

Abstract
Mutations in Munc18-1/STXBP1 (syntaxin-binding protein 1) are linked to various severe early epileptic encephalopathies and neurodevelopmental disorders. Heterozygous mutations in the STXBP1 gene include missense, nonsense, frameshift, and splice site mutations, as well as intragenic deletions and duplications and whole-gene deletions. No genotype-phenotype correlation has been identified so far, and patients are treated by anti-epileptic drugs because of the lack of a specific disease-modifying therapy. The molecular disease mechanisms underlying STXBP1-linked disorders are yet to be fully understood, but both haploinsufficiency and dominant-negative mechanisms have been proposed. This review focuses on the current understanding of the phenotypic spectrum of STXBP1-linked disorders, as well as discusses disease mechanisms in the context of the numerous pathways in which STXBP1 functions in the brain. We additionally evaluate the available animal models to study these disorders and highlight potential therapeutic approaches for treating these devastating diseases.
Keywords: encephalopathy, epilepsy, Munc18–1, STXBP1, synapse, therapeutic approaches
1 |. INTRODUCTION
Mutations in Munc18–1/STXBP1 are associated with severe early onset epileptic encephalopathies, such as Ohtahara syndrome, West syndrome, and Dravet syndrome (Carvill et al. 2014; Otsuka et al. 2010; Saitsu et al. 2008), as well as non-syndromic epilepsies, atypical Rett syndrome, and severe intellectual disability without epilepsy (Hamdan et al. 2009; Hamdan et al. 2011; Olson et al. 2015; Romaniello et al. 2015). These devastating and often fatal infantile encephalopathies are all characterized by severe intellectual disability and cerebral dysfunction, and most include unremitting epileptic activity with eventual cognitive, sensory and/or motor function deterioration (Dravet & Oguni 2013; Hrachovy & Frost 2013; Yamatogi & Ohtahara 2002). Although these disorders vary in their age of onset, developmental outcome, etiology, and seizure type (Carvill et al. 2014; Hrachovy & Frost 2013; Ohtahara & Yamatogi 2006), a common feature is that they are refractory to standard antiepileptics (McTague & Cross 2013; Panayiotopoulos 2005), similar to most neonatal seizure disorders (Berg et al. 2001). Thus, the prognosis is very poor.
Herein, we summarize the recent findings on STXBP1 function and underlying disease mechanisms, as well as highlight potential therapeutic rescue strategies that have been identified in the past 2 years.
2 |. STXBP1-LINKED ENCEPHALOPATHIES
Mutations in STXBP1 were first identified in 2008 in five patients with a devastating early infantile epileptic encephalopathy known as Ohtahara syndrome (Saitsu et al. 2008). Patients with Ohtahara syndrome have intractable seizures with a characteristic suppression-burst pattern on EEG and severe psychomotor retardation; the prognosis is extremely poor, with a mortality rate of up to 50% (Beal et al. 2012). Several of the identified patients went on to progress to West Syndrome, which is characterized by hypsarrhythmia on EEG and exists on a continuum with Ohtahara syndrome (Saitsu et al. 2010). Soon after, patients with other severe early onset epileptic encephalopathies (EOEEs) were also identified, including ones who initially presented with West syndrome (Otsuka et al. 2010), Lennox-Gastaut syndrome (Allen et al. 2013), Dravet syndrome (Carvill et al. 2014), early myoclonic encephalopathy (Nicita et al. 2015), and numerous unclassified EOEEs (Barcia et al. 2013; Kwong et al. 2015; Keogh et al. 2015; Mastrangelo et al. 2013; Di Meglio et al. 2015; Romaniello et al. 2014; Sampaio et al. 2015; Vatta et al. 2012). EOEEs are characterized by severe seizures that are thought to greatly contribute to the profound developmental delay in these patients (Beal et al. 2012).
Importantly, however, mutations were soon identified in a cohort of patients without EOEE (Hamdan et al. 2009). Subsequently, many more patients without EOEE were identified, including those with mental retardation and non-syndromic epilepsy (Hamdan et al. 2009), atypical Rett syndrome (Cogliati et al. 2019; Olson et al. 2015; Romaniello et al. 2015; Yuge et al. 2018; Lopes et al. 2016), ataxia-tremor-retardation syndrome without epilepsy (Gburek-Augustat et al. 2016; Degerliyurt et al. 2019), and intellectual disability without epilepsy (Hamdan et al. 2011; Stamberger et al. 2016; Rauch et al. 2012). The identification of these patients without epilepsy has led to the proposal that STXBP1-associated syndromes be re-categorized from EOEEs to STXBP1 encephalopathies (STXBP1-E), a set of neurodevelopmental disorders including epilepsy, as this study of 147 STXBP1 patients did not find a correlation between seizure outcome and cognitive outcome, or a statistical difference between the groups with milder ID and those with severe ID with regard to age at seizure onset, age at seizure freedom, and duration of seizures (Stamberger et al. 2016). Since then, the Deciphering Developmental Disorders (DDD) study done in the United Kingdom, which had broad inclusion criteria in order to study undiagnosed patients with pediatric developmental disorders, identified 11 children with STXBP1 mutations and only one of them had EOEE (Suri et al. 2017). It is notable that of these 10 patients without EOEE, five of their mutations were previously identified in patients with EOEE. Furthermore, two of these five patients had no history of seizures at all. Thus, although STXBP1 has long warranted inclusion in epilepsy panels, STXBP1 can now be characterized more broadly as a developmental disorder gene. As such, STXBP1 is now found not only in epilepsy gene panels but in multiple gene panels for intellectual disability and developmental delay. Given that the prevalence of intellectual disability is roughly 1% in the United States, additional testing and awareness will likely lead to an increase in the number of future STXBP1 diagnoses (Zablotsky et al. 2017).
Overall, STXBP1 encephalopathies have a wide phenotypic spectrum, but all patients have some form of intellectual disability and 85% of patients have some form of epilepsy, which in the majority of patients occurs very early in life, with a median onset of 6 weeks (Stamberger et al. 2016) (Figure 1). Most patients experience severe to profound intellectual disability (88%), and autism/autistic-like features are seen in one-fifth of patients (Stamberger et al. 2016). The most frequent seizure types are epileptic spasms, in almost two-third of patients; motor disturbances include ataxia, hypotonia, dystonia, tremor, spasticity, and dyskinesia (Stamberger et al. 2016), and awake bruxism has been identified in 80% of patients (Rezazadeh et al. 2019). Several older patients with STXBP1 mutations have shown signs of Parkinsonism, including tremor, bradykinesia, and antecollis (Alvarez Bravo & Yusta Izquierdo 2018; Keogh et al. 2015).
FIGURE 1.

Phenotypic spectrum of all known STXBP1 patients. (a) Patients were categorized by their final clinical diagnosis (green = non-epileptic syndromes; blue = epileptic syndromes). (b–g) Phenotypic spectra subcategorized by mutations. Final patient diagnoses were separated by type of mutation into (b) missense mutations, (c) nonsense mutations, (d) frameshift mutations, (e) splice site mutations, (f) intragenic deletions/duplications, and (g) whole-gene deletions (ASD, autism spectrum disorder; ATR, ataxia-tremor-retardation syndrome; ID, intellectual disability; EE, epileptic encephalopathies; EOEE, early onset epileptic encephalopathy)
Through our review of case reports and patient studies published since 2008, we have identified a total of 282 patients with STXBP1 mutations and have analyzed their mutation types and clinical phenotypes (Figure 1 and Table S1). The majority of these patients have epilepsy (85%) and all have intellectual disability or developmental delay (Figure 1a). In terms of epileptic syndromes, most patients with STXBP1 mutations have West syndrome (25%) or an unclassified EOEE (24%) (Figure 1a). When missense mutations are compared to the other mutation types that affect only STXBP1, there is no difference between the percentage of epileptic versus non-epileptic patients in these groups (approximately 80% and 20%, respectively, for both missense mutations and for the other mutation types combined), that is, no clear genotype-phenotype correlation exists. However, among patients with whole-gene deletions, non-syndromic epilepsy and ID without epilepsy encompass over 50% of the patients, in comparison with fewer than 20% patients overall (Figure 1g). This discrepancy may be because of the fact that other genes besides STXBP1 are also impacted by these deletions, which we discuss further below (see “Disease-causing mutations in STXBP1” section). Importantly, some differences do exist between the mutational groups, although none are significant with the current number of identified patients. Nonsense mutations are associated with EOEE more than any other group, and fewer non-epileptic cases are seen in intragenic deletions/duplications. Finally, missense mutations lead to the most diverse phenotypes, which may suggest that different missense mutations cause different syndromes. Although Figure 1 represents our most current understanding of the spectrum of STXBP1 disorders, it is important to note, however, that patients enter the healthcare system at different ages and some may be more likely to receive genetic tests, depending on their presenting symptoms. For instance, broad genetic sequencing methods have high diagnostic yields particularly in early-life epilepsies and are recommended as part of these patients’ initial evaluation (Berg et al. 2017; Butler et al. 2017; Shellhaas et al. 2017). The same is not true for intellectual disability, which may present later and may not be flagged as quickly for genetic testing. Thus, although there is likely an overrepresentation of patients with epilepsy in this dataset, the ~20% of patients overall without epilepsy (Figure 1) again reinforces the fact that STXBP1 syndromes are a set of neurodevelopmental disorders that include epilepsy and not just EOEEs. As whole-exome sequencing costs continue to fall and more clinics have access to these diagnostic methods, it is likely that the numbers of STXBP1 patients without epilepsy will increase (de Ligt et al. 2012; Rauch et al. 2012).
Because genetic sequencing is now a first-line investigation for children presenting with epilepsy (Symonds & McTague 2020) and is beneficial in those with developmental delay in the absence of epilepsy (Vandersluis et al. 2020), more STXBP1 patients overall are expected to be identified in the near future. As such, we will be able to more fully appreciate the clinical phenotype of STXBP1 syndromes: an understanding of the number of patients with STXBP1 mutations without epilepsy, as well as an idea of disease progression and overall morbidity and mortality. It is even possible that a genotype-phenotype correlation may emerge. Although we focus here on the developmental syndromes associated with STXBP1 mutations, a recent review highlighted the connection between STXBP1 mutations and the progression of neurologic symptoms later in the disease course, with features similar to early onset parkinsonism. (Lanoue et al. 2019). As patients with STXBP1 mutations get older, it will be important to follow up with these patients to determine the nature of this connection and whether a genotype-phenotype correlation exists for these late complications. Finally, understanding how STXBP1 mutations lead to dysfunction will be particularly important in developing novel and effective therapies.
3 |. STXBP1 FUNCTION
SEC1/Munc18-like (SM) proteins are essential for secretion in yeast (SEC1) (Aalto et al. 1991), Caenorhabditis elegans (UNC-18) (Hosono et al. 1992), zebrafish (Stxbp1) (Grone et al. 2016), Drosophila melanogaster (ROP) (Harrison et al. 1994), and in mice (Munc18–1) (Verhage et al. 2000), and impairments in these SM proteins are linked to severe deficits in vesicle fusion.
Neurotransmitter release is governed by the formation of neuronal SNARE-complexes, a process tightly controlled to meet the unique regulatory requirements of a synapse (reviewed in Rizo & Südhof 2012). SNARE proteins are maintained by accessory proteins, such as syntaxin-1 by STXBP1 (Figure 2). STXBP1 interacts with syntaxin-1 in two different binding modes: Binding of STXBP1 to the Habc domain of syntaxin-1 in its “closed conformation” prevents syntaxin-1 from forming ectopic SNARE-complexes while trafficking to the cell surface (Medine et al. 2007), and from forming uncontrolled SNARE-complexes with its cognate binding partners at the synapse (Burkhardt et al. 2008; Hata et al. 1993; Ma et al. 2011; Misura et al. 2000; Pevsner et al. 1994; Rickman et al. 2007). In addition to this inhibitory role, binding of STXBP1 to the N-terminus of syntaxin-1 in its “open conformation” has been shown to facilitate SNARE-complex formation and neurotransmitter release (Deak et al. 2009; Dulubova et al. 2007; Khvotchev et al. 2007; Rathore et al. 2010; Rickman et al. 2007; Shen et al. 2007), possibly by binding directly to assembled SNARE-complexes (Rodkey et al. 2008; Shen et al. 2007). However, this role of STXBP1’s interaction with syntaxin-1’s N-peptide has been contradicted by others, as it also has been shown that STXBP1 mutations that inhibit this interaction still support normal synaptic transmission (Meijer et al. 2012). Importantly, work on understanding STXBP1’s function in synaptic vesicle release is still ongoing. Recent studies have demonstrated that STXBP1 functions with Munc13–1 to prevent the de-priming of synaptic vesicles by NSF (He et al. 2017), and multiple groups have shown that STXBP1 may act as a template for SNARE-complex assembly (Jiao et al. 2018; Parisotto et al. 2014; Sitarska et al. 2017; Wang et al. 2019), which may be a conserved role for SM proteins (Baker et al. 2015; Jiao et al. 2018).
FIGURE 2.
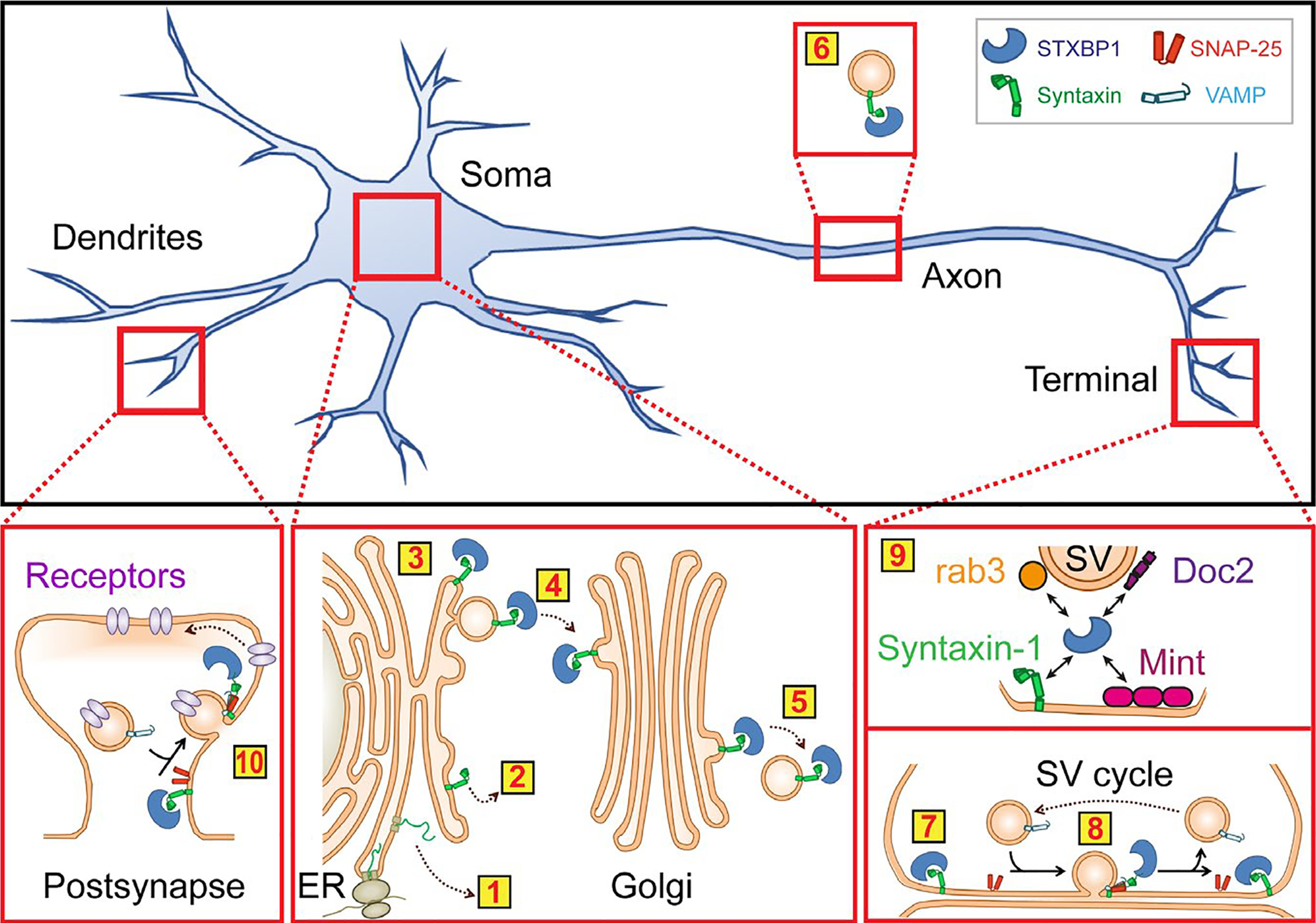
Proposed functions of STXBP1 within a neuron. Yellow boxes highlight cellular processes that mutations in STXBP1 may affect: Degradation (1) or promiscuous SNARE-complex formation (2) of syntaxin-1 during biosynthesis because of lack of STXBP1 binding (3); trafficking of syntaxin-1 from the ER to the Golgi (4) and to the synapse (5, 6); synaptic SNARE-complex formation with SNAP-25 and synaptobrevin-2/VAMP2 (7) and neurotransmitter release (8), via binding to rab3, Doc2, Mint1/2, and syntaxin-1 (9; note that the arrows here indicate experimental evidence for binding only); possibly SNARE-complex formation responsible for post-synaptic receptor trafficking (10). Note that the interaction of STXBP1 with syntaxin-1 and its effect on neurotransmitter release is the most established interaction, whereas the others have not been investigated to that detail. Yet, for completeness, these interactions are included here as well
Beyond syntaxin-1, the impact of STXBP1 on its other binding partners and their relevance to disease is not known. STXBP1 interacts with the synaptic proteins rab3 (Graham et al. 2008), Doc2 (Verhage et al. 1997), and Mint proteins (Okamoto & Südhof 1997) (Figure 2). The structural regions of STXBP1 required for these interactions as well as the functional implications of these interactions are relatively uncharacterized. Rab3 functions in vesicle docking (Fukuda 2008; Nonet et al. 1997; Schluter et al. 2004), and mice lacking rab3A or all four rab3 isoforms exhibit synaptic depression upon repeated stimulation (Geppert et al. 1994) or perinatal lethality and decreased release probability (Schluter et al. 2004), respectively. Doc2 proteins bind STXBP1 in competition with syntaxin-1 which may regulate the STXBP1/syntaxin-1 interaction during vesicle docking (Verhage et al. 1997). Doc2a/b translocate to the plasma membrane in the presence of elevated intracellular calcium (Groffen et al. 2004), and have been proposed to have both calcium-dependent and calcium-independent roles in various forms of neurotransmitter release (Groffen et al. 2006; Groffen et al. 2010; Pang et al. 2011; Yao et al. 2011) and synaptic plasticity (Sakaguchi et al. 1999; Xue et al. 2018; Yao et al. 2011). In addition, Doc2a and Doc2b have been shown to be differentially expressed in glutamatergic and GABAergic neurons (Courtney et al. 2018), respectively, suggesting that a potential impact of mutant STXBP1 on Doc2a and Doc2b could perturb the balance between excitation and inhibition often associated with seizures. Mint1 and Mint2 bind to the STXBP1/syntaxin-1 complex (Okamoto & Südhof 1997), and may recruit STXBP1 to the active zone (Biederer & Südhof 2000). Mint-1 knockout mice exhibit a marked deficit in GABAergic synaptic transmission (Ho et al. 2003). No studies have yet addressed the role of these STXBP1 binding partners in STXBP1-E, but their important functions in the synapse merit investigation of their pathogenic contributions. Inactivation of STXBP1 in mice and ROP in flies causes early lethality and leads to loss of neurotransmitter release and neurodegeneration (Harrison et al. 1994; Peng et al. 2015; Verhage et al. 2000), and inactivation of unc18 in worms causes severe paralysis (Miller et al. 1996), emphasizing the need for STXBP1 in neuronal communication. In addition, knockdown of syntaxin-1 phenocopies STXBP1 null mice (de Wit et al. 2006; Voets et al. 2001), STXBP1 knockout triggers a 40% reduction in Doc2 levels (Toonen et al. 2005; Voets et al. 2001), and Mint1/2/3 knockout causes a 33% increase in STXBP1 levels (Ho et al. 2006), demonstrating the intricate relationships between STXBP1 and its binding partners.
However, STXBP1 is not restricted to axons and axon terminals, but is also present in cell bodies. In the CA3 region of mice expressing Venus-tagged STXBP1, STXBP1 localized primarily to the cell body and axon (Cijsouw et al. 2014). The somatic localization suggests that STXBP1 might have also non-synaptic roles. STXBP1 was shown to regulate post-Golgi transport of vesicles to the plasma membrane, as well as subsequent vesicle fusion at the cell surface, for proper distribution of proteins crucial for excitatory neuron migration during corticogenesis, as well as other pre-synaptic proteins (Hamada et al. 2017; Law et al. 2016). Neurons lacking STXBP1 showed mislocalization of syntaxin-1 at the Golgi (Santos et al. 2017), suggesting that, without STXBP1, syntaxin-1 export from the Golgi apparatus to the plasma membrane and synapses is impaired (Figure 2). Last, STXBP1 has been shown to chaperone alpha-synuclein, a protein causatively linked to Parkinson’s disease (Polymeropoulos et al. 1997; Spillantini et al. 1997), which controls the self-replicating aggregation of alpha-synuclein (Chai et al. 2016).
Although knockout of STXBP1 in mice has been reported to not affect brain development (Verhage et al. 2000), it has been recently suggested that STXBP1 is crucial for radial migration of cortical neurons, as well as neurite extension during brain development (Hamada et al. 2017; Yamashita et al. 2016). In addition, studies in the fly suggest that the STXBP1-homolog ROP is a key regulator of dendrite development via interaction with the exocyst subunit Sec6 (Peng et al. 2015). This is particularly interesting since dendrite defects have been shown to be the strongest pathological correlate of intellectual disability (Kaufmann & Moser 2000).
Finally, STXBP1 has been shown to also interact with other members of the syntaxin protein family, and some of these syntaxin proteins have been shown to play important roles in the trafficking of post-synaptic neurotransmitter receptors (Arendt et al. 2015; Bin et al. 2018; Duan et al. 2020; Gu & Huganir 2016; Jurado et al. 2013; Kennedy et al. 2010) (Figure 2). With its synaptic and axonal subcellular distribution, it is unlikely that STXBP1 plays a role also in post-synaptic exocytosis, but this cannot be excluded until detailed studies investigate this possibility. In addition, it remains unknown if mutations in STXBP1 affect its possible post-synaptic function.
In summary, STXBP1 interacts with a variety of synaptic proteins, which highlights the central role of STXBP1 in neurotransmitter release. Early work focused on the now well-established importance of STXBP1’s interaction with the SNARE protein syntaxin-1, both in trafficking syntaxin-1 to the synapse and in facilitating SNARE-complex formation, although the exact molecular role that STXBP1 plays in this facilitation remains unclear and will require clarification. Similarly, STXBP1’s other synaptic binding partners, such as Doc2 and Mint proteins, were identified early, but their relevance to STXBP1’s activity and disease pathogenicity has either only been poorly explored or not at all. Because of STXBP1’s non-synaptic localization, other roles for STXBP1 have been identified, such as protein trafficking, dendrite development and Golgi transport, but these findings are far more recent and most merit further study. Thus, how mutations in STXBP1 affect each of these interactions and the physiological activity of STXBP1 and its interaction partners, however, remains unknown.
4 |. DISEASE-CAUSING MUTATIONS IN STXBP1
Disease-causing mutations impacting STXBP1 include missense, nonsense, frameshift, and splice-site mutations, as well as intragenic, whole gene, and multi-gene deletions (Stamberger et al. 2016) (Figure 3 and Table S1).
FIGURE 3.

Disease-causing missense mutations in STXBP1. Top: Primary sequence of STXBP1 with indication of its domain structure and positions of disease-linked missense mutations. Bottom: Localization of disease-causing missense mutations of STXBP1 in its tertiary structure (PDB code 4JEU1 (Colbert et al. 2013)). Mutated residues are highlighted in magenta in three different views. Protein structure images were generated using the PDB file and PyMOL (Schrödinger)
Missense mutations have been identified throughout the protein structure, with no domain either particularly spared or susceptible (Figure 3). Most missense mutations studied to date destabilize the STXBP1 protein, causing aggregation and degradation (Chai et al. 2016; Guiberson et al. 2018; Kovacevic et al. 2018; Martin et al. 2014; Saitsu et al. 2008). Missense mutations have been shown to co-aggregate wild-type STXBP1 when co-expressed (Chai et al. 2016; Guiberson et al. 2018). A misfolded protein that is prone to degradation and aggregation may also destabilize and co-aggregate the proteins it is directly bound to. Interestingly, co-aggregation of mutant STXBP1 with alpha-synuclein has been proposed (Chai et al. 2016), which may explain the Parkinsonian symptoms seen in some older patients (Lanoue et al. 2019). It remains to be seen whether this effect is also present in other types of mutations in which part of the protein, presumably containing the motif that is responsible for binding of STXBP1 to its interactors, remains intact. Other missense mutations have an additional impact on STXBP1 function. For example, the P335A/L mutation exhibits a hyper-secretion phenotype that is driven by a structural change in STXBP1 that is thought to promote SNARE complex formation and increased neurotransmitter release (Guiberson et al. 2018; Han et al. 2014; Munch et al. 2016; Parisotto et al. 2014; Park et al. 2017; Zhu et al. 2020). Although it was believed that only heterozygous mutations can be tolerated, two cases with Lennox-Gastaut syndrome were recently reported that carried a homozygous STXBP1 mutation (Lammertse et al. 2020). Interestingly, the associated missense mutation L446F was shown to have a milder impact on protein levels than other identified missense mutations, and to cause a gain-of-function phenotype regarding synaptic transmission.
Several mutations result in an early stop codon (Table S1). These range from relatively early in the protein sequence such as L36X (Stamberger et al. 2016) and E53X (Saitsu et al. 2010) to quite late (W522X) (Stamberger et al. 2016). Premature stop codons can result in a truncated protein, which may retain residual function depending on which features of the structure are preserved and to what degree the truncated protein is unstable and susceptible to degradation. A relatively stable, albeit truncated, STXBP1 protein could interact with STXBP1 and its binding partners, impeding their function and possibly destabilizing or co-aggregating them, similar to what is seen in cases with missense mutations. However, no study has found that a truncated STXBP1 protein is present at detectable levels in cells, and even small truncations disrupt syntaxin-1 binding (Hata & Südhof 1995), suggesting that a full-length STXBP1 protein may be required to bind some of its effectors.
It has been proposed that some STXBP1 transcripts bearing nonsense mutations (Table S1) may be degraded by nonsense-mediated decay. For a splice-site mutation that caused the loss of exon 8, mutant transcripts were reduced to ~ 30% of wild-type transcript levels (Saitsu et al. 2010; Saitsu & Matsumoto 2011). It remains unknown whether the remaining transcript is translated into protein. However, if the remaining mutant transcript is translated, this could lead to similarly low STXBP1 levels as shown for several missense mutations (Guiberson et al. 2018), with potentially deleterious effects on wild-type STXBP1 and STXBP1-binding partners, or put stress on protein quality control resources within the cell.
In contrast, some of the transcripts bearing nonsense mutations may undergo stop codon read-through, in which the premature stop codon is substituted for another amino acid, creating an effective missense mutation (Blanchet et al. 2015; Namy et al. 2004; Schueren et al. 2014). How deleterious this mutation may be depends on the original residue that was mutated, what amino acid the stop codon was substituted for, and how tolerable that particular substitution is at that particular position within the structure. Whether stop codon read-through occurs for nonsense mutations in STXBP1 remains unknown. However, enhancing premature stop codon read-through may be an attractive potential therapy for certain nonsense mutations that if bypassed, would result in a full-length and stable protein (Dabrowski et al. 2018).
Frameshift mutations also have been found throughout the STXBP1 sequence (Stamberger et al. 2016) (Table S1). Some are caused by indels quite early in the protein sequence, whereas most are in the middle to end of the sequence. Whether these frameshift mutations result in truncated proteins or are degraded as transcripts remains unclear.
There are also several deletions that encompass part or all of STXBP1 (Table S1). Partial deletions include those that eliminate a single exon (Stamberger et al. 2016; Saitsu et al. 2012b) and those that eliminate as many as half the exons (Deprez et al. 2010; Stamberger et al. 2016). These are predicted to form truncated proteins, but it remains unclear whether the resultant transcripts would be translated. Larger deletions that encompass the entire STXBP1 gene have been identified (Stamberger et al. 2016). However, these deletions often encompass many more genes than just STXBP1, including SPTAN1 (Campbell et al. 2012; Saitsu et al. 2008; Saitsu et al. 2010), mutations in which cause early infantile epileptic encephalopathy type 5. Two patients with deletions including SPTAN1 but not STXBP1 presented with intellectual disability, hypotonia, severe speech impairment, and brain abnormalities such as delayed myelination and a small pituitary (Campbell et al. 2012). In a cohort of five patients exhibiting intellectual disability, psychomotor developmental delay, delayed or absent speech, muscular hypotonia, and other neurological symptoms, and who had deletions in the chromosomal region 9q33.3-q34.11, the minimal region of overlap of these deletions did not include STXBP1, but rather RALGPS1 and GARNL3. Of these, a point mutation in GARNL3 has been identified in Autosomal Recessive Intellectual Disability (ARID) (Santos-Cortez et al. 2018). Taken together, this suggests that the patient phenotypes observed in heterozygous deletions that include multiple genes besides STXBP1 should not be ascribed to STXBP1 alone, but rather be viewed as multiple effects from the loss of multiple genes that may be additive or even independent of one another. It is currently unclear whether any of the reported whole-gene deletions affect only STXBP1.
5 |. HAPLOINSUFFICIENCY AND DOMINANT-NEGATIVE EFFECTS
The presence of deletion, nonsense, frameshift, splice site, and missense mutations throughout the sequence of STXBP1 has led to the assumption that the primary mechanism of disease is haploinsufficiency. Consequently, knockout and heterozygous models in worms, flies, and zebrafish have been developed, as well as heterozygous neurons from IPSCs and patient fibroblasts. Additionally, several heterozygous mouse models of STXBP1 have been generated and characterized to mixed results.
In C. elegans, knocking out the STXBP1 homolog unc-18 causes severe paralysis and profoundly reduced neurotransmitter release (Miller et al. 1996; Brenner 1974), but unlike knockouts in flies and mice, unc-18 knockout worms are viable. Several C. elegans models of STXBP1-E exist, and can be grouped according to whether the disease-causing mutation has replaced the endogenous unc-18 gene (Guiberson et al. 2018) or whether the mutation in the human STXBP1 gene is expressed exogenously, usually on an unc-18 null background (Zhu et al. 2020). These two approaches have complementary advantages and drawbacks. For example, because human STXBP1 rescues the unc-18 null phenotype, worms have been generated that express STXBP1 with disease-causing mutations to determine if and to what degree they are able to rescue the unc-18 null phenotype (Zhu et al. 2020). However, most STXBP1 missense mutations studied to date cause the protein to misfold and to be degraded. Two major factors in this process are temperature and expression level. The amino acid sequence of human STXBP1 has evolved such that the protein structure is thermostable at 37°C, whereas C. elegans are usually maintained at 20°C. The effects of missense mutations that cause misfolding and degradation in human cells may thus be masked or mitigated by the reduction of ambient thermal folding stress in worms. Furthermore, over-expression of human STXBP1 in worms may result in an artifactual exacerbation of a misfolding effect by overwhelming endogenous chaperones, or an opposite problem, the masking of a detrimental effect by expressing a high enough level of partially functional mutant protein that is sufficient to rescue normal synaptic function. Directly editing the endogenous unc-18 gene sidesteps these issues. Yet, although unc-18 and STXBP1 have high sequence similarity, and in 92% of patients with missense mutations the wild-type amino acid residue that is substituted is conserved between worms and humans, there are some patients whose mutations are less well suited for study in unc-18, and may thus need to be studied in the context of the human STXBP1.
In Drosophila, inactivation of the STXBP1 homolog Rop (Salzberg et al. 1993) causes embryonic lethality (Harrison et al. 1994). Heterozygous null mutations and two missense mutations in Rop mutant flies are viable but reveal decreased synaptic release (Wu et al. 1998). Knocking out the zebrafish homologs of STXBP1, stxbp1a and stxbp1b, causes immobility and early death or a seizure-like phenotype, respectively (Grone et al. 2016). Whereas heterozygous stxbp1a knockout had a mildly impaired dark flash response, heterozygous stxbp1b knockout zebrafish were behaviorally normal (Grone et al. 2016).
Neurons induced from stem cells have been used more recently to establish two models of haploinsufficiency. A 2015 model used homologous recombination to mutagenize the STXBP1 gene in human embryonic stem cells and thus create heterozygous lines to compare to isogenic wild-type control lines (Patzke et al. 2015). They found a 30% reduction in STXBP1 and syntaxin-1 protein levels in heterozygous induced neurons (similar to early mouse models of haploinsufficiency), normal neuronal development and survival, but a decrease in pre-synaptic neurotransmitter release. Interestingly, two groups have used fibroblasts from STXBP1 patients with Ohtahara syndrome to create induced neurons. One group found a 50% reduction in STXBP1 and syntaxin-1 proteins levels when compared to family derived control induced neurons and a defect in neurite extension (Yamashita et al. 2016). Another group established a protocol to induce pluripotent stem cell lines from postmortem tissue following the death of a patient with a STXBP1 mutation (Yamamoto et al. 2019). Induced neurons from patient fibroblasts may hold promise in understanding effects of mutations on neural development in human neurons, which may be more susceptible to reduction in STXBP1 than animal models. However, further study will require an increased number of patient samples to draw any conclusions.
Original mouse models failed to recapitulate the disease phenotypes seen in patients with STXBP1 nonsense or truncating mutations. Two early studies characterized the heterozygous mouse strain originally generated to study STXBP1 null mice, in which exons 2–6 were deleted and translation from amino acid 14 was prevented (Hager et al. 2014; Orock et al. 2018; Verhage et al. 2000). The first study of these STXBP1+/− mice noted that heterozygous mice had anxiety-like behavior, but did not find any evidence of cognitive impairment, motor dysfunction or epileptic seizures (Hager et al. 2014). The second study found some spatial learning and memory deficits but no evidence of seizures, perhaps because of only a 25% decrease in STXBP1 protein levels (Orock et al. 2018). Another group designed new STXBP1+/− mice, with exon 3 deleted, but these also only had a 25% reduction in STXBP1 in the cortex, although they did note a 50% reduction in the hippocampus (Miyamoto et al. 2017). There was no evidence of seizures, but cognitive deficits, elevated anxiety, and increased aggression was found (Miyamoto et al. 2017).
The two most recent models of STXBP1+/− mice have demonstrated phenotypes more similar to what is seen in patients. A 2018 study generated four new STXBP1+/− mouse models: in three of them, mice developed an epilepsy-like phenotype of twitches and jumps during sleep, as well as anxiety and mild cognitive impairments (Kovacevic et al. 2018). Importantly, the most recent model of STXBP1+/− mice recapitulates the disease phenotype very closely (Chen et al. 2020). The mice show a 40–50% reduction in STXBP1 protein in most brain regions at 3 months, and exhibit severe cognitive impairment, elevated anxiety and repetitive behaviors, repeated abnormal spike-wave discharges on EEGs as well as frequent myoclonic seizures, and motor disturbances including hindlimb clasping, which is suggestive of dystonia and impaired motor coordination (Chen et al. 2020). Furthermore, they identified impairments in cortical inhibition by both parvalbumin and somatostatin interneurons as a potential cause of cortical hyperexcitability, echoing a previous result that STXBP1 heterozygosity in GABAergic neurons specifically led to early lethality in a subset of mice and very strong epileptiform activity in the surviving subset (Chen et al. 2020; Kovacevic et al. 2018). This model of STXBP1+/− mice replicates the disease phenotype well and will hopefully allow for testing of potential treatments, which so far have only been tested in vitro and in C. elegans in vivo models (Guiberson et al. 2018).
However, a dominant-negative effect has also been observed with several missense mutations (Chai et al. 2016; Guiberson et al. 2018). We previously showed that five missense mutants are unstable, aggregate, and are rapidly degraded. Upon co-expression with wild-type STXBP1, mutant STXBP1 destabilized and co-aggregated wild-type STXBP1 as well, likely via direct binding. A missense disease model is thus also important to develop. Although no genotype-phenotype correlation has been identified yet, we may begin to identify differences between patients with missense and truncating mutations as more patients are diagnosed (Stamberger et al. 2016).
In summary, considering all of the known mutations, it is clear that 50% STXBP1 expression is insufficient for normal function, but that in some cases there is an additional dominant-negative effect present. However, so far, patients carrying missense mutations do not appear to have symptoms that are more severe than those who have deletions or truncating mutations that preclude the production of a STXBP1 protein, nor do patients with missense mutations appear to show progressive symptoms that distinguish them from patients with deletions or truncations. How can these observations be reconciled? If missense mutations do not exert a dominant-negative effect on wild-type STXBP1, the remaining mutant STXBP1 may retain some functionality, which would result in a functional STXBP1 pool above 50% and presumably milder or even a lack of symptoms. In support of this, several studies have demonstrated the apparent functionality of mutant STXBP1: expression of missense mutants in C. elegans partially or completely rescued the paralysis of the unc-18 knockout worms (Guiberson et al. 2018; Zhu et al. 2020), and expression of missense mutants in primary mouse neurons rescued cell viability, neurotransmitter release, and morphological defects associated with STXBP1 knockout (Guiberson et al. 2018; Kovacevic et al. 2018). However, given that missense mutations are just as severe as frameshift and truncation mutations, the dominant-negative activity of missense mutants likely further depletes the functional STXBP1 pool to a point at which symptoms are indistinguishable from those caused by a heterozygous deletion. In addition, it remains to be seen whether long-term, age-dependent differences emerge as more patients are identified and grow older. Importantly, recent work has implicated STXBP1 mutants in the co-aggregation the Parkinson’s disease-related protein alpha-synuclein (Chai et al. 2016), and some older patients have displayed Parkinsonian symptoms including tremor, bradykinesia, and antecollis (Alvarez Bravo & Yusta Izquierdo 2018; Keogh et al. 2015).
6 |. TREATMENT OPTIONS FOR STXBP1 ENCEPHALOPATHIES
Treatment options for STXBP1 encephalopathies are very limited and are currently focused on seizure control, although like for most neonatal seizure disorders, drug resistance is very common (Berg et al. 2001). The most commonly used anti-epileptic drugs (AEDs) are phenobarbital, valproic acid, and vigabatrin, and over half of the patients use more than three AEDs (Khaikin & Mercimek-Mahmutoglu 2016; Stamberger et al. 2016). Other treatments that have been effective are levetiracetam (Dilena et al. 2016; O’Brien et al. 2019; Stamberger et al. 2016; Vatta et al. 2012), ACTH (Liu et al. 2018), cannabidiol (Alvarez Bravo & Yusta Izquierdo 2018), and in rare cases, surgery (Otsuka et al. 2010; Weckhuysen et al. 2013). Over one-third of patients become seizure free, but 40% of patients still experience seizures, and of this group, almost 40% had them more than once a week (Stamberger et al. 2016). Furthermore, no treatments currently exist for the intellectual disability, motor or behavioral disturbances, which exert a significant impact on the quality of life of both the patients and their caregivers.
Our recent study suggests a novel therapeutic intervention for these syndromes. We identified three chemical chaperones, trehalose, sorbitol, and 4-phenylbutyrate (4-PB), which are able to restore STXBP1 protein levels, rescue synaptic deficits, and reduce aggregation in primary mouse neurons and in C. elegans models (Guiberson et al. 2018). Chemical chaperones can be divided into osmolytes and hydrophobic compounds: osmolytes like sorbitol and trehalose act by altering solvent properties, and hydrophobic compounds like 4-PB are thought to interact with the exposed hydrophobic regions of unfolded proteins and protect them from aggregation (Cortez & Sim 2014). Although these compounds are not specific to STXBP1, they were found to reverse every deficit caused by the mutant protein in our models and hold promise as a potential therapy. Importantly, all three compounds were able to increase levels of mutant and wild-type STXBP1 as well (Guiberson et al. 2018), hopefully enabling their use also for patients with nonsense and frameshift mutations. As in vivo testing of these compounds has only been done in missense C. elegans models, further testing will be required in heterozygous animal models to demonstrate in vivo effects for cases of nonsense and frameshift mutations. However, 4-PB has shown particular promise as a potential therapy, as it crosses the blood-brain barrier in non-human primates, is orally available, and FDA-approved for treatment of urea-cycle disorders (Iannitti & Palmieri 2011, Maestri et al. 1996). Currently, a clinical pilot trial of 4-PB in a small group of STXBP1 patients is set to begin in 2020, and it will be the first trial of a disease-modifying therapy in this patient population.
Although 4-PB is a promising potential therapy, a target-specific pharmacological chaperone that binds specifically to STXBP1 may be an important next step in developing therapeutic options. The high concentrations required by chemical chaperones increase the risk of off-target and side effects, which will be reduced by the use of pharmacological chaperones that can be used at much lower concentrations. However, identifying small molecules that can stabilize pathogenic proteins that are not enzymes, channels, or receptors has proven difficult, as these proteins do not have easily identifiable and functional binding sites, although several studies have been successful in other fields (Li et al. 2020; Mecozzi et al. 2014; Pena-Diaz et al. 2019). Further studies are required to screen for and identify such molecules that are effective for both wild-type and mutant STXBP1.
Other treatments that have been proposed include small molecules to prevent formation of non-productive synaptic SNARE complexes to increase monomeric syntaxin-1, and anti-sense oligonucleotide therapies to target the miRNAs that bind to STXBP1 mRNA and repress their expression (Hussain 2014; Stamberger et al. 2017). A recent study has identified miR-218 and miR-424 as responsible for regulating STXBP1 expression. Inhibition of their interaction with STXBP1 by antagomirs or steric blocking oligonucleotides led to an increase in STXBP1 protein levels (Bogush et al. 2019). Although anti-sense oligonucleotides have been successful for neuronal syndromes in the past, notably spinal muscular atrophy, they generally do not cross the blood brain barrier, and delivery remains a challenge (Wurster & Ludolph 2018). Furthermore, miR-218 and miR-424 likely regulate the expression of other genes, so off-target effects, both within neurons and other cell types, are a potential problem.
In summary, with an immediate need for treatment strategies, it is encouraging that multiple different strategies are currently being pursued. Strategies like those described above, which work to increase STXBP1 protein levels either via direct stabilization or increased protein translation, will only work for cases with loss-of-function mutations, which seem to be the vast majority of mutations. However, the recently published case of a homozygous missense mutation, L466F, that was shown to cause a gain-of-function, will not benefit from these therapies (Lammertse et al. 2020). Further work will be needed, first to predict how mutations affect STXBP1 function, as the clinical phenotype seems to be the same regardless of whether a mutation is loss- or gain-of function, and to develop therapies for these gain-of-function mutations. Furthermore, although it remains unclear if there is a critical period for treatment and when treatment should be started, it is promising that activation of mutant gene expression led to robust phenotypic reversal also in mature adult animals in Rett syndrome (Guy et al. 2007), and a similar strategy is currently pursued in a phase III clinical trial for Angelman syndrome (Bird et al. 2019).
7 |. CONCLUSIONS
Since the initial discovery of STXBP1 mutations in Ohtahara patients in 2008, our understanding of both STXBP1 function and dysfunction has greatly improved. The phenotypic spectrum of the disease has grown to include patients without epilepsy, and as new patients are identified by increased genetic screening, a more cohesive picture of the clinical syndrome will soon emerge, as well as information that may point to a potential genotype-phenotype correlation. Simultaneously, we now understand that STXBP1 may not function only at the pre-synapse, as was previously thought, and its role may include general protein trafficking, interactions with other syntaxin proteins, and functions at the post-synapse. However, it remains to be seen how STXBP1’s non-synaptic role is disturbed in disease. Furthermore, as new animal models are being developed — most significantly, a mouse model that truly recapitulates the disease — therapy development is now easier than before. A clinical trial for the first disease-modifying therapy in this patient population will soon be underway, and further therapeutic strategies are being investigated. Importantly, treatment options will likely need to focus on STXBP1 directly, to have an impact on all the complex neuronal pathways that STXBP1 affects.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by T32GM007739 (Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD PhD Program for D.A.), the Markey Graduate School of Medical Science Fellowship (N.G.L.G.), R01-NS113960 and R01-NS102181 (J.B.).
Funding information
Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD PhD Program, Grant/Award Number: T32GM007739; National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01-NS102181 and R01-NS113960
Abbreviations:
- 4-PB
4-phenylbutyrate
- ACTH
adrenocorticotropic hormone
- AED
anti-epileptic drug
- ASD
autism spectrum disorder
- ATR syndrome
ataxia-tremor-retardation syndrome
- EEG
electroencephalogram
- EOEE
early onset epileptic encephalopathies
- ER
endoplasmic reticulum
- ID
intellectual disability
- NSF
N-ethylmaleimide sensitive fusion protein
- SM proteins
SEC1/Munc18-like proteins
- SNAP
synaptosomal-associated protein
- SNARE
soluble N-ethylmaleimide sensitive factor attachment protein receptor
- STXBP1
syntaxin-binding protein 1
- STXBP1-E
STXBP1 encephalopathies
- SV
synaptic vesicle
- VAMP
vesicle-associated membrane protein
Footnotes
CONFLICTS OF INTEREST
The authors state that there was no conflict of interest in the preparation of this review.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- Aalto MK, Ruohonen L, Hosono K, & Keranen S (1991). Cloning and sequencing of the yeast Saccharomyces cerevisiae SEC1 gene localized on chromosome IV. Yeast, 7, 643–650. [DOI] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, … Winawer MR (2013). De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Bravo G, & Yusta Izquierdo A (2018). The adult motor phenotype of Dravet syndrome is associated with mutation of the STXBP1 gene and responds well to cannabidiol treatment. Seizure, 60, 68–70. [DOI] [PubMed] [Google Scholar]
- Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, & Chen L (2015). Retinoic acid and LTP recruit postsynaptic AMPA receptors using distinct SNARE-dependent mechanisms. Neuron, 86, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, & Hughson FM (2015). A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science, 349, 1111–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia G, Barnerias C, Rio M, Siquier-Pernet K, Desguerre I, Colleaux L, Munnich A, … Nabbout R (2013). A novel mutation in STXBP1 causing epileptic encephalopathy (late onset infantile spasms) with partial respiratory chain complex IV deficiency. European Journal of Medical Genetics, 56, 683–685. [DOI] [PubMed] [Google Scholar]
- Beal JC, Cherian K, & Moshe SL (2012). Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatric Neurology, 47, 317–323. [DOI] [PubMed] [Google Scholar]
- Berg AT, Coryell J, Saneto RP, Grinspan ZM, Alexander JJ, Kekis M, … Koh S (2017). Early-life epilepsies and the emerging role of genetic testing. JAMA Pediatr, 171, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Shinnar S, Levy SR, Testa FM, Smith-Rapaport S, & Beckerman B (2001). Early development of intractable epilepsy in children: a prospective study. Neurology, 56, 1445–1452. [DOI] [PubMed] [Google Scholar]
- Biederer T, & Südhof TC (2000). Mints as adaptors. Direct binding to neurexins and recruitment of munc18. Journal of Biological Chemistr, 275, 39803–39806. [DOI] [PubMed] [Google Scholar]
- Bin NR, Ma K, Harada H, Tien C-W, Bergin F, Sugita K … Sugita S (2018). Crucial role of postsynaptic Syntaxin 4 in mediating basal neurotransmission and synaptic plasticity in hippocampal CA1 neurons. Cell Rep, 23, 2955–2966. [DOI] [PubMed] [Google Scholar]
- Bird LM, Ochoa-Lubinoff C, & Tan WH (2019) STARS: Results from a Safety and Efficacy Study of OV101 (Gaboxadol) in Adults and Adolescents with Angelman Syndrome. In: 71st American Academy of Neurology Annual Meeting. Philadelphia, PA, USA. [Google Scholar]
- Blanchet S, Rowe M, Von der Haar T, Fabret C, Demais S, Howard MJ, & Namy O (2015). New insights into stop codon recognition by eRF1. Nucleic Acids Research, 43, 3298–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogush AI, Cheng C, Helbig I, Davidson BL, & Posser BL (2019). Targeting miRNAs as novel therapy for STXBP1 epileptic encephalopathy. STXBP1 Investigator and Family Meeting. Philadelphia (PA): [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt P, Hattendorf DA, Weis WI, & Fasshauer D (2008). Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J, 27, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KM, da Silva C, Alexander JJ, Hegde M, & Escayg A (2017). Diagnostic yield from 339 epilepsy patients screened on a clinical gene panel. Pediatric Neurology, 77, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yatsenko SA, Hixson P, Reimschisel T, Thomas M, Wilson W, … Scaglia F (2012). Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A. Genetics in Medicine, 14, 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Moller RS, Hjalgrim H, … Mefford HC (2014). GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology, 82, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai YJ, Sierecki E, Tomatis VM, Gormal RS, Giles N, Morrow IC, … Meunier FA (2016). Munc18–1 is a molecular chaperone for alpha-synuclein, controlling its self-replicating aggregation. The Journal of Cell Biology, 214, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Cai ZL, Chao ES, Chen H, Longley CM, Hao S, … Xue M (2020). Stxbp1/Munc18–1 haploinsufficiency impairs inhibition and mediates key neurological features of STXBP1 encephalopathy. eLife, 9, e48705. 10.7554/eLife.48705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cijsouw T, Weber JP, Broeke JH, Broek JA, Schut D, Kroon T, Saarloos I, Verhage M, & Toonen RF (2014). Munc18–1 redistributes in nerve terminals in an activity- and PKC-dependent manner. The Journal of Cell Biology, 204, 759–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati F, Giorgini V, Masciadri M, Bonati MT, Marchi M, Cracco I, … Russo S (2019). Pathogenic variants in STXBP1 and in genes for GABAa receptor subunities cause atypical Rett/Rett-like phenotypes. International Journal of Molecular Sciences, 20(15), 3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert KN, Hattendorf DA, Weiss TM, Burkhardt P, Fasshauer D, & Weis WI (2013). Syntaxin1a variants lacking an N-peptide or bearing the LE mutation bind to Munc18a in a closed conformation. Proceedings of the National Academy of Sciences, 110, 12637–12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez L, & Sim V (2014). The therapeutic potential of chemical chaperones in protein folding diseases. Prion, 8(2), 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney NA, Briguglio JS, Bradberry MM, Greer C, & Chapman ER (2018). Excitatory and inhibitory neurons utilize different Ca(2+) sensors and sources to regulate spontaneous release. Neuron, 98(977–991), e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Bukowy-Bieryllo Z, & Zietkiewicz E (2018). Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Molecular Medicine, 24, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, … Vissers LELM (2012). Diagnostic exome sequencing in persons with severe intellectual disability. New England Journal of Medicine, 367, 1921–1929. [DOI] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, & Verhage M (2006). Docking of secretory vesicles is syntaxin dependent. PLoS One, 1, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Südhof TC, & Rizo J (2009). Munc18–1 binding to the neuronal SNARE complex controls synaptic vesicle priming. Journal of Cell Biology, 184, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degerliyurt A, Kesen GG, & Ceylaner S (2019). Ataxia, tremor, intellectual disability: A case of STXBP1 encephalopathy with a new mutation. The Turkish Journal of Pediatrics, 61, 757–759. [DOI] [PubMed] [Google Scholar]
- Deprez L, Weckhuysen S, Holmgren P, Suls A, Van Dyck T, Goossens D, … De Jonghe P (2010). Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology, 75, 1159–1165. [DOI] [PubMed] [Google Scholar]
- Di Meglio C, Lesca G, Villeneuve N, Lacoste L, Abidi F, Cacciagli P, … Milh M (2015). Epileptic patients with de novo STXBP1 mutations: Key clinical features based on 24 cases. Epilepsia, 56, 1931–1940. [DOI] [PubMed] [Google Scholar]
- Dilena R, Striano P, Traverso M, Viri M, Cristofori G, Tadini L, … Zara F (2016). Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain and Development, 38, 128–131. [DOI] [PubMed] [Google Scholar]
- Dravet C, & Oguni H (2013). Dravet syndrome (severe myoclonic epilepsy in infancy). Handbook of Clinical Neurology, 111, 627–633. [DOI] [PubMed] [Google Scholar]
- Duan XL, Guo Z, He YT, Li YX, Liu YN, Bai HH, … Suo ZW (2020). SNAP25/syntaxin4/VAMP2/Munc18–1 complexes in spinal dorsal horn contributed to inflammatory pain. Neuroscience, 429, 203–212. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, & Rizo J (2007). Munc18–1 binds directly to the neuronal SNARE complex. Proceedings of the National Academy of Sciences, 104, 2697–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M (2008). Regulation of secretory vesicle traffic by Rab small GTPases. Cellular and Molecular Life Sciences, 65, 2801–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gburek-Augustat J, Beck-Woedl S, Tzschach A, Bauer P, Schoening M, & Riess A (2016). Epilepsy is not a mandatory feature of STXBP1 associated ataxia-tremor-retardation syndrome. European Journal of Paediatric Neurology, 20, 661–665. [DOI] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, & Sudhof TC (1994). The role of Rab3A in neurotransmitter release. Nature, 369, 493–497. [DOI] [PubMed] [Google Scholar]
- Graham ME, Handley MT, Barclay JW, Ciufo LF, Barrow SL, Morgan A, & Burgoyne RD (2008). A gain-of-function mutant of Munc18–1 stimulates secretory granule recruitment and exocytosis and reveals a direct interaction of Munc18–1 with Rab3. Biochemical Journal, 409, 407–416. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Brian EC, Dudok JJ, Kampmeijer J, Toonen RF, & Verhage M (2004). Ca(2+)-induced recruitment of the secretory vesicle protein DOC2B to the target membrane. Journal of Biological Chemistry, 279, 23740–23747. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Friedrich R, Brian EC, Ashery U, & Verhage M (2006). DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. Journal of Neurochemistry, 97, 818–833. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Diez Arazola R, Cornelisse LN, Lozovaya N, de Jong APH, … Verhage M (2010). Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science, 327, 1614–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grone BP, Marchese M, Hamling KR, Kumar MG, Krasniak CS, Sicca F, … Baraban SC (2016). Epilepsy, behavioral abnormalities, and physiological comorbidities in syntaxin-binding protein 1 (STXBP1) mutant zebrafish. PLoS One, 11, e0151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, & Huganir RL (2016). Identification of the SNARE complex mediating the exocytosis of NMDA receptors. Proceedings of the National Academy of Sciences, 113, 12280–12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiberson NGL, Pineda A, Abramov D, Kharel P, Carnazza KE, Wragg RT, Dittman JS, & Burré J (2018). Mechanism-based rescue of Munc18–1 dysfunction in varied encephalopathies by chemical chaperones. Nature Communications, 9, 3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, & Bird A (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager T, Maroteaux G, Pont P, Julsing J, van Vliet R, & Stiedl O (2014). Munc18–1 haploinsufficiency results in enhanced anxiety-like behavior as determined by heart rate responses in mice. Behavioural Brain Research, 260, 44–52. [DOI] [PubMed] [Google Scholar]
- Hamada N, Iwamoto I, Tabata H, & Nagata KI (2017). MUNC18–1 gene abnormalities are involved in neurodevelopmental disorders through defective cortical architecture during brain development. Acta Neuropathologica Communications, 5, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Dobrzeniecka S, Lortie A, Mottron L, Vanasse M, … Michaud JL (2011). Intellectual disability without epilepsy associated with STXBP1 disruption. European Journal of Human Genetics, 19, 607–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, … Michaud JL (2009). De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Annals of Neurology, 65, 748–753. [DOI] [PubMed] [Google Scholar]
- Han GA, Park S, Bin NR, Jung CH, Kim B, Chandrasegaram P (2014). A pivotal role for pro-335 in balancing the dual functions of Munc18–1 domain-3a in regulated exocytosis. Journal of Biological Chemistry, 289, 33617–33628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, & Rubin GM (1994). Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron, 13, 555–566. [DOI] [PubMed] [Google Scholar]
- Hata Y, Slaughter CA, & Südhof TC (1993). Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature, 366, 347–351. [DOI] [PubMed] [Google Scholar]
- He E, Wierda K, van Westen R, Broeke JH, Toonen RF, Cornelisse LN, & Verhage M (2017). Munc13–1 and Munc18–1 together prevent NSF-dependent de-priming of synaptic vesicles. Nature Communications, 8, 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Morishita W, Hammer RE, Malenka RC, & Sudhof TC (2003). A role for Mints in transmitter release: Mint 1 knockout mice exhibit impaired GABAergic synaptic transmission. Proceedings of the National Academy of Sciences, 100, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Morishita W, Atasoy D, Liu X, Tabuchi K, Hammer RE, Malenka RC, & Südhof TC (2006). Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. Journal of Neuroscience, 26, 13089–13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono R, Hekimi S, Kamiya Y, Sassa T, Murakami S, Nishiwaki K, … Kodaira KI (1992). The unc-18 gene encodes a novel protein affecting the kinetics of acetylcholine metabolism in the nematode Caenorhabditis elegans. Journal of Neurochemistry Journal of Neurochemistry, 58, 1517–1525. [DOI] [PubMed] [Google Scholar]
- Hrachovy RA, & Frost JD Jr (2013). Infantile spasms. Handbook of Clinical Neurology, 111, 611–618. [DOI] [PubMed] [Google Scholar]
- Hussain S (2014). Developing a PPI inhibitor-based therapy for STXBP1 haploinsufficiency-associated epileptic disorders. Frontiers in Molecular Neuroscience, 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannitti T, & Palmieri B (2011). Clinical and experimental applications of sodium phenylbutyrate. Drugs R D, 11, 227–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, He M, Port SA, Baker RW, Xu Y, Qu H, Xiong Y … Zhang Y (2018). Munc18–1 catalyzes neuronal SNARE assembly by templating SNARE association. eLife, 7, e41771. 10.7554/eLife.41771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Südhof TC, & Malenka RC (2013). LTP requires a unique postsynaptic SNARE fusion machinery. Neuron, 77, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, & Moser HW (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex, 10, 981–991. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, & Ehlers MD (2010). Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell, 141, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MJ, Daud D, Pyle A, Duff J, Griffin H, He L, … Chinnery PF (2015). A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late-onset juvenile-onset parkinsonism. Neurogenetics, 16, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaikin Y, & Mercimek-Mahmutoglu S (2016) STXBP1 Encephalopathy with Epilepsy. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. 2016 December 1. [Google Scholar]
- Khvotchev M, Dulubova I, Sun J, Dai H, Rizo J, & Südhof TC (2007). Dual modes of Munc18–1/SNARE interactions are coupled by functionally critical binding to syntaxin-1 N terminus. Journal of Neuroscience, 27, 12147–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacevic J, Maroteaux G, Schut D, Loos M, Dubey M, Pitsch J, … Verhage M (2018). Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain, 141, 1350–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AK, Ho AC, Fung CW, & Wong VC (2015). Analysis of mutations in 7 genes associated with neuronal excitability and synaptic transmission in a cohort of children with non-syndromic infantile epileptic encephalopathy. PLoS One, 10, e0126446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammertse HCA, van Berkel AA, Iacomino M, Toonen RF, Striano P, Gambardella A, Verhage M, & Zara F (2020). Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain, 143, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue V, Chai YJ, Brouillet JZ, Weckhuysen S, Palmer EE, Collins BM, & Meunier FA (2019). STXBP1 encephalopathy: Connecting neurodevelopmental disorders with alpha-synucleinopathies? Neurology, 93, 114–123. [DOI] [PubMed] [Google Scholar]
- Law C, Schaan Profes M, Levesque M, Kaltschmidt JA, Verhage M, & Kania A (2016). Normal molecular specification and neurodegenerative disease-like death of spinal neurons lacking the SNARE-associated synaptic protein Munc18–1. Journal of Neuroscience, 36, 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JG, Chiu J, Ramanjulu M, Blass BE, & Pratico D (2020). A pharmacological chaperone improves memory by reducing Abeta and tau neuropathology in a mouse model with plaques and tangles. Molecular Neurodegeneration, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wang L, Cai XT, Zhou H, Yu D, & Wang Z (2018). Therapeutic benefits of ACTH and levetiracetam in STXBP1 encephalopathy with a de novo mutation: A case report and literature review. Medicine (Baltimore), 97, e0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes F, Barbosa M, Ameur A, Soares G, de Sá J, Dias AI, … Maciel P (2016). Identification of novel genetic causes of Rett syndrome-like phenotypes. Journal of medical genetics, 53, 190–199. [DOI] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, & Rizo J (2011). Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nature Structural & Molecular Biology, 18, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestri NE, Brusilow SW, Clissold DB, & Bassett SS (1996). Long-term treatment of girls with ornithine transcarbamylase deficiency. New England Journal of Medicine, 335, 855–859. [DOI] [PubMed] [Google Scholar]
- Martin S, Papadopulos A, Tomatis VM, Sierecki E, Malintan NT, Gormal RS, … Meunier FA (2014). Increased polyubiquitination and proteasomal degradation of a Munc18–1 disease-linked mutant causes temperature-sensitive defect in exocytosis. Cell Reports, 9, 206–218. [DOI] [PubMed] [Google Scholar]
- Mastrangelo M, Peron A, Spaccini L, Novara F, Scelsa B, Introvini P, Raviglione F, Faiola S, & Zuffardi O (2013). Neonatal suppression-burst without epileptic seizures: Expanding the electroclinical phenotype of STXBP1-related, early-onset encephalopathy. Epileptic Disorders, 15, 55–61. [DOI] [PubMed] [Google Scholar]
- McTague A, & Cross JH (2013). Treatment of epileptic encephalopathies. CNS Drugs, 27, 175–184. [DOI] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, … Small SA (2014). Pharmacological chaperones stabilize retromer to limit APP processing. Nature Chemical Biology, 10, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medine CN, Rickman C, Chamberlain LH, & Duncan RR (2007). Munc18–1 prevents the formation of ectopic SNARE complexes in living cells. Journal of Cell Science, 120, 4407–4415. [DOI] [PubMed] [Google Scholar]
- Meijer M, Burkhardt P, de Wit H, Toonen RF, Fasshauer D, & Verhage M (2012). Munc18–1 mutations that strongly impair SNARE-complex binding support normal synaptic transmission. EMBO J, 31, 2156–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, & Rand JB (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proceedings of the National Academy of Sciences, 93, 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, & Weis WI (2000). Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature, 404, 355–362. [DOI] [PubMed] [Google Scholar]
- Miyamoto H, Shimohata A, Abe M, Abe T, Mazaki E, Amano K, … Yamakawa K (2017). Potentiation of excitatory synaptic transmission ameliorates aggression in mice with Stxbp1 haploinsufficiency. Human Molecular Genetics, 26, 4961–4974. [DOI] [PubMed] [Google Scholar]
- Munch AS, Kedar GH, van Weering JR, Vazquez-Sanchez S, He E, Andre T, … Sorensen JB (2016). Extension of helix 12 in Munc18–1 induces vesicle priming. Journal of Neuroscience, 36, 6881–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Rousset JP, Napthine S, & Brierley I (2004). Reprogrammed genetic decoding in cellular gene expression. Molecular Cell, 13, 157–168. [DOI] [PubMed] [Google Scholar]
- Nicita F, Ulgiati F, Bernardini L, Garone G, Papetti L, Novelli A, & Spalice A (2015). Early myoclonic encephalopathy in 9q33-q34 deletion encompassing STXBP1 and SPTAN1. Annals of Human Genetics, 79, 209–217. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, & Meyer BJ (1997). Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. The Journal of Neuroscience, 17, 8061–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien S, Ng-Cordell E, Astle DE, Scerif G, & Baker K (2019). STXBP1-associated neurodevelopmental disorder: a comparative study of behavioural characteristics. Journal of Neurodevelopmental Disorders, 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtahara S, & Yamatogi Y (2006). Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Research, 70(Suppl 1), S58–67. [DOI] [PubMed] [Google Scholar]
- Okamoto M, & Südhof TC (1997). Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. Journal of Biological Chemistry, 272, 31459–31464. [DOI] [PubMed] [Google Scholar]
- Olson HE, Tambunan D, LaCoursiere C, Goldenberg M, Pinsky R, Martin E, … Poduri A (2015). Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. American Journal of Medical Genetics Part A, 167A, 2017–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orock A, Logan S, & Deak F (2018). Munc18–1 haploinsufficiency impairs learning and memory by reduced synaptic vesicular release in a model of Ohtahara syndrome. Molecular and Cellular Neuroscience, 88, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka M, Oguni H, Liang JS, Ikeda H, Imai K, Hirasawa K, … Yamamoto T (2010). STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome-result of Japanese cohort study. Epilepsia, 51, 2449–2452. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP (2005). Syndromes of idiopathic generalized epilepsies not recognized by the International League Against Epilepsy. Epilepsia, 46(Suppl 9), 57–66. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, & Sudhof TC (2011). Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron, 70, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, … Sollner TH (2014). An extended helical conformation in domain 3a of Munc18–1 provides a template for SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex assembly. Journal of Biological Chemistry, 289, 9639–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Bin NR, Yu B, Wong R, Sitarska E, Sugita K, & Sugita S (2017). UNC-18 and tomosyn antagonistically control synaptic vesicle priming downstream of UNC-13 in caenorhabditis elegans. The Journal of Neuroscience, 37, 8797–8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke C, Han Y, Covy J, Yi F, Maxeiner S, Wernig M, & Südhof TC (2015). Analysis of conditional heterozygous STXBP1 mutations in human neurons. Journal of Clinical Investigation, 125, 3560–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Diaz S, Pujols J, Conde-Gimenez M, Čarija A, Dalfo E, García J, … Ventura S (2019). ZPD-2, a small compound that inhibits alpha-synuclein amyloid aggregation and its seeded polymerization. Frontiers in Molecular Neuroscience, 12, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Lee J, Rowland K, Wen Y, Hua H, Carlson N, Lavania S, Parrish JZ, & Kim MD (2015). Regulation of dendrite growth and maintenance by exocytosis. Journal of Cell Science, 128, 4279–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, & Scheller RH (1994). n-Sec1: a neural-specific syntaxin-binding protein. Proceedings of the National Academy of Sciences, 91, 1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E Ide SE, Dehejia A, Dutra A, … Nussbaum RL (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, & Shen J (2010). Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proceedings of the National Academy of Sciences, 107, 22399–22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, … Strom TM (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet, 380, 1674–1682. [DOI] [PubMed] [Google Scholar]
- Rezazadeh A, Uddin M, Snead OC 3rd, Lira V, Silberberg A, Weiss S, … Andrade DM (2019). STXBP1 encephalopathy is associated with awake bruxism. Epilepsy & Behavior, 92, 121–124. [DOI] [PubMed] [Google Scholar]
- Rickman C, Medine CN, Bergmann A, & Duncan RR (2007). Functionally and spatially distinct modes of munc18-syntaxin 1 interaction. Journal of Biological Chemistry, 282, 12097–12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, & Südhof TC (2012). The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices-guilty as charged? Annual Review of Cell and Developmental Biology, 28, 279–308. [DOI] [PubMed] [Google Scholar]
- Rodkey TL, Liu S, Barry M, & McNew JA (2008). Munc18a scaffolds SNARE assembly to promote membrane fusion. Molecular Biology of the Cell, 19, 5422–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaniello R, Saettini F, Panzeri E, Arrigoni F, Bassi MT, & Borgatti R (2015). A de-novo STXBP1 gene mutation in a patient showing the Rett syndrome phenotype. Neuroreport, 26, 254–257. [DOI] [PubMed] [Google Scholar]
- Romaniello R, Zucca C, Tenderini E, Arrigoni F, Ragona F, Zorzi G, … Borgatti R (2014). A novel mutation in STXBP1 gene in a child with epileptic encephalopathy and an atypical electroclinical pattern. Journal of Child Neurology, 29, 249–253. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, … Matsumoto N (2008). De novo mutations in the gene encoding STXBP1 (MUNC18–1) cause early infantile epileptic encephalopathy. Nature Genetics, 40, 782–788. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Okada I, Orii KE, Higuchi T, Hoshino H, … Matsumoto N (2010). STXBP1 mutations in early infantile epileptic encephalopathy with suppression-burst pattern. Epilepsia, 51, 2397–2405. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Shimono M, Senju A, Tanabe S, Kimura T, … Matsumoto N (2012b). Association of genomic deletions in the STXBP1 gene with Ohtahara syndrome. Clinical Genetics, 81, 399–402. [DOI] [PubMed] [Google Scholar]
- Saitsu H, & Matsumoto N (2011). De novo mutations in epilepsy. Developmental Medicine & Child Neurology, 53, 806–807. [DOI] [PubMed] [Google Scholar]
- Sakaguchi G, Manabe T, Kobayashi K, Orita S, Sasaki T, Naito A, … Takai Y (1999). Doc2alpha is an activity-dependent modulator of excitatory synaptic transmission. The European journal of neuroscience, 11, 4262–4268. [DOI] [PubMed] [Google Scholar]
- Salzberg A, Cohen N, Halachmi N, Kimchie Z, & Lev Z (1993). The Drosophila Ras2 and Rop gene pair: A dual homology with a yeast Ras-like gene and a suppressor of its loss-of-function phenotype. Development, 117, 1309–1319. [DOI] [PubMed] [Google Scholar]
- Sampaio M, Rocha R, Biskup S, & Leao M (2015). Novel STXBP1 mutations in 2 patients with early infantile epileptic encephalopathy. Journal of Child Neurology, 30, 622–624. [DOI] [PubMed] [Google Scholar]
- Santos-Cortez RLP, Khan V, Khan FS, Mughal Z-U-N, Chakchouk I, Lee K, … Leal SM (2018). Novel candidate genes and variants underlying autosomal recessive neurodevelopmental disorders with intellectual disability. Human Genetics, 137, 735–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos TC, Wierda K, Broeke JH, Toonen RF, & Verhage M (2017). Early golgi abnormalities and neurodegeneration upon loss of presynaptic proteins Munc18–1, Syntaxin-1, or SNAP-25. The Journal of Neuroscience, 37, 4525–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter OM, Schmitz F, Jahn R, Rosenmund C, & Südhof TC (2004). A complete genetic analysis of neuronal Rab3 function. Journal of Neuroscience, 24, 6629–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueren F, Lingner T, George R, Hofhuis J, Dickel C, Gartner J, & Thoms S (2014). Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. Elife, 3, e03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shellhaas RA, Wusthoff CJ, Tsuchida TN, Glass HC, Chu CJ, Massey SL, … Cilio MR (2017). Profile of neonatal epilepsies: Characteristics of a prospective US cohort. Neurology, 89, 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, & Melia TJ (2007). Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell, 128, 183–195. [DOI] [PubMed] [Google Scholar]
- Sitarska E, Xu J, Park S, Liu X, Quade B, Stepien K, … Rizo J (2017). Autoinhibition of Munc18–1 modulates synaptobrevin binding and helps to enable Munc13-dependent regulation of membrane fusion. eLife, 6, e24278. 10.7554/eLife.24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, & Goedert M (1997). Alpha-synuclein in Lewy bodies. Nature, 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, … Møller RS (2016). STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology, 86, 954–962. [DOI] [PubMed] [Google Scholar]
- Stamberger H, Weckhuysen S, & De Jonghe P (2017). STXBP1 as a therapeutic target for epileptic encephalopathy. Expert Opinion on Therapeutic Targets, 21, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Suri M, Evers JMG, Laskowski RA, O’Brien S, Baker K, Clayton-Smith J, … Wright CF (2017). Protein structure and phenotypic analysis of pathogenic and population missense variants in STXBP1. Molecular Genetics & Genomic Medicine, 5, 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds JD, & McTague A (2020). Epilepsy and developmental disorders: Next generation sequencing in the clinic. European Journal of Paediatric Neurology, 24, 15–23. [DOI] [PubMed] [Google Scholar]
- Toonen RF, de Vries KJ, Zalm R, Südhof TC, & Verhage M (2005). Munc18–1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. Journal of Neurochemistry, 93, 1393–1400. [DOI] [PubMed] [Google Scholar]
- Vandersluis S, Li CM, Cheng L, Gajic-Veljanoski O, Wells D, & Holubowich C (2020). Genome-wide sequencing for unexplained developmental disabilities or multiple congenital anomalies: A health technology assessment. Ontario Health Technology Assessment Series, 20, 1–178. [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Tennison MB, Aylsworth AS, Turcott CM, Guerra MP, Eng CM, & Yang Y (2012). A novel STXBP1 mutation causes focal seizures with neonatal onset. Journal of Child Neurology, 27, 811–814. [DOI] [PubMed] [Google Scholar]
- Verhage M, de Vries KJ, Roshol H, Burbach JP, Gispen WH, & Südhof TC (1997). DOC2 proteins in rat brain: complementary distribution and proposed function as vesicular adapter proteins in early stages of secretion. Neuron, 18, 453–461. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, … Südhof TC (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science, 287, 864–869. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, & Verhage M (2001). Munc18–1 promotes large dense-core vesicle docking. Neuron, 31, 581–591. [DOI] [PubMed] [Google Scholar]
- Wang S, Li Y, Gong J, Ye S, Yang X, Zhang R, & Ma C (2019). Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nature Communications, 10, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckhuysen S, Holmgren P, Hendrickx R Jansen AC, Hasaerts D, Dielman C, … Suls A (2013). Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia, 54, e74–e80. [DOI] [PubMed] [Google Scholar]
- Wu MN, Littleton JT, Bhat MA, Prokop A, & Bellen HJ (1998). ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. The EMBO Journal, 17, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurster CD, & Ludolph AC (2018). Antisense oligonucleotides in neurological disorders. Therapeutic Advances in Neurological Disorders, 11, 1756286418776932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue R, Ruhl DA, Briguglio JS, Figueroa AG, Pearce RA, & Chapman ER (2018). Doc2-mediated superpriming supports synaptic augmentation. Proceedings of the National Academy of Sciences, 115, E5605–E5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Otsu M, Okumura T, Horie Y, Ueno Y, Taniguchi H, (2019). Generation of three induced pluripotent stem cell lines from postmortem tissue derived following sudden death of a young patient with STXBP1 mutation. Stem Cell Research, 39, 101485. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Chiyonobu T, Yoshida M, Maeda H, Zuiki M, Kidowaki S, & Hosoi H (2016). Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia, 57, e81–e86. [DOI] [PubMed] [Google Scholar]
- Yamatogi Y, & Ohtahara S (2002). Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain and Development, 24, 13–23. [DOI] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, & Chapman ER (2011). Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell, 147, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuge K, Iwama K, Yonee C, Matsufuji M, Sano N, Saikusa T, … Matsuishi T (2018). A novel STXBP1 mutation causes typical Rett syndrome in a Japanese girl. Brain and Development, 40, 493–497. [DOI] [PubMed] [Google Scholar]
- Zablotsky B, Black LI, & Blumberg SJ (2017). Estimated prevalence of children with diagnosed developmental disabilities in the United States, 2014–2016. NCHS Data Brief. (291), 1–8. [PubMed] [Google Scholar]
- Zhu B, Mak JCH, Morris AP, Marson AG, Barclay JW, Sills GJ, & Morgan A (2020). Functional analysis of epilepsy-associated variants in STXBP1/Munc18–1 using humanized Caenorhabditis elegans. Epilepsia, 61(4), 810–821. 10.1111/epi.16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.