

Abstract
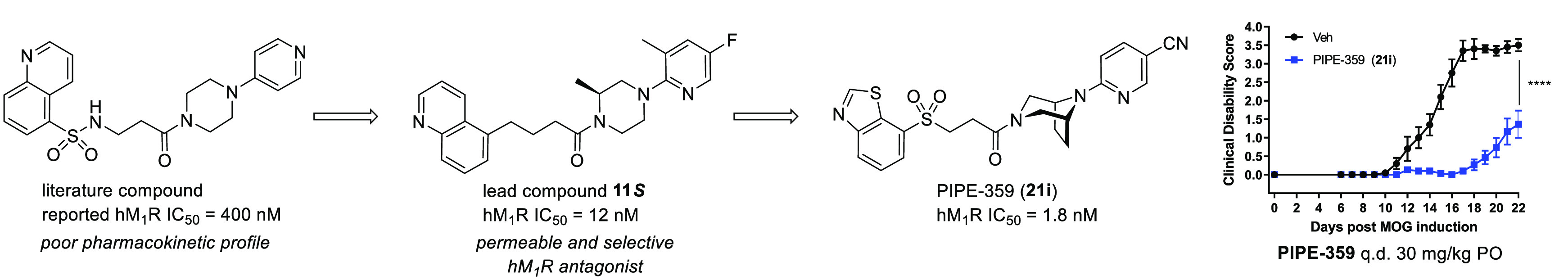
The discovery of PIPE-359, a brain-penetrant and selective antagonist of the muscarinic acetylcholine receptor subtype 1 is described. Starting from a literature-reported M1 antagonist, linker replacement and structure–activity relationship investigations of the eastern 1-(pyridinyl)piperazine led to the identification of a novel, potent, and selective antagonist with good MDCKII-MDR1 permeability. Continued semi-iterative positional scanning facilitated improvements in the metabolic and hERG profiles, which ultimately delivered PIPE-359. This advanced drug candidate exhibited robust efficacy in mouse myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalitis (EAE), a preclinical model for multiple sclerosis.
Keywords: M1-selective, antimuscarinic, M1 antagonist, EAE, remyelination
Multiple sclerosis (MS) is an immune-mediated disorder characterized by destruction of the insulating myelin that surrounds the axons of neurons in the central nervous system (CNS).1−3 The result of demyelination is an impairment of conduction along the affected nerve, which can manifest itself in a variety of neurological symptoms from mild to severe. A 2017 study4 estimated that nearly 1 million individuals in the U.S. are living with MS. Fortunately, there have been tremendous breakthroughs in pharmacotherapies for the treatment of MS in the last few decades.5,6 A vast majority of these drugs dampen the peripheral immune response, resulting in the reduction of relapses and the delay of overall progression of disabilities.7 However, a true “cure”, which would require the repair and restoration of nerve function, is not currently available.
Remyelination represents an attractive avenue to facilitate repair of nerve function within the CNS in patients diagnosed with MS.8 Remyelination involves differentiation of oligodendrocyte progenitor cells (OPCs) into mature oligodendrocytes,9 the neuroglia responsible for creating the myelin sheath. High-throughput screening efforts have identified nonselective antimuscarinics as myelin-regenerative compounds.10,11 Subsequently benztropine (Figure 1), a CNS-penetrant antimuscarinic, and clemastine, an antihistamine that possesses anticholinergic activity, have both demonstrated efficacy in mouse myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalitis (MOG-EAE),10,12 a widely used preclinical model for multiple sclerosis. As a reduction in clinical score in EAE can be attributed to either an immunomodulatory effect, remyelination, or some combination thereof, Chan and co-workers used M1 muscarinic receptor (M1R) knockout mice to demonstrate that remyelination itself is sufficient.12 Additional support for the M1R as a target for remyelination is provided by the phase II ReBUILD trial, where clemastine fumarate showed encouraging results in promoting visual-pathway remyelination in patients with MS as determined by reduced visual-evoked potential (VEP) P100 latency as the primary outcome measure.13 The effect is thought to be the result of off-target antagonism of muscarinic receptors.12,14 Taken together, these results show that selective M1R antagonism represents a truly differentiated approach to the treatment of MS through remyelination. Accordingly, details of the discovery of a CNS-penetrant M1R antagonist, PIPE-359, are provided herein.
Figure 1.

Structures of the nonselective antimuscarinics benztropine and scopolamine and the mixed antihistamine–antimuscarinic clemastine.
There are five subtypes (M1–M5) of muscarinic acetylcholine receptors (mAChRs), a subclass of G-protein-coupled receptors (GPCRs), which are widely distributed to varying degrees in the CNS and periphery that elicit a diverse range of biological functions.15 The orthosteric binding site is highly conserved across all five subtypes,16 and consequently, very few selective small-molecule M1R antagonists have been identified to date.17−20 Conversely, there are myriad nonselective antagonists, many of which are FDA-approved medications.21 Commonly prescribed antimuscarinics include benztropine (Cogentin) for symptoms of Parkinson’s disease, tiotropium (Spiriva) for chronic obstructive pulmonary disease (COPD) or asthma, and oxybutynin (Ditropan) for overactive bladder (OAB). Despite the prevalence of pan-antimuscarinics, a selective M1R antagonist is desirable to reduce unwanted side effects associated with nonselective compounds.21,22 For example, the selective M1R antagonist VU0255035 (Figure 2) demonstrated inhibition of pilocarpine-induced seizures in mice at 10 mg/kg ip but did not result in the cognitive impairment observed with the nonselective M1R antagonist scopolamine in a hippocampus-dependent learning model.17 A related molecule, VU0415248,18 served as the starting point for our structure–activity relationship (SAR) study.
Figure 2.

M1R-selective antagonist VU0255035 and initial SAR development from VU0415248.
A structural breakdown of VU0415248 is shown in Figure 2. As the target is a centrally expressed M1R, we initially focused on replacing the sulfonamide linker with a less polar carbon-based linker, a design element intended to facilitate brain permeation, as topological polar surface area (tPSA) and hydrogen-bond donor (HBD) count are known to negatively correlate with brain exposure.23−25 Direct replacement of the sulfonamide group of VU0415248 with a methylene unit gave compound 1, which came with a surprising 5-fold increase in antagonist potency on the human M1R (hM1R). Despite the decrease in tPSA and lack of an HBD, compound 1 suffered from considerable PgP-mediated efflux in the MDCKII-MDR1 permeability assay,26 likely due to the presence of a highly basic eastern pyridyl ring. Lessening of the basicity by the addition of a fluorine atom (compound 2) significantly reduced the efflux but unfortunately came with a cost in potency.
To identify a viable lead with appreciable potency and minimal PgP efflux, a small library of weakly basic (calculated pKa ≤ 7)27 analogues (3–17) were synthesized by coupling readily available monopyridinyl cyclodiamines with 4-(quinolin-5-yl)butanoic acid. Human M1R IC50 values are shown in Table 1. IC50 values ranging from 6.3 to 69 nM were observed for compounds 5, 9, 11, 15, and 16, indicating that a strongly basic nitrogen atom was not required for potency. Potency was achieved with the pyridyl nitrogen either ortho (9 and 11), meta (15), or para (5 and 16) to the piperazine substitution. Replacing the piperazine with a 2,6-diazaspiro[3.3]heptane (12 vs 6), 2,5-diazabicyclo[2.2.1]heptane (13 vs 6), or homopiperazine (17 vs 9) decreased the potency. Comparisons of 1-(5-fluoropyridin-2-yl)piperazine amide 6 with analogues that possessed methyl substitutions on the piperazine ring (7 and 10) revealed potency losses of roughly half an order of magnitude. However, potency increases were observed when a methyl group was added to the fluoropyridine ring ortho to the piperazine substitution (9 vs 6, 8 vs 7, and 11 vs 10). In fact, “magic dimethyl” analogue 11 (IC50 = 6.3 nM) was approximately 40-fold, 10-fold, and 200-fold more potent than the desmethyl (6), methylpyridyl (9), and methylpiperazyl (10) analogues, respectively. The enantiomers of compound 11 were synthesized individually, and their functional activities, measured across the human M1–4 receptors, are shown in Table 2. In-house data for benztropine are included for reference. Both enantiomers 11R and 11S were potent antagonists of the hM1R and exhibited selectivities superior to that of benztropine. Excellent selectivities (>75-fold) versus the hM2 and hM3 receptors were observed, and the S isomer (11S) displayed better selectivity versus the hM4R (ΔpEC50 = 1.3) than did the R isomer (11R) (ΔpEC50 = 0.8). On the basis of the activity and selectivity profile of 11S, the compound was selected for profiling in the MDCKII-MDR1 assay, which revealed good permeability (8.0 × 10–6 cm/s) and low PgP-mediated efflux (efflux ratio = 0.5). Further optimizations would be based on this promising early lead (11S).
Table 1. IC50a and pIC50 Values for Various 4-(Quinolin-5-yl)butanoic Amide Analogues.

IC50 values were taken as dose-dependent decreases in the EC80 acetylcholine response determined in CHO-K1 cells expressing the hM1R.
Table 2. Human Muscarinic Receptor (hMnR) Functional Potencies (pIC50) and Selectivities (ΔpIC50) for Compounds 11R, 11S, and Benztropine.
| hMn pIC50 (ΔpIC50[M1 – Mn]) |
||||
|---|---|---|---|---|
| compd | M1 | M2 | M3 | M4 |
| 11R | 8.2 | 6.2 (2.0) | 6.3 (1.9) | 7.4 (0.8) |
| 11S | 7.9 | 5.7 (2.2) | 5.9 (2.0) | 6.6 (1.3) |
| benztropine | 8.6 | 7.2 (1.4) | 7.7 (0.9) | 8.3 (0.3) |
While a desirable level of hM1R potency was achieved with a number of 4-(quinolin-5-yl)butanoic amides, selected compounds 5, 9, 11, 15, and 16 all lacked metabolic stability when incubated in rat liver microsomes (<1% parent remaining after 15 min of incubation). An initial attempt to improve the metabolic stability involved decreasing the overall lipophilicity28 of the molecule via replacement of the quinoline ring. A series of carbon-based amide analogues were designed in which the western bicyclic heteroarene contained a ring nitrogen atom in a similar or adjacent position to that of the quinoline,18 while the eastern end of the scaffold was fixed as the (S)-1-(5-fluoro-3-methylpyridin-2-yl)-3-methylpiperazine of compound 11S. Results are shown in Table 3. The less lipophilic quinazoline 18a and hydroxymethyl quinoline 18c were less potent (83 and 170 nM, respectively) than compound 11S and failed to significantly improve the metabolic stability versus 11S in rat liver microsome incubations. While not less lipophilic (as calculated) than 11S, the moderately potent benzo[d]thiazole 18d displayed a >25-fold increase in stability. Adding an additional amino substituent to the benzo[d]thiazole (compound 18e) or swapping the sulfur atom for an oxygen (benzo[d]oxazole 18f) did not further improve the stability.
Table 3. Positional Scan of the Western Bicyclic Heteroarene.

Calculated for pH 7.4 using the ChemAxon logD plugin.
Percentage of parent compound remaining after 15 min of incubation (at 1 μM initial concentration) in rat liver microsomes (0.5 mg/mL).
Although benzo[d]thiazole 18d had increased metabolic stability versus 11S in vitro, the improvement was not significant enough to warrant further in vivo profiling. Attention subsequently turned toward modification of the linker. Based on 18d, a limited scan of more polar heteroatom-containing linkers was performed. Of the four linker variants shown in Table 4, only 19c containing a 3-sulfonylamide linker retained appreciable potency. While introduction of the sulfone functionality further protected the scaffold from oxidative metabolism (vs 18d), further optimization was necessary.
Table 4. Positional Scan of Heteroatom-Containing Linkers.

At this point in our discovery program, reoptimization of the eastern 1-(pyridinyl)cyclodiamine end of the molecule was carried out while fixing the western aromatic and linker as the more stable 3-(benzo[d]thiazol-7-ylsulfonyl)propanamide. An emphasis was placed on designing compounds with logD values significantly lower than that of sulfone 19c (calcd logD = 2.7). In this vein, we initially limited the cyclodiamine core to the less lipophilic unsubstituted piperazine. Coupling of several 1-(pyridinyl)piperazines with 3-(benzo[d]thiazol-7-ylsulfonyl)propanoic acid produced derivatives 20a–h shown in Table 5. Better potencies were achieved when the pyridine nitrogen atom was ortho to the piperazine substitution (compounds 20e–g) versus being meta (20h) or para (20a–d). Of particular note, nicotinonitrile 20e (IC50 = 5.8 nM) exhibited potency on par with the most potent compound (11R, IC50 = 6.3 nM) in the carbon-based series. Additionally, 20e was found to be considerably more stable in rat liver microsome incubations (59% parent remaining at 15 min) compared with the initial sulfone lead 19c (9.6% remaining at 15 min). For this reason, 20e was selected for further profiling in vivo. At 2 h after oral dose (10 mg/kg), the average unbound plasma and brain concentrations in rats (n = 2) of 20e were 13 and 2.2 nM, respectively (Table 6). The presence of free 20e in the rat CNS after oral administration at a concentration approaching its in vitro functional potency prompted us to assess M1R engagement in vivo. Unfortunately, a preliminary receptor occupancy experiment29 performed in mice gave underwhelming results, as a 30 mg/kg po dose achieved only ∼50% receptor occupancy at 2 h, suggesting that the compound would be unsuitable for evaluation in the EAE model. In addition, further characterization of 20e revealed a potential hERG liability (69% inhibition at 3 μM).
Table 5. Positional Scan of the Eastern Pyridyl.

Table 6. Rat NeuroPK and hERG Inhibition Data for Compound 20e.
| compd | Cp (nM) | fu,p | Cp,u (nM) | Cb,u (nM) | Kp,uu | hERG @ 3 μM |
|---|---|---|---|---|---|---|
| 20e | 89 | 0.15 | 13 | 2.2 | 0.17 | 69% |
The next set of optimizations involved fixing the niconitrile and western 3-(benzo[d]thiazol-7-ylsulfonyl)propanamide sections of 20e and performing a scan of the piperazine core (Table 7). The goal was to achieve improvements in PK and/or M1R potency, either of which should translate into increased M1R occupancy in vivo. To improve brain permeation via the reduction of polar surface area, tetrahydropyridine 21a and piperidine 21b were prepared. Unfortunately, both 21a (80 nM) and 21b (19 nM) had decreased potencies compared with 20e (5.8 nM). Methylation of the piperazine ring was preferred closer to the cyanopyridine (21d vs 21c), and potency was maintained with an additional methyl substitution on the piperazine ring (21g), but only when it was placed adjacent to the anilinic nitrogen (21g vs 21f). When two methyl groups were placed adjacent to the amide nitrogen atom of the piperazine (compounds 21e and 21h), the potency dropped significantly. Also included in the scan were bridged bicyclic variants 21i–l, all of which exhibited reasonable hM1R antagonism. Comparison of symmetrical azatropanes 21i (IC50 = 1.8 nM) and 21j (IC50 = 8.7 nM) revealed a preference for the bridge to be proximal to the cyanopyridine, similar to the SAR observed with dimethylated analogues 21g and 21h. Gratifyingly, readouts for potent azatropane 21i in both rat microsomal stability (67% remaining at 15 min) and hERG inhibition (36% inhibition at 3 μM) revealed improvements versus 20e.
Table 7. Positional Scan of the Central Azacycle.

Further profiling of compound 21i, designated as PIPE-359, was carried out, and the results are shown in Table 8. Radioligand binding affinity (Ki) measurements with [3H]-N-methyl scopolamine revealed exceptionally high affinity for the hM1R (Ki = 0.14 nM). Furthermore, PIPE-359 exhibited good-to-excellent selectivities versus the hM2R, hM3R, and hM4R in both functional and binding30 settings. PIPE-359 had moderate intrinsic clearance when incubated with both rat and mouse liver microsomes (extraction ratios of 0.42 and 0.47, respectively). In rat PK (2/10 mg/kg iv/po), a total clearance of 56 mL min–1 kg–1 was observed, along with an oral bioavailability of 4.5%. More importantly, an unbound brain concentration (Cb,u) of 3.4 nM was reached 2 h after 10 mg/kg oral administration of PIPE-359, a concentration several fold over the measured hM1R Ki.
Table 8. Profile of PIPE-359 (21i).

| Human mAChR Activity | |
|---|---|
| IC50 (pIC50) | ΔpIC50[M1 – Mn] |
| M1: 1.8 nM (8.8) | |
| M2: 200 nM (6.7) | 2.1 |
| M3: 55 nM (7.3) | 1.5 |
| M4: 22 nM (7.7) | 1.1 |
| Ki (pKi) | ΔpKi[M1 – Mn] |
| M1: 0.14 nM (9.8) | |
| M2: 19 nM (7.7) | 2.1 |
| M3: 0.84 nM (9.1) | 0.7 |
| M4: 6.5 nM (8.2) | 1.6 |
| In Vitro ADMET |
|---|
| microsomal stability |
| Clint (μL/min/mg): 42 (human), 23 (rat), 21 (mouse) |
| predicted extraction ratio: 0.72 (h), 0.42 (r), 0.47 (m) |
| tissue binding |
| PPB fu: 0.20 (r), 0.18 (m) |
| BTB fu: 0.13 (m) |
| MDCK-MDR1 permeability |
| Papp (A-B): 7.7 × 10–6 cm/s |
| efflux ratio: 3.9 |
| hERG inhibition (patch clamp) |
| IC50: 5.6 μM |
| CYP inhibition |
| IC50: > 10 μM (2C9, 2D6, 3A4) |
| Rat PK |
|---|
| iv/po 2/10 mg/kg |
| Cl/Clu: 56/273 mL min–1 kg–1 |
| t1/2 (iv): 0.9 h |
| AUC/AUCu: 100/21 h ng/mL |
| F: 4.5% |
| NeuroPK 10 mg/kg po 2 h after dose |
| Cb/Cb,u: 26/3.4 nM |
| Cp/Cp,u: 80/16 nM |
| Kp/Kp,uu: 0.33/0.21 |
To assess the candidacy and dosing regimen of PIPE-359 in the MOG-EAE model, mouse M1R occupancy studies were performed (Figure 3). In an initial experiment, PIPE-359 was dosed orally at 0.1, 1.0, 10, and 30 mg/kg (n = 3/group). At 2 h, a dose-dependent increase in M1R occupancy was observed, with full occupancy achieved at 30 mg/kg (Figure 3a). A follow-up time-course M1R occupancy study with a 30 mg/kg po dose of PIPE-359 revealed that full receptor occupancy was maintained for up to 8 h (Figure 3b) with a drop to 40% occupancy at 24 h. Bioanalysis of the 8 h plasma and forebrain concentrations of PIPE-359 revealed an average unbound plasma concentration (Cp,u) of 117 nM and an average Cb,u of 4.9 nM (8 h Kp,uu = 0.04), the latter being well above the functional potency (IC50 = 1.8 nM) and binding affinity (Ki = 0.14 nM) of PIPE-359. While the 24 h brain concentrations of PIPE-359 were below the limit of quantification, extrapolation from the average unbound plasma concentration (Cp,u at 24 h = 2.9 nM) using a Kp,uu value of 0.04 yields a Cb,u at 24 h of 0.12 nM, a value consistent with the 40% occupancy observed. On the basis of these results, PIPE-359 was evaluated in the mouse MOG-EAE model by dosing orally qd at 30 mg/kg. As shown in Figure 3c, separation of clinical scores between drug- and vehicle-treated animals emerged on day 11, increased through to the peak of disease (day 17), and persisted throughout the chronic phase of the study. To our knowledge, this is the first demonstration of efficacy in EAE with a selective M1R antagonist.
Figure 3.
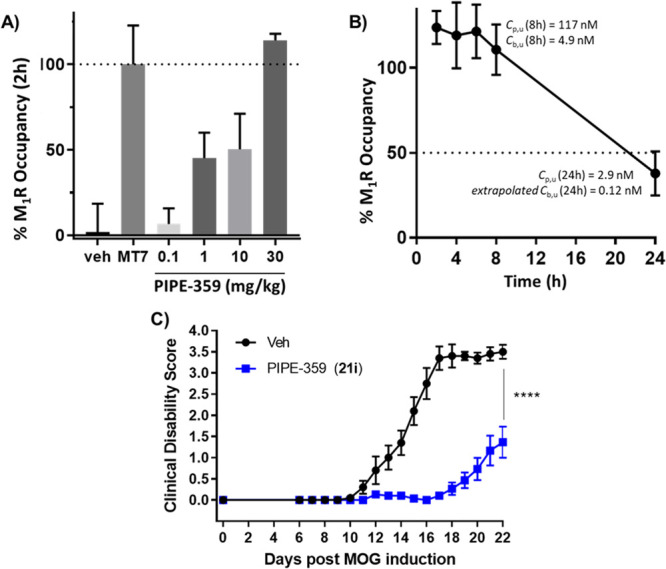
Mouse receptor occupancy and MOG-EAE experiments with PIPE-359 (21i). (A) Dose–response receptor occupancy 2 h after po dose. [3H]-Pirenzepine (300 nM) was added ex vivo as a radiotracer. Occupancy was measured as % response of hM1R-selective antagonist muscarinic toxin 7 (MT7)31 added ex vivo (300 nM). (B) Receptor occupancy time course of PIPE-359 at 30 mg/kg po. (C) Prophylactic treatment (qd 30 mg/kg po) with PIPE-359 in MOG-EAE performed in C57BL/6 mice (n = 10–15/group). EAE induction was performed on day 0 followed by once daily administration (qd) of PIPE-359 at 30 mg/kg po or vehicle for 21 days. Clinical scores were recorded daily, and changes were observed starting on day 6 and continued through day 22. ****, p < 0.0001 based on Sidak’s multiple comparison tests respective to vehicle controls.
In summary, PIPE-359 (21i), a brain-penetrant and selective hM1R antagonist with remarkable efficacy in mouse MOG-EAE was discovered. A first round of optimization from a known weakly active hM1R-selective antagonist delivered a permeable, potent, and selective molecule (compound 11S). Subsequent rounds of positional scanning improved the pharmacokinetics and hERG liabilities, ultimately delivering PIPE-359. The demonstration of efficacy with PIPE-359 in EAE provides a path forward for the use of selective hM1R antagonists as treatments for MS patients. Continued optimizations, which involve improving target selectivity and brain permeation, are ongoing and will be reported in due course.
Acknowledgments
The authors thank Yunlong Yang (WuXi AppTec), Yu Xue (Pharmaron, Inc.), and their colleagues for synthetic contributions, and Danxi Li (Pharmaron, Inc.) and her colleagues for providing in vitro ADMET data. We also thank John Frazer (Beacon Discovery) for providing radioligand binding data for compound 21i.
Glossary
Abbreviations
- MS
multiple sclerosis
- MOG-EAE
myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalitis
- CNS
central nervous system
- OPC
oligodendrocyte progenitor cell
- VEP
visual-evoked potential
- M1R
muscarinic receptor subtype 1
- mAChR
muscarinic acetylcholine receptor
- GPCR
G-protein-coupled receptor
- CHO
Chinese hamster ovary
- rMS
rat microsomal stability
- PgP
P-glycoprotein 1
- MDCKII-MDR1
Madin-Darby canine kidney II-multidrug resistance protein
- PK
pharmacokinetic
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- ip
intraperitoneal
- po
per os (oral)
- iv
intravenous
- qd
quaque die (once daily)
- SAR
structure–activity relationship
- HBD
hydrogen-bond donor
- tPSA
topological polar surface area
- PPB
plasma protein binding
- fu
fraction unbound
- BTB
brain tissue binding
- Cb,u
unbound brain concentration
- Cp,u
unbound plasma concentration
- Kp,uu
ratio of unbound brain compound concentration to unbound plasma compound concentration
- hERG
human ether-à-go-go-related gene
- CYP
cytochrome P450.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00626.
Conditions for in vitro biological assays, in vitro ADMET assays, synthetic procedures, analytical data for all compounds, and full characterization data for compounds 11S, 20e, and 21i (PDF)
Author Contributions
† T.O.S. and Y.X. contributed equally. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lemus H. N.; Warrington A. E.; Rodriguez M. Multiple Sclerosis: Mechanisms of Disease and Strategies for Myelin and Axonal Repair. Neurol. Clin. 2018, 36, 1–11. 10.1016/j.ncl.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman E. M.; Racke M. K.; Raine C. S. Multiple Sclerosis-The Plaque and Its Pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- National Multiple Sclerosis Society . What Is MS? https://www.nationalmssociety.org/What-is-MS (accessed 2020-12-10).
- Wallin M. T.; Culpepper W. J.; Campbell J. D.; Nelson L. M.; Langer-Gould A.; Marrie R. A.; Cutter G. R.; Kaye W. E.; Wagner L.; Tremlett H.; Buka S. L.; Dilokthornsakul P.; Topol B.; Chen L. H.; LaRocca N. G. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology 2019, 92, e1029–e1040. 10.1212/WNL.0000000000007035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello K.; Halper J.; Kalb R.; Skutnik L.; Rapp R.. The Use of Disease-Modifying Therapies in Multiple Sclerosis: Principles and Current Evidence. A Consensus Paper by the Multiple Sclerosis Coalition, updated September 2019. https://www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/DMT_Consensus_MS_Coalition.pdf (accessed 2020-12-10).
- Goldschmidt C.; McGinley M. P. Advances in the Treatment of Multiple Sclerosis. Neurol. Clin. 2021, 39, 21–33. 10.1016/j.ncl.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommer P. S.; Milo R.; Han M. H.; Satyanarayan S.; Sellner J.; Hauer L.; Illes Z.; Warnke C.; Laurent S.; Weber M. S.; Zhang Y.; Stuve O. Immunological aspects of approved MS therapeutics. Front. Immunol. 2019, 10, 1564. 10.3389/fimmu.2019.01564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassari L. E.; Feng J.; Clayton B. L. L.; Oh S.-H.; Sakaie K.; Tesar P. J.; Wang Y.; Cohen J. A. Developing therapeutic strategies to promote myelin repair in multiple sclerosis. Expert Rev. Neurother. 2019, 19, 997–1013. 10.1080/14737175.2019.1632192. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Deshmukh V.; Tardif V.; Lyssiotis C. A.; Green C. C.; Kerman B.; Kim H. J.; Padmanabhan K.; Swoboda J. G.; Ahmad I.; Kondo T.; Gage F. H.; Theofilopoulos A. N.; Lawson B. R.; Schultz P. G.; Lairson L. L. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F.; Fancy S. P. J.; Shen Y. A.; Niu J.; Zhao C.; Presley B.; Miao E.; Lee S.; Mayoral S. R.; Redmond S. A.; Etxeberria A.; Xiao L.; Franklin R. J. M.; Green A.; Hauser S. L.; Chan J. R. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. 10.1038/nm.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F.; Lehmann-Horn K.; Shen Y. A.; Rankin K. A.; Stebbins K. J.; Lorrain D. S.; Pekarek K.; Sagan S. A.; Xiao L.; Teuscher C.; von Büdingen H.-C.; Wess J.; Lawrence J. J.; Green A. J.; Fancy S. J. P.; Zamvil S. S.; Chan J. R. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 2016, 5, e18246 10.7554/eLife.18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A. J.; Gelfand J. M.; Cree B. A.; Bevan C.; Boscardin W. J.; Mei F.; Inman J.; Arnow S.; Devereux M.; Abounasr A.; Nobuta H.; Zhu A.; Friessen M.; Gerona R.; von Büdingen H. C.; Henry R. G.; Hauser S. L.; Chan J. R. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. 10.1016/S0140-6736(17)32346-2. [DOI] [PubMed] [Google Scholar]
- Tesar P. J.; Cohen J. A. Clemastine fumarate for promotion of optic nerve remyelination. Lancet 2017, 390, 2421–2422. 10.1016/S0140-6736(17)32639-9. [DOI] [PubMed] [Google Scholar]
- Muscarinic Receptors; Fryer A. D., Christopoulos A., Nathanson N. M., Eds.; Handbook of Experimental Pharmacology, Vol. 208; Springer: Berlin, 2012. [Google Scholar]
- Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; Kobilka T. S.; Sexton P. M.; Kobilka P. K.; Christopoulos A. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler D. J.; Williams R.; Bridges T. M.; Xiang Z.; Kane A. S.; Byun N. E.; Jadhav S.; Mock M. M.; Zheng F.; Lewis L. M.; Jones C. K.; Niswender C. M.; Weaver C. D.; Lindsley C. W.; Conn P. J. A Novel Selective Muscarinic Acetylcholine Receptor Subtype 1 Antagonist Reduces Seizures without Impairing Hippocampus-Dependent Learning. Mol. Pharmacol. 2009, 76, 356–368. 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Lamers A. P.; Bridges T. M.; Sulikowski G. A.; Utley T. J.; Sheffler D. J.; Noetzel M. J.; Morrison R. D.; Daniels J. S.; Niswender C. M.; Jones C. K.; Conn P. J.; Lindsley C. W.; Wood M. R. Development of a more highly selective M1 antagonist from the continued optimization of the MLPCN Probe ML012. Bioorg. Med. Chem. Lett. 2012, 22, 1044–1048. 10.1016/j.bmcl.2011.11.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Utley T. J.; Sevel C.; Mattmann M. E.; Cheung Y.-Y.; Bridges T. M.; Morrison R. D.; Sheffler D. J.; Niswender C. M.; Daniels J. S.; Conn P. J.; Lindsley C. W.; Wood M. R. Development of novel M1 antagonist scaffolds through the continued optimization of the MLPCN probe ML012. Bioorg. Med. Chem. Lett. 2012, 22, 5035–5040. 10.1016/j.bmcl.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller N. R.; Daniels R. N.; Lee P.; Conn P. J.; Lindsley C. W. Synthesis and SAR of N-(4-(4-alklylpiperazin-1-yl)phenyl)benzamides as muscarinic acetylcholine receptor subtype 1 (M1) antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 2174–2177. 10.1016/j.bmcl.2010.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew M. L.; Mulsant B. H.; Pollock B. G.; Lehman M. E.; Greenspan A.; Mahmoud R. A.; Kirshner M. A.; Sorisio D. A.; Bies R. R.; Gharabawi G. Anticholinergic activity of 107 medications commonly used by older adults. J. Am. Geriatr. Soc. 2008, 56, 1333–1341. 10.1111/j.1532-5415.2008.01737.x. [DOI] [PubMed] [Google Scholar]
- Wagg A.; Verdejo C.; Molander U. Review of cognitive impairment with antimuscarinic agents in elderly patients with overactive bladder. Int. J. Clin. Pract. 2010, 64, 1279–1286. 10.1111/j.1742-1241.2010.02449.x. [DOI] [PubMed] [Google Scholar]
- Dichiara M.; Amata B.; Turnaturi R.; Marrazzo A.; Amata E. Tuning Properties for Blood–Brain Barrier (BBB) Permeation: A Statistics-Based Analysis. ACS Chem. Neurosci. 2020, 11, 34–44. 10.1021/acschemneuro.9b00541. [DOI] [PubMed] [Google Scholar]
- Gupta M.; Lee H.-J.; Barden C. J.; Weaver D. F. The Blood–Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. 10.1021/acs.jmedchem.9b01220. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving Beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1, 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The MDR1-MDCKII cell line was obtained from The Netherlands Cancer Institute (Amsterdam) Borst Laboratory. For functional comparisons between the widely used Borst and National Institutes of Health (NIH) MDR1-MDCKII cell lines as well as relevant experimental procedures, see:Feng B.; West M.; Patel N. C.; Wager T.; Hou X.; Johnson J.; Tremaine L.; Liras J. Validation of Human MDR1-MDCK and BCRP-MDCK Cell Lines to Improve the Prediction of Brain Penetration. J. Pharm. Sci. 2019, 108, 2476–2483. 10.1016/j.xphs.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Calculated with the ChemAxon pKa plugin.
- Hopkins A. L.; Keserü M. G.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Compound 20e was dosed orally at 30 mg/kg in mice 2 h prior to brain collection and dissection. The forebrain tissues were homogenized and incubated with 3H-pirenzepine for 10 min. After washing, the residual radioactivity in the forebrain homogenate was quantified and normalized. For an additional application of 3H-pirenzepine, see:; Valuskova P.; Farar V.; Forczek S.; Krizova I.; Myslivecek J. Autoradiography of 3H-pirenzepine and 3H-AFDX-384 in Mouse Brain Regions: Possible Insights into M1, M2, and M4 Muscarinic Receptors Distribution. Front. Pharmacol. 2018, 9, 124. 10.3389/fphar.2018.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reported binding affinities (Ki values) of several pan-antimuscarinics across human M1–5 receptors, see:Casarosa P.; Bouyssou P.; Germeyer S.; Schnapp A.; Gantner A.; Pieper M. Preclinical Evaluation of Long-Acting Muscarinic Antagonists: Comparison of Tiotropium and Investigational Drugs. J. Pharmacol. Exp. Ther. 2009, 330, 660–668. 10.1124/jpet.109.152470. [DOI] [PubMed] [Google Scholar]
- Maeda S.; Xu J.; Kadji F. M. N.; Clark M. J.; Zhao J.; Tsutsumi N.; Aoki J.; Sunahara R. K.; Inoue A.; Garcia K. C.; Kobilka B. K. Structure and selectivity engineering of the M1 muscarinic receptor toxin complex. Science 2020, 369, 161–167. 10.1126/science.aax2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.