

Abstract
Background:
The resistance against antimalarial drugs represents a global challenge in the fight and control of malaria. The Brazilian biodiversity can be an important tool for research and development of new medicinal products. In this context, toxinology is a multidisciplinary approach on the development of new drugs, including the isolation, purification, and evaluation of the pharmacological activities of natural toxins. The present study aimed to evaluate the cytotoxicity, as well as the antimalarial activity in silico and in vitro of four compounds isolated from Rhinella marina venom as potential oral drug prototypes.
Methods:
Four compounds were challenged against 35 target proteins from P. falciparum and screened to evaluate their physicochemical properties using docking assay in Brazilian Malaria Molecular Targets (BraMMT) software and in silico assay in OCTOPUS® software. The in vitro antimalarial activity of the compounds against the 3D7 Plasmodium falciparum clones were assessed using the SYBR Green I based assay (IC50). For the cytotoxic tests, the LD50 was determined in human pulmonary fibroblast cell line using the [3(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay.
Results:
All compounds presented a ligand-receptor interaction with ten Plasmodium falciparum-related protein targets, as well as antimalarial activity against chloroquine resistant strain (IC50 = 3.44 μM to 19.11 μM). Three of them (dehydrobufotenine, marinobufagin, and bufalin) showed adequate conditions for oral drug prototypes, with satisfactory prediction of absorption, permeability, and absence of toxicity. In the cell viability assay, only dehydrobufotenin was selective for the parasite.
Conclusions:
Dehydrobufotenin revealed to be a potential oral drug prototype presenting adequate antimalarial activity and absence of cytotoxicity, therefore should be subjected to further studies.
Keywords: Bufadienolides, Antimalarial drug Docking, Natural compounds
Background
Malaria is an important human parasitic disease, occurring in tropical and subtropical areas of the planet [1]. The malaria parasites resistance to ancient antimalarials consists of the biggest hurdles to malaria control [2]. Because of the resistance to antimalarials, artemisinin and its derivatives have been the first-line antimalarial agents against Plasmodium falciparum [3,4]. Artemisinin-based combination therapies (ACT) are the most effective regimens for the first-line treatment for P. falciparum infections. Despite of the WHO recommendations for using and prescribing the ACTs, pharmacokinetic and pharmacodynamic studies with P. falciparum strains have already demonstrated the development of resistance to these compounds [5,6,7]. This phenomenon is responsible to increase the mortality in endemic areas contributing to the appearance and expansion of new outbreaks of P. falciparum malaria. Thus, new strategies are required to prevent increased resistance to ACTs. In addition, a potential strategy would be to add a third drug with independent antiparasitic activity [6].
Natural products have providing a great contribution to the development of new drugs [8]. In fact, many of the antimalarial drugs commercially available are derivatives of phytoconstituents [9]. In addition to the plant-derived remedies, animal extracts, products, and even secretions are also a source of a plethora of therapeutical agents [10].
The venoms secreted by the paratoid glands of amphibians from the order Anura is the first line of defense against predators and microorganisms [11,12]. The toads of Bufonidae family have been widely studied due to the bioactive properties found in the Rhinella marina venom, which have already shown antitumor [13,14,15], antiviral [16,17], and antiparasitic activities [18]. The cholesterol-derived steroid structures called bufadienolides are major active compounds in the venom of Bufonidae family and are considered a promising source of bioproducts [19,20]. Furthermore, the alkaloids dehydrobufotenin and bufotenine also identified in R. marina venom have demonstrated to possess antiproliferative and antiviral activity, respectively [20,21, 22, 23].
The development of malaria drugs is slower than that involving the antibacterial drugs [24]. However, this process can be speeded up with the aid of computational drug planning tools, known as molecular modeling or docking, to design new compounds and to study their respective protein targets [25, 26]. The docking is a robust tool for investigating the chemical interactions of ligands and receptors and to explore the structural factors related to the biological effect [27, 28].
To date, there are almost no studies investigating compounds isolated from the bufonides venom as potential new antimalarial drugs. Therefore, present study aimed to evaluate the cytotoxicity, as well as the antimalarial activity in silico and in vitro of four compounds isolated from Rhinella marina venom as potential oral drug prototypes.
Methods
Sample collection
The animals (R. marina) were collected in the Branca de Neve Community, Mato Grosso, Brazil (Latitude 11°51'51.59 "S/Longitude 55°22'47.99" W), from January to March of 2015, in the municipality of Sinop, Mato Grosso state, North-Western Brazil. The vegetation where the individuals were found is classified as dense humid forest and the climate of the region is tropical with an average temperature of 24° C, relative air humidity of ~80%, and average annual rainfall of 2,034 mm.
The amphibians were captured and identified by the biologist (D. J. Rodrigues - 95 IBAMA, SISBIO: number 30034-1). The secretions were obtained by manual compression of the parotoid macrogland and the animals were returned to nature after this procedure. The voucher specimens (R. marina - ABAM-H 2256) were collected and deposited in the zoological collection (Acervo Biológico da Amazônia Meridional) of the Federal University of Mato Grosso located at Sinop city (the collection permit was issued by the Chico Mendes Institute for Biodiversity Conservation).
All experiments were performed according to internationally accepted guidelines for the care and use of laboratory animals and were previously approved by the Federal University of Mato Grosso Institutional Animal Care and Use Committee (Protocol 23108.700260/14-7) and National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SisGen AE 19081).
Extraction of R. marina Venom samples and isolation
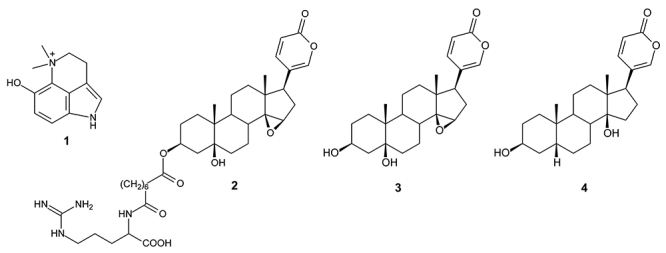
R. marina toad venoms were dried, powdered and extracted three times (3 x 20 mL) with 100% methanol (MeOH) in ultrasound waves for 10 minutes at room temperature [14]. The extract was fractionated on Sephadex LH-20 column using methanol as eluent. Four fractions were obtained: CRV-6 (783.8 mg); CRV-28 (102.9 mg); CRV-52 (315.8 mg) and CRV-70 (394.1 mg). The structure of the isolated compounds marinobufotoxin, dehydrobufotenin, marinobufagin, and bufalin are presented in Figure 1 [29].
Figure 1. Molecular structures of R. marina venom fractions. (1) Dehydrobufotenine (CRV - 28), (2) marinobufotoxin (CRV-6-21-58), (3) marinobufagin (MB-1) and (4) bufalin (MB-3).
Evaluation of molecular docking
The compounds were designed using MarvinSketch® software (ChemAxon, Cambridge, MA, USA) and the molecular structures were refined through MOPAC® software (Stewart Computational Chemistry, Colorado Springs, CO, USA) using the PM7 semi-empirical method. The compounds dehydrobufotenine, marinobufotoxin, marinobufagin and bufalin were submitted to the molecular docking calculations in the AutoDock Vina® program [30] using OCTOPUS® platform [31] and the configuration files were determined through a re-docking step [32]. Thus, the virtual screening of antimalarial drugs was performed using the BraMMT data bank according [33]. From the generated binding energy values, Δ (binding energy of the crystallographic ligand - binding energy of the compound) values were calculated. Thus, Δ values greater than “0”, show that it has higher binding energy than the crystallographic ligand, suggesting greater interaction with the target. Table 1 lists the molecular targets used to build the BraMMT platform.
Table 1. Molecular targets, location and enzymatic class of the 35 molecular targets obtained from the Tropical Disease Research (TDR target database) for building the Brazilian Malaria Molecular Targets (BraMMT).
| PDB Code | Name | Enzymatic class | Location |
|---|---|---|---|
| 1LF3 | Plasmepsin II | Hydrolase | Digestive vacuole |
| 1LYX | Triosephosphate Isomerase (PfTIM)-Phosphoglycolate | Isomerase | Cytoplasm |
| 1NHW | Enoyl-acyl-carrier-protein reductase | Oxidoreductase | Apicoplast |
| 1O5X | Triosephosphate Isomerase | Isomerase | Cytoplasm |
| 1QNG | Peptidyl-prolil cis-trans isomerase | Isomerase | Cytoplasm |
| 1RL4 | Formylmethionine deformylase | Hydrolase | Apicoplast |
| 1TV5 | Dihydroorotate dehydrogenase | Oxidoreductase | Cytoplasm e Nucleus |
| 1U4O | L-lactate dehydrogenase | Oxidoreductase | Cytoplasm |
| 1YWG | glyceraldehyde-3-phosphate dehydrogenase | Oxidoreductase | Cytoplasm |
| 2AAW | Glutathione s-transferase | Transferase | Cytoplasm |
| 2ANL | Plasmepsin IV | Hidrolase | Digestive vacuole |
| 2OK8 | Putative ferredoxin--NADP reductase | Oxidoreductase | Apicoplast |
| 2PML | Ser/Thr protein kinase | Transferase | Cytoplasm |
| 2Q8Z | Orotidine-monophosphate-descarboxylase | Liase | Nucleus |
| 2VFA | Hypoxantine-guanine phosphoribosyltransferase | Transferase | Apicoplast |
| 2VN1 | 70 KDA peptidylprolyl isomerase | Isomerase | Nucleus |
| 2YOG | Thymidylate kinase | Transferase | Nucleus |
| 3AZB | Beta-hydroxyacyl-ACP dehydratase | Lyase | Cytoplasm |
| 3BPF | Falcipain II | Hydrolase | Digestive vacuole |
| 3CLV | Rab5 Protein | Signaling protein | Cytoplasm |
| 3FNU | HAP Protein | Hydrolase | Digestive vacuole |
| 3K7Y | Aspartate aminotransferase | Transferase | Cytoplasm |
| 3N3M | Orotidine 5’-phosphate decarboxylase | Lyase | Apicoplast |
| 3PHC | Purine nucleoside phosphorylase | Transferase | Nucleus |
| 3QS1 | Plasmepsin I | Hydrolase | Digestive vacuole |
| 3T64 | Deoxyuridine 5’-triphosphate nucleotidohydrolase | Hydrolase | Nucleus |
| 3TLX | Adenylate kinase 2 | Transferase | Cytoplasm and mitochondria |
| 4B1B | Thioredoxin reductase | Oxidoreductase | Cytoplasm |
| 4C81 | 22-C-Methyl-D-Erythritol 2,4-Cyclodiphosphate synthase | Lyase | Apicoplast |
| 4J56 | Thioredoxin reductase 2 | Oxidoreductase | Cytoplasm |
| 4N0Z | Glutaredoxin | Oxidoreductase | Cytoplasm |
| 4P7S | Macrophage migration inhibitory factor-like protein | Cytokine inhibitor | Cytoplasm |
| 4QOX | Calcium-dependent protein kinase 4 | Transferase | Cytoplasm |
| PfATP6 | Calcium pump ortholog ATPase | Transporter | Membrane |
|
PfHT
(10.5452/ma-aej21) |
Hexose carrier protein | Transporter | Membrane |
Evaluation of physicochemical and ADMET properties
The physicochemical and ADMET properties of the compounds dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin (CRV-28, CRV-6-28-51, MB-1 and MB-3, respectively) were analyzed using DataWarrior® software and SwissADME website [34]. The properties of molecular mass, partition coefficient (ClogP), number of hydrogen donor groups, and number of hydrogen acceptor groups were predicted. The toxicological characteristics of the ligand, such as mutagenicity, tumogenicity, and irritability, were analyzed [35]. Finally, the pharmacokinetic processes of absorption, distribution, metabolism, excretion, and toxicity were estimated [34].
In vitro culture of P. falciparum
P. falciparum W2 strain (chloroquine resistant) [36, 37, 38] was cultured in blood stage culture to test the antiplasmodial efficacy of toad venom compounds (1, 2, 3, and 4). P. falciparum continuous culture was maintained as previously described [36,39] with minor modifications. Parasites were maintained at 5% hematocrit using type O+ human erythrocytes in RPMI 1640 medium (Sigma-Aldrich®, St. Louis, Missouri, USA) supplemented with 25 mM NaHCO3, 1.0% albumax, 45 mg/L hypoxanthine, 40 μg/mL gentamycin and incubated at 37º C under approximately 5% of CO2. The parasites at early stages were synchronized at ring stage by sorbitol treatment [40]. Initial parasitemia was adjusted to 0.5% with 2% hematocrit in all experiments.
In vitro antiplasmodial activity
In vitro antiplasmodial activity of the bufadienolides (compounds dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin) was done in 96 well plates [41]. The growth inhibition of intraerythrocytic forms and parasite morphology in culture was assessed by microscopic observation of the Giemsa-stained thin blood films. Ring stage parasites (0.5% parasitemia and 2% hematocrit) were added to each well of 96-well microculture plates. The compounds (dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin) were dissolved in DMSO and diluted to concentrations ranging from 0.78 to 100 μg/mL using complete medium and stored a 4º C. After incubation at 37º C for 48 hours, P. falciparum growth inhibition was assessed in Giemsa-stained smears by observing 5,000 erythrocytes per 1 thin blood film in triplicate. The culture medium was replaced with fresh medium with or without test samples/control drugs. Chloroquine (CQ) was used as a reference antimalarial. The activity of the compounds (dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin) was expressed as the percentage reduction in parasitemia relative to controls without drugs. All experiments were performed in triplicate. The results were expressed as the mean of the IC50 (Drug concentration that reduced parasite viability in 50%).
In vitro cytotoxicity
In vitro cytotoxicity of each compound was assessed on WI-26VA4 (ATCC CCL-95.1, USA) human pulmonary fibroblast cells. The cells were cultured in RPMI-1640 (Sigma-Aldrich®, St. Louis, Missouri, USA) medium supplemented with 10% heat-inactivated fetal bovine serum and 100 μg/mL of gentamycin in a 5% CO2 atmosphere at 37º C. The cells were washed with culture medium, trypsinized, distributed in a flat-bottomed 96-well plate (5×103 cells/well), and incubated for 18 hours at 37º C for cell adherence [42]. The compounds (20 μL) were diluted in different concentrations ranging from 0.2 - 200 μg/mL and incubated with the cells for 24 hours in a 5% CO2 atmosphere at 37º C.
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL; 20 μL/well) was added to evaluate mitochondrial viability; after a further 3 hours incubation, the supernatants were carefully removed, 100 μL of DMSO was added to each well, and the reactions were mixed to solubilize the formazan crystals. The optical density was determined at 540 nm to measure the signal and background, respectively (Spectra Max340PC384, Molecular Devices, Sunnyvale, California, USA) [43, 44, 45, 46, 47, 48]. The cell viability was expressed as a percentage of the control absorbance in the untreated cells after subtracting the appropriate background.
The minimum lethal dose for 50% of the cells (LD50) was determined as described [49].
Selectivity index (SI)
A selectivity index (SI) corresponding to the ratio between the cytotoxic and antiplasmodial activities of each compound tested. The values greater than 10 were considered indicative of lack of toxicity, whereas the substances with values below 10 were considered toxic [38]. The SI index was calculated as follow:
Statistical analysis
The concentrations of compounds able to inhibit 50% of parasite growth (IC50) were determined based on the equation of the curve obtained by plotting the % of parasitemia regression vs the log of the concentration of compound. The coefficients of regression of these curves were calculated using the method of least squares. The LD50 were determined based on the equation of the curve obtained by plotting the % of cellular death versus the concentration of compound (GraphPad Prism Software, version 5.0 for Windows, San Diego, California, USA). The average IC50 and LD50 were compared using ANOVA. Statistical significance was defined at the 5% level (P<0.05).
Results
Compounds
Compound 1 (CRV - 28)
Dehydrobufotenin - molecular formula: C15H12N2O; IT-ESI-MS [M+H]+ 203.1; 1H NMR (CD3OD- 600 MHz): δ 7.11(s, 1H), δ 6.81(d, J = 8.6 Hz, 1H), δ 7.29(d, J = 8.7 Hz, 1H), δ 3.29(d, J = 5.8 Hz, 2H), δ 4.1(t, J = 5.9 Hz, 2H) and δ 3.68(s, 6H). 13C NMR (CD3OD - 150 MHz): δ 122.5, δ 120.6, δ 104.6, δ 121.1, δ 149.0, δ 115.0, δ 118.9, δ 128.9, δ 20.0, δ 69.6 and δ 54.0.
The CRV-6 fraction was submitted to the Sephadex LH-20 column with MeOH. The sub-fractions CRV-6-28 was further fractionated by silica gel column, eluted in CHCl3/MeOH with an increasing polarity gradient system. The subgroup obtained was CRV-6-28-51 (35.1 mg) and through NMR analysis and mass spectrometry was identified as marinobufotoxin (2).
Compound 2 (CRV-6-28-51)
3-(N-suberoylargininyl) marinobufagin (marinobufotoxin) - molecular formula: C38H56N4O9; IT-ESI-MS [M+H]+ 713.5; 1H NMR (CD3OD - 600 MHz): δ 5.14 (m), δ 3.68 (s, 1H), δ 2.56 (d, J= 9.9 Hz, 1H), δ 0.73 (s, 3H), δ 0.94 (s, 3H), δ 7.46 (d, J = 1.8 Hz, 1H), δ 7.90 (dd, J = 9.8 and 2.4 Hz, 1H), δ 6.28 (t, J = 9.6 Hz, 1H), δ 4.28 (dd, J = 8.4 and 4.9 Hz, 1H), δ 1.88 (m, 2H), δ 1.61 (m, 2H), δ 3.20 (m, 2H), δ 2.20-2.38 (m, 4H), δ 1.71 (m, 4H) and δ 1.36 (m, 4H). 13C NMR (CD3OD - 150 MHz) δ (ppm): 26.4, 25.7, 72.2, 36.4, 74.3, 36.1, 24.3, 33.9, 43.2, 41.6, 22.5, 39.7, 46.1, 75.7, 61.1, 27.7, 48.3, 17.0, 17.1, 124.5, 150.6, 149.7, 115.4, 164.6, 55.2, 31.1, 25.8, 42.0, 174.8/175.7, 35.4/37.3, 26.3/26.6, 29.4/30.0, 178.8 and 158.6.
The CRV-70 fraction (394.1 mg) was fractionated in silica gel column. The CRV-70-04 sub-fraction was analyzed by NMR and mass spectrometry, and its majority compound was identified as marinobufagin (3). Subsequently, this sub-fraction was submitted to purification by High Performance Liquid Chromatography (HPLC) using ultrapure water (eluent A) and acetonitrile (eluent B), the system was eluted in isocratic mode with 60% eluent B, obtaining the group MB-3 (7.9 mg), which was identified as bufalin (4). The spectral data of the isolated compounds are in accordance with the literature [25, 46, 47, 48] and described below. The structures are shown in Figure 1.
Compound 3 (MB-1)
Marinobufagin - molecular formula: C24H32O5; IT-ESI-MS [M+H]+ 401.3; 1H NMR (CDCl3 - 600 MHz): δ 4.16-4.19 (m), δ 3.49 (s, 1H), δ 2.46 (d, J= 10.1Hz, 1H), δ 0.77 (s, 3H), δ 0.97 (s, 3H), δ 7.23 (d, J = 9.8 Hz, 1H), δ 7.76 (dd, J = 9.8 and 2.5 Hz, 1H) and δ 6.24 (dd, J = 9.8 and 0.8 Hz, 1H). 13C NMR (CDCl3 - 150 MHz): 24.9, 28.1, 68.1, 39.5, 74.7, 34.8, 23.4, 32.7, 42.8, 41.0, 21.6, 39.5, 45.2, 74.7, 59.9, 32.4, 47.7, 16.9, 16.9, 122.4, 149.8, 147.0, 115.4 and 162.2.
Compound 4 (MB-3)
Bufalin - molecular formula: C24H34O4; IT-ESI-MS [M+H]+ 387.3; 1H NMR (CDCl3 - 600 MHz): δ 4.13-4.18 (m), δ 2.56 (dd, J = 9.7 and 6.6 Hz, 1H), δ 0.69 (s, 3H), δ 0.94 (s, 3H), δ 7.22 (d, J = 1.8 Hz, 1H), δ 7.84 (dd, J = 9.7 and 2.6 Hz, 1H) and δ 6.26 (d, J = 9.7 Hz, 1H) 13C NMR (CDCl3 - 150 MHz): 29.8, 28.0, 66.9, 33.4, 36.1, 26.6, 21.5, 42.5, 35.8, 35.5, 21.5, 41.0, 48.5, 85.5, 32.8, 28.8, 51.3, 16.7, 23.9, 122.8, 148.6, 146.9, 115.5 and 162.6.
In silico: Virtual screening
Table 1 shows the molecular targets, location, and enzymatic class of the 35 molecular targets obtained from the Tropical Disease Research (TDR targets database) for building the Brazilian Malaria Molecular Targets (BraMMT). The compounds (dehydrobufotenin, marinobufotoxin, marinobufagin, bufalin) were assayed for the docking methodology in the BraMMT data bank. Virtual screening performed against the 35 molecular targets in the database using OCTOPUS software presented 10 potential targets for all compounds tested (Table 2). These results were found when the binding energy values are lower than the crystallographic control.
Table 2. Binding energies between compounds tested and molecular targets (kcal.mol-1).
| Compounds | Binding energy (kcal.mol-1) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1NHW | Δ | 1O5X | Δ | 2OK8 | Δ | 2VFA | Δ | 3AZB | Δ | 3BPF | Δ | 4N0Z | Δ | 4P7S | Δ | PfATP6 | Δ | PfHT | Δ | |
| Crystallographic | -8.3 | -5.3 | -2.0 | -5.8 | -6.3 | -6.3 | -4.3 | -6.0 | -7.2 | -5.7 | ||||||||||
| Dehydrobufotenine | -6.9 | -1.4 | -5.6 | 0.3 | -2.6 | 0.6 | -2.1 | -3.7 | -0.4 | -5.9 | -5.8 | -0.5 | -3.9 | -0.4 | -5.0 | -1.0 | -6.6 | -0.6 | -5.8 | 0.1 |
| Marinobufotoxin | -9.9 | 1.6 | -5.9 | 0.6 | -7.9 | 5.9 | -9.2 | 3.4 | -6.8 | 0.5 | -7.5 | 1.2 | -6.6 | 2.3 | -7.3 | 1.3 | -9.3 | 2.1 | -8.8 | 3.1 |
| Marinobufagin | -10.9 | 2.6 | -5.8 | 0.5 | -8.4 | 6.4 | -9.0 | 3.2 | -6.6 | 0.3 | -7.7 | 1.4 | -6.6 | 2.3 | -7.9 | 1.9 | -8.2 | 1.0 | -9.9 | 4.2 |
| Bufalin | -10.8 | 2.5 | -6.1 | 0.8 | -7.9 | 5.9 | -9.4 | 3.6 | -7.2 | 0.9 | -7.1 | 0.8 | -7.1 | 2.8 | -8.6 | 2.6 | -8.3 | 1.1 | -9.3 | 3.6 |
| D-glucose | - | - | - | - | - | - | - | - | - | -4.3 | 0.2 | |||||||||
PfHT is characterized as glucose transporter of P. falciparum. The docking, QM/MM and molecular dynamics simulations were already performed by our group [49, 50, 51]. The Figures 2, 3, 4, 5 and 6 shows 2D ligand-receptor interactions maps with PfHT. The figures show the chemicals bonds that occurred between the compound and the target, which enables the identification of pharmacophoric groups and possible structural improvements for better oral permeability, absorption, and bioavailability. The 3D binding diagram of the compounds is show in Figure 7. Docking with D-Glucose and PfHT was performed as a control.
Figure 2. Residues in the active site of PfHT target interacting with the compound marinobufagin (MB-1).
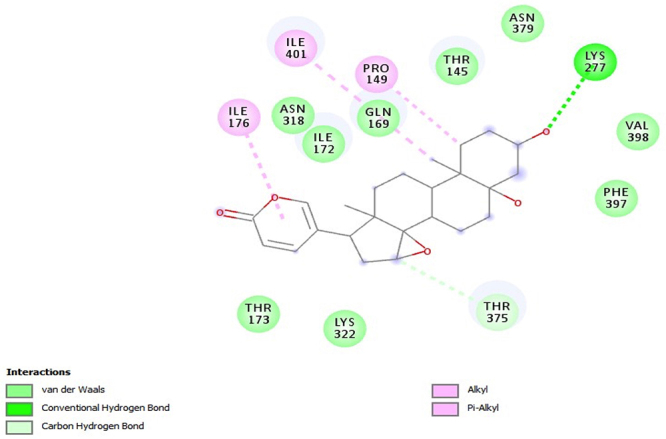
Figure 3. Residues in the active site of PfHT target interacting with compound the marinobufotoxin (CRV-6-21-58).
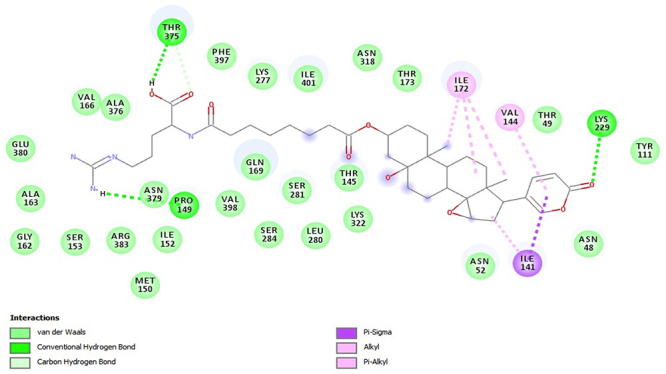
Figure 4. Intermolecular interactions of the compound bufalin (MB-3) with PfHT.
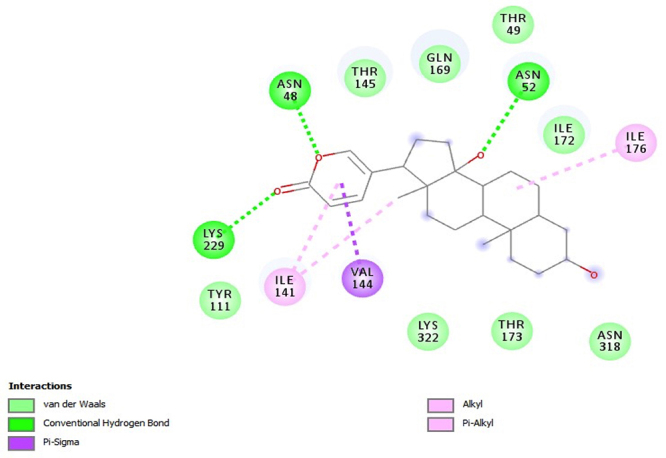
Figure 5. Intermolecular interactions of the compound dehydrobufotenine (CRV-28) with PfHT.
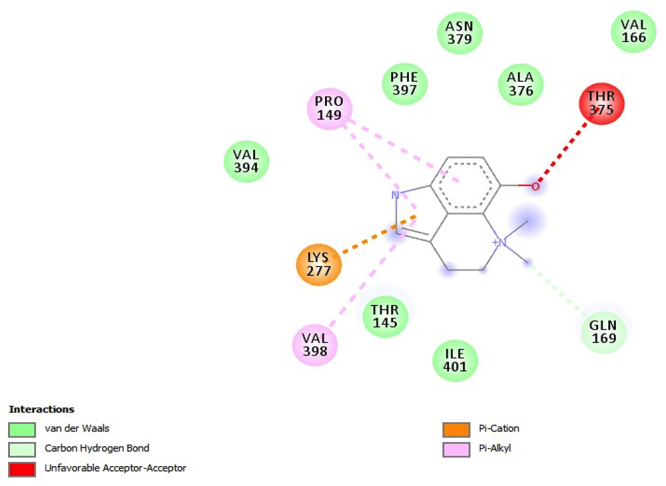
Figure 6. Intermolecular interactions of D-glucose with PfHT.
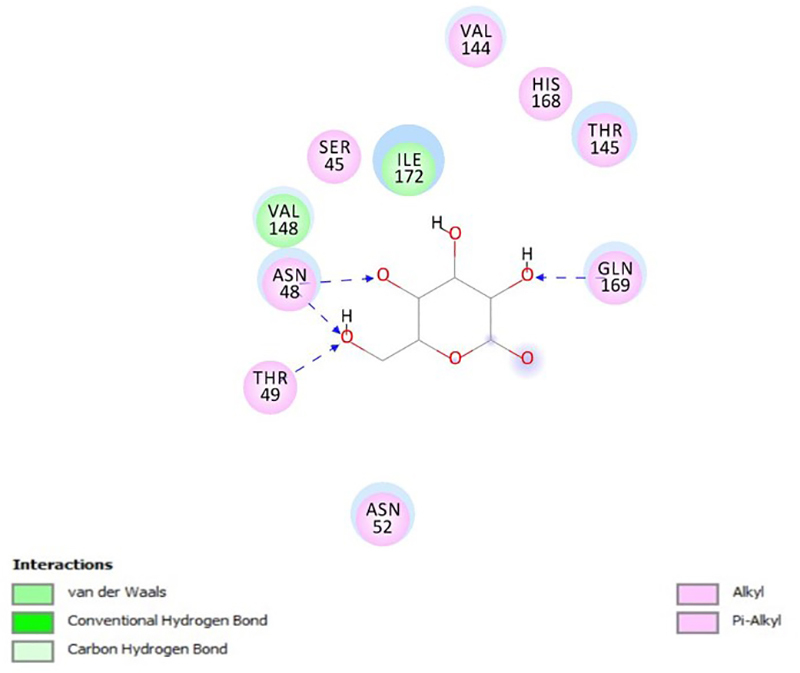
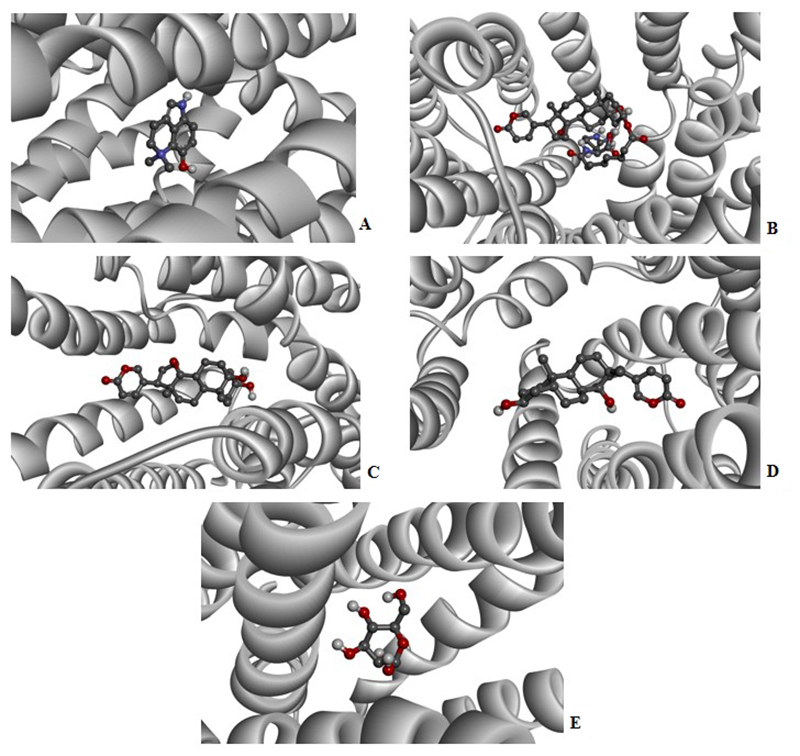
Figure 7. Three-dimensional structure of PfHT complexed with of R. marina venom fractions. (A) Dehydrobufotenine (CRV-28), (B) marinobufotoxin (CRV-6-21-58), (C) marinobufagin (MB-1), (D) bufalin (MB-3) and (E) D-glucose.
Marinobufagin binds to 2-pyrone group in PfHT protein by residue ILE-176 while the perhydrophenanthrene nucleus binds by residues ILE-401 and PRO-149 (Figure 2). Marinobufotoxin binds to 2-pyrone group by residue VAL-144 while the perhydrophenanthrene nucleus binds by residues ILE-172 (Figure 3). Bufalin interacts with the protein electrostatically from the same active sites with residues ILE-141 and ILE-176 (Figure 4).
The compound dehydrobufotenin is an alkaloid derivative with consequent presence of the quinoline ring like the quinine and its analogs. Among the interactions involving the binding to the PfHT protein, the quinoline nucleus performs electrostatic bonding with the PRO149 residue (Figure 5). Although there is an unfavorable interaction between compound dehydrobufotenin and PfHT, this negative event is offset by other interactions.
D-glucose interacts in the same biding site of PfHT with electrostatic bonding in the residues GLN169 and THR145 (Figure 6).
Table 3 presents the information obtained from the physical-chemical properties by the DataWarrior® software. The compound marinobufotoxin presented CLogP < 5 (2.80), molecular mass was greater than 500 (712.88 g/mol), the hydrogen accepting groups that performed interactions are more than 10 (13 groups), and the hydrogen donor groups are more than 5 (6 groups). Thus, based on the rules of Lipinski, compound marinobufotoxin is expected to present unfavorable pharmacokinetics properties (absorption, distribution, metabolization, excretion, and toxicity).
Table 3. Molecular mass (g/mol), partition coefficient (CLogP), number of hydrogen donor groups and number of hydrogen acceptor groups of tested compounds.
| Compounds | Molecular mass | CLogP | H+ Acceptors | H+ Donors |
|---|---|---|---|---|
| Dehydrobufotenine | 203.26 | 0.51 | 3 | 2 |
| Marinobufotoxin | 712.88 | 2.80 | 13 | 6 |
| Marinobufagin | 400.51 | 1.87 | 5 | 2 |
| Bufalin | 386.53 | 2.99 | 4 | 2 |
The compounds dehydrobufotenin, marinobufagin and bufalin presented molecular mass below 500 (203.26 g/mol; 400.51 g/mol and 383.53 g/mol, respectively), CLogP <5 (0.51; 1.87 and 2.99, respectively), less than 10 hydrogen acceptor groups (3 groups; 5 groups and 4 groups, respectively) and less than 5 hydrogen donor groups (2 groups). Thus, these results, based on the rules of Lipinski, the compounds dehydrobufotenin, marinobufagin and bufalin have sufficiently acceptable ADMET properties.
Gleeson [52] suggests in his study that compounds with CLogP less than 4 and molecular weight less than 400 have a more favorable ADMET profile than those suggested by Lipinski. Following the Gleeson theory, all compounds have CLogP < 3 and compounds dehydrobufotenin, marinobufagin and bufalin have molecular mass below 400.
Toxicological characteristics of the four compounds using the DataWarrior® software, factors such as mutagenicity, tumogenicity or irritability were not evidenced (Table 4).
Table 4. Toxicological characteristics of compounds obtained from R. marina venom.
| Compounds | Mutagenicity | Tumorgenicity | Irritability |
|---|---|---|---|
| Dehydrobufotenine | Absent | Absent | Absent |
| Marinobufotoxin | Absent | Absent | Absent |
| Marinobufagin | Absent | Absent | Absent |
| Bufalin | Absent | Absent | Absent |
In vitro antiplasmodial activity
The compounds dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin diluted in DMSO were assayed for antiplasmodial activity against chloroquine-resistant P. falciparum W2. Table 5 shows the antiplasmodial activity of dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin in two different experiments. Starting from 100 μg/mL, the compounds were diluted to various concentrations (0.78-100 μg/mL) to calculate the IC50 values. The samples (dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin) showed IC50 values ranged from 3.44 to 19.11 μM (Table 5). The marinobufagin and bufalin had the IC50 values close to chloroquine, the antimalarial used as a positive control.
Table 5. The lethal drug concentration that reduced parasite viability in 50% (IC50), lethal drug concentration that reduced WI-26VA4 cells viability in 50% (LD50), and selectivity index (SI) values obtained from in vitro tests with venom fractions from R. marina venom, and chloroquine (CQ) against P. falciparum W2 strain.
| Compounds | IC50 ± SD (μM)* | LD50 a ± SD (μM)* | SIa |
|---|---|---|---|
| Dehydrobufotenine | 19.11 ± 0.20 | 235.76 ± 4.03 | 12.33 |
| Marinobufotoxin | 5.31 ± 0.25 | 8.89 ± 2.66 | 1.67 |
| Marinobufagin | 3.89 ± 0.42 | 3.04 ± 0.25 | 0.78 |
| Bufalin | 3.44 ± 0.43 | 25.9 ± 7.04 | 7.52 |
| Chloroquine (CQ) | 1.04 ± 0.21 | >100 | >100 |
aLD50 and SI values were obtained with MTT cytotoxic test in human pulmonary fibroblast cells (WI-26VA4).
*Mean and standard deviation (SD) of triplicate experiments.
Cytotoxic activity on human pulmonary fibroblast cells
To evaluate the cytotoxic activity of the compounds dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin, the MTT assay conducted in human pulmonary fibroblast cells (WI-26VA4). It was observed that the compounds marinobufotoxin, marinobufagin and bufalin showed high cytotoxicity to this cell line with low LD50 values (marinobufotoxin = 8.89 μM; marinobufagin = 3.04 μM and bufalin = 25.9 μM, respectively) while dehydrobufotenin showed low cytotoxic with high LD50 value (235.76 μM) (Table 5).
Evaluating the selectivity index (SI), although compounds marinobufotoxin, marinobufagin and bufalin have shown potentially active, only the compound dehydrobufotenin showed high selectivity for the parasites when analyzed by MTT assay (SI>10) (Table 5).
ADMET likeness properties
Pharmacokinetic behavior of a compound can determine the success or failure of its biological activity [53]. New potential antimalarial candidates must present good oral bioavailability and good membrane permeability as properties that can lead the in vivo experiments to reach success [54]. In this sense, SwissADME web tool [30] allows an in silico inference of the main physical-chemical and pharmacokinetic properties of the compounds. In Table 6 are presented the SwissADME profile of the four compounds.
Table 6. Physicochemical properties of dehydrobufotenin, marinobufotoxin, marinobufagin, bufalin and the antimalarial chloroquine according to SwissADME web tool.
| PHYSICOCHEMICAL PROPERTIES | Dehydrobufotenine | Marinobufotoxin | Marinobufagin | Bufalin | Chloroquine |
|---|---|---|---|---|---|
| Formula | C12H15N2O | C38H56N4O9 | C24H32O5 | C24H34O4 | C18H26ClN3 |
| Molecular weight | 203.26 g/mol | 712.87 g/mol | 400.51 g/mol | 386.52 g/mol | 319.87 g/mol |
| Num. heavy atoms | 15 | 51 | 29 | 28 | 22 |
| Num. arom heavy atoms | 9 | 6 | 6 | 6 | 10 |
| Fractions Csp3 | 0.33 | 0.76 | 0.79 | 0.79 | 0.50 |
| Num. rotable bonds | 0 | 18 | 1 | 1 | 8 |
| Num. H-bond acceptors | 2 | 10 | 5 | 4 | 2 |
| Num. H-bond donors | 2 | 6 | 2 | 2 | 1 |
| Molar Refractivity | 66.18 | 190.16 | 108.86 | 109.86 | 97.41 |
| TPSA | 36.02 Ų | 217.57 Ų | 83.20 Ų | 70.67 Ų | 28.16 Ų |
| LIPOPHILICITY | |||||
| Log P olw (ILOGP) | -1.27 | 3.47 | 3.27 | 3.34 | 3.95 |
| Log P olw (XLOGP3) | 1.64 | 3.23 | 2.50 | 3.2 | 4.63 |
| Log P olw (WLOGP) | 1.62 | 4.10 | 3.37 | 4.24 | 4.62 |
| Log P olw (MLOGP) | -2.19 | 2.12 | 2.75 | 3.58 | 3.20 |
| Log P olw (SILICOS-IT) | 2.15 | 4.89 | 3.82 | 3.99 | 4.32 |
| Consensus Log P olw | 0.39 | 3.56 | 3.14 | 3.67 | 4.15 |
| WATER SOLUBILITY | |||||
| Log S (ESOL) | -2.58 | -5.19 | -3.99 | -4.35 | -4.55 |
| Solubility | 5.38e-01 mg/mL; 2.65e-03 mol/L | 4.56e-03 mg/mL; 6.40e-06 mol/L | 4.14e-02 mg/mL; 1.03e-04 mol/L | 1.75e-02 mg/mL; 4.52e-05 mol/L | 9.05e-03 mg/mL; 2.83e-05 mol/L |
| Class | Soluble | Moderately soluble | Soluble | Moderately soluble | Moderately soluble |
| Log S (Ali) | -2.01 | -7.47 | -3.89 | -4.36 | -4.95 |
| Solubility | 1.99e+00 mg/mL; 9.78e-03 mol/L | 2.40e-05 mg/mL; 3.37e-08 mol/L | 5.13e-02 mg/mL; 1.28e-04 mol/L | 1.70e-02 mg/mL; 4.41e-05 mol/L | 3.61e-03 mg/mL; 1.13e-05 mol/L |
| Class | Soluble | Poorly soluble | Soluble | Moderately soluble | Moderately soluble |
| Log S (SILICOS-IT) | -4.09 | -7.46 | -4.73 | -5.2 | -6.92 |
| Solubility | 1.63e-02 mg/mL; 8.04e-05 mol/L | 2.45e-05 mg/mL; 3.44e-08 mol/L | 7.42e-03 mg/mL; 1.85e-05 mol/L | 3.68e-03 mg/mL; 9.53e-06 mol/L | 3.86e-05 mg/mL; 1.21e-07 mol/L |
| Class | Moderately soluble | Poorly soluble | Moderately soluble | Moderately soluble | Poorly soluble |
| PHARMACOKINETICS | |||||
| GI absorption | High | Low | High | High | High |
| BBB permeant | Yes | No | No | Yes | Yes |
| P-gp substrate | Yes | Yes | Yes | Yes | No |
| CYP1A2 inhibitor | Yes | No | No | No | Yes |
| CYP2C19 inhibitor | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | Yes | No | Yes |
| CYP3A4 inhibitor | No | Yes | No | No | Yes |
| Log Kp (skin permeation) | -6.38 cm/s | -8.36 cm/s | -6.97 cm/s | -6.39 cm/s | -4.96 cm/s |
| DRUGLIKENESS | |||||
| Lipinski | Yes; 0 violation | No; 3 violations: MW>500, NorO>10, NHorOH>5 | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose | Yes | No; 3 violations: MW>500, NorO>10, NHorOH>5 | Yes | Yes | Yes |
| Veber | Yes | No; 2 violations: Rotors>10, TPSA>140 | Yes | Yes | Yes |
| Egan | Yes | No; 1 violation: TPSA>131.6 | Yes | Yes | Yes |
| Muegge | Yes | No; 4 violations: MW>600, TPSA>150, Rotors>15, H-don>5 | Yes | Yes | Yes |
| Biovailability Score | 0.55 | 0.17 | 0.55 | 0.55 | 0.55 |
| MEDICINAL CHEMISTRY | |||||
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert |
| Brenk | 1 alert quaternary_nitrogen_2 | 3 alerts: Three-membered_heterocycle, imine_1, imine_2 | 1 alert: Three-membered_heterocycle | 0 alert | 0 alert |
| Leandlikeness | No; 1 violation: MW<250 | No; 2 violations: MW>350, Rotors>7 | No; 1 violation: MW>350 | No; 1 violation: MW>350 | No; 2 violations: Rotors>7, XLOGP3>3.5 |
| Synthetic accessibility | 2.14 | 7.71 | 6.07 | 5.56 | 2.76 |
Bufalin, the most active compound, exhibits numbers of hydrogen bond acceptors (NHA) and hydrogen bond donors (NHD) in accordance with the rule of five by Lipinski. The LogS predicition of bufalin is -5.2, comparable with chloroquine (-6.92), indicating a good solubility. Although, the predicted polar surface area (PSA) of 70.67 Å2 for bufalin suggests that the polarity of this compound is a limiting factor for oral bioavailability [55]. In counterpoint, the synthetic accessibility of bufalin (5.56) is within the range of a non-complicated synthetic accessibility. bufalin could be a potential template for new antimalarial candidates.
Discussion
The drug discovery process is a major challenge in the pharmaceutical science due the time and money employed in all the phases of developing of a new drug entity [31]. Aiming to reduce cost and time in this process, structure-based virtual screening is an important in silico technique for drug design [56].
In this context, BraMMT database with 35 molecular targets of Plasmodium falciparum was used in this work. Table 1 shows all the targets and location of the proteins that were used for in silico binding assays with compounds isolated from R. marina. Three compounds interacted significantly with 10 potential targets (Table 2).
Of all 35 potential targets of BraMMT, the hexose transporter of Plasmodium falciparum (PfHT) interacted significantly with all tested compounds (Table 2). The target PfHT is a membrane protein of the parasite responsible for glucose transport. During the biological development of the parasites in the host´s red blood cells, the plasmodium requires glucose whose uptake is driven by carrier proteins. In red blood cells infected by P. falciparum glucose consumption is increased provided by PfHT. Inhibition of glucose transport to infected red blood cells impairs the parasite's metabolism, leading to death. Therefore, compounds that inhibit PfHT can be considered promising in the development of new bioactive compounds capable of treating malaria infections [53]. Additional potential targets analyzed are related to other structures of the parasite, such as apicoplast, cytoplasm, digestive vacuole, and sarcoplasmic reticulum.
The PfHT protein is a P. falciparum hexose transporter. Figures 2, 3, 4, 5 and 6 show the interaction of test compounds with PfHT. It is possible to visualize in the figures the chemical bonds that occurred between the compound and the target. All compounds interact at the same PfHT binding site of D-glucose [53] with residues GLN169 and THR145. The compounds interact with the PfHT receptor mainly through Van der Walls interactions, hydrogen bonds, and electrostatic bonds.
Figures 2, 3 and 4 shows that the electrostatic bonds interact with the PfHT receptor by the 2-pyrone group and the perhydrophenanthrene nucleus, demonstrating that the expression of antimalarial activity is associated with the presence of these structures. These groups are common to bufodianolides suggesting that the expression of antimalarial activity is associated with the presence of these structures.
One of the parameters introduced in the rational development of new drugs is Lipinski's rules “rule of five”. These parameters include molecular weight (M.M.) ≤ 500 g/mol, number of hydrogen bonding donor atoms ≤ 5, number of hydrogen bond acceptor atoms ≤ 10, and calculated octanol/water partition coefficient (cLogP) ≤ 5 [42]. The partition coefficient (CLogP) is a measure of the lipophilicity of a substance related to the interaction of the compound with the medium. This is an important tool regarding the study of absorption and transport. Furthermore, the program for the evolution of hazardous compounds recommends this measure, as it also provides estimates of toxicological factors [57].
Gleeson [58] suggests in his study that compounds with CLogP less than 4 and molecular weight less than 400 have a more favorable ADMET profile than those suggested by Lipinski. Following the Gleeson theory, all compounds have CLogP < 4 and compounds dehydrobufotenin, marinobufagin and bufalin have molecular mass below 400. Therefore, these three compounds (dehydrobufotenin, marinobufagin and bufalin) also fit Gleeson's theory. Therefore, compounds dehydrobufotenin, marinobufagin and bufalin showed sufficiently acceptable absorption, distribution, metabolism, excretion, and toxicity properties, according to Lipinski's rule and Gleeson's theory.
Compounds with high molecular weight and with an excessive number of hydrogen acceptor and donor groups, have greater difficulty in crossing the lipid bilayer of cell membranes. This is because such characteristics increase the lipophilicity of the compound, hindering solubility, and therefore impacting and the drug oral bioavailability [59]. Based in all these definitions, among the four compounds investigated in this study, the dehydrobufotenin was the one that presented the most favorable ADMET properties.
Secretions from 2 toad species, R. marina and R. guttatus, were chemically investigated previously. Two extracts and a pure substance (telocinobugagin) presented potential antimalarial activity [18]. When analyzed the IC50 values of all compounds tested ensure that IC50 values for the tested compounds ranged from 3.44 μM to 19.11 μM (dehydrobufotenin, marinobufotoxin, marinobufagin and bufalin).
According Mahmoudi [60], a potentially effective antimalarial compound possess an IC50 than 10 μM. The results published by Torres [52], indicated that alkaloids isolated from different parts of the Aspidosperma ulei plants, were moderately active against P. falciparum. These compounds presented IC50 values close to 20 μM. Based on this theories, the compounds marinobufotoxin (5.31 μM), marinobufagin and bufalin (3.89 μM and 3.44 μM, respectively) were considered potentially active while dehydrobufotenin (19.11 μM) expresses moderate activity.
The compounds marinobufotoxin, marinobufagin and bufalin showed cytotoxic activities against human pulmonary fibroblast cells (WI-26VA4) in MTT assay (LD50 = 8.89 μM; 3.04 μM and 25.9 μM, respectively). These results corroborate with previous studies that have reported a higher cytotoxic activity of venom extracts from R. marina in comparison to those from R. guttatus due the presence of 2 other bufadienolides (telocinobufagin, and resibufogenin) [13]. Similarly, extracts of R. marina venom from Peruvian Amazon with different compositions showed higher cytotoxic activity in antiproliferative tests with different tumor cell lines [21].
In our study the dehydrobufotenin compound showed the highest LD50 value (235.76 μM), indicating no cytotoxic effect against human pulmonary fibroblast cells. Low cytotoxicity of the bufadienolides fractions (telocinobufagin) against cancer cell lines (HL-60, SF-295, MDA-MB-435, and HCT-8) was also demonstrated [61]. However, to date, this was the first time that isolated dehydrobufotenine molecule was evaluated in cytotoxic test.
According to Bézivin [62], values higher than 10 (SI>10) is indicative of high selectivity values, whereas values below 10 (SI<10) are considered as low selectivity. In this study, although compounds 2, 3 and 4 were shown to be potentially active, only compound 1 was selective for the parasite, as it presented a selectivity index value greater than 10 (IS> 200).
In this work, it was important to assess the cytotoxic activity and evaluate the selectivity index for testing natural compounds with possible antimalarial potential. The exclusive observation of the IC50 values would result in wrong conclusions about the antimalarial potential of the compounds.
Conclusions
In summary, in docking assay all compounds tested promoted interaction between ligand-receptor with 10 targets of P. falciparum. Although in silico assays predicted good absorption, permeability, and absence of toxicity for three test compounds, in vitro assays demonstrated that only one compound expressed antimalarial activity and absence of cytotoxicity. The compound dehydrobufotenin can serve as a prototype molecule for the development of more active compounds.
Abbreviations
BraMMT: Brazilian Malaria Molecular Targets; DMSO: dimethyl sulfoxide; IBAMA: Brazilian Institute of Environment and Renewable Natural Resources; SISBIO: System for authorization of collection of biological material.
Footnotes
Availability of data and materials: All data generated or analyzed during this study are included in this published article.
Funding: This work was financially supported by the State of Mato Grosso Research Foundation (FAPEMAT), resource from the Universal Call for Proposals no. 042/2016 and State of Minas Gerais Research Foundation (FAPEMIG). The present work was carried out with the support of the Coordination for the Improvement of Higher Education Personnel (CAPES), financing code 001. The authors thank CAPES postdoctoral fellowship of the Graduate Program in Pharmaceutical Sciences. They are also thankful for the financial support of CNPq and CNPq/INCTBioNat (465637/2014-0), and CAPES (1776790/2017-0).
Ethics approval: All experiments were performed according to internationally accepted guidelines for the care and use of laboratory animals. The present study was approved by the Federal University of Mato Grosso Institutional Animal Care and Use Committee (protocol 23108.700260/14-7) and National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SisGen AE 19081).
Consent for publication: Not applicable.
References
- White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014 Feb 22;383(9918):723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- Naß J, Efferth T. Development of artemisinin resistance in malaria therapy. Pharmacol Res. 2019 Aug;146:104275. doi: 10.1016/j.phrs.2019.104275. [DOI] [PubMed] [Google Scholar]
- Nosten F, White NJ. Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg. 2007 Dec;77(6):181–192. [PubMed] [Google Scholar]
- Ocan M, Akena D, Nsobya S, Kamya MR, Senono R, Kinengyere AA, et al. Persistence of chloroquine resistance alleles in malaria endemic countries: a systematic review of burden and risk factors. Malar J. 2019 Mar 12;18(1):76. doi: 10.1186/s12936-019-2716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. Antimalarial drug resistance and combination chemotherapy. Philos Trans R Soc Lond B Biol Sci. 1999 Apr 29;354(1384):739–749. doi: 10.1098/rstb.1999.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White NJ. Antimalarial drug resistance. J Clin Invest. 2004 Apr 15;113(8):1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhiet AMA, Abdelraheem MH, Kheir A, Omer S, Gismelseed L, Abdel-Muhsin AA, et al. Evolution of Plasmodium falciparum drug resistance genes following artemisinin combination therapy in Sudan. Trans R Soc Trop Med Hyg. 2019 Nov 01;113(11):693–700. doi: 10.1093/trstmh/trz059. [DOI] [PubMed] [Google Scholar]
- Veiga-Júnior VF, Pinto AC, Maciel MAM. Plantas Medicinais: Cura segura? Quím Nova. 2005 May-Jun;28(3):519–528. [Google Scholar]
- World Health Organization . The African health monitor: African traditional medicine day. 2019. [12 September 2019]. Available from: http://apps.who.int/medicinedocs/documents/s21374en/s21374en.pdf. [Google Scholar]
- Lev E. Traditional healing with animals (zootherapy): medieval to present-day Levantine practice. J Ethnopharmacol. 2003 Mar;85(1):107–118. doi: 10.1016/s0378-8741(02)00377-x. [DOI] [PubMed] [Google Scholar]
- Kowalski K, Marciniak P, Rosinski G, Richlik L. Toxic activity and protein identification from the parotoid gland secretion of the common toad Bufo bufo. Comp Biochem Physiol C Toxicol Pharmacol. 2018 Feb;205:43–52. doi: 10.1016/j.cbpc.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Shibao PYT, Cologna CT, Morandi-Filho R, Wiezel GA, Fujimura PT, Ueira-Vieira C, et al. Deep sequencing analysis of toad Rhinella schneideri skin glands and partial biochemical characterization of its cutaneous secretion. J Venom Anim Toxins incl Trop Dis. 2018;24:36. doi: 10.1186/s40409-018-0173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PM, Lima DJ, Debiasi BW, Soares BM, Machado Kda C, Noronha Jda C, et al. Antiproliferative activity of Rhinella marina and Rhaebo gutattus venom extracts from Southern Amazon. Toxicon. 2013 Sep;72:43–51. doi: 10.1016/j.toxicon.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Sousa LQ, Machado KDC, Oliveira SF, Araújo LD, Monção-Filho ED, Melo-Cavalcante AA, et al. Bufadienolides from amphibians: A promising source of anticancer prototypes for radical innovation, apoptosis triggering and Na+/K+-ATPase inhibition. Toxicon. 2017 Mar 01;127:63–76. doi: 10.1016/j.toxicon.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Machado KDC, Sousa LQ, Lima DJB, Soares BM, Cavalcanti BC, Maranhão SS, et al. Marinobufagin, a molecule from poisonous frogs, causes biochemical, morphological and cell cycle changes in human neoplasms and vegetal cells. Toxicol Lett. 2018 Mar 15;285:121–131. doi: 10.1016/j.toxlet.2017.12.018. [DOI] [PubMed] [Google Scholar]
- Wang DL, Qi FH, Tang W, Wang FS. Chemical constituents and bioactivities of the skin of Bufo bufo gargarizans Cantor. Chem Biodivers. 2011 Apr;8(4):559–567. doi: 10.1002/cbdv.201000283. [DOI] [PubMed] [Google Scholar]
- Vigerelli H, Sciani JM, Jared C, Antoniazzi MM, Caporale GM, da Silva Ade C, et al. Bufotenine is able to block rabies virus infection in BHK-21 cells. J Venom Anim Toxins incl Trop Dis. 2014 Oct 13;20(1):45. doi: 10.1186/1678-9199-20-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfi FF, Guedes KS, Andrighetti CR, Aguiar AC, Debiase BW, Noronha JC, et al. Antiplasmodial and Cytotoxic Activities of Toad Venoms from Southern Amazon, Brazil. Korean J Parasitol. 2016 Aug;54(4):415–421. doi: 10.3347/kjp.2016.54.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff J, Noronha JC, Bonfilio R, Sinhorin AP, Rodrigues DJ, Chaves MH, et al. Quantification of bufadienolides in the poisons of Rhinella marina and Rhaebo guttatus by HPLC-UV. Toxicon. 2016 Sep 01;119:311–318. doi: 10.1016/j.toxicon.2016.07.003. [DOI] [PubMed] [Google Scholar]
- Vigerelli H, Sciani JM, Pereira PMC, Lavezo AA, Silva ACR, Collaço RCO, et al. Bufotenine, a tryptophan-derived alkaloid, suppresses the symptoms and increases the survival rate of rabies-infected mice: the development of a pharmacological approach for rabies treatment. J Venom Anim Toxins incl Trop Dis. 2020 Feb 03;26:e20190050. doi: 10.1590/1678-9199-jvatitd-2019-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeda-Hirschmann G, Quispe C, Arana GV, Theoduloz C, Urra FA, Cárdenas C. Antiproliferative activity and chemical composition of the venom from the Amazonian toad Rhinella marina (Anura: Bufonidae) Toxicon. 2016 Oct;121:119–129. doi: 10.1016/j.toxicon.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Abdelfatah S, Lu X, Schmeda-Hirschmann G, Efferth T. Cytotoxicity and antimitotic activity of Rhinella schneideri and Rhinella marina venoms. J Ethnopharmacol. 2019 Oct 05;:112049. doi: 10.1016/j.jep.2019.112049. [DOI] [PubMed] [Google Scholar]
- Cunha Neto Rdos S, Vigerelli H, Jared C, Antoniazzi MM, Chaves LB, da Silva Ade C, et al. Synergic effects between ocellatin-F1 and bufotenine on the inhibition of BHK-21 cellular infection by the rabies virus. J Venom Anim Toxins Incl Trop Dis. 2015 Dec 02;21:50. doi: 10.1186/s40409-015-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Swaminathan S, Hyder KA, Christophel EM, Pendse RN, Sreenivas AN, et al. Drug resistance in malaria, tuberculosis, and HIV in South East Asia: biology, programme, and policy considerations. BMJ. 2017 Sep 05;358:j3545. doi: 10.1136/bmj.j3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro EJ, Fraga CAM, Miranda ALP, Rodrigues CR. A química medicinal de N-acilidrazonas: Novos compostos-protótipos de fármacos analgésicos, anti-inflamatórios e antitrombóticos. Quím Nova. 2002;25(1):129–148. [Google Scholar]
- Asokkumar K, Prathyusha LT, Umamaheshwari M, Sivashanmugam T, Subhadradevi V, Jagannath P, et al. Design, ADMET and docking studies on some novel chalcone derivatives as soluble epoxide hydrolase enzyme inhibitors. J Chil Chem Soc. 2012;57(4):1442–1446. [Google Scholar]
- Carvalho I, Pupo MT, Borges ÁDL. Introdução a modelagem molecular de fármacos no curso experimental de química farmacêutica. Quím Nova. 2003 May-Jun;26(3):428–438. [Google Scholar]
- Guantai E, Chibale K. How can natural products serve as a viable source of lead compounds for the development of new/novel anti-malarials? Malar J. 2011 Mar 15;10(1):S2. doi: 10.1186/1475-2875-10-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia IJP, Oliveira GC, Valadares JMM, Banfi FF, Andrade SN, Freitas TR, et al. New bufadienolides extracted from Rhinella marina inhibit Na,K-ATPase and induce apoptosis by activating caspases 3 and 9 in human breast and ovarian cancer cells. Steroids. 2019 Dec;152:108490. doi: 10.1016/j.steroids.2019.108490. [DOI] [PubMed] [Google Scholar]
- Jaghoori MM, Bleijlevens B, Olabarriaga SD. 1001 ways to run AutoDock Vina for virtual screening. J Comput Aided Mol Des. 2016 Mar;30(3):237–249. doi: 10.1007/s10822-016-9900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia EHB, Medaglia LR, Silva AM, Taranto AG. Molecular architect: A user-friendly workflow for virtual screening. ACS Omega. 2020 Mar 20;5:6628–6640. doi: 10.1021/acsomega.9b04403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia EH, Campos VA, Dos Reis Santos B, Costa MS, Lima IG, Greco SJ, et al. Octopus: a platform for the virtual high-throughput screening of a pool of compounds against a set of molecular targets. J Mol Model. 2017 Jan;23(1):26. doi: 10.1007/s00894-016-3184-9. [DOI] [PubMed] [Google Scholar]
- Nunes RR, Fonseca ALD, Pinto ACS, Maia EHB, Silva AMD, Varotti FP, et al. Brazilian malaria molecular targets (BraMMT): selected receptors for virtual high-throughput screening experiments. Mem Inst Oswaldo Cruz. 2019 Feb 25;114:e180465. doi: 10.1590/0074-02760180465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 Mar 03;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander T, Freyss J, Von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015 Feb 23;55(2):460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Campbell CC, Collins WE, Nguen-Dinh P, Barber A, Broderson JR. Plasmodium falciparum gametocytes from culture in vitro develop to sporozoites that are infectious to primates. Science. 1982 Sep 10;217(4564):1048–1050. doi: 10.1126/science.7051285. [DOI] [PubMed] [Google Scholar]
- Ranford-Cartwright LC, Hayton KL, Ferdig MT. Chapter 6 - Plasmodium Experimental Genetic Crosses. In: Carlton JM, Perkins SL, Deitsch KW, editors. Malaria Parasites: Comparative Genomics, Evolution, and Molecular Biology. Horizon Scientific Press; New York: 2013. pp. 127–144. [Google Scholar]
- Bell C, Hall JE, Kyle DE, Grogl M, Ohemeng KA, Allen MA, et al. Structure-activity relationships of analogs of pentamidine against Plasmodium falciparum and Leishmania mexicana amazonensis. Antimicrob Agents Chemother. 1990 Jul;34(7):1381–1386. doi: 10.1128/aac.34.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976 Aug 20;193(4254):673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979 Jun;65(3):418–420. [PubMed] [Google Scholar]
- Carvalho LH, Brandão MG, Santos-Filho D, Lopes JL, Krettli AU. Antimalarial activity of crude extracts from Brazilian plants studied in vivo in Plasmodium berghei-infected mice and in vitro against Plasmodium falciparum in culture. Braz J Med Biol Res. 1991;24(11):1113–1123. [PubMed] [Google Scholar]
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986 May 22;89(2):271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Twentyman PR, Fox NE, Rees JK. Chemosensitivity testing of fresh leukaemia cells using the MTT colorimetric assay. Br J Haematol. 1989 Jan;71(1):19–24. doi: 10.1111/j.1365-2141.1989.tb06268.x. [DOI] [PubMed] [Google Scholar]
- Valsalam S, Agastian P, Esmail GA, Ghilan AM, Al-Dhabi NA, Arasu MV. Biosynthesis of silver and gold nanoparticles using Musa acuminata colla flower and its pharmaceutical activity against bacteria and anticancer efficacy. J Photochem Photobiol B. 2019 Dec;201:111670. doi: 10.1016/j.jphotobiol.2019.111670. [DOI] [PubMed] [Google Scholar]
- Karkehabadi H, Yousefifakhr H, Zadsirjan S. Cytotoxicity of endodontic irrigants on human periodontal ligament cells. Iran Endod J. 2018;13(3):390–394. doi: 10.22037/iej.v13i3.20438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DB, Junior, Araújo JSCA, Oliveira LM, Neri FSM, Moreira POL, Taranto AG, et al. Identification of novel antiplasmodial compound by hierarquical virtual screening and assays. J Biomol Struct Dyn. 2020 May 13;:1–9. doi: 10.1080/07391102.2020.1763837. [DOI] [PubMed] [Google Scholar]
- Neri FSM, Costa DB, Júnior, Froes TQ, Silva PBG, Egito MS, Moreira POL, et al. Antileishmanial activity evaluation of thiazolidine-2,4-dione against Leishmania infantum and Leishmania braziliensis. Parasitol Res. 2020 Jul;119(7):2263–2274. doi: 10.1007/s00436-020-06706-3. [DOI] [PubMed] [Google Scholar]
- Pessôa MTC, Valadares JMM, Rocha SC, Silva SC, McDermott JP, Sánchez G, et al. 21-Benzylidene digoxin decreases proliferation by inhibiting the EGFR/ERK signaling pathway and induces apoptosis in HeLa cells. Steroids. 2020 Mar;155:108551. doi: 10.1016/j.steroids.2019.108551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Céu de Madureira M, Paula-Martins A, Gomes M, Paiva J, Proença da Cunha A, do Rosário V. Antimalarial activity of medicinal plants used in traditional medicine in S. Tomé and Príncipe islands. J Ethnopharmacol. 2002 Jun;81(1):23–29. doi: 10.1016/s0378-8741(02)00005-3. [DOI] [PubMed] [Google Scholar]
- Stoffman EJL, Clive DLJ. Synthesis of 4-haloserotonin derivatives and synthesis of the toad alkaloid dehydrobufotenine. Tetrahedron. 2010 Jun 19;66(25):4452–4461. [Google Scholar]
- Yoshika M, Komiyama Y, Konishi M, Akizawa T, Kobayashi T, Date M, et al. Novel digitalis-like factor, marinobufotoxin, isolated from cultured Y-1 cells, and its hypertensive effect in rats. Hypertension. 2007 Jan;49(1):209–214. doi: 10.1161/01.HYP.0000250433.64202.78. [DOI] [PubMed] [Google Scholar]
- Torres ZES, Silveira ER, Rocha e Silva LF, Lima ES, De Vasconcellos MC, De Andrade Uchoa DE, et al. Chemical composition of Aspidosperma ulei Markgr. and antiplasmodial activity of selected indole alkaloids. Molecules. 2013 Jun;18(6):6281–6297. doi: 10.3390/molecules18066281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca AL, Nunes RR, Braga VML, Comar-Jr M, Alves RJ, Varotti F de P, et al. Docking, QM/MM, and molecular dynamics simulations of the hexose transporter from Plasmodium falciparum (PfHT) J Mol Graph Model. 2016 May;66:174–186. doi: 10.1016/j.jmgm.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Kirk K, Horner HA, Kirk J. Glucose uptake in Plasmodium falciparum-infected erythrocytes is an equilibrative not an active process. Mol Biochem Parasitol. 1996 Nov 25;82(2):195–205. doi: 10.1016/0166-6851(96)02734-x. [DOI] [PubMed] [Google Scholar]
- Woodrow CJ, Burchmore RJ, Krishna S. Hexose permeation pathways in Plasmodium falciparum-infected erythrocytes. Proc Natl Acad Sci U S A. 2000 Aug 29;97(18):9931–9936. doi: 10.1073/pnas.170153097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto CN. Homology models in docking and high-throughput docking. Curr Top Med Chem. 2011;11(12):1528–1534. doi: 10.2174/156802611795860951. [DOI] [PubMed] [Google Scholar]
- Silva LR, Ferreira MMC. Estudo do coeficiente de partição octanol-água de bifenilas policloradas (PCBs) utilizando parâmetros topológicos. Quim Nova. 2003 May-Jun;26(3):312–318. [Google Scholar]
- Gleeson MP. Generation of a set of simple, interpretable ADMET rules of thumb. J Med Chem. 2008 Feb 28;51(4):817–834. doi: 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001 Mar 01;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Mahmoudi N, Julian-Ortiz JV, Ciceron L, Gálvez J, Mazier D, Danis M. Identification of new antimalarial drugs by linear discriminant analysis and topological virtual screening. J Antimicrob Chemother. 2006 Mar;57(3):489–497. doi: 10.1093/jac/dki470. [DOI] [PubMed] [Google Scholar]
- Cunha-Filho GA, Resck IS, Cavalcanti BC, Pessoa CO, Moraes MO, Ferreira JRO, et al. Cytotoxic profile of natural and some modified bufadienolides from toad Rhinella schneideri parotoid gland secretion. Toxicon. 2010 Sep 01;56(3):339–348. doi: 10.1016/j.toxicon.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Bézivin C, Tomasi S, Lohézic-Le Dévéhat F, Boustie J. Cytotoxic activity of some lichen extracts on murine and human cancer cell lines. Phytomedicine. 2003;10(6-7):499–503. doi: 10.1078/094471103322331458. [DOI] [PubMed] [Google Scholar]