

Abstract
Interleukin-1 receptor associated kinase 4 (IRAK4) is a promising therapeutic target for diffuse large B-cell lymphoma driven by MYD88 L265P mutant, acting both as a kinase and a scaffolding protein for downstream signaling molecules. While previous efforts to modulate IRAK4 activity with kinase inhibitors alone displayed moderate efficacy, protein degradation may offer a solution to blocking both IRAK4 kinase activity and scaffolding capabilities. To this end, the potent IRAK4 degrader 9 was discovered, and it effectively inhibited the activation of downstream NF-κB signaling and outperformed the parent compound 1. In addition, compound 9 displayed a substantial advantage in reduction of the viability of OCI-LY10 and TMD8 cells over the parent compound 1. These results underline the potential that eliminating both the kinase and scaffolding functions of IRAK4 may result in superior and broader efficacy than inhibiting the kinase activity alone.
Keywords: IRAK4, PROTAC, protein degrader, scaffolding role, diffuse large B-cell lymphoma
In activated B cell-like diffuse large B-cell lymphoma (ABC-DLBCL) cells, an L265P mutation of myeloid differentiation primary response 88 (MYD88) induces receptor-activation-independent oligomerization of the Myddosome, which comprises MYD88 and members of the interleukin-1 receptor associated kinase (IRAK) family. Then formation of the Myddosome leads to an increase in the kinase activity of IRAK4, resulting in phosphorylation of the IκB kinase (IKK) complex and constitutive nuclear factor kappa B (NF-κB) activation to promote B-cell proliferation and survival.1,2 IRAK4 is an essential mediator to promote tumor growth in MYD88 mutant lymphoma.1 Several IRAK4 kinase inhibitors targeting DLBCL have been discovered, but these selective IRAK4 kinase inhibitors with great inhibitory potency had merely modest or very little antitumor activity as single agents against ABC-DLBCL in vitro or in vivo.3−6 The poor activity indicates that inhibition of IRAK4 kinase activity alone may not be sufficient to block MYD88-driven signaling because of a nonkinase function of IRAK4. Meanwhile, Vollmer et al. reported that the catalytic activity of downstream IRAK1 was triggered by an allosteric mechanism induced by its interaction with IRAK4, indicating a potential scaffolding function of IRAK4.7 Besides, several studies also demonstrated that specific inhibition of IRAK4 kinase activity only partially inhibited NF-κB and mitogen-activated protein kinase (MAPK) signaling because the Myddosome formation was stabilized in the absence of IRAK4 kinase activity.8−10 These results indicate that the scaffolding function of IRAK4 plays an important role in the MYD88–IRAK4–NF-κB signaling pathway.
Targeted protein degradation using the proteolysis targeting chimera (PROTAC) technology has emerged as a novel therapeutic modality in drug discovery.11−15 A PROTAC is a heterobifunctional molecule that consists of three key structural components: a protein of interest (POI) ligand, an E3 ubiquitin ligase recruiting ligand, and a linker connecting these two ligands.16 PROTACs mediate the degradation of selected POIs by hijacking the activity of E3 ubiquitin ligases for POI ubiquitination and subsequent degradation by the proteasome system.17 Unlike conventional tactics that only inhibit IRAK4 kinase activity, PROTAC degraders of IRAK4 may eliminate both the kinase and scaffolding functions, presumably providing a more potential approach to blocking MYD88–IRAK4–NF-κB signal transduction.
Recently, Nunes and co-workers identified an IRAK4 PROTAC with potent degradation of IRAK4 in peripheral blood mononuclear cells (PBMCs), concentrating on autoimmune diseases.18 To date, some work on PROTACs targeting BTK for DLBCL has been described.19,20 Herein we disclose a series of PROTAC degraders of IRAK4 and focus on the antiproliferative effects against ABC-DLBCL cell lines and the suppression of signaling molecules downstream of IRAK4.
Herein, to prepare IRAK4 PROTAC molecules, we chose to incorporate IRAK4 inhibitor 1 (Figure 1a),21 which potently binds to the IRAK4 kinase domain (IRAK4 IC50 = 70.0 ± 10.5 nM in our hands), and the cereblon (CRBN) E3 ligase ligand pomalidomide into one compound. According to the results of the docking study, compound 1 showed a similar binding mode as the original ligand22 (Figure S1), and the 2,2-dimethylpentanoate moiety was the optimal position to attach the linkers such that critical binding interactions with IRAK4 were not disrupted (Figure 1b).We synthesized a set of PROTAC molecules by conjugating compound 1 to pomalidomide via flexible linkers of various lengths, including hydrophilic polyethylene glycol (PEG) and hydrophobic all-carbon chains (Table 1).
Figure 1.
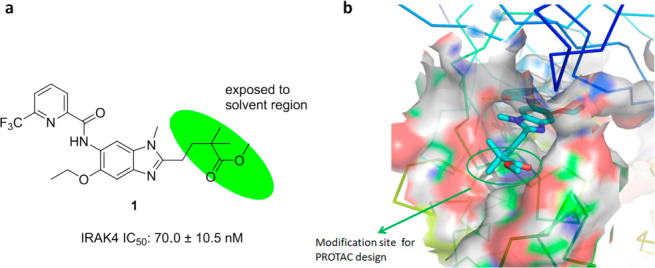
(a) Chemical structure of potent IRAK4 inhibitor 1. (b) Binding mode of compound 1 with the IRAK4 protein (PDB entry 6UYA), showing the 2,2-dimethylpentanoate moiety as a solvent-exposed vector.
Table 1. Chemical Structures of Compounds 2–9 and 9-Me.


The synthetic route to access PROTACs 2–9 and 9-Me is summarized in Scheme 1. First, amine 11 was synthesized by a ring-closure reaction of 10 with 5-methoxy-4,4-dimethyl-5-oxopentanoic acid followed by removal of Boc. Then EDCI/HOBT-mediated amidation of amine 11 with 6-(trifluoromethyl)picolinic acid afforded 1, which was subsequently hydrolyzed with lithium hydroxide to give acid 12. Thalidomide analogues 14a and 14b were prepared from 3-fluorophthalic anhydride by a general procedure.23 Nucleophilic aromatic substitution of fluoro-substituted thalidomide 14a or its N-methyl analogue 14b with mono-Boc-protected diamines followed by removal of Boc afforded linker-attached thalidomide analogues 15a–i. Finally, PROTAC molecules 2–9 and 9-Me were prepared by coupling of acid 12 with amines 15a–i in the presence of EDCI and HOBt.
Scheme 1. Synthesis of Compounds 2–9 and 9-Me.

Reagents and conditions: (a) (i) 5-methoxy-4,4-dimethyl-5-oxopentanoic acid, HATU, DIPEA, DMF, rt; (ii) CH3COOH, rt; (b) TFA, DCM, rt; (c) 6-(trifluoromethyl)picolinic acid, EDCI, HOBT, DIPEA, DMF, rt; (d) LiOH·H2O, THF/H2O, 50 °C; (e) amine 15, EDCI, HOBT, DIPEA, DMF, rt; (f) 2,6-dioxopiperidine-3-ammonium chloride, NaOAc, CH3COOH, reflux; (g) MeI, K2CO3, DMF, 60 °C; (h) mono-Boc-protected diamine, DIPEA, NMP, microwave, 90 °C.
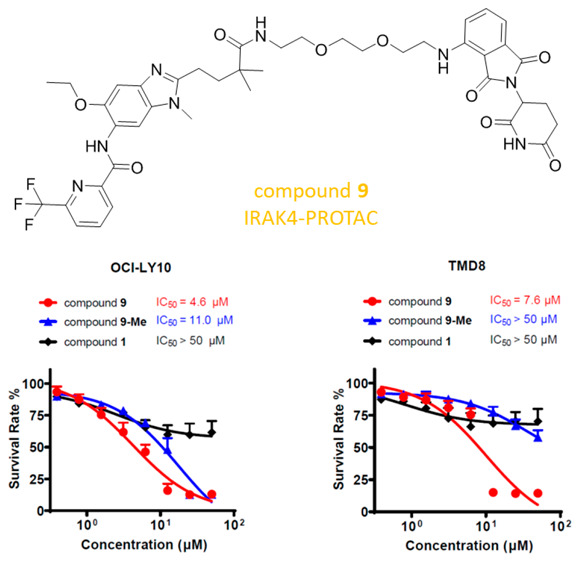
We chose two ABC-DLBCL cell lines, OCI-LY10 and TMD8, harboring the MYD88 L265P mutation, to test the cellular activity of compounds 2–9. We initially evaluated the IRAK4 degradation effect by immunoblotting analysis after 24 h treatment with the compounds at a concentration of 1 μM. The results showed that compounds 2–8 with shorter linkers afforded almost no IRAK4 degradation in OCI-LY10 cells (Figure 2a). In contrast, compound 9, a PROTAC with a PEG2 linker, vastly decreased the IRAK4 protein level. Similarly, compound 9 also potently reduced the IRAK4 protein level in TMD8 cells. Meanwhile, compound 7 displayed moderate but weaker degradation activity compared with compound 9. In addition, compounds 2–6 and 8 scarcely demonstrated IRAK4 degradation, which revealed the superiority of compound 9 (Figure 2b). There are many potential reasons why these shorter PROTACs did not degrade IRAK4. The most likely reason may be that the length of the shorter linker could not facilitate the formation of a stable “target–PROTAC–ligase” ternary complex. The longer linker promoted ternary complex formation and promoted PROTAC efficacy.
Figure 2.
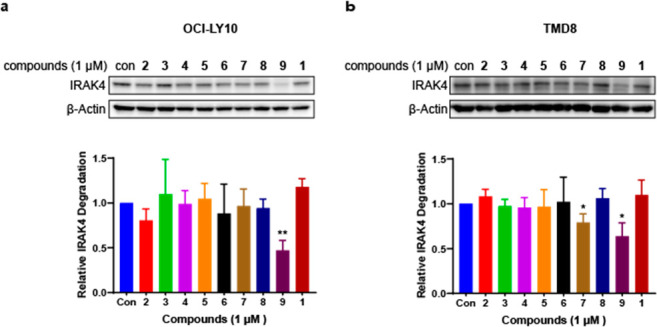
IRAK4 protein levels of (a) OCI-LY10 and (b) TMD8 cells treated with compounds 2–9 for 24 h were determined by Western blot and quantified relative to the DMSO control. All Western blot figures represent the results from at least three independent experiments. Data are shown as mean ± standard deviation (SD), and significance of differences was determined by Student’s t test (*, p < 0.05; **, p < 0.01).
We then measured the dynamic effects of compound 9 on IRAK4 degradation in tumor cells. A dose–response study showed that compound 9 effectively reduced the IRAK4 protein level in a concentration-dependent manner in OCI-LY10 (Figure 3a) and TMD8 cells (Figure 3b). Moreover, a time–response study revealed that compound 9 rendered a time-dependent decrease in IRAK4 level at a concentration of 1 μM in these cells (Figure 3c,d).
Figure 3.
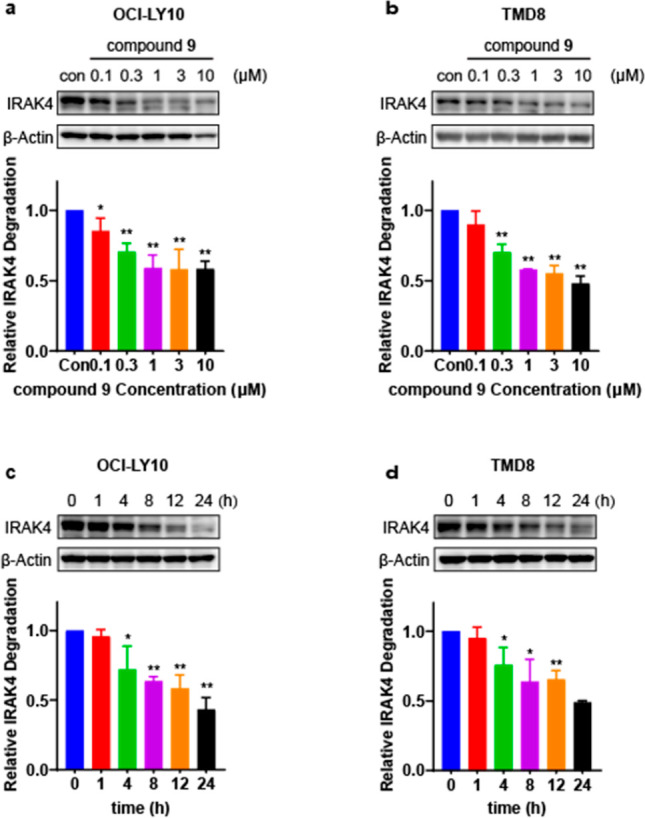
Compound 9 induced concentration- and time-dependent depletion of IRAK4 protein in OCI-LY10 and TMD8 cells. (a, b) OCI-LY10 and TMD8 were incubated for 24 h with various concentrations of 9. (c, d) OCI-LY10 and TMD8 were incubated with 1 μM 9 for different periods of time. Protein levels were visualized by immunoblotting and quantified relative to the DMSO control. All Western blot figures represent the results from at least three independent experiments. Data were showed as mean ± standard deviation (SD), and significance of differences was determined by Student’s t test (*p < 0.05, **p < 0.01).
To confirm that the IRAK4 degradation caused by compound 9 was proteasome-dependent, we designed the negative control compound 9-Me by attaching a methyl group to the key imide NH in 9, which could presumably block the binding of thalidomide analogues to CRBN E3 ligase.24 As expected, in contrast to the efficient IRAK4 degradation induced by compound 9, no IRAK4 protein degradation was detected after 9-Me treatment in either OCI-LY10 or TMD8 cells (Figure 4a). To explore whether the proteasome system contributed to compound 9’s IRAK4 degradation activity, we preincubated OCI-LY10 and TMD8 cells with the proteasome inhibitor MG-132 and then detected the target protein abundance upon treatment with compound 9. As shown in Figure 4b, pretreatment with the proteasome inhibitor MG-132 (1 μM) for 2 h significantly abolished the IRAK4 degradation induced by compound 9. Furthermore, we assumed that the CRBN recruiting ligand and IRAK4 inhibitor could competitively affect the degradation activity of compound 9. Therefore, we investigated the IRAK4 degradation effect of compound 9 alone and upon cotreatment with excess pomalidomide, which recruits CRBN, or excess IRAK4 inhibitor 1 (Figure 4c). The results showed that pomalidomide (10 μM) or parent compound 1 (10 μM) were effective at abrogating the IRAK4 degradation induced by compound 9, indicating that degradation of the IRAK4 protein by PROTAC compound 9 required competitive binding to CRBN or IRAK4. Overall, these results together with the data for control compound 9-Me demonstrate that compound 9 degrades IRAK4 protein through a CRBN- and proteasome-dependent mechanism and that CRBN or IRAK4 inhibitors can antagonize the degradation at higher concentrations.
Figure 4.

Evaluation of the mechanism of action of IRAK4 degradation induced by compound 9 in OCI-LY10 and TMD8 cells. (a) The negative control compound 9-Me showed no IRAK4 degradation in OCI-LY10 and TMD8 cells. (b) Western blot for IRAK4 after pretreatment with 1 μM MG-132 in OCI-LY10 and TMD8 cells. (c) Western blot for IRAK4 after cotreatment with 10 μM compound 1 and pomalidomide in OCI-LY10 and TMD8 cells.
Harboring the MYD88 L265P mutation triggers tumor growth through activation of the IRAK4–NF-κB signaling pathway in the ABC subtype of DLBCL.1 We then studied the effects of compound 9 on phosphorylation of IRAK4 (p-IRAK4) and the downstream molecules IKK and NF-κB. Compound 9 displayed concentration-dependent inhibition of p-IRAK4 and its downstream signaling, p-IKKα/β and p-NF-κB, in OCI-LY10 cells (Figure 5a). Besides, compound 9 exhibited more potent inhibitory effects on p-NF-κB than compound 9-Me at the same concentration (Figure S3). Analogously, similar inhibition patterns of compound 9 were observed in TMD8 cells (Figure 5b).
Figure 5.
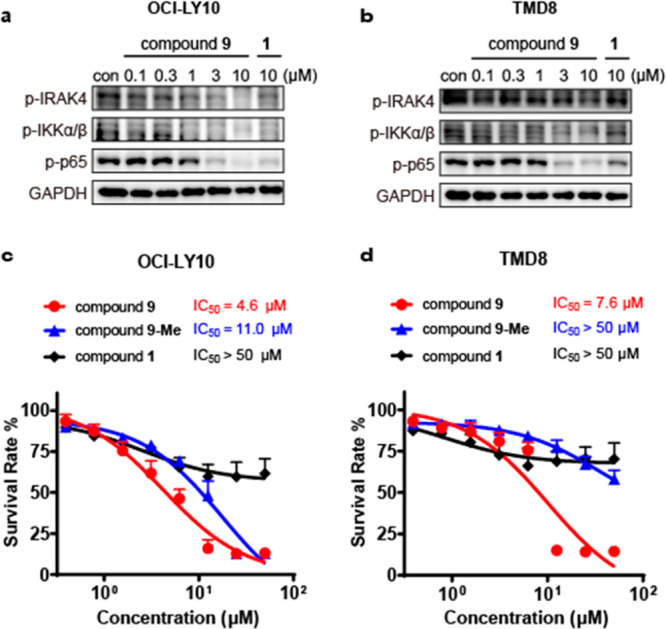
Compound 9 significantly inhibited the growth of MYD88 L265P mutant cell lines and inhibited the signaling molecules downstream of IRAK4 in a concentration-dependent manner. (a, b) Western blotting analysis of signaling molecules downstream of IRAK4 treated with compound 9 in (a) OCI-LY10 and (b) TMD8 cells. (c, d) Antiproliferative activity of compound 9 in (c) OCI-LY10 and (d) TMD8 cells. Data are shown as mean ± SD.
We next compared the antiproliferative effects of compounds 1, 9, and 9-Me in OCI-LY10 (Figure 5c) and TMD8 cells (Figure 5d). Treatment with parent compound 1 as a single agent led to only a small antiproliferative effect (IC50 > 50 μM) against these MYD88 L265P mutant cells. In comparison, compound 9 significantly inhibited proliferation of the cells with IC50 values of 4.6 and 7.6 μM, respectively. On the other hand, the control compound 9-Me was much less active at inhibiting cell growth in these cells (IC50 = 11 μM and IC50 > 50 μM, respectively). On the basis of the above results, the IRAK4 degrader displayed more potent antiproliferative activity toward the DLBCL cells than IRAK4 kinase inhibitor 1. Moreover, compound 9 showed cellular selectivity for ABC-DLBCL cells with mutant MYD88 L265P over wild-type MYD88 (Figure S2). The removal of the IRAK4 kinase activity and scaffold function caused complete reduction of the NF-κB signaling pathway and thus potently inhibited the growth of ABC-DLBCL cells harboring the MYD88 L265P mutant. Although we confirmed the correlation between cytotoxicity against the MYD88 L265P mutant cell line and degradation of IRAK4, a proteomic analysis to measure changes in protein abundance would help assess the selectivity and off targets of degrader 9 in the future.
In conclusion, we have reported a PROTAC molecule, compound 9, that potently degrades kinase IRAK4 in OCI-LY10 and TMD8 cells in a concentration- and time-dependent manner. The PROTAC-induced IRAK4 degradation was dependent on binding to CRBN and was reversed upon blocking of proteasome activity. In addition, compound 9 efficiently blocked the IRAK4–NF-κB signaling pathway and displayed a substantial advantage in inhibiting the growth of cell lines expressing the MYD88 L265P mutant compared with parent compound 1. PROTACs targeting kinase IRAK4 may be worthy of further investigation for the treatment of the MYD88-driven B-cell lymphoma, which is associated with constitutively active NF-κB signaling.
Acknowledgments
This work was supported by funds from the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA12020228), the Science and Technology Commission of Shanghai Municipality (18431907100), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (2018ZX09711002-011-016), and the National Natural Science Foundation of China (21702220).
Glossary
Abbreviations
- ABC
activated B cell-like
- Boc
tert-butoxycarbonyl
- DLBCL
diffuse large B cell lymphoma
- IKK
IκB kinase
- IRAK1
interleukin-1 receptor associated kinase 1
- IRAK4
interleukin-1 receptor associated kinase 4
- MAPK
mitogen-activated protein kinase
- MYD88
myeloid differentiation primary response gene 88
- NF-κB
nuclear factor kappa B
- PBMC
peripheral blood mononuclear cell
- POI
protein of interest
- PROTAC
proteolysis targeting chimera
- TLR
toll-like receptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00474.
Synthetic methods, characterization of key compounds, biological assay protocols, and data (PDF)
Author Contributions
# Y.C. and Y.N. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Ngo V. N.; Young R. M.; Schmitz R.; Jhavar S.; Xiao W.; Lim K.-H.; Kohlhammer H.; Xu W.; Yang Y.; Zhao H.; Shaffer A. L.; Romesser P.; Wright G.; Powell J.; Rosenwald A.; Muller-Hermelink H. K.; Ott G.; Gascoyne R. D.; Connors J. M.; Rimsza L. M.; Campo E.; Jaffe E. S.; Delabie J.; Smeland E. B.; Fisher R. I.; Braziel R. M.; Tubbs R. R.; Cook J. R.; Weisenburger D. D.; Chan W. C.; Staudt L. M. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.-H.; Romero D. L.; Chaudhary D.; Robinson S. D.; Staudt L. M. IRAK4 kinase as a novel therapeutic target in the ABC subtype of diffuse large B cell lymphoma. Blood 2012, 120, 62–62. 10.1182/blood.V120.21.62.62. [DOI] [Google Scholar]
- Chen Y.; Bai G.; Ning Y.; Cai S.; Zhang T.; Song P.; Zhou J.; Duan W.; Ding J.; Xie H.; Zhang H. Design and synthesis of imidazo[1,2-b]pyridazine IRAK4 inhibitors for the treatment of mutant MYD88 L265P diffuse large B-cell lymphoma. Eur. J. Med. Chem. 2020, 190, 112092. 10.1016/j.ejmech.2020.112092. [DOI] [PubMed] [Google Scholar]
- Kelly P. N.; Romero D. L.; Yang Y.; Shaffer A. L. III; Chaudhary D.; Robinson S.; Miao W.; Rui L.; Westlin W. F.; Kapeller R.; Staudt L. M. Selective interleukin-1 receptor–associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. 10.1084/jem.20151074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degorce S. L.; Anjum R.; Bloecher A.; Carbajo R. J.; Dillman K. S.; Drew L.; Halsall C. T.; Lenz E. M.; Lindsay N. A.; Mayo M. F.; Pink J. H.; Robb G. R.; Rosen A.; Scott J. S.; Xue Y. Discovery of a series of 5-azaquinazolines as orally efficacious IRAK4 targeting MyD88 L265P mutant diffuse large B cell lymphoma. J. Med. Chem. 2019, 62, 9918–9930. 10.1021/acs.jmedchem.9b01346. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; Degorce S. L.; Anjum R.; Culshaw J.; Davies R. D. M.; Davies N. L.; Dillman K. S.; Dowling J. E.; Drew L.; Ferguson A. D.; Groombridge S. D.; Halsall C. T.; Hudson J. A.; Lamont S.; Lindsay N. A.; Marden S. K.; Mayo M. F.; Pease J. E.; Perkins D. R.; Pink J. H.; Robb G. R.; Rosen A.; Shen M.; McWhirter C.; Wu D. Discovery and optimization of pyrrolopyrimidine inhibitors of interleukin-1 receptor associated kinase 4 (IRAK4) for the treatment of mutant MYD88 L265P diffuse large B-cell lymphoma. J. Med. Chem. 2017, 60, 10071–10091. 10.1021/acs.jmedchem.7b01290. [DOI] [PubMed] [Google Scholar]
- Vollmer S.; Strickson S.; Zhang T.; Gray N.; Lee K. L.; Rao V. R.; Cohen P. The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists. Biochem. J. 2017, 474, 2027–2038. 10.1042/BCJ20170097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing L.; Stochaj W.; Siegel M.; Czerwinski R.; Dower K.; Wright Q.; Hirschfield M.; Casanova J. L.; Picard C.; Puel A.; Lin L. L.; Rao V. R. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J. Biol. Chem. 2014, 289, 10865–10875. 10.1074/jbc.M113.544809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D.; Balka K. R.; Cardona Gloria Y.; Rao V. R.; Latz E.; Masters S. L. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J. Biol. Chem. 2018, 293, 15195–15207. 10.1074/jbc.RA118.003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S.; Karim F.; Kiessu E.; Cushing L.; Lin L. L.; Ghandil P.; Hoarau C.; Casanova J. L.; Puel A.; Rao V. R. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J. Biol. Chem. 2018, 293, 15208–15220. 10.1074/jbc.RA118.003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis targeting chimeras (PROTACs) in ’beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. Proteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discovery Today: Technol. 2019, 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Sun X.; Gao H.; Yang Y.; He M.; Wu Y.; Song Y.; Tong Y.; Rao Y. PROTACs: great opportunities for academia and industry. Signal Transduction Targeted Ther. 2019, 4, 64. 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies R. J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]
- Gao H.; Sun X.; Rao Y. PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. 10.1021/acsmedchemlett.9b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva S. L.; Crews C. M. Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes J.; McGonagle G. A.; Eden J.; Kiritharan G.; Touzet M.; Lewell X.; Emery J.; Eidam H.; Harling J. D.; Anderson N. A. Targeting IRAK4 for degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. 10.1021/acsmedchemlett.9b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. H.; Zhao X. W.; Ding N.; Gao H. Y.; Wu Y.; Yang Y. Q.; Zhao M.; Hwang J.; Song Y. Q.; Liu W. L.; Rao Y. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y. R.; Starr J.; Borzilleri K.; Smith J.; Zhu H. Y.; Farley K. A.; Ding W. D.; Schiemer J.; Feng X. D.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J. S.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X. Y.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S.; Bothe U.; Nubbemeyer R.; Bömer U.; Gunther J.; Schmidt N.; Andres D.; Siebeneicher H.; Sutter A.. Substituted benzimidazoles, pharmaceutical preparations containing same, and use of same to produce drugs. WO 2018060174 A1, April 5, 2018.
- Rajapaksa N. S.; Gobbi A.; Drobnick J.; Do S.; Kolesnikov A.; Liang J.; Chen Y.; Sujatha-Bhaskar S.; Huang Z.; Brightbill H.; Francis R.; Yu C.; Choo E. F.; DeMent K.; Ran Y.; An L.; Emson C.; Maher J.; Wai J.; McKenzie B. S.; Lupardus P. J.; Zarrin A. A.; Kiefer J. R.; Bryan M. C. Discovery of Potent Benzolactam IRAK4 Inhibitors with Robust in Vivo Activity. ACS Med. Chem. Lett. 2020, 11, 327–333. 10.1021/acsmedchemlett.9b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C.-Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018, 61, 462–481. 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.