

Abstract

Potent JNK3 isoform selective inhibitors were developed from a thiophenyl-pyrazolourea scaffold. Through structure activity relationship (SAR) studies utilizing enzymatic and cell-based assays, and in vitro and in vivo drug metabolism and pharmacokinetic (DMPK) studies, potent and highly selective JNK3 inhibitors with oral bioavailability and brain penetrant capability were developed. Inhibitor 17 was a potent and isoform selective JNK3 inhibitor (IC50 = 35 nM), had significant inhibition to only JNK3 in a panel profiling of 374 wild-type kinases, had high potency in functional cell-based assays, had high stability in human liver microsome (t1/2 = 66 min) and a clean CYP-450 inhibition profile, and was orally bioavailable and brain penetrant. Moreover, cocrystal structures of compounds 17 and 27 in human JNK3 were solved at 1.84 Å, which showed that these JNK3 isoform selective inhibitors bound to the ATP pocket, had interactions in both hydrophobic pocket-I and hydrophobic pocket-II.
Keywords: JNK3, Kinase Inhibitor, Alzheimer’s Diseases (AD), Pyrazolourea, Neurodegeneration
There are three isoforms for the mitogen-activated protein kinase c-Jun N-terminal kinases: JNK1, JNK2, and JNK3.1 Among them, JNK1 and JNK2 are ubiquitously expressed,2,3 while JNK3 is mainly expressed in the central nervous system (CNS).1,2 JNK3 has been found to be related to various CNS and/or neurodegenerative diseases, such as Alzheimer’s disease (AD),4−7 Parkinson’s disease (PD),8−10 Huntington disease (HD),11 stroke,12,13 epilepsy,14 and spinal muscular atrophy (SMA).15 Therefore, development of brain penetrant and potent JNK3 inhibitors has been the focus of many academic laboratories and pharma over the past decades.16−25 In addition, since JNK1 is found to be not obviously related to neurodegenerative diseases, identification and development of isoform selective JNK3 inhibitors are of particular interest for drug discovery. Despite numerous efforts to develop JNK3 inhibitors, so far none of the small molecule JNK3 compounds have entered clinical trials. Therefore, novel JNK3 inhibitors with excellent DMPK properties are still needed for CNS applications.
In our previous studies, we discovered that compounds derived from a pyrazole-urea scaffold could yield potent and isoform selective JNK3 inhibitors, as exemplified by compounds 1 and 2 in Figure 1.16 These JNK3 inhibitors had high selectivity against JNK1 and the closely related kinase p38α. Compound 2 even had high selectivity against JNK2 (>500-fold).16 Compounds from this scaffold also possessed high general kinase selectivity. In a panel profiling study against a panel size of 464 kinases, at 10 μM, 1 showed significant inhibition (>80% inh.) to only 4 off-targets (Clk2, haspin, Mek3, and Ysk4). Both inhibitors 1 and 2 were clean in P450 inhibitions and also gave fair PK properties by iv dosing. In addition, 1 demonstrated fair brain penetration in mice.16
Figure 1.

Two lead structures from the pyrazole-urea scaffold and strategies for further optimizations.
The favored properties encouraged us to carry out further optimizations for this pyrazole-urea scaffold in order to obtain better JNK3 inhibitors for CNS applications. The major liability was the low oral bioavailability; compounds we previously reported gave none or very low oral bioavailability. In addition, the general in vivo PK properties also needed to be improved.16
To obtain JNK3 inhibitors having higher drugability for CNS applications, our optimizations will focus on two areas, as shown in Figure 1: the left-side N-phenyl amide group (Area-A) and the right-side aniline phenyl moiety (Area-B). Based on the cocrystal structure of 2 in JNK3,16 small substitutions (R1) (other than the 2-Cl) to the aniline phenyl group (Area-B) could be tolerated. We reasoned that substitutions by electron-withdrawing groups might be able to improve PK properties and reduce potential metabolic toxicity. Our previous SAR studies demonstrated that the left-side amide NH moiety is indispensable for a high JNK3 affinity. Based on the crystal structure of 2 in JNK3, the phenyl ring of the benzamide provided hydrophobic interactions with surrounding protein residues and thus could be replaced by other aromatic structures without significantly affecting the JNK3 inhibition activity.
We herein report the SAR optimizations based on these two areas of lead compounds 1 and 2. Highly potent and selective (including isoform selectivity against JNK1) JNK3 inhibitors were obtained from the optimizations. In addition, the optimized novel JNK3 inhibitors exhibited oral bioavailability and good brain penetration. The synthesis, purification, and characterization of all JNK3 inhibitors (analogues 3–28) were following similar procedures as those described in our previous publication (also see the Supporting Information).
The first step in our optimizations was to replace the phenyl ring in Area-A with a 5-membered aromatic moiety. We started by using a thiazole ring. As shown in Figure 2, the thiazole analogue 3 had no inhibitions to either JNK3 or JNK1. We reasoned that the H-bond acceptor nitrogen in the thiazole ring might have disturbed the optimal hinge-binding patterns for the inhibitor to JNK3. We then switched to no-nitrogen-containing thiophene moieties for a replacement. Indeed, much better JNK3 inhibitory activities were observed. The 2,6-disubstitution pattern analogue 4 gave a submicromolar IC50 JNK3 inhibition (0.8 μM) while still keeping a good isoform selectivity against JNK1. Even higher JNK3 inhibition (and isoform selectivity against JNK1) was observed for the 2,4-disubstitution analogue 5 (IC50: 0.2 μM). The best JNK3 inhibitor was obtained when a 3,5-disubstituted thiophene ring was applied (analogue 6), which exhibited a JNK3 inhibition IC50 of 0.05 μM and had high isoform selectivity against JNK1 (IC50: 3.6 μM, 72-fold selectivity). Moreover, both 5 and 6 had almost no inhibitions to their closely related kinase p38α (IC50 > 20 μM). Therefore, the chemotype incorporating a 3,5-disubstituted thiophene ring (as in 6) will be used for further optimizations.
Figure 2.

Optimization of Area-A: replacement of the benzamide phenyl ring by a 5-membered aromatic moiety.
We then attempted to optimize the amide moiety in compound 6. As shown in Table 1, various amides were prepared to optimize the JNK3 inhibitory potency and isoform selectivity against JNK1 and JNK2 (assessed by the ratio of the IC50 value of JNK1 or JNK2 over JNK3). In terms of JNK3 inhibitory activity, simple alkyl amides (7 and 8) resulted in a reduction of JNK3 inhibition compared to the heterocyclic ring-containing amide 6. Replacing the azetidine with an R-configured pyrrolidine (9) also reduced the JNK3 inhibition. N-Methylation to the ring-nitrogen (10) gained some activity. Interestingly, deuteration to the N-methyl group (11) did not affect the activity at all. On the other hand, changing the chirality of the pyrrolidine ring from R to S (12) led to an increase in JNK3 inhibitory activity. Moreover, application of a bulkier N-substitution (13) further increased the JNK3 inhibition. Further increasing the ring size led to a slight decrease in JNK3 inhibition (14 vs 12). However, replacing the ring nitrogen with an oxygen (15) could gain JNK3 inhibitory activity. In addition, replacing the 2H-pyran in 15 by a tetrahydrofuran ring (16) led to a compound with a very high JNK3 inhibitory activity (IC50: 8 nM). Further decreasing the ring size by applying an oxetane moiety (17) resulted in some loss of JNK3 inhibitory activity, but the IC50 of 17 was still comparable to or slightly better than that of lead 6. Interestingly, α-methyl substitution to the oxetane ring (18) did not significantly affect the JNK3 inhibition. Insertion of a methylene group between the heterocyclic ring and the amine moiety (compounds 19 to 24) led to deteriorated JNK3 inhibitors. Generally, these compounds had lower JNK3 inhibitions as compared to their corresponding analogues, such as 19 vs 14, 21 vs 12, and 22 vs 16. The only exception is compound 24. Its IC50 value was about half that of its corresponding analogue 15.
Table 1. SAR Studies for the Amide Moietya.


aIC50 is the mean of ≥2 experiments with errors within 30% of the mean. bNot determined. cR: ratio of IC50 values for JNK1, JNK2 over JNK3
Similar to lead inhibitors 5 and 6, the IC50 values against p38α for compounds listed in Table 1 were all greater than 20 μM, indicating a generally excellent selectivity of this pyrazole-urea based scaffold against kinase p38α (the selectivity for most compounds is >100-fold). Ιn terms of the isoform selectivity against JNK1, some compounds showed a fair selectivity (R value between 20 and 50), such as compounds 9, 14, 15, and 19–22; many compounds exhibited a good selectivity (R value between 50 and 100), such as compounds 6, 8, 10–14, 18, and 24; and a few compounds demonstrated an excellent selectivity (R value greater than 100), such as compounds 7, 16, 17, and 23. In summary, amides with a nitrogen-containing heterocyclic ring gave lower selectivity, and amides with a methylene moiety inserted between the heterocycle and the amino group exhibited worse isoform selectivity against JNK1. The rationales for isoform selectivity of JNK3 over JNK1 have been elucidated in our previous publications.26
In our previous studies, we demonstrated that, by crystallography and sequence residue mutation studies, isoform selectivity against JNK2 is not easy to achieve.26 Nevertheless, we still observed fair to excellent isoform selectivity against JNK2 in some of our lead compounds. For example (Table 1), compounds 14, 15, and 24 all had a JNK2 IC50 value in the micromolar range and gave an isoform selectivity vs JNK3 between 20- and 30-fold; compounds 8, 12, and 17 exhibited an even better selectivity of around 40-fold. An exceptionally high isoform selectivity was observed for compound 16, which is >250-fold. Although the residues inside and around the binding pockets are almost identical, we believe that the sequence difference between JNK2 and JNK3 can still yield different sizes and shapes for their binding pockets, and that might be the reason for the observed isoform selectivity of JNK3 against JNK2 for some of our lead compounds.
In order to identify lead compounds for further optimizations (Area-B, for example), in vitro DMPK and cytotoxicity assays were performed to evaluate selected compounds in Table 1. As shown in Table 2, these JNK3 inhibitors were clean in CYPs inhibitions and they exhibited low inhibitory activities against the four select P450 isozymes. Stabilities in human and mouse microsomes were also fair to good for most compounds listed in Table 2. In the human microsome, amides with a nitrogen-containing heterocyclic ring gave better stability than those compounds with an oxygen-containing heterocyclic ring (12/14 vs 15/16). Interestingly, the opposite trend was observed in cytotoxicity assays; compounds with an oxygen-containing heterocyclic ring demonstrated lower toxicity than compounds with a nitrogen-containing heterocyclic ring (16/17 vs 12/14). Although compound 24 was very potent for JNK3 and had isoform selectivity, it had serious liability in human microsomal stability. Therefore, based on overall consideration of JNK3 inhibitory potency, isoform selectivity (against JNK1/JNK2), in vitro DMPK properties, and cytotoxicity, compounds 16 and 17 were selected as new leads for further optimizations.
Table 2. Data for P450 Inhibition, Microsomal Stability, and MTT Cytotoxicity Assays for Selected JNK3 Inhibitors.
| CYP-450% inh.a | mic.
stability t1/2 (min) |
|||
|---|---|---|---|---|
| cmpd | 1A2/2C9/2D6/3A4 | human | mouse | MTT cell viability at 10 μMb |
| 8 | –2/–4/–29/–45 | 49 | 35 | 103% |
| 12 | –1/–9/20/–18 | 54 | 20 | 94% |
| 14 | –6/–19/–5/–34 | 92 | 15 | 89% |
| 15 | 32/7/0/–35 | 32 | 16 | 97% |
| 16 | 17/38/1/–20 | 33 | 13 | 112% |
| 17 | 6/8/–15/–34 | 66 | 24 | 104% |
| 24 | 36/64/32/21 | 8 | 15 | 95% |
% inh. at 10 μM.
Data were the average of ≥2 experiments performed in SHSY5Y cells.
In our previous SAR studies for this pyrazole-urea scaffold, we reported that, among monosubstituted (on the aniline moiety) analogues, the 2-Cl substitution was the best in terms of JNK3 inhibitory potency and selectivity.16 The crystal structure of lead 2 in JNK3 indicated that Area-B (see Figure 1) of compounds derived from this chemotype binds to the selectivity pocket of JNK3, and only small substitutions (other than the 2-Cl) to the phenyl ring could be tolerated.16 Therefore, F-substitution will be applied to further optimize Area-B of leads 16 and 17. This extra F-substitution is expected to stabilize the aniline moiety, thus could lead to improved DMPK properties. In addition, the electron-withdrawing character of an F-substituent might also be able to reduce the potential metabolic toxicities associated with anilines.
As shown in Figure 3, the 6-F substituted analogue 25 gave a similar JNK3 inhibitory potency, while the 4-F substituted analogue 26 exhibited a slightly lower potency as compared to lead 17. Performing α-methylation to the thiophene ring of 25 resulted in a compound (27) which also had a slightly reduced JNK3 inhibitory potency. On the other hand, applications of both a 6-F substitution and an α-methylation (on the thiophene ring) to 16 resulted in a compound (28) which had a 7-fold reduction in JNK3 inhibitions. It is important to point out that new compounds incorporating these structural modifications still had high selectivity against p38α and still kept a high isoform selectivity against JNK1 (all >100-folds) and JNK2.
Figure 3.

Further optimizations to lead inhibitors 16 and 17 (Area-B optimizations and methylation to the thiophene ring).
As shown in Table 3, the fluorinated analogues were also clean in CYP inhibitions, and their cytotoxicity were still low at 10 μM. As expected, the microsomal stabilities (in both human and mouse microsomes) were much higher as compared to leads 16 and 17. Interestingly, α-methylation to the thiophene ring resulted in a lower microsomal stability in mice (27 vs 25).
Table 3. Data for P450 Inhibition, Microsomal Stability, and MTT Cytotoxicity Assays for Optimized JNK3 Inhibitors.
| CYP-450% inh.a | mic.
stability t1/2 (min) |
|||
|---|---|---|---|---|
| cmpd | 1A2/2C9/2D6/3A4 | human | mouse | MTT cell viability at 10 μMb |
| 25 | 36/48/31/24 | 100 | 72 | 102% |
| 26 | 38/47/30/–5 | >120 | 68 | 104% |
| 27 | 22/33/1/16 | 110 | 57 | 111% |
| 28 | 34/35/17/26 | 40 | 38 | 114% |
% inh. at 10 μM.
Data were the average of ≥2 experiments performed in SHSY5Y cells.
Kinase panel profiling studies were applied to evaluate the general selectivity of lead JNK3 inhibitors. Thus, 17 and 25 at 5 μM and 27 at 10 μM (the profiling concentration was set to be ≥100-fold of the JNK3 IC50 value of each compound) were subjected to profiling studies against a panel of ∼374 wild-type kinases (Reaction Biology Corporation, www.reactionbiology.com). Data (see Supporting Information) showed that all three compounds had significant inhibition (>80% inhibition) against only JNK3 at the profiled concentrations. These results demonstrated that JNK3 inhibitors derived from this thiophenyl-pyrazolourea scaffold had not only isoform selectivity (vs JNK1/2) but also a broadly high selectivity against other kinases. Moreover, the profiling data revealed that replacement of the phenyl ring in our previously published inhibitors (leads 1 and 2) by a thiophenyl ring favored JNK3 inhibition and had further increased the general kinase selectivity.
Crystallography studies were applied to reveal rationales for the favored JNK3 inhibition of this thiophenyl pyrazolourea chemotype. Thus, cocrystal structures of inhibitors 17, 25 and 27 (shown in the TOC Graphic) with human JNK3 were pursued. The 1.84 Å crystal structure of 17 (Figure 4) showed that, in the ATP-binding pocket, the pyrazole nitrogen atom and the amide NH moiety H-bonded to hinge residue M149 of JNK3. Another key H-bonding interaction from this mode was the water-bridged H-bonds between the aniline-urea NH and the side-chain NH2 of residue K93. Water-bridged H-bonds were also observed for the amide carbonyl group with nearby protein residues. The amide heterocyclic ring pointed to the outside of the binding pocket, and toward the solvent. The aniline moiety bound to hydrophobic pocket-I (the selectivity pocket), which is responsible mainly for the isoform selectivity against JNK1. The thiophenyl moiety positioned around hydrophobic pocket-II and its aromatic ring was a little twisted (∼30°) with the central pyrazole ring. Hydrophobic interactions in both hydrophobic pocket-I and hydrophobic pocket-II and that around the pyrazole ring all contributed to the high JNK3 affinity.
Figure 4.
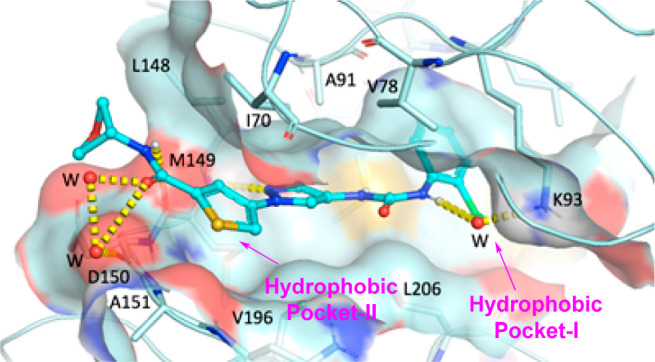
Overlay X-ray crystal structure of inhibitor 17 in human JNK3 (2.0 Å).
This crystal structure binding mode provides the rationales for the high general selectivity of the thiophene-pyrazolo-urea based JNK3 inhibitors. Basically, there are two major reasons: (1) binding to the Selectivity Pocket which is unique to JNK3;26 (2) the existence of a urea moiety in these compounds, located right under the P-loop. In most other ATP-competitive kinase inhibitors, the corresponding position is normally a large aromatic ring, which renders strong hydrophobic interactions and not much differentiation among various kinases.
We previously reported that JNK3 phosphorylates APP and facilitates its rapid endocytosis and processing, producing Aβ42.4 Aβ42 then activates AMP-activated protein kinase (AMPK) in neurons, which inhibits the mTOR translational pathway by phosphorylating Raptor. This translational block leads to ER stress, which activates JNK3 again, forming a positive feedback loop that accelerates Aβ42 production. To assess the feasibility of applying our newly developed JNK3 inhibitors to reduce and/or to slow down this process, we evaluated the cell-based inhibition capability of selected compounds on the phosphorylation of APPT668. As shown in Figure 5 (A,B,C), JNK3 inhibitors 17, 26, and 28 significantly inhibited the phosphorylation of residue T668 of APP, even at a concentration of as low as 10 nM.
Figure 5.

Cell-based inhibition of APPT668 phosphorylation in primary neurons in the presence or absence of JNK3 inhibitors: (A) 17, (B) 26, and (C) 28.
Finally, in vivo brain exposure experiments were performed for compound 17 in mice to assess the oral bioavailability and brain penetration capability of lead JNK3 inhibitors. As shown in Table 4, po dosing for compound 17 exhibited significant brain presence of the JNK3 inhibitor, indicating that 17 is brain penetrant and has oral bioavailability.
Table 4. Brain and Plasma Concentration of Inhibitors 17 in Micea.
| cmpd (dosing) | plasma concentration | brain concentration |
|---|---|---|
| 17 (po, 10 mg/kg) | 0.96 μM | 1.08 μM |
Data were generated from more than three determinations, and samples collected 1 h after dosing. Formulation: DMSO/PEG400/PBS: 1:4:5 by volume.
In summary, we have developed a class of highly selective (including isoform selectivity) and potent JNK3 inhibitors based on a thiophenyl pyrazolourea chemotype. The optimized inhibitors had significant inhibition to only JNK3 in a panel of 374 wild-type kinases, and were potent in inhibiting the phosphorylation of APPT668 of primary cortical and hippocampal neurons. Compared to the starting lead compound 2, the replacement of the phenyl ring (as in 2) by a thiophenyl ring had significantly improved the general selectivity against other kinases. In addition, the newly optimized JNK3 inhibitors had good in vitro and in vivo DMPK properties, and more importantly had oral bioavailability and were brain penetrant, which are important for CNS applications. Compared to other reported JNK3 inhibitors, our thiophenyl pyrazolourea based JNK3 lead compounds have both high selectivity (including isoform selectivity) and excellent in vivo PK properties. Future research for these optimized JNK3 inhibitors will mainly focus on pharmacodynamics and in vivo efficacy studies in animal models against various neurodegenerative diseases, such as AD and PD. These studies will be reported in due course.
Acknowledgments
This work was supported by NIH/NIA grant RO1AG055059 (S.O.Y and Y.F) and ADDF (S.O.Y).
Glossary
Abbreviations
- JNK
c-jun N-terminal kinases
- ATP
adenosine triphosphate
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ALS
Amyotrophic lateral sclerosis
- SAR
structure–activity relationship
- DMPK
drug metabolism and pharmacokinetics.
Biographies
Yangbo Feng is Director of Medicinal Chemistry at Reaction Biology Corporation (RBC). His research interests are in small molecule drug discovery focusing on CNS and oncology diseases. Yangbo received his Ph.D degree from University of California, San Diego under the guidance of Professor Murray Goodman. Subsequently he joined Professor Kevin Burgess’s group at Texas A&M University, College Station to pursue postdoctoral researches. Prior to joining RBC, Yangbo was Associate Scientific Director of Medicinal Chemistry in the Translational Research Institute of Scripps Florida, where he directed a research group focusing on chemical biology, medicinal chemistry, and combinatorial chemistry.
Sung Ok Yoon is a professor in the Department of Biological Chemistry and Pharmacology at the Ohio State University Medical School. She directs a research group that focuses on understanding neurodegenerative diseases, such as Alzheimer’s disease. A goal of her team is to utilize the research toward developing inhibitors to treat these diseases.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00533.
Details for the synthesis of 3–28; 1H NMR data; LC-MS data; procedures for bioassays and DMPK studies; kinase panel profiling data for 17, 25, and 27; cocrystal structures of 17, 25, and 27 in human JNK3 (PDF)
The authors declare no competing financial interest.
Notes
The coordinates of JNK3:17, JNK3:25, and JNK3:27 complexes were deposited under PDB ID 7KSK, 7KSJ and 7KSI, respectively.
Supplementary Material
References
- Derijard B.; Hibi M.; Wu I. H.; Barrett T.; Su B.; Deng T. L.; Karin M.; Davis R. J. Jnk1 - a Protein-Kinase Stimulated by Uv-Light and Ha-Ras That Binds and Phosphorylates the C-Jun Activation Domain. Cell 1994, 76, 1025–1037. 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Barrett T.; Whitmarsh A. J.; Cavanagh J.; Sluss H. K.; Derijard B.; Davis R. J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. 10.1002/j.1460-2075.1996.tb00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Shi X. Q.; Zhang Q. J.; Hampong M.; Paddon H.; Wahyuningsih D.; Pelech S. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. J. Biol. Chem. 2002, 277, 43648–43658. 10.1074/jbc.M203214200. [DOI] [PubMed] [Google Scholar]
- Yoon S. O.; Park D. J.; Ryu J. C.; Ozer H. G.; Tep C.; Shin Y. J.; Lim T. H.; Pastorino L.; Kunwar A. J.; Walton J. C.; Nagahara A. H.; Lu K. P.; Nelson R. J.; Tuszynski M. H.; Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 2012, 75, 824–37. 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Fei Z.; Luo S.; Wang H. MiR-335–5p Inhibits beta-Amyloid (Abeta) Accumulation to Attenuate Cognitive Deficits Through Targeting c-jun-N-terminal Kinase 3 in Alzheimer’s Disease. Curr. Neurovasc Res. 2020, 17, 93–101. 10.2174/1567202617666200128141938. [DOI] [PubMed] [Google Scholar]
- Morishima Y.; Gotoh Y.; Zieg J.; Barrett T.; Takano H.; Flavell R.; Davis R. J.; Shirasaki Y.; Greenberg M. E. beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourmaud S.; Paquet C.; Dumurgier J.; Pace C.; Bouras C.; Gray F.; Laplanche J. L.; Meurs E. F.; Mouton-Liger F.; Hugon J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J. Psychiatry Neurosci 2015, 40, 151–61. 10.1503/jpn.140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker C. E.; Khan S.; Cameron M. D.; Robertson H. A.; Robertson G. S.; LoGrasso P. JNK Inhibition Protects Dopamine Neurons and Provides Behavioral Improvement in a Rat 6-Hydroxydopamine Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 207–212. 10.1021/cn1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S.; Vila M.; Teismann P.; Davis R. J.; Hirsch E. C.; Przedborski S.; Rakic P.; Flavell R. A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 665–70. 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V.; Silva R. M.; Oo T. F.; Cheng H. C.; Rzhetskaya M.; Kholodilov N.; Flavell R. A.; Kuan C. Y.; Rakic P.; Burke R. E. JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. J. Neurochem. 2008, 107, 1578–88. 10.1111/j.1471-4159.2008.05713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G. A.; You Y. M.; Pollema S. L.; Kaminska A.; Liu K.; Yoshioka K.; Bjorkblom B.; Coffey E. T.; Bagnato C.; Han D.; Huang C. F.; Banker G.; Pigino G.; Brady S. T. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–71. 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D. D.; Kuan C. Y.; Whitmarsh A. J.; Rincon M.; Zheng T. S.; Davis R. J.; Rakic P.; Flavell R. A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Kuan C. Y.; Whitmarsh A. J.; Yang D. D.; Liao G.; Schloemer A. J.; Dong C.; Bao J.; Banasiak K. J.; Haddad G. G.; Flavell R. A.; Davis R. J.; Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15184–9. 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lemos L.; Junyent F.; Verdaguer E.; Folch J.; Romero R.; Pallas M.; Ferrer I.; Auladell C.; Camins A. Differences in activation of ERK1/2 and p38 kinase in Jnk3 null mice following KA treatment. J. Neurochem. 2010, 114, 1315–22. 10.1111/j.1471-4159.2010.06853.x. [DOI] [PubMed] [Google Scholar]
- Genabai N. K.; Ahmad S.; Zhang Z.; Jiang X.; Gabaldon C. A.; Gangwani L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 6986–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K.; Iqbal S.; Hernandez P.; Park H.; LoGrasso P. V.; Feng Y. Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives. J. Med. Chem. 2014, 57, 10013–30. 10.1021/jm501256y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K. Pyridopyrimidinone Derivatives as Potent and Selective c-Jun N-Terminal Kinase (JNK) Inhibitors. ACS Med. Chem. Lett. 2015, 6, 413–8. 10.1021/ml500474d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graczyk P. P.; Khan A.; Bhatia G. S.; Palmer V.; Medland D.; Numata H.; Oinuma H.; Catchick J.; Dunne A.; Ellis M.; Smales C.; Whitfield J.; Neame S. J.; Shah B.; Wilton D.; Morgan L.; Patel T.; Chung R.; Desmond H.; Staddon J. M.; Sato N.; Inoue A. The neuroprotective action of JNK3 inhibitors based on the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole scaffold. Bioorg. Med. Chem. Lett. 2005, 15, 4666–70. 10.1016/j.bmcl.2005.07.076. [DOI] [PubMed] [Google Scholar]
- Jung H.; Aman W.; Hah J. M. Novel scaffold evolution through combinatorial 3D-QSAR model studies of two types of JNK3 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2139–2143. 10.1016/j.bmcl.2017.03.063. [DOI] [PubMed] [Google Scholar]
- Tuffaha G. O.; Hatmal M. M.; Taha M. O. Discovery of new JNK3 inhibitory chemotypes via QSAR-Guided selection of docking-based pharmacophores and comparison with other structure-based pharmacophore modeling methods. J. Mol. Graphics Modell. 2019, 91, 30–51. 10.1016/j.jmgm.2019.05.015. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; Laughlin J. D.; Park H.; LoGrasso P. V.; Patricelli M.; Nomanbhoy T. K.; Sorger P. K.; Alessi D. R.; Gray N. S. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X.; Huang H.; Li Y.; Jiang L.; Wang Y.; Jin H.; Jiao N.; Zhang L.; Zhang L.; Liu Z. Multistage Screening Reveals 3-Substituted Indolin-2-one Derivatives as Novel and Isoform-Selective c-Jun N-terminal Kinase 3 (JNK3) Inhibitors: Implications to Drug Discovery for Potential Treatment of Neurodegenerative Diseases. J. Med. Chem. 2019, 62, 6645–6664. 10.1021/acs.jmedchem.9b00537. [DOI] [PubMed] [Google Scholar]
- Dou X.; Huang H.; Jiang L.; Zhu G.; Jin H.; Jiao N.; Zhang L.; Liu Z.; Zhang L. Rational modification, synthesis and biological evaluation of 3,4-dihydroquinoxalin-2(1H)-one derivatives as potent and selective c-Jun N-terminal kinase 3 (JNK3) inhibitors. Eur. J. Med. Chem. 2020, 201, 112445. 10.1016/j.ejmech.2020.112445. [DOI] [PubMed] [Google Scholar]
- Oh Y.; Jang M.; Cho H.; Yang S.; Im D.; Moon H.; Hah J. M. Discovery of 3-alkyl-5-aryl-1-pyrimidyl-1H-pyrazole derivatives as a novel selective inhibitor scaffold of JNK3. J. Enzyme Inhib. Med. Chem. 2020, 35, 372–376. 10.1080/14756366.2019.1705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning A. M.; Davis R. J. Targeting JNK for therapeutic benefit: From JuNK to gold?. Nat. Rev. Drug Discovery 2003, 2, 554–565. 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Park H.; Iqbal S.; Hernandez P.; Mora R.; Zheng K.; Feng Y.; LoGrasso P. Structural Basis for JNK2/3 Isoform Selective Aminopyrazoles. Sci. Rep. 2015, 5, 8047. 10.1038/srep08047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.