

Abstract

Phosphoinositide 3-kinases (PI3Ks) are a family of enzymes that control a wide variety of cellular functions such as cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking. PI3Kγ plays a critical role in mediating leukocyte chemotaxis as well as mast cell degranulation, making it a potentially interesting target for autoimmune and inflammatory diseases. We previously disclosed a novel series of PI3Kγ inhibitors derived from a benzothiazole core. The truncation of the benzothiazole core led to the discovery of a structurally diverse alkynyl thiazole series which displayed high PI3Kγ potency and subtype selectivity. Further medicinal chemistry optimization of the alkynyl thiazole series led to identification of compounds such as 14 and 32, highly potent, subtype selective, and CNS penetrant PI3Kγ inhibitors. Compound 14 showed robust inhibition of PI3Kγ mediated neutrophil migration in vivo.
Keywords: PI3Kγ, benzothiazole, alkynylthiazole, neutrophil migration
The phosphoinositide 3-kinases (PI3Ks) are lipid kinases that phosphorylate the 3-hydroxyl group of the inositol ring of phosphatidylinositol (PI) substrates within the plasma membrane and intracellular compartments.1 PI3Ks control a wide variety of cellular signaling pathways, including migration, proliferation, differentiation, and vesicular trafficking.2 Based on sequence homology and lipid substrate specificity, PI3Ks are divided into three classes. The class I PI3Ks (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ) are the most extensively studied and are further divided into two subclasses: 1A and 1B.3 Class IA PI3Ks (α, β, δ) contain p110α, p110β, and p110δ as catalytic subunits and are activated by tyrosine kinase receptor signaling while class IB PI3Ks contain only p110γ as catalytic subunit and are activated by GPCRs via its βγ regulatory subunits.3,4 PI3Kα and PI3Kβ are ubiquitously expressed and play a key role in cell growth, survival, and proliferation; hence, their inhibition has been mainly a strategy that targets cancer therapy.1 PI3Kγ is widely expressed in granulocytes, monocytes, and macrophages, whereas the PI3Kδ isoform is also found in B and T-cells.1 PI3Kγ and PI3Kδ represent key modulators of innate and adaptive immune responses. PI3Kγ knockout mice showed reduced chemoattractant-induced neutrophil migration to infection sites and respiratory burst.5−8 Furthermore, PI3Kδ and γ deficiency blocks the recruitment of neutrophils to inflammation sites.9,10 PI3Kγ plays a critical role in mediating leukocyte chemotaxis as well as mast cell degranulation, making it a potentially interesting drug target for autoimmune and inflammatory diseases.11,12 The discovery of PI3Kγ specific inhibitors has faced a daunting challenge due to the high sequence homology at the active site of class I PI3Ks and the conserved topology of the ATP-binding site of the closely related protein kinases such as mTOR and DNA-PK.13 Compounds with good isoform selectivity are critical tools to study the function of the PI3K pathway and enable the development of treatments that maximize the therapeutic indices.13b We previously reported three new classes of potent and isoform selective PI3Kγ inhibitors derived from benzothiazole 1,14 thiazolopiperidine 2,15 and isoindoline 3(16) scaffolds as potential therapy for multiple sclerosis (MS) (Figure 1). In an effort to further expand the structural diversity of our potent, selective, and CNS penetrant PI3Kγ inhibitors for the treatment of CNS disorders such as MS, we sought to exploit an alternative scaffold derived from alkynyl thiazole as exemplified by compound 4. In this paper, we describe the design and synthesis of a highly potent, isoform-selective, and CNS penetrant PI3Kγ inhibitor derived from an alkynylthiazole core (Figure 1).
Figure 1.

Structures of our previously reported potent and selective PI3Kγ inhibitors.
As we reported previously,14 benzothiazole urea 1 is a very potent PI3Kγ inhibitor with good isoform selectivity. Our initial design strategy involved the truncation of the benzothiazole to form the alkynyl thiazole core, generating a structurally diverse molecule while maintaining similar potency and selectivity vectors of the benzothiazole core (Figure 2). We discovered that fluoroalkylimidazoles were generally more isoform selective compared to the corresponding alkylimidazoles during the exploration of the thiazolopiperidine series,15 and thus, we incorporated the difluoromethyl imidazole moiety in the alkynylthiazole series.
Figure 2.

Evolution of the benzothiazole into alkynylthiazole
The alkyne functionality has been broadly exploited in drug discovery programs, and it brings a number of advantages and disadvantages. The acetylene group has been successfully employed as a nonclassical bioisostere of a wide range of functional groups, including chlorine, iodine, and the nitrile group,17 but the terminal alkynyl group can also negatively impact drug metabolism through irreversible inhibition of CYP450 enzymes and the formation of GSH conjugates.17 Alkynyl groups have been previously used in the PI3Kγ inhibitor field. The alkynyl pyrazole-containing 5 (IPI-549) (Figure 3) demonstrated improved potency and high isoform selectivity, with excellent metabolic stability and pharmacokinetic properties.18
Figure 3.
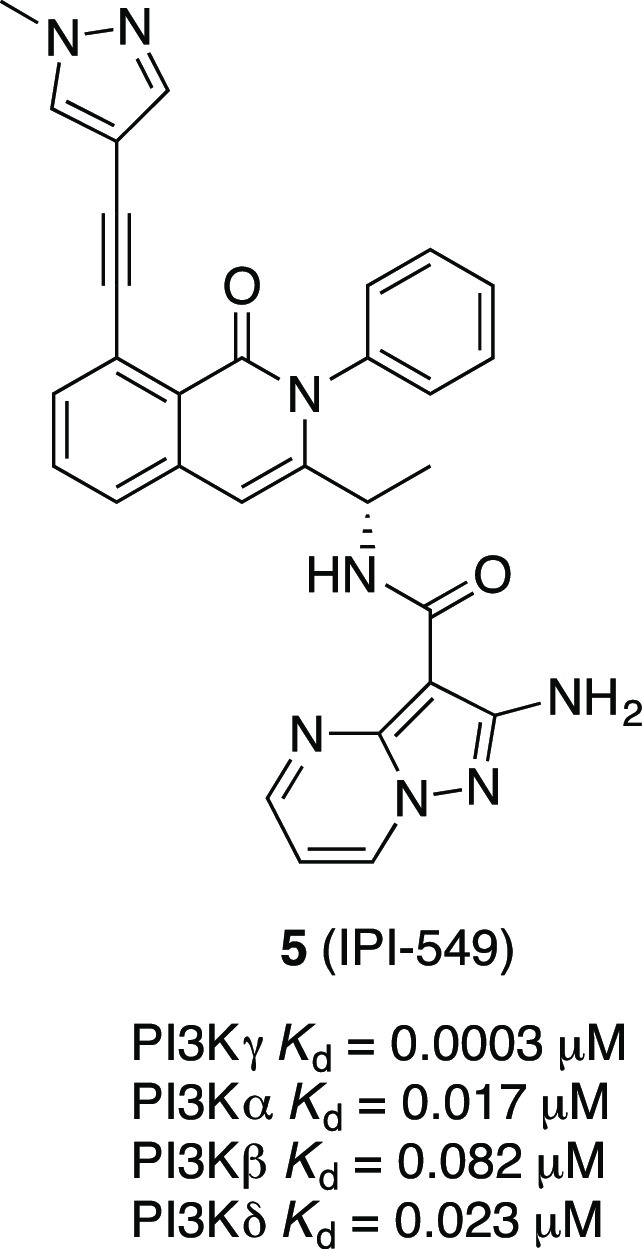
Chemical structure of the PI3Kγ inhibitor IPI-549.
To optimize the potency and the overall profile of this new series, we performed docking of the proposed new alkynyl thiazole analogs such as compound 4 with PI3Kγ. Using the crystal structure of 1 bound to PI3Kγ (PDB ID 4PS3), we analyzed if it would be possible to maintain or improve the contacts with the protein. The docking results suggested that compound 4 would likely bind in a similar manner to 1, maintaining a similar conformation and the majority of the contacts. The change in vector space around the methoxypyridine may preclude compound 4 from participation in a water-mediated hydrogen bond network to Tyr867 and Asp841 as seen in several public crystal structures14 (Figure 4). The loss of the methoxy group on compound 4 also corresponded to a decrease in docking score and resulted in an inversion of the pyridine nitrogen orientation. The “ring flip” observed in combination with the altered projection of the alkyne from the thiazole core allows for a hydrogen bond between the pyridine nitrogen of 4 and Lys833. Overall, the docking results suggest that the core binding should be maintained and that potency can be gained through a number of interactions.
Figure 4.
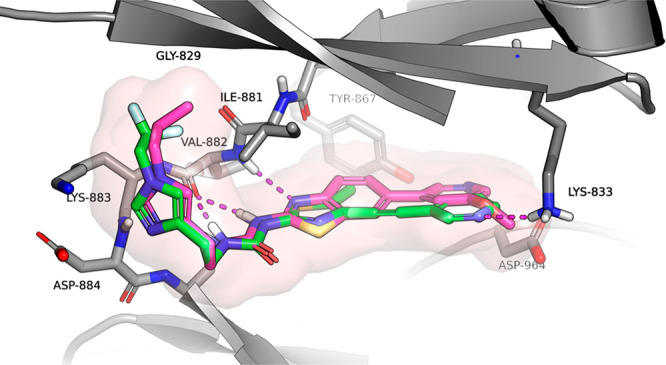
Docking model for compound 4 (green) bound to PI3Kγ and comparison to compound 1 (purple).
We first explored the optimization of the urea substituents on the western side of the molecule and the pyridine substitution on the eastern side in the context of the alkynylthiazole core (Table 1). Most of these new analogs exhibit excellent PI3Kγ potency and good selectivity against PI3Kα. Imidazole compounds 4 and 6 showed good selectivity against PI3Kβ (>28-fold). We have previously reported14 that an introduction of phenoxyethyl urea in the benzothiazole series can increase the PI3Kα selectivity due to unfavorable interaction of the phenyl group with the Asp residue of PI3Kα. However, replacement of the imidazole moiety with an O-aryl group (7) failed to improve PI3Kγ potency or PI3Kα selectivity. Replacement of the imidazole moiety with an alkoxy group increased the PI3Kγ potency while maintaining PI3Kα selectivity (10, 11, and 12) whereas compounds with short alkoxyethyl ureas (8, 9) showed poor PI3Kα selectivity. All compounds tested in Table 1 showed a noticeable decrease in cellular potency which is frequently impacted by numerous factors including cell membrane permeability, serum protein binding, off-target activity, and ATP concentration.19 Imidazole urea compounds (4 and 6) and alkoxy urea compounds (10 and 12) exhibited sub-micromolar cellular potencies. However, compounds 4 and 6 exhibited very poor CNS exposure (B/P ratio < 0.01) as the imidazole is a structural feature that increases the potential for P-gp-mediated efflux (ER 10 and 67 for compounds 4 and 6, respectively). From this list, we selected compound 12 over 11 for further analog generation considering in vitro human and rat liver microsomal stability data and cellular potency. Compound 12 displayed better microsomal stability (human and rat) than compound 11 (% remaining after 30 min HLM/RLM 12 92%/85% vs 11 66% /58%), and increased cellular potency (12 = 0.34 μM vs 11 = 0.64 μM). Compound 12 showed low efflux in MDR1-MDCK assay with ER = 1.4.
Table 1. SAR of Alkynylthiazoles with Varying Pyridine and Urea Substitutions.

THP-1 cell assay involves the capacity of the compounds to inhibit PI3Kγ-stimulated phosphorylation of endogenous AKT at Ser-473.
To better understand the binding mode of the compounds from this new series, an X-ray cocrystal structure of human PI3Kγ in complex with compound 11 (PDB ID 7KKE) was obtained (Figure 5). The 3,3-dimethylbutoxy linker of compound 11 occupies a hydrophobic pocket adjacent to the ATP-binding site while the linker oxygen makes an H-bond interaction with Asp884. The thiazole nitrogen and the NH of the urea form a bidentate hinge-binding interaction with the backbone carbonyl (C=O) of Val882. As the pyridine atom shifts its location, the pyridine nitrogen no longer participates in the water-mediated H-bond network with Tyr867 and Asp841 as seen in the benzothiazole series.14 Instead, the pyridine nitrogen makes a favorable H-bond (2.83 Å) interaction with Lys833.
Figure 5.
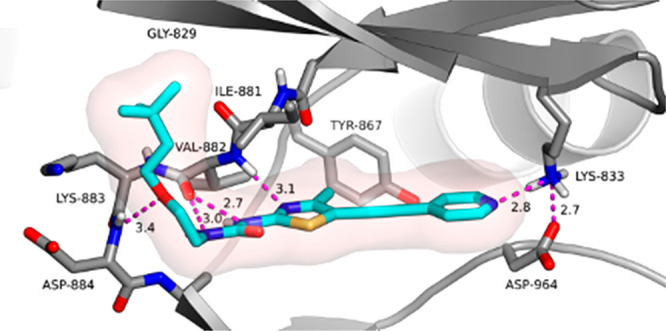
X-ray cocrystal structure of 11 bound to PI3Kγ (PDB ID 7KKE).
We next explored the SAR of the eastern pyridine group while keeping the O-methylcylopropoxyethyl-substituted urea from 12 (Table 2). The methoxy-substituted 3- and 4-pyridines 13, 14, and 15 and 2-fluorine and 2-chlorine substituted 4-pyridines 17 and 18 displayed good PI3Kγ potency and selectivity over the α isoform, but with no improvement in selectivity toward the β or δ subtypes. The methoxy-substituted 2-pyridine (16) did not improve PI3Kγ potency and selectivity over the α isoform. A pyridine analog with a strong electron withdrawing group such as CF3, 19, showed reduced PI3Kγ potency. Dimethoxy substituted pyrazine analog 20 showed good PI3Kγ potency and α isoform selectivity. Using fused bicycles as replacements for the pyridine in the alkynyl thiazoles led to a consistent reduction in PI3Kγ potency, except for 22 and 26. Most of these bicyclic-substituted compounds also exhibited reduced PI3Kα selectivity except for compound 23.
Table 2. SAR of the Pyridine Substitutions.
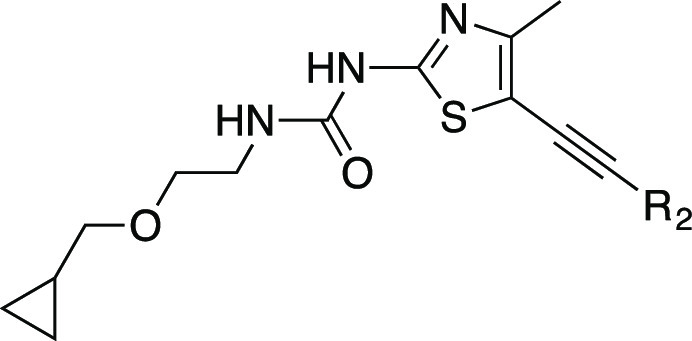

We next introduced chirality to the alkoxy ethyl urea linker aiming to further improve PI3Kγ potency and subtype selectivity (Table 3). Compounds with α-substituted ureas demonstrated dramatically reduced PI3Kγ potency (12 vs 27, 28). It was observed that the chirality plays a role in the potency gain as the (R)-1-methyl ethoxyurea 27 is 4-fold more potent than the (S)-1-methyl ethoxyurea analogue 28. Interestingly, β-substituted ethoxyurea maintained good PI3Kγ potency, specifically with the (S)-2-methyl alkoxyurea analogues (30, 31, 32, and 33). We envisioned that alkyl substituents in the linker adjacent to the urea might shield its H-bond donor ability and prevent an efficient interaction with Val887 in the hinge region. We observed that several (S)-2-methyl alkoxyurea analogues exhibited excellent PI3Kγ potency and α selectivity (30, 31, 32, and 33). Compound 31 showed the best PI3Kα selectivity (663-fold) and PI3Kδ selectivity (34-fold) while maintaining single-digit nanomolar PI3Kγ potency. The replacement of the cyclopropyl group with a cyclobutyl group slightly decreased PI3Kγ affinity but increased PI3Kα selectivity (30 vs 34). However, 34 is 9-fold less potent than 30 in the THP-1 (MCP-1) cellular assay.
Table 3. SAR of the Urea Linker Substitutions.
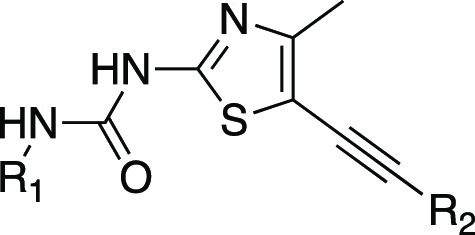

We selected a few compounds based on enzymatic potency, cellular potency, and isoform selectivity within the series for further in vitro profiling and identification of suitable compounds for pharmacokinetic evaluation (Table 4). However, most of these compounds showed lower potency in our THP-1 cellular assay, which may be due to poor solubility or cell permeability. Our ultimate goal was to identify CNS-penetrant PI3Kγ inhibitors for the potential treatment of CNS disorders such as multiple sclerosis.16 Therefore, It is very important to maintain low P-gp-mediated efflux, good B/P ratio, and good cell permeability. Our top compounds in Table 4 showed CNS multiparameter optimization (MPO) desirability scores20 greater than 4. Compounds 14, 31, and 32 showed good MDCK-MDR1 cell permeability.21 We expected reduced CNS exposure for compounds 4 and 23 which are P-gp substrates (ER = 10 and 12, respectively, in rats). The replacement of the difluoromethyl imidazole side chain with a cyclopropylmethoxy group dramatically decreased the efflux ratio (14, 31, and 32). Compound 32 had the best brain-to-plasma ratio in rats (B/P) as a result of the low MDCK-MDR1 efflux ratio (0.9) and high permeability. The higher lipophilicity (cLogD 3.2) attributed compound 32 accounts for good permeability. Additionally, most of these compounds showed good microsomal stability in human and rats except for compound 31. One of the key challenges in developing inhibitors of PI3K is cross-inhibition of other protein kinases, most notably mTOR and DNA-PK.13 All lead compounds in Table 4 exhibited high DNA-PK selectivity profiles, and most of the top compounds did not display hERG liabilities.
Table 4. Overall Profiles for Compounds 4, 14, 23, 31, and 32.
| 4 | 14 | 23 | 31 | 32 | |
|---|---|---|---|---|---|
| MW, clogD, PSA | 416, 2.7, 85 | 416, 3.0, 95 | 395, 2.7, 91 | 400, 3.0, 85 | 431, 3.2, 95 |
| THP-1 (MCP-1) IC50 (μM) | 0.13 | 0.18 | 0.60 | 0.38 | 0.20 |
| MDCK-MDRI E.R./PappA-B | 10/8 | 2/10 | 12/4 | 2/16 | 0.9/34 |
| Rat B/Pa | 0.013 | 0.1 | 0.3 | ||
| CNS MPO | 5.0 | 4.6 | 5.1 | 4.9 | 4.3 |
| CYP 2C9 IC50 (μM) | 0.49 | 7 | 11 | 11 | 15 |
| Microsome stabilityb H/R | 92/100 | 89/69 | 100/73 | 56/51 | 79/100 |
| Protein binding % H/R | 98.11/98.88 | 94.12/92.96 | 98.66/>99 | >99/>99 | |
| DNA-PK Ki (μM) | 3.4 | 2.8 | >7 | >7 | 3.5 |
| hERG (Planar patch) IC50 (μM) | 13 | >30 | >30 | 28 | >30 |
| Kinase selectivity | >4 μM for 13 kinases | >4 μM for 13 kinases | >4 μM for 13 kinases | >4 μM for 13 kinases | >4 μM for 13 kinases |
Determined from 1 mg/kg oral dose, 1 h time point.
% Remaining after 30 min.
Compounds 14, 23, and 32 were selected for pharmacokinetic (PK) studies in rats (Table 5). Compounds 14 and 32 exhibited good oral PK profiles with high exposure and excellent bioavailability in rats. However, both of these compounds showed moderate clearance and short half-life in intravenous PK studies. In contrast, compound 23 showed the best intravenous PK profile with the best clearance (12 mL/min/kg) and good half-life (2.6 h) in rats. However, we did not pursue compound 23 further as it was a P-gp substrate (ER = 12).
Table 5. IV and PO PK Profiles of Compounds 14, 23, and 32.
| 14 | 23 | 32 | |
|---|---|---|---|
| Rat IV PK (1mg/kg) | |||
| C1 (mL/min/kg)/Clu | 40/3571 | 12/170 | 34/3400 |
| T1/2 (h) | 1 | 2.6 | 1 |
| Vss(L/kg) | 3 | 2.3 | 1.7 |
| Rat PO PK (5mg/kg) | |||
| AUCo-inf (μg·h/mL) | 7.1 | 6.8 | |
| Cmax (μg/mL) | 2.1 | 1.1 | |
| T1/2 (h) | 1.3 | 2.7 | |
| % F | 100 | 100 | |
Based on PK properties in rats, we selected compound 14 to investigate the effect of PI3Kγ inhibition activity in vivo. Chemoattractant, IL-8 stimulated neutrophil migration into air pouches in mice has previously been shown to be dependent on PI3Kγ.18 In response to the inflammatory process, PI3Kγ activated immune cells such as neutrophils migrate to the sites of inflammation.22 To demonstrate PI3Kγ inhibition activity of compound 14 in vivo, we evaluated the effect of orally administered compound 14 (50 mg/kg) on IL-8 stimulated neutrophil migration into the air pouches on mice. Compound 14 significantly reduced (85%) neutrophil recruitment into the air pouches at a 50 mg/kg dose and exhibited good brain exposure with a B/P of 0.43 at 4 h postdosing (Figure 6). This result demonstrates that orally administered compound 14 can inhibit PI3Kγ activation in vivo.
Figure 6.
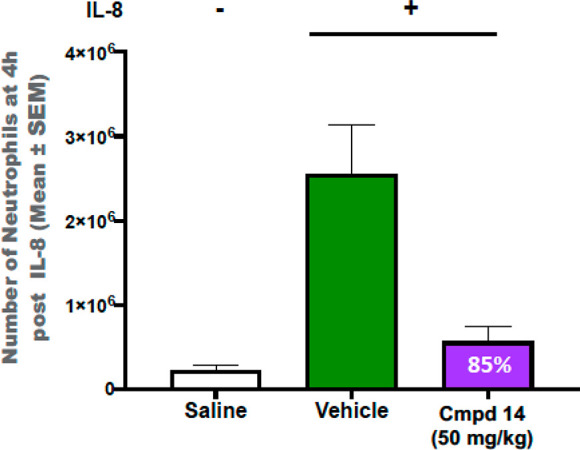
Effect of compound 14 on neutrophil migration in the mouse air pouch model.
The general syntheses of alkynylthiazoles, exemplified by compounds 4, 13, and 17, are shown in Schemes 1 and 2. In Scheme 1, the five-step sequence starts with the commercially available thiazole amide 35. The iodination of 35 with NIS followed by Sonagashira coupling with 3-ethynylpyridine afforded compound 37 in good yield. The removal of the acetyl group of 37 with hydrazine hydrate afforded compound 38 in 77% yield. Activation of compound 38 with CDI followed by coupling with 2-(1-(2,2-difluoroethyl)-1H-imidazol-4-yl)ethan-1-amine afforded target compound 4 in moderate yield.
Scheme 1. Synthesis of Alkynylthiazole 4.

Reagents and conditions: (a) NIS, DCM, r.t, 10 min, 66%; (b) 3-ethynylpyridine, Pd(PPh3)4, CuI, Et3N, dioxane, r.t., 1.5 h, 77%; (c) hydrazine hydrate, THF, 85 °C, 6 h, 77%; (d) CDI, DMF, 70 °C, 2 h, 92%; (e) 2-(1-(2,2-difluoroethyl)-1H-imidazol-4-yl)ethan-1-amine, Et3N, THF, r.t., 20 h, 58%.
Scheme 2. Synthesis of Alkynylthiazoles 13 and 17.

Reagents and conditions: (a) CDI, DCM, 2-(cyclopropylmethoxy)ethanamine, 60 °C, 2 h, 45%; (b) 3-methoxy-5-((trimethylsilyl)ethynyl)pyridine, PdCl2(PPh3)2, CuI, TEA, TBAF, THF, 64 °C, 1 h, 41%; (c) ethynyl(trimethyl)silane, CuI, Pd(PPh3)2, TEA, THF, 65 °C, 1 h, 58%; (d) 2-fluoro-3-iodopyridine, CuI, Pd(PPh3)2, TEA, TBAF, THF, 65 °C,1 h.
Compounds 13 and 17 were synthesized as shown in Scheme 2 (routes 1 and 2). Activation of compound 40 with CDI followed by coupling with 2-(cyclopropylmethoxy)ethanamine afforded compound 41 in 45% yield. Sonagashira coupling with 41 with 3-methoxy-5-((trimethylsilyl)ethynyl)pyridine in the presence of TBAF afforded compound 13 in 41% yield. The Sonagashira coupling of 41 with ethynyl(trimethyl)silane compound 42 gave 58% yield. The second Sonagashira coupling of 42 with 2-fluoro-3-iodopyridine in the presence of TBAF afforded compound 17 in 50% yield.
In summary, we have designed and synthesized a new series of highly potent and isoform selective inhibitors of PI3Kγ using structure-guided drug design based on our earlier reported work in this area. The SAR study focused on optimizing the in vitro potency, the isoform selectivity, and the ADME profile of the compounds in order to find lead compounds. The lead compounds 14 and 32 showed good in vitro metabolic stability in rat and human liver microsomes and good in vivo pharmacokinetic properties in rat, along with good CNS exposure. Compound 14 showed in vivo efficacy in a mouse air pouch model by reducing neutrophil migration in 85% at 50 mg/kg dose. The potent PI3Kγ inhibitors identified within this series can thus serve as suitable in vivo tool compounds.
Acknowledgments
The authors thank Greg May and Ron Grey for analytical chemistry support, Caroline Chandra Tjin and Pedro Garcia-Barrantes for useful comments, suggestions, and editing, and Sara Swift for modeling assistance.
Glossary
Abbreviations
- CNS
central nervous system
- MCP-1
monocyte chemoattractant protein 1
- MDCK
Madin Darby
- MDR1
multidrug resistance mutation 1
- GPCR
G-protein-coupled receptor
- PgP
P-glycoprotein-1
- ER
efflux ratio
- NIS
N-iodosuccinimide
- CDI
N,N′-carbonyldiimidazole
- DCM
dichloromethane
- TBAF
tetra-n-butylammonium fluoride
- THF
tetrahydrofuran
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00573.
Experimental details for synthesis, protein expression, assay methods, and crystallography (PDF)
Author Present Address
† T.M.C.: Tango Therapeutics, 100 Binney St, Suite 700, Cambridge, Massachusetts 02142, United States
The authors declare no competing financial interest.
Notes
All authors are current or former employees of Vertex Pharmaceuticals Incorporated.
Supplementary Material
References
- Banham-Hall E.; Clatworthy M. R.; Okkenhaug K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 245–258. 10.2174/1874312901206010245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L. C. The phosphoinositide 3-kinase pathway. Science 2002, 296 (5573), 1655–1657. 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Guillermet-Guibert J.; Graupera M.; Bilanges B. The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Leevers S. J.; Ahmadi K.; Timms J.; Katso R.; Driscoll P. C.; Woscholski R.; Parker P. J.; Waterfield M. D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Sasaki T.; Irie-Sasaki J.; Jones R. G.; Oliveira-Dos-Santos A. J.; Stanford W. L.; Bolon B.; Wakeham A.; Itie A.; Bouchard D.; Kozieradzki I.; Joza N.; Mak T. W.; Ohashi P. S.; Suzuki A.; Penninger J. M. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Hirsch E.; Katanaev V. L.; Garlanda C.; Azzolino O.; Pirola L.; Silengo L.; Sozzani S.; Mantovani A.; Altruda F.; Wymann M. P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Li Z.; Jiang H.; Xie W.; Zhang Z.; Smrcka A. V.; Wu D. Roles of PLC-beta2 and-beta3 and PI3Kgamma in chemoattractant mediated signal transduction. Science 2000, 287, 1046–1049. 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Condliffe A. M.; Davidson K.; Anderson K. E.; Ellson C. D.; Crabbe T.; Okkenhaug K.; Vanhaesebroeck B.; Turner M.; Webb L.; Wymann M. P.; Hirsch E.; Ruckle T.; Camps M.; Rommel C.; Jackson S. P.; Chilvers E. R.; Stephens L. R.; Hawkins P. T. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 2005, 106, 1432–1440. 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- Hirsch E.; Katanaev V. L.; Garlanda C.; Azzolino O.; Pirola L.; Silengo L.; Sozzani S.; Mantovani A.; Altruda F.; Wymann M. P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–53. 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Puri K. D.; Doggett T. A.; Douangpanya J.; Hou Y.; Tino W. T.; Wilson T.; Graf T.; Clayton E.; Turner M.; Hayflick J. S.; Diacovo T. G. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood 2004, 103, 3448–56. 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- Camps M.; Rückle T.; Ji H.; Ardissone V.; Rintelen F.; Shaw J.; Ferrandi C.; Chabert C.; Gillieron C.; Francon B.; Martin T.; Gretener D.; Perrin D.; Leroy D.; Vitte P. A.; Hirsch E.; Wymann M. P.; Cirillo R.; Schwarz M. K.; Rommel C. Blockade of PI3K., suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. (N. Y., NY, U. S.) 2005, 11, 936–943. 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Wymann M. P.; Bjoerkloef K.; Calvez R.; Finan P.; Thomast M.; Trifilieff A.; Barbier M.; Altruda F.; Hirsch E.; Laffargue M. Phosphoinositide 3-kinase.,: a key modulator in inflammation and allergy. Biochem. Soc. Trans. 2003, 31, 275–280. 10.1042/bst0310275. [DOI] [PubMed] [Google Scholar]
- Sundstrom T. J.; Anderson A. C.; Wright D. L. Inhibitors of phosphoinositide-3-kinase: a structure-based approach to understanding potency and selectivity. Org. Biomol. Chem. 2009, 7, 840–850. 10.1039/b819067b. [DOI] [PubMed] [Google Scholar]
- Collier P. N.; Martinez-Botella G.; Cornebise M.; Cottrell K. M.; Doran J. D.; Griffith J. P.; Mahajan S.; Maltais F.; Moody C. S.; Huck E. P.; Wang T.; Aronov A. M. Structural basis for isoform selectivity in a class of benzothiazole inhibitors of phosphoinositide 3- kinase γ. J. Med. Chem. 2015, 58, 517–521. 10.1021/jm500362j. [DOI] [PubMed] [Google Scholar]
- Collier P. N.; Messersmith D.; Le Tiran A.; Bandarage U. K.; Boucher C.; Come J.; Cottrell K. M.; Damagnez V.; Doran J. D.; Griffith J. P.; Khare-Pandit S.; Krueger E. B.; Ledeboer M. W.; Ledford B.; Liao Y.; Mahajan S.; Moody C. S.; Roday S.; Wang T.; Xu J.; Aronov A. M. Discovery of highly isoform selective thiazolopiperidine inhibitors of of phosphoinositide 3-kinaseb.. J. Med. Chem. 2015, 58, 5684–5688. 10.1021/acs.jmedchem.5b00498. [DOI] [PubMed] [Google Scholar]
- Come J. H.; Collier P. N.; Henderson J. A.; Pierce A. C.; Davies R. J.; Le Tiran A.; O’Dowd H.; Bandarage U. K.; Cao J.; Deininger D.; Grey R.; Krueger E. B.; Lowe D. B.; Liang J.; Liao Y.; Messersmith D.; Nanthakumar S.; Sizensky E.; Wang J.; Xu J.; Chin E. Y.; Damagnez V.; Doran J. D.; Dworakowski W.; Griffith J. P.; Jacobs M. D.; Khare-Pandit S.; Mahajan S.; Moody C. S.; Aronov A. M. Design and Synthesis of a Novel Series of Orally Bioavailable, CNS-Penetrant, Isoform Selective Phosphoinositide 3-KinaseK (PI3K) Inhibitors with Potential for the Treatment of Multiple Sclerosis (MS). J. Med. Chem. 2018, 61, 5245–5256. 10.1021/acs.jmedchem.8b00085. [DOI] [PubMed] [Google Scholar]
- Talele T. T. Acetylene Group, Friend or Foe in Medicinal Chemistry. J. Med. Chem. 2020, 63, 5625–5663. 10.1021/acs.jmedchem.9b01617. [DOI] [PubMed] [Google Scholar]
- Liu T.; Lescarbeau A.; Nair S. J.; Grenier L.; Pradeilles J. A.; Glenadel Q.; Tibbitts T.; Rowley A. M.; DiNitto J. P.; Brophy E. E.; O’Hearn E. L.; Ali J.; Evans C. AA.; Winkler D. G.; Goldstein S. I.; O’Hearn P.; Martin C. M.; Hoyt J. G.; Soglia J. R.; Cheung C.; Pink M. M.; Proctor J. L.; Palombella V. J.; Tremblay M. R.; Castro A. C. Discovery of a selective phosphoinositide-3-kinase (PI3K)-gamma inhibitor (IPI-549) as an immuno-oncology clinical candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. 10.1021/acsmedchemlett.6b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaid A. G.; Cornella-Taracido I. Causes and Significance of Increased Compound Potency in Cellular or Physiological Contexts. J. Med. Chem. 2018, 61, 1767–1773. 10.1021/acs.jmedchem.7b00762. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe D. A.; et al. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med. Chem. 2011, 3, 2063–2077. 10.4155/fmc.11.149. [DOI] [PubMed] [Google Scholar]
- Sasaki T.; Irie-Sasaki U.; Jones R. G.; Oliveira-Do-Santos A. J.; Stanford W. L.; Bolon B.; Wakeham A.; Itie A.; Bouchard D.; Kozieradzki I.; Joza N.; Mak T. W.; Ohashi P. S.; Suzuki A.; Penninger J. M. Function of PI3K in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.