

Abstract
Repurposing E3 ubiquitin ligases for targeted protein degradation via customized molecular glues or proteolysis-targeting chimeras (PROTACs) is an increasingly important therapeutic modality. Currently, a major limitation in the design of suitable molecular glues and PROTACs is our fragmentary understanding of E3 ligases and their ligand space. We here describe a quantitative assay for the discovery and characterization of E3 ligase ligands that is based on the thermophoretic behavior of a custom reporter ligand. Thereby, it is orthogonal to commonly employed fluorescence-based assays and less affected by the optical properties of test compounds. It can be employed for the high-throughput screening of compound libraries for a given ligase but also for hit validation, which we demonstrate with the identification of unexpected well-binders and non-binders, yielding new insights into the ligand space of cereblon (CRBN).
Keywords: Molecular Glue, IMiD, PROTAC, VHL, Cereblon, MST
The contemporary concept of targeted protein degradation is exponentially gaining attention as an important strategy in various clinical settings, most prominently in cancer therapy.1 One of the most promising approaches in this area is to target proteins of interest (POI) for ubiquitination via E3 ubiquitin ligases.2 The human genome encodes for several hundreds of such ligases, which are specialized in their substrate specificity and their spatiotemporal expression. Of these hundreds of ligases, only a handful are characterized, of which the VHL (von Hippel–Lindau) and CRBN (Cereblon) substrate receptors are among those that have gained the most attention.3−6
For repurposing E3 ligases to ubiquitinate POIs, two different major strategies are currently being employed, which are based on two conceptually different classes of small molecules. The first approach relies on small molecules called molecular glues, which mediate interactions between proteins that would not interact without the glue molecule.7 For CRBN, such molecules are typically based on the thalidomide scaffold and constitute the class of immunomodulatory drugs termed IMiDs.8 While natural substrates of CRBN are still largely elusive, the binding of IMiDs is known to modulate CRBN substrate specificity to recognize “neo-substrates”, in particular a group of zinc-finger transcription factors, which is thought to be the main driving force for the efficacy of the FDA-approved IMiD lenalidomide in multiple myeloma.9 Here, the substrate specificity can be adjusted by small changes to the IMiD structure.10,11 However, the specificity of IMiDs appears to be rather broad, and also largely unrelated small molecules that are able to bind to CRBN can yield degradation profiles and induce in vivo effects comparable to those of IMiDs.12−14
The other major strategy is the proteolysis targeting chimera (PROTAC) approach, which was coined by Crews and co-workers in 2001.15 PROTACs are rather large bifunctional molecules, in which a ligand for an E3 ligase is connected to a specific binder of a POI via a flexible linker. While their larger molecular weight may pose a disadvantage, their chimeric nature allows for high target specificity. Their E3 ligase ligand is typically derived from the natural recognition motif of the ligase, e.g., a hydroxyproline-containing peptide for VHL, which can bind to a groove on its surface (Figure 1)16 or, when the natural ligand is unknown, from a small-molecule binder like thalidomide, which binds to a conserved tri-tryptophan pocket in CRBN (Figure 1).12,17 The warhead for the POI can often be derived from an existing binder or inhibitor of the POI. The third component, the linker, is the least constrained part and can be of any length that brings the POI into an orientation that allows for ubiquitination via the E3 ligase.1 In practice, the development of a PROTAC for efficient ubiquitination of a certain POI can involve extensive screening of different linker types and of different E3 ligands for different E3 ligases, until a functional combination is identified.18
Figure 1.

Overall structure and ligand binding modes for E3 ligase substrate receptors VHL and CRBN. (A) Structure of VHL-Elongin B-Elongin C (VBC) complex with bound HIF1-α peptide (PDB: 1LM8). (B) Illustrative surface representation of the VHL binding groove with bound 11mer reporter, based on the same structure, highlighting the position of the fluorophore and hydroxyproline (Hyp). (C) Structure of the bacterial CRBN homologue MsCI4 bound to thalidomide (PDB: 4V2Y). (D) Illustrative surface representation of MsCI4 bound to BODIPY-uracil reporter. (E) Proposed clash of nitrofurantoin in the binding pocket of CRBN. Nitrofurantoin (green) was aligned with a structure of hydantoin in complex with MsCI4 (PDB: 5OH7) and shown in comparison to thalidomide (red). The protruding moiety of nitrofurantoin comes in close proximity to W99 of MsCI4. (F) Depiction of the pharmacophore-based nomenclature for 5- and 6-membered rings. R refers to the protruding moiety with the canonical exit vector.
Currently, both the molecular glue/IMiD and the PROTAC approach are limited by the number of available E3 ligases and ligands, pointing at the need for the characterization of further ligases and the discovery of new ligands. For the latter, only a limited number of methods can be used to distinguish between unspecific interactions and the specific binding of a ligand to the specific binding pocket of the ligase. This is often realized by following the behavior of a fluorescent reporter ligand, which competes for the same binding site. For VHL, this has been implemented with a FAM-labeled substrate-peptide reporter that is monitored in a fluorescence polarization (FP) assay.19 While FAM FP is constant while the reporter is bound to VHL, its displacement by a test compound leads to a drop in FP, which can be used to determine affinities in a titration experiment. The same approach has also been used for CRBN, with thalidomide coupled to a Cy5 fluorophore as a reporter.17 An alternative specific for CRBN is a FRET (Förster resonance energy transfer) assay exploiting the presence of three strictly conserved tryptophan residues that are lining the ligand binding pocket. Here, the reporter ligand MANT-uracil carries a MANT (N-methyl-anthraniloyl) fluorophore, which forms a FRET pair with these tryptophans, linked to uracil as the actual binding moiety, such that the displacement of MANT-uracil from the binding pocket can be followed by the loss of the FRET effect.20
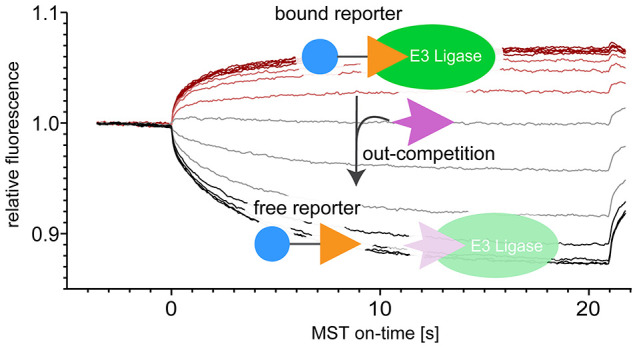
As both the FP and the FRET assay rely on changes of fluorescent properties that are initiated by the displacement of the respective reporter ligand, these assays can be hampered by overlapping optical properties such as autofluorescence of the test compounds. We therefore sought to establish a universal competitive assay that is orthogonal to these approaches, based on microscale thermophoresis (MST). A competitive MST-based assay relies on the behavior of a reporter ligand in a thermal gradient that is induced at a defined time using an infrared laser. This behavior is dominated by the thermal migration of the reporter along (positive thermophoresis) or against the temperature gradient (negative thermophoresis) and by a temperature-related intensity change (TRIC) effect, both of which can differ greatly between the free and the E3 ligase-bound state.21,22 While also in this MST assay the reporter is monitored via its fluorescence, the assay is not based on fluorescence changes initiated by the displacement of the reporter from the ligase during sample preparation but on changes transiently induced by the application of the thermal gradient during the experiment.23 Here, we implemented this assay both for an established VHL reporter ligand and for a CRBN reporter that we customized for this study.
For VHL, we employed the complex consisting of VHL, elongin B, and elongin C, together with two FAM-coupled reporter peptides with a length of 11 and 19 amino acids, which correspond to the reporter ligands previously used in FP experiments (Figure 2, see also Figure 1).19 To determine their affinity, we performed initial titration experiments, varying the VHL concentration. The normalized MST traces show that the reporters exhibit positive thermophoretic behavior, i.e., move out of the heated spot induced during the measurement (Figure 2). Progressive addition of, and thus the binding of the reporter to, VHL dampens this effect, yielding a sigmoidal dose–response curve corresponding to Kd values of 434 and 53 nM for the short and long reporter peptide, respectively. For comparison, we also performed FP experiments with these reporters, obtaining Kd values of 280 and 20 nM, which is also in reasonable agreement with the values reported previously for comparable FP experiments (560 and 36 nM).19 Having determined the affinity of the reporter peptides, we performed competition experiments with unlabeled versions of the same peptides, in which the latter were titrated as test ligands against a constant concentration of VHL and reporter, for each reporter peptide. The normalized MST traces show that the progressive addition of the test ligands, and thus the out-competition and dissociation of the reporters from VHL, leads to a restoration of their thermophoretic behavior in unbound state; a fit of the resulting sigmoidal dose–response curves yields Ki values of 535 or 604 nM for the short and 167 or 203 nM for the long unlabeled peptide (Figure 2). These experiments show that the assay can yield meaningful results using established fluorescent reporters. However, optimized reporters can improve the sensitivity dramatically, as we demonstrate in the following.
Figure 2.

MST traces and dose–response curves for Kd determination of short (FAM-11mer) and long (FAM-19mer) reporter peptide to VHL, and competition experiments with the short (11mer) and long (19mer) peptides. IC50 and derived Ki values are shown in blue and black, respectively, together with their confidence intervals. All values are in nM. FAM, fluorescein amidite; Hyp, hydroxyproline.
For the adaptation for CRBN, we designed the custom reporter ligand BODIPY-uracil, which has a BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) 493/503 fluorophore linked to uracil as the CRBN-binding moiety (Figure 3, see also Figure 1), inspired by the MANT-uracil reporter used in the FRET assay.20 As the ligase construct, we tested both the human CRBN (hCRBN) thalidomide binding domain (hTBD), which can be expressed in E. coli with high yields,24 as well as the bacterial CRBN homologue Magnetospirillum gryphiswaldense cereblon isoform 4 (MsCI4) that we previously employed in the FRET assay.20 Initial titration experiments of these CRBN constructs against the reporter revealed several advantages of this customized setup. First, the initial fluorescence of the reporter was quenched by CRBN binding, which allowed for the determination of the dissociation constant prior to the MST measurement to Kd = 3.8 μM for MsCI4 and 11 μM for hTBD (Figure 3). However, more important are two major advantages concerning the actual MST experiment. The first one lies in the unusual thermophoretic behavior of the BODIPY-uracil::CRBN system, which changes sign between complexation states. While the free BODIPY-uracil shows positive thermophoresis, it is negative for both the BODIPY-uracil::MsCI4 and the BODIPY-uracil::hTBD complex: The MST traces reveal that the reporter is depleted from the heated spot in unbound state (decreasing fluorescence), but enriched when in complex with the ligase (increasing fluorescence). The second major advantage is a strong TRIC effect of the BODIPY fluorophore, which manifests in a fast initial response of the fluorophore at the onset of the measurement when the jump in temperature is applied, long before thermophoresis is approaching equilibrium.22 The MST traces of the BODIPY-uracil::MsCI4 and BODIPY-uracil::hTBD titrations reveal that both the thermophoretic behavior and the TRIC effect change sign in the same direction between free and complexed state, and thus are cumulative, leading to an exceptional sensitivity (Figure 3).
Figure 3.

MST traces and affinities of the BODIPY-uracil reporter and reference compounds to the bacterial MsCI4 and the thalidomide binding domain of human CRBN (hTBD). The affinity of BODIPY-uracil was determined via initial fluorescence measurements. The MST traces show that binding or unbinding of the reporter yields an inversion of the thermophoretic behavior. The affinities of DMSO, succinimide (without DMSO), and thalidomide (with 0.5% DMSO) were determined via out-competition of the reporter in MST measurements, for which the MST traces and derived dose–response curves are shown. IC50 and derived Ki values are shown in blue and black, respectively, together with their confidence intervals. All values are in μM.
In a next step, we determined the affinities of a set of reference compounds in competitive titrations, which includes thalidomide, succinimide, and DMSO. The latter is of special importance, as DMSO itself is competitive with CRBN binders.6,20 In the MST assay with MsCI4, the competitive DMSO titration yielded a Ki of 11 mM, corresponding to a concentration of 0.08% (v/v) DMSO, which is in good agreement with the value obtained in the FRET assay (5.7 mM/0.06%). In contrast, the DMSO titration with the hTBD yielded a Ki of 137 mM, corresponding to a concentration of 1.0% (v/v) DMSO, which is virtually identical to the Ki obtained in the FRET assay for a similar hTBD construct, and significantly higher than that for MsCI4.20 This led us to test the applicability of the assay in the absence and presence of DMSO. To this end, we performed and evaluated competitive titration experiments with the water-soluble binder succinimide without DMSO, and with thalidomide at a constant DMSO concentration of 0.5% (v/v). For succinimide, in the absence of DMSO, we obtained ideal MST traces for both constructs and Ki values of 0.64 and 8.2 μM for MsCI4 and hTBD, respectively (Figure 3). For thalidomide, however, the difference in DMSO tolerance between the two constructs was readily apparent from the traces. For MsCI4, the characteristic reversal of the thermophoretic behavior was not observed, because the 0.5% DMSO were well above the Ki value of the DMSO::MsCI4 interaction, such that most of the reporter was already out-competed by DMSO at the lowest thalidomide concentrations; a quantitative analysis was not attempted (Figure 3). In contrast, the experiment with hTBD yielded largely unperturbed traces and a Ki value of 8.5 μM; a detailed analysis and discussion of the impact of DMSO competition on the characterization of CRBN ligands can be found in the Supporting Information.
These results indicate that the hTBD-based MST assay is significantly more robust and HTS-amenable than the MsCI4-based FRET assay. Therefore, we tested the assay’s applicability for the HTS of compound libraries, which relies on single-concentration measurements instead of titration series. To this end, we determined the Z′ factor of the assay in the presence of 0.5% DMSO, once focused on the TRIC effect (on-time 5s) and once including the full MST behavior (on-time 20s) (Figure 4). The two evaluations resulted in Z′ factors of 0.85 and 0.83, suggesting that the evaluation of the TRIC effect with 5s per sample is sufficient for HTS,25 which can be performed with the commercially available MST- or TRIC-based devices that use a 384-well SBS plate format.
Figure 4.

Determination of Z′ factor for hTBD. MST traces of solvent (0.5% DMSO) and positive control thalidomide (top panel) and scatter plot and resulting Z′ values at on-time 5s and on-time 20s (bottom panel). Means of positive and negative controls are shown by solid lines; dashed lines indicate 3-fold standard deviations.
With the established hTBD-based assay, we sought to characterize the binding of a number of compounds, with a focus on those that were not measurable in the MsCI4-based FRET assay due to overlapping optical properties or solubility issues. This includes the third generation IMiD avadomide (Figure S1) as well as a number of only remotely related pharmaceuticals. We had previously identified lactams and cyclic imides as minimal CRBN-binding moieties and predicted several pharmaceuticals based on these moieties as CRBN binders, some of which could not be examined in the FRET assay due to solubility issues. This includes the antidepressant γ-lactam rolipram and the antiviral uracil dasabuvir, of which only the binding of rolipram could be validated so far via crystallography.13 Further, we included the antibiotic hydantoin nitrofurantoin, which was previously predicted but not yet tested as a binder. All these mentioned pharmaceuticals and IMiDs are single-branched in the same position of their core binding moieties. We had previously defined this as position a of the binding moiety, and define everything attached to this a position the “protruding” moiety13 (Figure 1). Consequently, this position marks the canonical exit vector from the binding pocket, which is an important determinant when assembling linkers and ligase ligands in PROTAC design.18,26
We first ran a series of experiments to determine the affinities of the established binders, the IMiDs lenalidomide, pomalidomide, avadomide, and iberdomide and also rolipram. For the IMiDs, the resulting affinities are in the expected range, with iberdomide having the highest affinity (Ki = 4.9 μM)27 (Figure 5). Rolipram, for which the binding had not been quantified so far, bound with a relatively low affinity of Ki = 102 μM, substantiating the notion that cyclic imides pose higher-affinity CRBN-binding moieties than lactams.13 While these measurements were of rather confirmatory nature, we were surprised by the results we obtained for nitrofurantoin and dasabuvir.
Figure 5.

Chemical structures, dose–response curves and affinity values for (A) IMiDs and rolipram, (B) small hydantoins and hydantoins branched via hydrazo groups, and (C) uracils to hTBD. IC50 and derived Ki values are shown in blue and black, respectively, together with their confidence intervals. All values are in μM. n.b., no binding; 2-NP-AHD, 1-(2-nitrobenzylideneamino)hydantoin; NF-DB, 1-(3-(5-nitrofuran-2-yl)allylidene)amino)hydantoin.
Unexpectedly, nitrofurantoin did not show any binding in the MST assay, a result that we could also confirm in the MsCI4-based FRET assay (Figures 5 and S1). To understand the reasons for this lack of affinity, we tested a number of related N1-substituted hydantoins, i.e., hydantoins branched in position a. We started with the minimal binder hydantoin (Ki = 216 μM) itself, N1-methylhydantoin (68 μM) and N1-aminohydantoin (229 μM). The higher affinity of N1-methylhydantoin supports our previous finding that single-branching in position a leads to increased affinity, while the result for N1-aminohydantoin indicates that this does not hold true for the branching with an amino group, possibly due to the protonation of the latter. We continued with commercially available hydantoins with larger protruding moieties, which were all branched via a hydrazo group as in nitrofurantoin (Figure 5). Intriguingly, all showed the same lack of affinity to hTBD; for two of them, 1-benzylideneaminohydantoin and 1-(2-nitrobenzylideneamino)hydantoin, we also confirmed this absence of affinity in the FRET assay with MsCI4, ruling out that it is due to an artifact of one specific assay or protein construct. Consequently, the reasons for the lack of binding are presumably to be ascribed to the hydrazo group, which is coplanar with the hydantoin ring due to the resonance of the free electron pairs of the nitrogen atoms in the hydantoin moiety. With this particular geometry, binding is presumably precluded as it would potentially force the hydrazo carbon into a steric clash with one of the tryptophans of the binding pocket (Figure 1).
Furthermore, as a part of the hydantoin series, we tested the importance of the carbonyl in position b (see Figure 1) in the context of hTBD by comparing the affinity of hydantoin to that of 4-imidazolidinone, in which this group is absent, and 2-thiohydantoin, in which it is replaced with a thiocarbonyl. As reported for MsCI4,13 the lack of the group led to a reduction, while the thiocarbonyl variant led to an increase of affinity, suggesting that branching in this position should be tested more systematically (Figure 5).
The surprising result for dasabuvir, a large uracil branched in position a,28 was an unexpectedly high affinity (Ki = 4.7 μM) that is competitive with that of the third generation IMiD iberdomide and the highest affinity of all hTBD binders determined in this study (Figure 5). Consequently, we tested a range of related N1-substituted uracils, starting with parent uracil (212 μM). As expected, single branching with a methyl group in position a led to an improved affinity for N1-methyluracil (31 μM), while the larger substitution in uridine led to a drop (390 μM). The further addition of a phosphate group to that protruding moiety in uridine-5′-monophosphate (UMP) even abolished binding completely. As the 5′-OH group of uridine is outside the binding pocket and not recognized in a specific manner,12 this suggests that a negative charge on the linking or protruding moiety as the phosphate group in UMP could generally be detrimental for binding. For confirmation, we also tested UMP binding in the MsCI4-based FRET assay, where uridine showed a high affinity,13 and could indeed also not see any residual affinity for UMP (Figure S1). Finally, we tested sofosbuvir, another large uracil that is chemically distant to dasabuvir. Although binding to hTBD was observable, the affinity to the human domain is very weak (>1 mM). We had observed a significantly higher affinity of sofosbuvir for MsCI4 in the FRET assay (22 μM),13 which we could also confirm in the MST assay using MsCI4 (27 μM). This is the first time that we observe a larger discrepancy in the affinity of compounds between MsCI4 and hTBD. As sofosbuvir carries a complex protruding moiety, for which secondary interactions more distant to the canonical binding pocket may not be properly reflected by the bacterial homologue, this points at limitations of MsCI4 as a surrogate system for hCRBN.
In conclusion, we have shown that the competitive MST-based assay poses a promising tool for the characterization of the ligand space of potentially any ligase for which a suitable reporter ligand can be synthesized. It is a complementary alternative to currently employed fluorescence-based assays that is largely unaffected by possibly overlapping optical properties of test ligands. As demonstrated for VHL, the assay can be performed with an existing reporter ligand, but the careful selection of the fluorophore can significantly improve sensitivity, as demonstrated for CRBN, which also makes it promising for HTS. Here, the BODIPY-uracil::hTBD system has proven to be very robust, especially as the association or dissociation of this particular reporter::ligase complex yields an inversion of the thermophoretic behavior; we could use it to evaluate a number of compounds that were not manageable in the MsCI4-based FRET assay, and for a systematic analysis of the influence of DMSO on CRBN binding assays. Thus, we have gained novel insight into the CRBN ligand space, revealing steric and charge restraints for the branching at the canonical branching position a of the core binding moiety, and suggesting to further explore branching at position b, which could yield novel exit vectors for the design of IMiDs and PROTACs. Further, with dasabuvir, we discovered a largely unrelated FDA-approved pharmaceutical that competes even with the affinity of the third generation IMiD iberdomide. In fact, dasabuvir is the highest-affinity hTBD binder we have tested so far. It seems likely that it benefits from additional interactions of its rather bulky protruding moiety with the immediate surroundings of the core binding pocket, which may encourage a more systematic screening for such interactions for the design of protruding or linking moieties that contribute to the affinity of IMiDs and CRBN-based PROTACs.
Acknowledgments
We thank Andrei Lupas for continuing support, Iuliia Boichenko for initial contributions, Reinhard Albrecht for the cloning of hTBD, and Kerstin Bär and Maxim Drömer for excellent technical assistance.
Glossary
Abbreviations
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
- CRBN
cereblon
- FAM
fluorescein amidite
- FP
fluorescence polarization
- FRET
Förster resonance energy transfer
- IMiD
immunomodulatory drug
- MANT
N-methyl-anthraniloyl
- MsCI4
Magnetospirillum gryphiswaldense cereblon isoform 4
- MST
microscale thermophoresis
- POI
protein of interest
- PROTAC
proteolysis-targeting chimera
- TBD
thalidomide binding domain
- TRIC
temperature-related intensity change
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00440.
Analysis and discussion of the impact of DMSO competition on the characterization of CRBN ligands; analysis and discussion of the influence of fluorescence quenching and autofluorescent test compounds; experimental procedures; FRET assay data for avadomide, nitrofurantoin and UMP; influence of DMSO; influence of initial fluorescence on MST behavior (PDF)
Author Contributions
† S.M. and C.H. contributed equally.
This work was supported by institutional funds of the Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P. P.; Hamann L. G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15 (10), 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18 (12), 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Steinebach C.; Kehm H.; Lindner S.; Vu L. P.; Kopff S.; Marmol A. L.; Weiler C.; Wagner K. G.; Reichenzeller M.; Kronke J.; Gutschow M. PROTAC-mediated crosstalk between E3 ligases. Chem. Commun. 2019, 55 (12), 1821–1824. 10.1039/C8CC09541H. [DOI] [PubMed] [Google Scholar]
- Girardini M.; Maniaci C.; Hughes S. J.; Testa A.; Ciulli A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27 (12), 2466–2479. 10.1016/j.bmc.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. D.; Boichenko I.; Coles M.; Lupas A. N.; Hernandez Alvarez B. Structural dynamics of the cereblon ligand binding domain. PLoS One 2015, 10 (5), e0128342 10.1371/journal.pone.0128342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor-Ruiz C.; Bauer S.; Brand M.; Kozicka Z.; Siklos M.; Imrichova H.; Kaltheuner I. H.; Hahn E.; Seiler K.; Koren A.; Petzold G.; Fellner M.; Bock C.; Muller A. C.; Zuber J.; Geyer M.; Thoma N. H.; Kubicek S.; Winter G. E. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16 (11), 1199–1207. 10.1038/s41589-020-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Handa H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int. J. Hematol. 2016, 104 (3), 293–299. 10.1007/s12185-016-2073-4. [DOI] [PubMed] [Google Scholar]
- Kronke J.; Udeshi N. D.; Narla A.; Grauman P.; Hurst S. N.; McConkey M.; Svinkina T.; Heckl D.; Comer E.; Li X.; Ciarlo C.; Hartman E.; Munshi N.; Schenone M.; Schreiber S. L.; Carr S. A.; Ebert B. L. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343 (6168), 301–5. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thoma N. H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532 (7597), 127–30. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Kronke J.; Fink E. C.; Hollenbach P. W.; MacBeth K. J.; Hurst S. N.; Udeshi N. D.; Chamberlain P. P.; Mani D. R.; Man H. W.; Gandhi A. K.; Svinkina T.; Schneider R. K.; McConkey M.; Jaras M.; Griffiths E.; Wetzler M.; Bullinger L.; Cathers B. E.; Carr S. A.; Chopra R.; Ebert B. L. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523 (7559), 183–8. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. D.; Boichenko I.; Coles M.; Zanini F.; Lupas A. N.; Hernandez Alvarez B. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J. Struct. Biol. 2014, 188 (3), 225–32. 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Boichenko I.; Bar K.; Deiss S.; Heim C.; Albrecht R.; Lupas A. N.; Hernandez Alvarez B.; Hartmann M. D. Chemical Ligand Space of Cereblon. ACS Omega 2018, 3 (9), 11163–11171. 10.1021/acsomega.8b00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C.; Pliatsika D.; Mousavizadeh F.; Bar K.; Hernandez Alvarez B.; Giannis A.; Hartmann M. D. De-Novo Design of Cereblon (CRBN) Effectors Guided by Natural Hydrolysis Products of Thalidomide Derivatives. J. Med. Chem. 2019, 62 (14), 6615–6629. 10.1021/acs.jmedchem.9b00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J. H.; Yang H.; Ivan M.; Gertler F.; Kaelin W. G. Jr.; Pavletich N. P. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 2002, 296 (5574), 1886–9. 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Bohm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E.; Harper J. W.; Jenkins J. L.; Thoma N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512 (7512), 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Sosic I.; Lindner S.; Bricelj A.; Kohl F.; Ng Y. L. D.; Monschke M.; Wagner K. G.; Kronke J.; Gutschow M. A MedChem toolbox for cereblon-directed PROTACs. MedChemComm 2019, 10 (6), 1037–1041. 10.1039/C9MD00185A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 2012, 134 (10), 4465–8. 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boichenko I.; Deiss S.; Bar K.; Hartmann M. D.; Hernandez Alvarez B. A FRET-Based Assay for the Identification and Characterization of Cereblon Ligands. J. Med. Chem. 2016, 59 (2), 770–4. 10.1021/acs.jmedchem.5b01735. [DOI] [PubMed] [Google Scholar]
- Duhr S.; Braun D. Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (52), 19678–82. 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. J.; Duhr S.; Baaske P.. Microscale Thermophoresis (MST). In Encyclopedia of Biophysics; Roberts G., Watts A., Eds.; Springer: Berlin, Heidelberg, 2018; pp 1–5. [Google Scholar]
- Seidel S. A.; Dijkman P. M.; Lea W. A.; van den Bogaart G.; Jerabek-Willemsen M.; Lazic A.; Joseph J. S.; Srinivasan P.; Baaske P.; Simeonov A.; Katritch I.; Melo F. A.; Ladbury J. E.; Schreiber G.; Watts A.; Braun D.; Duhr S. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59 (3), 301–15. 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akuffo A. A.; Alontaga A. Y.; Metcalf R.; Beatty M. S.; Becker A.; McDaniel J. M.; Hesterberg R. S.; Goodheart W. E.; Gunawan S.; Ayaz M.; Yang Y.; Karim M. R.; Orobello M. E.; Daniel K.; Guida W.; Yoder J. A.; Rajadhyaksha A. M.; Schonbrunn E.; Lawrence H. R.; Lawrence N. J.; Epling-Burnette P. K. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J. Biol. Chem. 2018, 293 (16), 6187–6200. 10.1074/jbc.M117.816868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. P.; Rees S.; Kalindjian S. B.; Philpott K. L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162 (6), 1239–49. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Ng Y. L. D.; Sosic I.; Lee C. S.; Chen S. R.; Lindner S.; Vu L. P.; Bricelj A.; Haschemi R.; Monschke M.; Steinwarz E.; Wagner K. G.; Bendas G.; Luo J.; Gutschow M.; Kronke J. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11 (13), 3474–3486. 10.1039/D0SC00167H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Zhang W.; Man H. W.; Muller G.; Khambatta G.; Baculi F.; Hickman M.; LeBrun L.; Pagarigan B.; Carmel G.; Lu C. C.; Lu G.; Riley M.; Satoh Y.; Schafer P.; Daniel T. O.; Carmichael J.; Cathers B. E.; Chamberlain P. P. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61 (2), 535–542. 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- Gentile I.; Buonomo A. R.; Borgia G. Dasabuvir: A Non-Nucleoside Inhibitor of NS5B for the Treatment of Hepatitis C Virus Infection. Rev. Recent Clin. Trials 2014, 9 (2), 115–23. 10.2174/1574887109666140529222602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.