have crucial roles in maintaining normal cellular functions such as calcium homeostasis, lipid biosynthesis, and intracellular trafficking (Ballabio and Gieselmann, 2009). Enzyme deficiencies associated with lysosomal storage disorders such as MPS I and VII impair the degradation of certain materials, leading to undegraded storage not only in the lysosomes but also in the ER, Golgi bodies, and other organelles (Ballabio and Gieselmann, 2009). In addition to endocytosis, phagocytosis, and direct transport, cytoplasmic components destined for degradation also reach lysosomes via autophagy by fusion of autophagosomes and lysosomes. This process can be induced upon cellular stress, including ER stress and mitochondrial damage (Rashid et al., 2015; de la Mata et al., 2016). Impaired autophagy has been reported in the brains of MPS IIIA and IIIC mice, in human MPS VI fibroblasts, in chondrocytes of MPS VII mice, and other types of lysosomal storage disorders (LSD) (Settembre et al., 2008; Tessitore et al., 2009; Fraldi et al., 2010; Pshezhetsky, 2016; Bartolomeo et al., 2017); however, direct links between lysosomal GAG accumulation, cellular stress, and impaired autophagy in MPS remain poorly understood.
The objective of this study was to conduct a quantitative ultrastructural and histological investigation of the major skeletal cell types in MPS I and VII dog vertebrae at the onset of postnatal growth. Specifically, relative lysosomal storage, alterations to other organelles (ER, mitochondria, Golgi, and nuclei), and the broader consequences on cell health and function, including autophagy and apoptosis, were examined.
2. MATERIALS AND METHODS
2.1. Animals and tissue collection
All animal work was undertaken with approval from the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. MPS I and VII dogs were raised and housed at the University Of Pennsylvania School Of Veterinary Medicine (accredited by the Association for Assessment and Accreditation of Laboratory Animal Care) under National Institutes of Health (NIH) and United States Department of Agriculture (USDA) guidelines for the care and use of animals in research as previously described (Peck et al., 2015). MPS I dogs have a G‐to‐A transition in the donor splice site in intron 1 of the IDUA gene, while MPS VII dogs have a missense mutation (R166H) in the GUSB gene, and both exhibit similar skeletal phenotypes to human patients (Spellacy et al., 1983; Haskins et al., 1984; Tandon et al., 1996; Ray et al., 1998; Haskins, 2007; Smith et al., 2010; Chiaro et al., 2013). The original MPS I dog was a Plott hound (Spellacy et al., 1983), while the original MPS VII dog was a German shepherd (Haskins et al., 1984), and both have since been outbred. Genotypes of control (homozygous normal), MPS I (homozygous), and MPS VII (homozygous) affected dogs were determined via blood cell PCR analysis at birth. Animals (n = 5 each for control, MPS I and MPS VII) were euthanized at 9 days of age via an overdose of sodium pentobarbital (80 mg/kg) in accordance with the American Veterinary Medical Association guidelines (Peck et al., 2015). This age was selected as it immediately precedes normal commencement of secondary ossification in the vertebrae and long bones of control dogs, and affected animals are yet to exhibit any tissue‐level or clinical skeletal disease manifestations (Peck et al., 2015). Sexes of the dogs were as follows: 3 females and 2 males (controls), 5 females (MPS I), and 2 females and 3 males (MPS VII). There are no reported effects of sex on skeletal phenotypes in either MPS subtype. Immediately following euthanasia, thoracic and lumbar spines were removed and individual vertebrae isolated for further analyses as outlined below.
2.2. Transmission electron microscopy
Thoracic vertebral bodies (T11) were fixed in 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH7.4, overnight and decalcified in formic acid and EDTA (Formical 2000; Statlab, Lewisville, USA) at 4°C for one week. Samples were then postfixed in 2% osmium tetroxide for 1 hour at room temperature and rinsed in distilled water prior to en bloc staining with 2% uranyl acetate. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed‐812 (Electron Microscopy Sciences). Ultrathin (80 nm) sections cut in coronal plane were stained with uranyl acetate and lead citrate, and examined using transmission electron microscopy (TEM) (JEOL 1010; JEOL Ltd; Tokyo, Japan; fitted with a Hamamatsu digital camera; Hamamatsu Corporation, Shizuoka, Japan; and AMT Advantage NanoSprint500 software; Advanced Microscopy Techniques, Woburn, MA, USA). Ultrastructural analyses were performed for the following cell types: the round, resting chondrocytes in the cartilaginous vertebral epiphyses; the flattened, columnar, proliferating chondrocytes, and the large, columnar hypertrophic chondrocytes in the growth plates; and osteoclasts, osteoblasts, and osteocytes in the adjacent, calcified metaphyseal bone. The following parameters were measured using ImageJ software (NIH, Bethesda, MD, USA): total cell area (μm2), cell area occupied by vacuoles (lysosomal storage, % of total cell area), rough ER lumen diameter (nm), and the number of mitochondria and Golgi bodies (all cell types), and the number of nuclei (osteoclasts only). For rough ER, six lumens were randomly chosen, and three measurements of the lumen diameter were taken on the two ends and the middle of the lumen (averaged). For each sample, measurements were performed for 3 randomly selected cells of each type, with results averaged prior to statistics.
2.3. In situ cell death assay and immunohistochemistry
Lumbar vertebral bodies (L1–L3) were fixed in 10% neutral buffered formalin for 1–2 weeks and then decalcified as above prior to paraffin processing and embedding. Mid‐sagittal, 5 µm sections were cut followed by deparaffinization and rehydration. Detection of apoptotic cells was carried out on these sections using an In Situ Cell Death Detection Kit, POD (TUNEL assay; Sigma‐Aldrich) following the manufacturer's instructions. To examine autophagy for epiphyseal and growth plate chondrocytes, expression of light chain 3 isoform B (LC3B), a marker for autophagosomes, was examined using immunohistochemistry. Antigen retrieval was carried out by incubating the sections with 10 mg/ml proteinase K (Roche Diagnostics, Basel, Switzerland) at 37°C for 4 mins. Sections were then treated with 3% hydrogen peroxide for 12 minutes to block endogenous peroxidase activity, followed by Background Buster (Innovex Biosciences, Richmond, VA, USA) for 10 minutes at room temperature to block non‐specific protein binding. Sections were then incubated with an anti‐LC3B antibody (ab48394, 1:400 dilution; Abcam; Cambridge, UK) or a rabbit IgG isotype control antibody (negative control, Figure S1) (#31235; 1:400 dilution; Thermo Fisher; Waltham, MA, USA) overnight at 4°C. Antibody staining was visualized using the Vectastain Elite ABC‐Peroxidase Kit (Vector Laboratories, Burlingame, CA, USA) and diaminobenzidine chromogen (Thermo Fisher) according to the manufacturer's protocols. Finally, sections were counterstained with hematoxylin QS (Vector Laboratories) and cover‐slipped with aqueous mounting medium (Agilent Technologies, Santa Clara, USA). For analysis, all slides were imaged under bright field light microscopy (Eclipse 90i; Nikon, Tokyo, Japan). Three randomly selected regions‐of‐interest within both the epiphyseal cartilage (0.15 mm2 per region) and the growth plate (each region 330 µm wide) were analyzed. For each region, the number of TUNEL‐positive or LC3B‐immunopositive cells was counted and normalized as a percentage of the total number of cells present. Cell counting for each region was performed by three individuals who were blinded to the study groups and averaged. The average result for all three regions was then determined and taken as a single biological replicate for statistical comparisons.
2.4. Statistical analyses
Data were initially tested for normality via Q‐Q plots using GraphPad Prism version 8.0 (GraphPad Software Inc.). Statistical differences were then established via Kruskal–Wallis non‐parametric tests with Dunn's pair‐wise post hoc analyses. Results were reported as median and interquartile range. Statistical significance was defined as p < 0.05.
3. RESULTS
3.1. Relative cell size and lysosomal storage for different skeletal cell types
Vacuoles indicative of lysosomal storage were clearly observed for most examined skeletal cell types in the cartilaginous epiphyses, growth plates, and calcified metaphyseal bone of MPS I and VII vertebrae (Figure 1; asterisks indicate storage vacuoles). Due to the complex internal structure of osteoclasts, accurate discrimination and quantification of storage vacuoles in these cells was not possible. With respect to overall cell size (Figure 2A), resting chondrocytes were not significantly larger in either MPS I or VII epiphyseal cartilage compared to those in control dogs. Proliferating chondrocytes were significantly larger in MPS I growth plates, but not in MPS VII growth plates, compared to those in control dogs (p = 0.03), while hypertrophic chondrocytes were not significantly larger in either MPS I or MPS VII dogs compared to controls. In the calcified metaphyseal bone, neither osteoblasts nor osteocytes were significantly larger in either MPS I or MPS VII dogs compared to controls, while MPS I osteoclasts were enlarged compared to controls but did not reach statistical significance (p = 0.07). With the exception of osteoclasts (3.87‐fold vs MPS VII, p < 0.01), the size of all skeletal cell types did not differ significantly between those obtained from MPS I and VII dogs.
Figure 1.
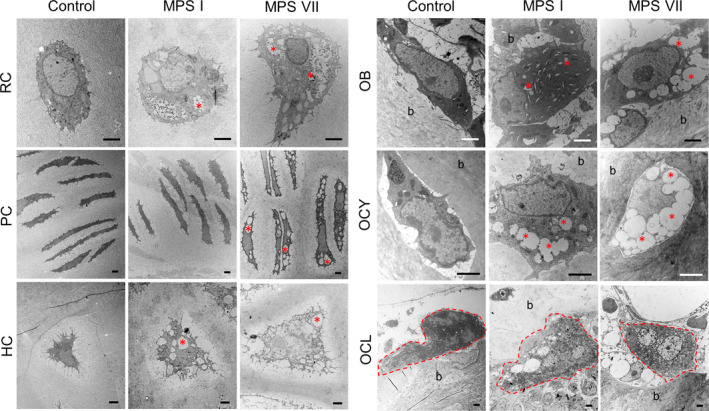
Representative TEM images of different skeletal cell types in 9‐day‐old control, MPS I and MPS VII dog vertebrae. RC: Resting chondrocytes in epiphyseal cartilage; PC and HC: proliferating and hypertrophic chondrocytes, respectively, in the growth plate; OB, OCY, and OCL: osteoblasts, osteocytes, and osteoclasts, respectively, in calcified metaphyseal bone. Red asterisks indicate examples of the locations of storage vacuoles. Osteoclasts are encircled with a red dashed line. (b) bone matrix. Scale = 2 µm
Figure 2.
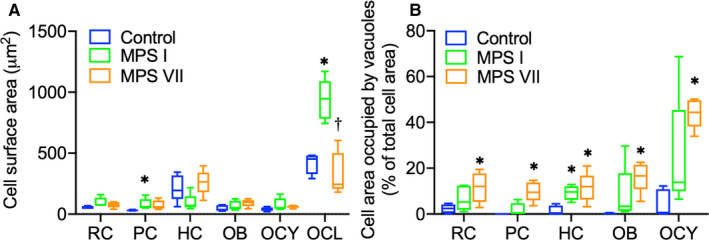
(A) Quantification of overall cell area and (B) percentage of cell area occupied by storage vacuoles for different skeletal cell types in 9‐day‐old control, MPS I and MPS VII dog vertebrae. RC: Resting chondrocytes in epiphyseal cartilage; PC and HC: proliferating and hypertrophic chondrocytes, respectively, in the growth plate; OB, OCY, and OCL: osteoblasts, osteocytes, and osteoclasts, respectively, in calcified metaphyseal bone. Median, interquartile range (box), and 10th and 90th percentiles (whiskers); *p < 0.05 vs control; †p < 0.05 vs MPS I; Kruskal–Wallis non‐parametric tests with Dunn's post hoc tests; n = 5
With respect to the percentage of the cell area occupied by storage vacuoles (Figure 2B), in epiphyseal cartilage, resting epiphyseal chondrocytes from MPS VII, but not MPS I dogs, exhibited significantly increased area occupied by vacuoles compared to controls (p = 0.04). In the growth plates, area occupied by vacuoles was significantly greater for proliferating chondrocytes from MPS VII (p < 0.01), but not MPS I dogs, while area occupied by vacuoles for hypertrophic chondrocytes was significantly increased in cells from both MPS I and VII dogs compared to those from control dogs (p = 0.04 and p = 0.02, respectively). Finally, in the metaphyseal bone, area occupied by vacuoles for MPS VII osteoblasts was significantly greater compared to controls (p < 0.01). Similarly, area occupied by vacuoles was significantly greater for MPS VII osteocytes compared to controls (p < 0.01), where vacuoles occupied 44.37% (median) of the overall cell area. Area occupied by vacuoles was more pronounced in MPS VII than MPS I for all cell types, but there were no significant differences between these groups.
3.2. Endoplasmic reticulum dilation
Rough ER dilation is an indicator of ER stress, which in turn may negatively impact protein synthesis, trafficking, and cell function (Boyce and Yuan, 2006; Chavez‐Valdez et al., 2016; Yip et al., 2019). Therefore, rough ER lumen dilation was measured using TEM (Figure 3). In MPS I vertebrae, the rough ER lumens of resting epiphyseal chondrocytes, growth plate proliferating and hypertrophic chondrocytes, and metaphyseal osteoblasts were all significantly dilated compared to those from control dogs (p = 0.02, p = 0.01, p < 0.01, p = 0.02, respectively). Rough ER dilation was most striking for resting chondrocytes in MPS I epiphyseal cartilage (2.01‐fold vs control, p < 0.01; Figure 3B). In MPS VII vertebrae, rough ER lumen dilation was similarly greatest for resting epiphyseal chondrocytes, but was not significantly different from control for any cell type.
Figure 3.
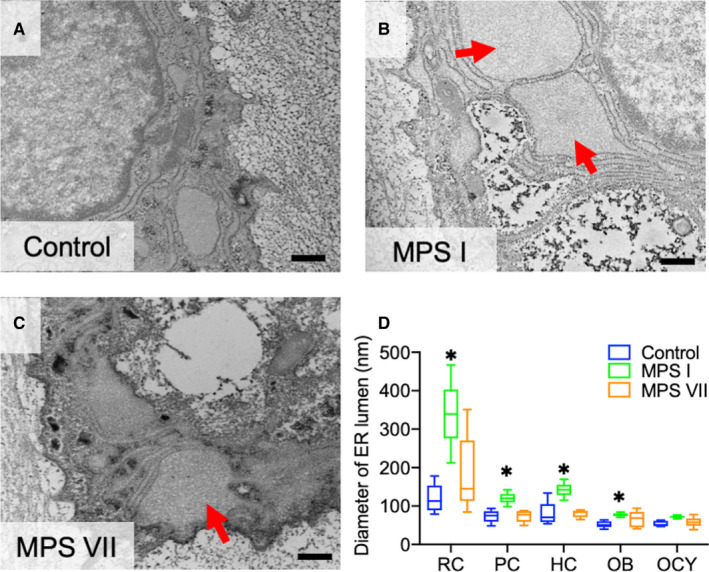
Rough ER lumen dilation in MPS I and VII skeletal cells. (A–C) Representative TEM images of resting epiphyseal cartilage chondrocytes from control, MPS I and MPS VII dog vertebrae. ER lumens in MPS I and MPS VII resting chondrocytes appear dilated (arrows) compared to controls. Scale = 500 nm. (D) Quantification of rough ER lumen diameters for different skeletal cell types in 9‐day‐old control, MPS I and MPS VII dog vertebrae. RC: Resting chondrocytes in epiphyseal cartilage; PC and HC: proliferating and hypertrophic chondrocytes, respectively, in the growth plate; OB and OCY: osteoblasts and osteocytes, respectively, in calcified metaphyseal bone. Median, interquartile range (box), and 10th and 90th percentiles (whiskers); *p < 0.05 vs control; Kruskal–Wallis non‐parametric tests with Dunn's post hoc tests; n = 5
3.3. Cell nuclei,golgi,and mitochondria
Fusion of mononuclear preosteoclasts to form multinuclear mature osteoclasts is a key step during osteoclastogenesis (Väänänen and Laitala‐Leinonen, 2008; Miyamoto, 2011). To assess osteoclast maturity, the number of nuclei in vertebral metaphyseal osteoclasts was determined using TEM. The number of osteoclast nuclei was lower in both MPS I (54.55% of control, p < 0.05) and MPS VII (50.00% of control, p = 0.07) vertebrae, suggesting impaired osteoclastogenesis (Figure 4A–D). The number and morphology of mitochondria and Golgi bodies were also assessed for each cell type (Figure 4E–H). No significant differences were found, with the exception of the number of mitochondria in MPS VII osteocytes, which was significantly lower than for controls (46.51%, p = 0.02). Additionally, larger and small cristae‐filled mitochondria were observed in approximately 40% of all MPS VII osteoclasts examined (Figure 4H).
Figure 4.
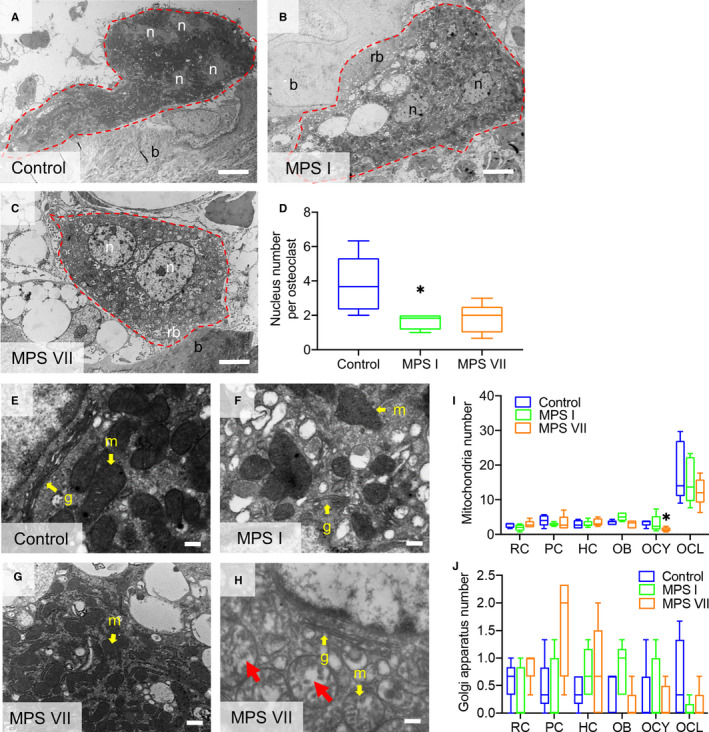
(A–C) Representative TEM images of osteoclasts in the calcified metaphyseal bone of 9‐day‐old control, MPS I and MPS VII dog vertebrae illustrating numbers of nuclei (n). Osteoclast borders are outlined (red dashed lines); b: bone matrix; and rb: ruffled border; Scale = 4 μm. (D) Quantification of the number of osteoclasts. (E–G) Higher magnification TEM images osteoclasts showing mitochondria (m) and Golgi bodies (G) (yellow arrows, examples). (H) A subset of mitochondria in MPS VII osteoclasts appeared swollen (red arrows, examples). (I) Quantification of the total numbers of mitochondria and (J) Golgi bodies in different skeletal cell types. RC: Resting chondrocytes in epiphyseal cartilage; PC and HC: proliferating and hypertrophic chondrocytes, respectively, in the growth plate; OB, OCY, and OCL: osteoblasts, osteocytes and osteoclasts, respectively, in calcified metaphyseal bone. Median, interquartile range (box), and 10th and 90th percentiles (whiskers); *p < 0.05 vs control; Kruskal–Wallis non‐parametric tests with Dunn's post hoc tests; n = 5
3.4. Autophagy and apoptosis
In previous work, we demonstrated that MPS VII chondrocytes exhibit impaired ability to progress through hypertrophic differentiation (Peck et al., 2015). Therefore, to provide further insights into how storage affects the stress response of these cells and contributes to abnormal function, the relative numbers of apoptotic chondrocytes in MPS I and VII vertebrae were examined using the TUNEL assay (Figure 5). The number of apoptotic resting chondrocytes in vertebral epiphyseal cartilage was significantly greater in both MPS I and VII compared to controls (1.52‐ and 1.43‐fold, respectively; p = 0.02 for both). In the growth plates, the numbers of apoptotic proliferating and hypertrophic chondrocytes were not significantly different from controls for either MPS I or VII.
Figure 5.
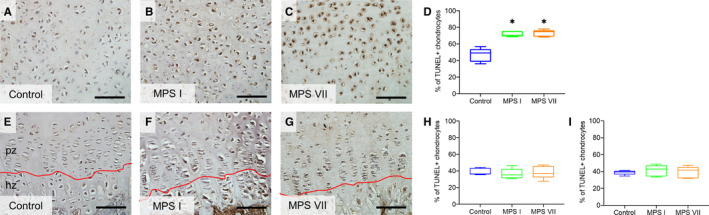
(A–C) Representative histological images; (D) quantification of apoptotic resting epiphyseal cartilage chondrocytes from 9‐day‐old control, MPS I and MPS VII dog vertebrae. (E–G) Representative histological images; (H) quantification of apoptotic proliferating and (I) hypertrophic growth plate chondrocytes. In situ TUNEL assay; pz: proliferating zone; hz: hypertrophic zone; scale = 100 µm; median, interquartile range (box), and 10th and 90th percentiles (whiskers); *p < 0.05 vs control; Kruskal–Wallis non‐parametric tests with Dunn's post hoc tests; n = 5
To provide insights into impaired autophagy of MPS I and VII chondrocytes, the number of these cells expressing the autophagy marker LC3B was examined using immunohistochemistry (Figure 6). LC3B is expressed in the early stages of autophagosome formation and interacts with autophagy receptors such as p62/SQSTM1 for autophagic degradation (Johansen and Lamark, 2011). Elevated LC3B expression is a marker of autophagosome accumulation, indicating impaired autophagosome‐lysosome fusion and degradation. The number of LC3B‐immunopositive resting chondrocytes was significantly greater for MPS VII vertebral epiphyseal cartilage compared to control dogs (2.57‐fold vs control, p = 0.01), but was not for MPS I vertebrae. Similarly, in the growth plate, the number of LC3B‐immunopositive proliferating chondrocytes was significantly elevated for MPS VII (5.53‐fold vs control, p = 0.02) but not MPS I vertebrae. The number of LC3B‐immunopositive hypertrophic chondrocytes was elevated in MPS I and VII vertebrae (2.54‐ and 2.90‐fold vs control, p = 0.03 and p = 0.05, respectively). There were no significant differences found for the number of LC3B‐immunopositive chondrocytes between MPS I and VII vertebrae.
Figure 6.
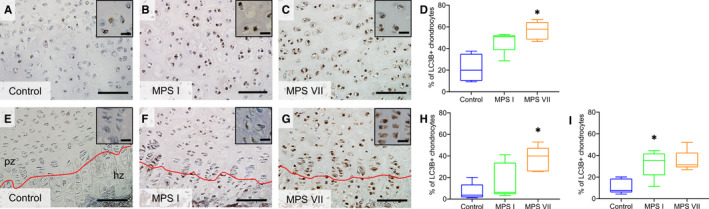
(A–C) Representative immunostaining and (D) quantification of LC3B‐positive resting epiphyseal cartilage chondrocytes from 9‐day‐old control, MPS I and MPS VII dog vertebrae. (E–G) Representative immunostaining and (H) quantification of LC3B‐positive proliferating and (I) hypertrophic growth plate chondrocytes. pz: proliferating zone; hz: hypertrophic zone; scale = 100 µm (inset scale = 20 µm); median, interquartile range (box), and 10th and 90th percentiles (whiskers); *p < 0.05 vs control; Kruskal–Wallis non‐parametric tests with Dunn's post hoc tests; n = 5
4. DISCUSSION
Skeletal manifestations are present in several MPS subtypes, including I and VII, and include impaired longitudinal bone growth, failed cartilage‐to‐bone conversion in secondary ossification centers, and diminished bone quality in primary ossification centers (Nuttall et al., 1999; Herati et al., 2008; Metcalf et al., 2009; Fung et al., 2010; Smith et al., 2012; Lin et al., 2013; Peck et al., 2015; Jiang et al., 2018; Jiang et al., 2020). Clinically, these manifestations are associated with short stature and progressive skeletal deformities that negatively impact patient quality of life (Zimet et al., 1997; Ross et al., 2004; Polgreen and Miller, 2010). In this study, we investigated how lysosomal storage differentially impacts the major skeletal cell types responsible for the pathogenesis of each of these manifestations at the onset of postnatal growth using clinically relevant canine models of two MPS subtypes (I and VII) that exhibit significant skeletal disease.
For all vertebral cell types for which quantitative analysis was possible (i.e., all except for osteoclasts), the area occupied by vacuoles was elevated in both MPS I and VII dogs at 9 days of age compared to controls dogs. Notably, at this early age neither MPS I nor VII animals exhibit obvious tissue‐level nor clinical abnormalities. Additionally, the area occupied by vacuoles was greater for MPS VII cells compared to those from MPS I dogs. One possible explanation for this may be the fact that MPS VII dogs accumulate chondroitin sulfate, whereas MPS I dogs do not, and chondroitin sulfate is the predominant extracellular GAG present in cartilaginous skeletal tissues (Mathews and Lozaityte, 1958). It may also explain why skeletal disease is generally more severe in MPS VII than in MPS I patients (Neufeld and Muenzer, 2001). Previously, we demonstrated that MPS VII dogs exhibit delayed commencement of secondary ossification in vertebrae and long bones, and that this can be traced to the failure of the chondrocytes that reside in the epiphyseal cartilage to proceed through hypertrophic differentiation at the requisite developmental stage (Peck et al., 2015). Our findings here show that this may be in part explained by the fact that these cells already exhibit significantly elevated storage immediately prior to commencement of secondary ossification and that this storage is associated with elevated levels of apoptosis and impaired autophagy. Interestingly, while MPS VII growth plate chondrocytes, and metaphyseal osteoblasts and osteocytes also exhibited increased storage, we showed previously that at 9 days of age growth plates and metaphyseal calcified bone are otherwise normal, with associated defects in longitudinal bone growth and poor bone quality, respectively, not manifesting until later stages of postnatal growth (Peck et al., 2015; Peck et al., 2016). Of all the cell types examined, osteocytes exhibited the greatest area occupied by vacuoles. These cells are former osteoblasts that become entombed in the bone matrix and function as master regulators of bone turnover (Goldring, 2015). Like epiphyseal chondrocytes, their location removes them from any direct blood supply, which likely makes them inaccessible to exogenous drugs and may in part explain why bone disease is recalcitrant to treatments such as enzyme replacement therapy.
Interestingly, although the area occupied by vacuoles in MPS I resting epiphyseal chondrocytes was not as great as in those from MPS VII dogs, these cells exhibited significantly dilated rough ER lumens compared to controls, which is a hallmark of ER stress (Yip et al., 2019). We previously found elevated mRNA expression levels of the transcription factor C/EBP homologous protein (CHOP or DDIT3), a factor activated during ER stress (Marciniak et al., 2004), in MPS VII vertebral cartilage using RNA‐seq analysis (Peck et al., 2019). Combined with our findings here of ER lumen dilation, this supports the hypothesis that GAG accumulation in MPS chondrocytes is associated with ER stress. To buffer ER stress, cells initiate the unfolded protein response (UPR) and enlarge the ER compartment to restore its normal function (Schroder and Kaufman, 2005); however, depending on the severity of ER stress, apoptotic cell death can be induced if the stress exceeds a threshold (Rashid et al., 2015). We found significantly increased numbers of TUNEL‐positive apoptotic epiphyseal chondrocytes in both MPS I and MPS VII, which is a potential consequence of ER stress. Further study is needed, however, to fully elucidate the role of ER stress signaling and its relationship to cell death and failed endochondral ossification in MPS.
We also demonstrated evidence of impaired autophagy by MPS I and VII epiphyseal and growth plate chondrocytes by elevated LC3B staining, which indicates accumulation of autophagosomes. LC3B, one of the five members of LC3 proteins that are used as endogenous autophagic markers, has a central role in autophagosome membrane structure (Koukourakis et al., 2015). Notably, similar findings have been reported in GUSB KO chondrocytes (mimicking MPS VII) (Bartolomeo et al., 2017), human cells, and animal models of other MPS types as well as other types of lysosomal storage disorders; some of which further demonstrated failure of autophagosome–lysosome fusion potentially due to deficient lysosomal degradation, suggesting impaired autophagy (Settembre et al., 2008; Tessitore et al., 2009; Fraldi et al., 2010; Pshezhetsky, 2016). Molecular connections exist between autophagy and apoptosis, including interactions between Beclin 1 and BCL‐2 (Liang et al., 1998); UVRAG and BAX (Yin et al., 2011); and caspase‐mediated cleavage of Beclin 1 (Wirawan et al., 2010). Thus, it is possible that the increased apoptosis by MPS I and VII chondrocytes is in part a consequence of impaired autophagy.
Effective bone resorption requires multinucleated osteoclasts with formation of ruffled membranes, abundant mitochondria, and normal lysosome function (Miyamoto, 2011, Väänänen and Laitala‐Leinonen, 2008, Lacombe et al., 2013). Our findings demonstrated significantly lower numbers of nuclei in the osteoclasts of MPS I and MPS VII vertebrae, suggesting impaired fusion of mononuclear precursor osteoclasts. Interestingly, osteoclasts in MPS I and VII vertebrae exhibited a normal ruffled boarder, indicating that metaphyseal bone resorption may not be significantly impacted at this very young age. A previous study found that osteoclasts from adult 8‐week‐old MPS VII mice were dissociated from the bone matrix, lack a distinct ruffled border, and had diminished function (Monroy et al., 2002). It is unclear whether impaired fusion of preosteoclasts can affect the function of mature osteoclasts at later ages for MPS dogs as pathology progressively develops.
This study had several limitations. Our findings are descriptive and based on observations of cellular morphology and expression of a limited number of markers. While molecular mechanisms can be inferred from these findings, in‐depth mechanistic studies focusing on the links between lysosomal storage, cell stress, and the broader dysfunction of MPS skeletal cells (such as impaired differentiation) are now needed. Additionally, in this study we only analyzed a single neonatal time point. Future studies will examine how the abnormalities observed continue to manifest during later stages of postnatal growth. Another limitation of this study is that the fixation method used may have resulted in some cell shrinkage artifact. In the future, this could be ameliorated through the use of other fixative solutions such as ruthenium hexamine trichloride to better maintain connection of the cell membrane to the pericellular matrix (Hunziker et al., 1983). Finally, the colorimetric TUNEL assay used in this study may have resulted in some non‐specific staining of cells due to endogenous peroxidase activity.
In summary, the results of this study provide important new insights into how GAG storage differentially impacts different skeletal cell types in two subtypes of MPS that exhibit progressive and debilitating skeletal abnormalities, and point to potential mechanisms, such as ER stress and impaired autophagy, underlying cellular dysfunction and increased apoptotic cell death. Our results lay the foundation for comprehensive mechanistic studies and highlight the importance of early intervention and treatment, as well as the challenges underlying effective drug delivery to skeletal cells.
AUTHOR CONTRIBUTIONS
ZJ contributed to conceptual design, performed experiments and drafted the manuscript; YKL and MW performed experiments; MLC contributed to conceptual design; and LJS contributed to conceptual design and drafted the manuscript. All authors reviewed and approved the manuscript prior to submission.
Supporting information
Figure S1
ACKNOWLEDGEMENTS
Funding for this study was received from the National Institutes of Health (R01AR071975), with additional support received from the Penn Center for Musculoskeletal Disorders (NIH P30AR069619). Technical support from staff at the Electron Microscopy Resource Laboratory at the University of Pennsylvania Perelman School of Medicine and animal care provided by staff at the Referral Center for Animal Models at the University of Pennsylvania School of Veterinary Medicine (NIH P40OD010939) are both gratefully acknowledged. The authors have no conflicts to declare.
Jiang Z, Lau YK, Wu M, Casal ML, Smith LJ. Ultrastructural analysis of different skeletal cell types in mucopolysaccharidosis dogs at the onset of postnatal growth. J. Anat.2021;238:416–425. 10.1111/joa.13305
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Aghajanian, P. & Mohan, S. (2018) The art of building bone: emerging role of chondrocyte‐to‐osteoblast transdifferentiation in endochondral ossification. Bone Research, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair, D. , Hein, L.K. , Hopwood, J.J. & Byers, S. (2006) Intra‐articular enzyme administration for joint disease in feline mucopolysaccharidosis VI: Enzyme dose and interval. Pediatric Research, 59, 538–543. [DOI] [PubMed] [Google Scholar]
- Ballabio, A. & Gieselmann, V. (2009) Lysosomal disorders: From storage to cellular damage. Biochimica et Biophysica Acta (BBA) ‐ Molecular Cell Research, 1793(4), 684–696. [DOI] [PubMed] [Google Scholar]
- Bartolomeo, R. , Cinque, L. , De Leonibus, C. , Forrester, A. , Salzano, A.C. , Monfregola, J et al (2017) mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. Journal of Clinical Investigation, 127(10), 3717–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, M. , Braun, S. , Coerdt, W. , Merz, E. , Young, E. & Sewell, A.C. (1992) Fetal presentation of Morquio disease type A. Prenatal Diagnosis, 12, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Boyce, M. & Yuan, J. (2006) Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death and Differentiation, 13, 363–373. [DOI] [PubMed] [Google Scholar]
- Chavez‐Valdez, R. , Flock, D.L. , Martin, L.J. & Northington, F.J. (2016) Endoplasmic reticulum pathology and stress response in neurons precede programmed necrosis after neonatal hypoxia‐ischemia. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 48, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaro, J.A. , Baron, M.D. , Del Alcazar, C.M. , O'Donnell, P. , Shore, E.M. , Elliott, D.M et al (2013) Postnatal progression of bone disease in the cervical spines of mucopolysaccharidosis I dogs. Bone, 55, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata, M. , Cotán, D. , Villanueva‐Paz, M. , de Lavera, I. , Álvarez‐Córdoba, M. , Luzón‐Hidalgo, R et al (2016) Mitochondrial dysfunction in lysosomal storage disorders. Diseases (Basel, Switzerland), 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, H.R. & Spiro, D. (1961) The fine structure of bone cells. The Journal of Biophysical and Biochemical Cytology, 11, 627–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraldi, A. , Annunziata, F. , Lombardi, A. , Kaiser, H.‐J. , Medina, D.L. , Spampanato, C et al (2010) Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. The EMBO journal, 29, 3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fung, E.B. , Johnson, J.A. , Madden, J. , Kim, T. & Harmatz, P. (2010) Bone density assessment in patients with mucopolysaccharidosis: A preliminary report from patients with MPS II and VI. Journal of Pediatric Rehabilitation Medicine, 3(1), 13–23. [PMC free article] [PubMed] [Google Scholar]
- Goldring, S.R. (2015) The osteocyte: key player in regulating bone turnover. RMD Open, 1, e000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskins, M.E. (2007) Animal models for mucopolysaccharidosis disorders and their clinical relevance. Acta Paediatrica Supplement, 96, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskins, M.E. , Desnick, R.J. , DiFerrante, N. , Jezyk, P.F. & Patterson, D.F. (1984) Beta‐glucuronidase deficiency in a dog: A model of human mucopolysaccharidosis VII. Pediatric Research, 18, 980–984. [DOI] [PubMed] [Google Scholar]
- Herati, R.S. , Knox, V.W. , O'Donnell, P. , D'Angelo, M. , Haskins, M.E. & Ponder, K.P. (2008) Radiographic evaluation of bones and joints in mucopolysaccharidosis I and VII dogs after neonatal gene therapy. Molecular Genetics and Metabolism, 95, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker, E.B. , Herrmann, W. & Schenk, R.K. (1983) Ruthenium hexammine trichloride (RHT)‐mediated interaction between plasmalemmal components and pericellular matrix proteoglycans is responsible for the preservation of chondrocytic plasma membranes in situ during cartilage fixation. Journal of Histochemistry and Cytochemistry, 31, 717–727. [DOI] [PubMed] [Google Scholar]
- Jiang, Z. , Derrick‐Roberts, A.L.K. , Jackson, M.R. , Rossouw, C. , Pyragius, C.E. , Xian, C et al (2018) Delayed development of ossification centers in the tibia of prenatal and early postnatal MPS VII mice. Molecular Genetics and Metabolism, 124, 135–142. [DOI] [PubMed] [Google Scholar]
- Jiang, Z. , Derrick‐Roberts, A.L.K. , Reichstein, C. & Byers, S. (2020) Cell cycle progression is disrupted in murine MPS VII growth plate leading to reduced chondrocyte proliferation and transition to hypertrophy. Bone, 132, 115195. [DOI] [PubMed] [Google Scholar]
- Johansen, T. & Lamark, T. (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy, 7, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukourakis, M.I. , Kalamida, D. , Giatromanolaki, A. , Zois, C.E. , Sivridis, E. , Pouliliou, S et al (2015) Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One, 10, e0137675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn, S.C. , Koehne, T. , Cornils, K. , Markmann, S. , Riedel, C. , Pestka, J.M et al (2015) Impaired bone remodeling and its correction by combination therapy in a mouse model of mucopolysaccharidosis‐I. Human Molecular Genetics, 24, 7075–7086. [DOI] [PubMed] [Google Scholar]
- Lacombe, J. , Karsenty, G. & Ferron, M. (2013) Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle, 12, 2744–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X.H. , Kleeman, L.K. , Jiang, H.H. , Gordon, G. , Goldman, J.E. , Berry, G et al (1998) Protection against fatal sindbis virus encephalitis by Beclin, a Novel Bcl‐2‐interacting protein. Journal of Virology, 72, 8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H.Y. , Shih, S.C. , Chuang, C.K. , Chen, M.R. , Niu, D.M. & Lin, S.P. (2013) Assessment of bone mineral density by dual energy x‐ray absorptiometry in patients with mucopolysaccharidoses. Orphanet Journal of Rare Diseases, 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie, E.J. , Ahmed, Y.A. , Tatarczuch, L. , Chen, K.S. & Mirams, M. (2008) Endochondral ossification: How cartilage is converted into bone in the developing skeleton. International Journal of Biochemistry and Cell Biology, 40, 46–62. [DOI] [PubMed] [Google Scholar]
- Marciniak, S.J. , Yun, C.Y. , Oyadomari, S. , Novoa, I. , Zhang, Y. , Jungreis, R et al (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes and Development, 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, M.B. & Lozaityte, I. (1958) Sodium chondroitin sulfate‐protein complexes of cartilage. I. Molecular weight and shape. Archives of Biochemistry and Biophysics, 74, 158–174. [DOI] [PubMed] [Google Scholar]
- Metcalf, J.A. , Zhang, Y. , Hilton, M.J. , Long, F. & Ponder, K.P. (2009) Mechanism of shortened bones in mucopolysaccharidosis VII. Molecular Genetics and Metabolism, 97, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, T. (2011) Regulators of osteoclast differentiation and cell‐cell fusion. Keio Journal of Medicine, 60, 101–105. [DOI] [PubMed] [Google Scholar]
- Monroy, M.A. , Ross, F.P. , Teitelbaum, S.L. & Sands, M.S. (2002) Abnormal osteoclast morphology and bone remodeling in a murine model of a lysosomal storage disease. Bone, 30, 352–359. [DOI] [PubMed] [Google Scholar]
- Nakashima, T. & Takayanagi, H. (2011) New regulation mechanisms of osteoclast differentiation. Annals of the New York Academy of Sciences, 1240, E13–E18. [DOI] [PubMed] [Google Scholar]
- Neufeld, E. & Muenzer, J. (2001) The Mucopolysaccharidoses In: Sciver C., Beaudet A., Sly W. & Valle D. (Eds.) The metabolic and molecular bases of inherited disease (pp. 3421–3452). New York, NY: McGraw hill. [Google Scholar]
- Nuttall, J.D. , Brumfield, L.K. , Fazzalari, N.L. , Hopwood, J.J. & Byers, S. (1999) Histomorphometric analysis of the tibial growth plate in a feline model of mucopolysaccharidosis type VI. Calcified Tissue International, 65, 47–52. [DOI] [PubMed] [Google Scholar]
- Peck, S.H. , Kang, J.L. , Chiaro, J.A. , Malhotra, N.R. , O'Donnell, P. , Haskins, M.E et al (2016) Progression of vertebral bone disease in mucopolysaccharidosis VII dogs from birth to skeletal maturity. In Orthopedic Research Society 2016 Annual Meeting). Orlando, FL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck, S.H. , O'Donnell, P.J.M. , Kang, J.L. , Malhotra, N.R. , Dodge, G.R. , Pacifici, M et al (2015) Delayed hypertrophic differentiation of epiphyseal chondrocytes contributes to failed secondary ossification in mucopolysaccharidosis VII dogs. Molecular Genetics and Metabolism, 116, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck, S.H. , Tobias, J.W. , Shore, E.M. , Malhotra, N.R. , Haskins, M.E. , Casal, M.L et al (2019) Molecular profiling of failed endochondral ossification in mucopolysaccharidosis VII. Bone, 128, 115042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgreen, L.E. & Miller, B.S. (2010) Growth patterns and the use of growth hormone in the mucopolysaccharidoses. Journal of Pediatric Rehabilitation Medicine, 3, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshezhetsky, A.V. (2016) Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy, 12, 1059–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid, H.‐O. , Yadav, R.K. , Kim, H.‐R. & Chae, H.‐J. (2015) ER stress: Autophagy induction, inhibition and selection. Autophagy, 11, 1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, J. , Bouvet, A. , DeSanto, C. , Fyfe, J.C. , Xu, D. , Wolfe, J.H et al (1998) Cloning of the canine beta‐glucuronidase cDNA, mutation identification in canine MPS VII, and retroviral vector‐mediated correction of MPS VII cells. Genomics, 48, 248–253. [DOI] [PubMed] [Google Scholar]
- Ross, J.L. , Sandberg, D.E. , Rose, S.R. , Leschek, E.W. , Baron, J. , Chipman, J.J et al (2004) Psychological adaptation in children with idiopathic short stature treated with growth hormone or placebo. Journal of Clinical Endocrinology and Metabolism, 89, 4873–4878. [DOI] [PubMed] [Google Scholar]
- Schroder, M. & Kaufman, R.J. (2005) ER stress and the unfolded protein response. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 569, 29–63. [DOI] [PubMed] [Google Scholar]
- Settembre, C. , Fraldi, A. , Jahreiss, L. , Spampanato, C. , Venturi, C. , Medina, D et al (2008) A block of autophagy in lysosomal storage disorders. Human Molecular Genetics, 17, 119–129. [DOI] [PubMed] [Google Scholar]
- Silveri, C.P. , Kaplan, F.S. , Fallon, M.D. , Bayever, E. & August, C.S. (1991) Hurler syndrome with special reference to histologic abnormalities of the growth plate. Clinical Orthopaedics and Related Research, 305–311. [PubMed] [Google Scholar]
- Smith, L.J. , Baldo, G. , Wu, S. , Liu, Y. , Whyte, M.P. , Giugliani, R et al (2012) Pathogenesis of lumbar spine disease in mucopolysaccharidosis VII. Molecular Genetics and Metabolism, 107, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, L.J. , Martin, J.T. , Szczesny, S.E. , Ponder, K.P. , Haskins, M.E. & Elliott, D.M. (2010) Altered lumbar spine structure, biochemistry, and biomechanical properties in a canine model of mucopolysaccharidosis type VII. Journal of Orthopaedic Research, 28, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellacy, E. , Shull, R.M. , Constantopoulos, G. & Neufeld, E.F. (1983) A canine model of human alpha‐L‐iduronidase deficiency. Proceedings of the National Academy of Sciences of the United States of America, 80, 6091–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon, V. , Williamson, J.B. , Cowie, R.A. & Wraith, J.E. (1996) Spinal problems in mucopolysaccharidosis I (Hurler syndrome). Journal of Bone and Joint Surgery. British Volume, 78, 938–944. [DOI] [PubMed] [Google Scholar]
- Tessitore, A. , Pirozzi, M. & Auricchio, A. (2009) Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. PathoGenetics, 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väänänen, H.K. & Laitala‐Leinonen, T. (2008) Osteoclast lineage and function. Archives of Biochemistry and Biophysics, 473, 132–138. [DOI] [PubMed] [Google Scholar]
- Wirawan, E. , Vande Walle, L. , Kersse, K. , Cornelis, S. , Claerhout, S. , Vanoverberghe, I et al (2010) Caspase‐mediated cleavage of Beclin‐1 inactivates Beclin‐1‐induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death and Disease, 1, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, E. , Fushimi, K. , Suzuki, Y. , Shimizu, K. , Takami, T. , Zustin, J et al (2013) Pathogenesis of Morquio A syndrome: an autopsied case reveals systemic storage disorder. Molecular Genetics and Metabolism, 109, 301–311. [DOI] [PubMed] [Google Scholar]
- Yin, X. , Cao, L. , Peng, Y. , Tan, Y. , Xie, M. , Kang, R et al (2011) A critical role for UVRAG in apoptosis. Autophagy, 7, 1242–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, R.K.H. , Chan, D. & Cheah, K.S.E. (2019) Mechanistic insights into skeletal development gained from genetic disorders. Current Topics in Developmental Biology, 133, 343–385. [DOI] [PubMed] [Google Scholar]
- Zimet, G.D. , Owens, R. , Dahms, W. , Cutler, M. , Litvene, M. & Cuttler, L. (1997) Psychosocial outcome of children evaluated for short stature. Archives of Pediatrics and Adolescent Medicine, 151, 1017–1023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.