

Abstract
Recent advances in the generation of microglia from human induced pluripotent stem cells (iPSCs) have provided exciting new approaches to examine and decipher the biology of microglia. As these techniques continue to evolve to encompass more complex in situ and in vivo paradigms, so too have they begun to yield novel scientific insight into the genetics and function of human microglia. As such, researchers now have access to a toolset comprised of three unique “flavors” of iPSC-derived microglia: in vitro microglia (iMGs), organoid microglia (oMGs), and xenotransplanted microglia (xMGs). The goal of this review is to discuss the variety of research applications that each of these techniques enables and to highlight recent discoveries that these methods have begun to uncover. By presenting the research paradigms in which each model has been successful, as well as the key benefits and limitations of each approach, it is our hope that this review will help interested researchers to incorporate these techniques into their studies, collectively advancing our understanding of human microglia biology.
Keywords: cerebral organoids, chimeric, induced pluripotent stem cells, microglia, microglia-like cells, xenotransplantation
1 |. INTRODUCTION
Microglia, the principle innate immune cells of the brain, play critical roles in development, homeostasis, and neurological disease. A great deal of what we currently know about microglial function has been informed by elegant studies of mouse microglia. Yet recent experiments have revealed important differences between murine and human microglia (Galatro et al., 2017; Gosselin et al., 2017; Smith & Dragunow, 2014), highlighting the need for new techniques that could allow researchers to better interrogate the genetics and function of human microglia. Fortunately, recent advances in the use of induced pluripotent stem cells (iPSCs) have provided exciting new approaches to examine and decipher human microglial biology. This review aims to discuss the development and application of these new techniques and provide a summary of key benefits and limitations of each approach. In doing so, we hope to encourage interested researchers to incorporate these methods into their studies, further advancing our collective understanding of microglia biology.
Microglia were first described by multiple researchers in the late 1800’s to early 1900’s, including Gluge, Nissl, Achúcarro, Merzbacher, and Cajal (Tremblay, Lecours, Samson, Sanchez-Zafra, & Sierra, 2015). However, it was Pio del Rio-Hortega that truly delineated these cells from other neuroglia by developing a new method of specifically labeling microglia, describing their mesodermal lineage, propensity to alter morphology in response to neuronal damage, and their appetite for lipids and cellular debris (Río-Hortega, 1919a, 1919b, 1919c, 1919d, 1939; Sierra et al., 2016). Yet, despite these clear descriptions from 100 years ago, the field’s understanding of microglia has only begun to truly expand in the past three decades as additional methods of examining these cells have been developed.
Just as del Rio-Hortega established novel histological techniques to visualize microglia, the development of a multitude of murine models have enabled many new techniques, including genetic alterations, fluorescent cell labeling, and multi-photon microscopy, which have together served as key drivers of this new era of microglia discovery. Indeed, the influential roles of microglia in brain development, plasticity, homeostasis, and their dynamic response to neurotrauma and disease were highly dependent on in vivo studies of mouse microglia (Davalos et al., 2005; Nimmerjahn, Kirchhoff, & Helmchen, 2005; Wolf, Boddeke, & Kettenmann, 2017). Likewise, del Rio-Hortega’s initial assertion of the possible mesodermal lineage of microglia was first clearly demonstrated by Ginhoux and colleagues, by definitively tracing the developmental ontogeny of murine microglia back to their yolk-sac progenitors (Ginhoux et al., 2010). Mouse models have also greatly advanced our understanding of how microglia respond to a variety of pathological environments. For example, transgenic models of Alzheimer’s disease (AD) were critical in clarifying the roles of the gene triggering receptor expressed on myeloid cells 2 (TREM2) in the microglial response to amyloid beta (Aβ) plaques. While initial genome-wide association study (GWAS) findings utilized human data to associate TREM2 gene variants with AD risk (Guerreiro, Wojtas, et al., 2013; Jonsson et al., 2013), the resulting decreases in migration and barrier formation in response to Aβ plaques were first identified in multiple mouse models of AD (Jay et al., 2015; Ulrich et al., 2014; Y. Wang et al., 2015). These findings were subsequently confirmed in human patients, demonstrating that this phenotype was relevant to microglia in AD while also lending support to the validity of the murine models (Yuan et al., 2016).
However, it is becoming increasingly evident that rodents cannot always recapitulate human genetics, a caveat that is abundantly clear in studies of polygenic diseases such as AD (Dawson, Golde, & Lagier-Tourenne, 2018; Friedman et al., 2018; Ueda, Gullipalli, & Song, 2016). For example, when examining the homology between the human and mouse proteins associated with gene loci that have been implicated in AD risk (Figure 1), over half of the proteins that are enriched in microglia (Figure 1, red text) display less than 70% homology between the species. Furthermore, mice lack any reliable orthologue for either of the risk genes CD33 or CR1. In addition to protein homology, differences in gene expression have also been reported between the species. For example, multiple studies have performed transcriptomic comparisons between human and murine microglia, reporting significant differences in the expression of complement system genes, several inflammatory cytokines, and genes related to neurodegenerative diseases such as AD and Parkinson’s disease (PD; Galatro et al., 2017; Gosselin et al., 2017; Smith & Dragunow, 2014). Overall, these species-specific differences likely contribute to the inability of mouse models to accurately mimic many human conditions, often hindering the translatability of preclinical research (Burns, Li, Mehta, Awad, & Morgan, 2015).
FIGURE 1.
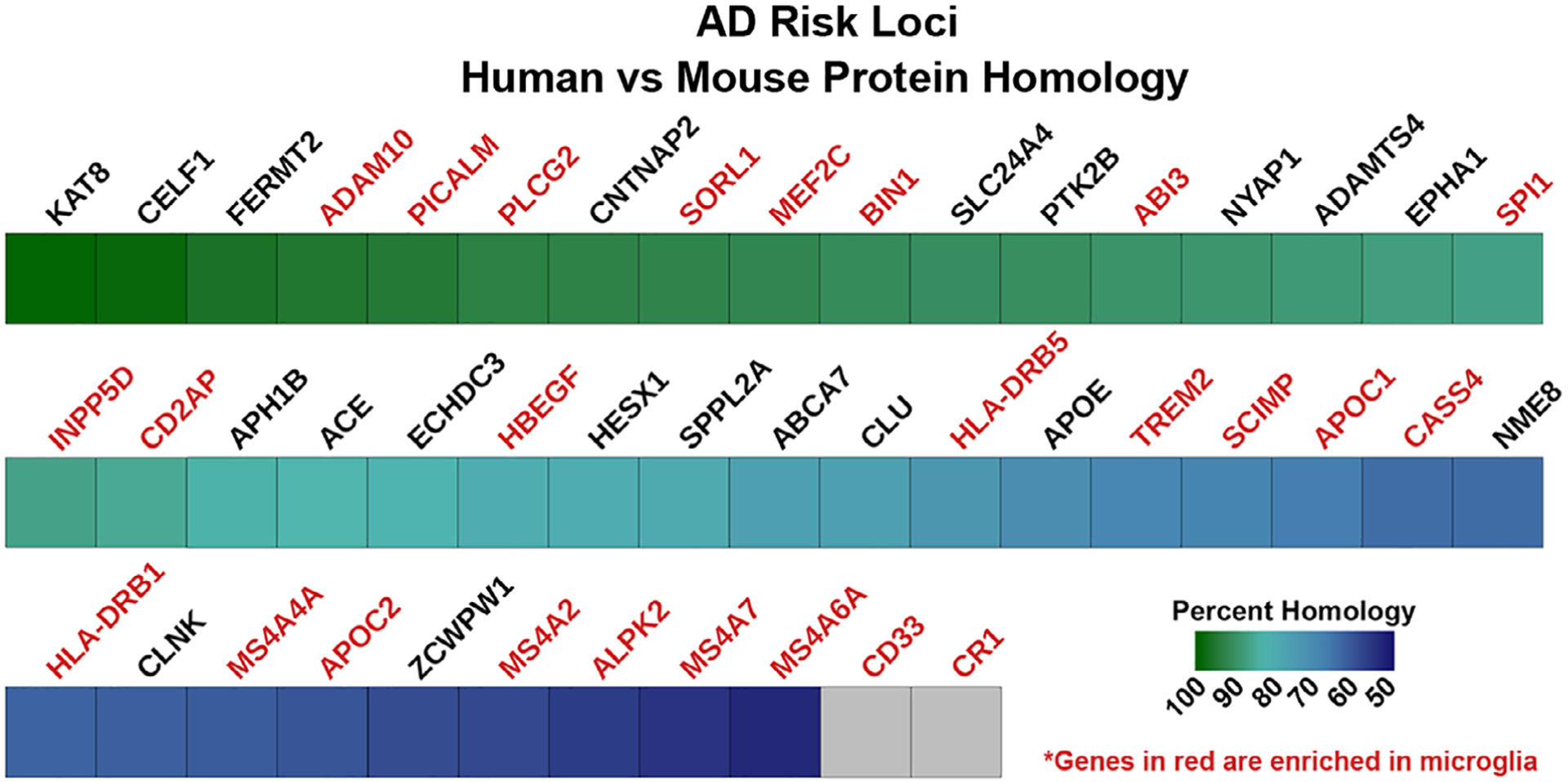
Homology between human and mouse proteins associated with Alzheimer’s disease risk. Human amino acid sequences for genes associated with Alzheimer’s disease GWAS-identified risk loci were compared to the homologous mouse proteins using NCBI’s Homologene database. Genes in red are either highly or specifically expressed in microglia and gray boxes denote genes with no reliable orthologues in the mouse (i.e., <50% homology with any possible murine orthologue)
Some researchers have sought to address this caveat by developing methods to isolate microglia from human tissue followed by maintenance in an in vitro environment. This challenging, yet important approach has indeed been highly informative. For example, studies of human brain-derived microglia have provided insight into the differential gene expression signatures between human microglia and macrophages across varying activation states (Healy et al., 2018). Likewise, the use of primary human microglia has uncovered important human-specific features, including their decreased propensity to produce the inflammatory molecule nitric oxide compared to murine microglia (Janabi, 2002), and differences in responsiveness to pharmacological agents, such as valproic acid and propentofylline, when compared to animal models (Gibbons et al., 2011; Landry, Jacobs, Romero-Sandoval, & DeLeo, 2012). However, this approach also comes with disadvantages that have hindered the widespread applicability of this approach. First, primary human microglia are notoriously difficult to obtain, especially in numbers adequate for well powered biological experiments. As such, there are a limited number of labs that have the expertise and access to tissue needed to routinely isolate human microglia. Second, it has recently been demonstrated that once microglia are removed from the brain, they undergo rapid transcriptomic and phenotypic changes that diminish the accuracy of these cells as a model for in vivo microglia (Bohlen et al., 2017; Butovsky et al., 2014; Gosselin et al., 2017). Finally, the tissue samples used in isolations are typically obtained postmortem or from patients under-going surgical resection of brain tumors or epileptic foci. This presents an important unresolved concern, as there has yet to be a study thoroughly examining the chronic effects of long-term exposure to pathological conditions in order to determine whether microglia isolated from disease-affected tissues are phenotypically normal, or if they possess aberrant behaviors and gene expression due to their environmental origins. Therefore, it is difficult to say with confidence that microglia isolated in this manner and subsequently cultured can be readily applied to experiments focused on homeostatic microglia function or disorders unrelated to the condition that had afflicted the host tissue.
However, these concerns have recently served as a catalyst for progress, resulting in another set of technical developments aimed at counteracting many of the issues associated with both murine microglia and primary human microglia. Primarily, this has come in the form of several new protocols devising and describing methods to generate large numbers of human microglia in vitro from iPSCs. Several applications of these techniques have now been described that further allow the cells to be studied in both in situ and in vivo environments, further addressing concerns with environmental dependencies.
In this review, we will discuss many of these applications. However, as the technical aspects of the protocols for generating iPSC-derived microglia have recently been reviewed in detail (Haenseler & Rajendran, 2019; Pocock & Piers, 2018; Timmerman, Burm, & Bajramovic, 2018), this article will only broadly discuss the developmental ontogeny of microglia and how most differentiation protocols aim to mimic this ontogeny within a dish (Figure 2). Instead, we will focus this review on the following three areas: (1) we will present a brief background for each environmental model along with the experimental validations that were performed. (2) We will review the applications of each technique with an emphasis on results that could not reliably be obtained using traditional methods. (3) We will address the benefits and limitations of each technique highlighting appropriate applications of each paradigm, the improvements offered over current methods, and areas of further development that may continue to enhance these techniques. By discussing the benefits and applications of each model, we aim to demonstrate to the reader that these techniques comprise a complementary toolset that allows for highly controlled experiments where preliminary discoveries from more high-throughput in vitro methods can subsequently be validated in more complex in situ or in vivo models. Our hope is that this will help interested researchers to understand the utility of these new methods and the type of unique and informative experiments that can now be readily pursued with human iPSC-microglia.
FIGURE 2.
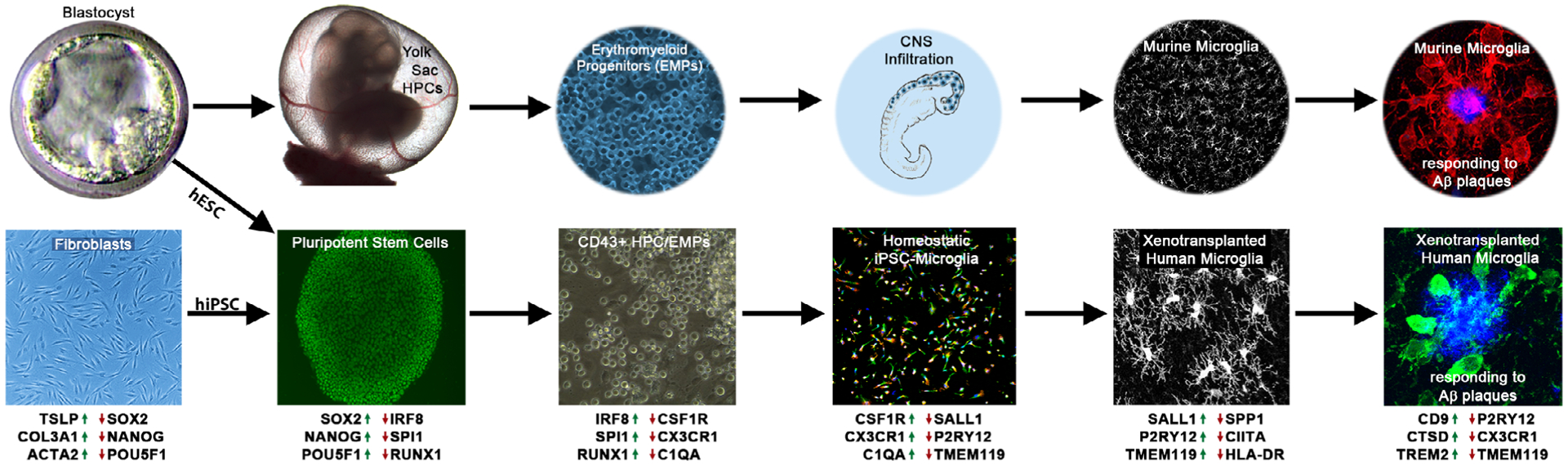
In vivo and in vitro microglia ontogeny. (Top) Lineage-tracing studies in mice have shown that microglia follow a distinct developmental pathway in vivo. Pluripotent embryonic stem cells (ESCs) within the inner cell mass of the blastocyst give rise to hypoblasts which in turn produce the extraembryonic endoderm of the yolk-sac. Hemogenic endothelium within the yolk-sac then gives rise to primitive hematopoietic progenitor cells (HPCs) that can further differentiate into EMPs. These cells then infiltrate into the developing central nervous system (CNS) where maturation under homeostatic conditions results in neatly tiled microglia that develop complex ramifications. In contrast, pathological conditions, such as the development of amyloid plaques in a murine model of Alzheimer’s disease (AD), induces robust migratory and morphological alterations. (Bottom) iPSC-derived microglia protocols seek to mimic in vivo ontogeny in order to generate cells that appropriately recapitulate endogenous human microglia. This is accomplished by obtaining either human ESCs (hESCs) or reprogramming human fibroblasts (or other cells) into induced pluripotent stem cells (hiPSC). These pluripotent stem cells can then be further differentiated into CD43+ microglial progenitors that resemble in vivo HPCs/EMPs. By providing additional key microglial growth factors and signaling molecules including CSF-1, IL34, and TGFβ, that mimic the homeostatic brain environment, researchers can promote the further differentiation and maturation of large numbers of iPSC-derived microglia (iMGs). Subsequent xenotransplantation of these microglia into the murine brain induces similar morphology and tiling patterns as endogenous murine microglia while retaining the ability to respond to AD pathology. Upregulated (green arrows) and downregulated (red arrows) genes are relevant to the human cellular identity at each differentiation step
Before we begin, we would like to briefly discuss a cohesive terminology for the cells generated using these developing techniques. We and others have referred to the in vitro flavor of these cells with a variety of names and acronyms including “iPSC-microglia-like cells,” “pluripotent microglia-like cells,” “iMGLs,” “pMGLs,” “hiMacs,” “hiMicros,” “iPSC-MG,” “iPS-MG,” and so on all to signify that no two protocols are identical and that these cells do not perfectly recapitulate the in vivo condition. While variation exists between each specific differentiation protocol and none of these methods offer an exact model of homeostatic, in vivo microglia, the same is true of more traditional microglia models. Mouse microglia possess substantial genetic variation when compared to humans; primary human microglia undergo significant transcriptomic alterations when removed from the brain and cultured; and ex vivo microglia are routinely isolated from tissue affected by diseases such as epilepsy and cancer and further confounded by isolation artifacts caused by enzymatic digestions and cell sorting. Yet each of these traditional approaches yield cells that are referred to as microglia, not microglia-like cells or macrophages. Therefore, for the sake of simplicity, we will refer to all in vitro iPSC-derived microglia as iMGs regardless of the differentiation protocol used to generate the cells. Additionally, we will refer to derivatives of this methodology as oMGs, for cerebral organoid microglia, and xMGs, for xenotransplanted microglia, as each of these terms have already been published in the literature (Hasselmann et al., 2019; Ormel et al., 2018; Xiang et al., 2018).
2 |. GENERATION OF MICROGLIA FROM HUMAN IPSCS
In an effort to address the difficulty of acquiring enough tissue to routinely isolate large amounts of human microglia, multiple scientists converged on a solution to a common problem by publishing six protocols for generating microglia from iPSCs between 2016 and 2017 (Abud et al., 2017; Douvaras et al., 2017; Haenseler, Sansom, et al., 2017; Muffat et al., 2016; Pandya et al., 2017; Takata et al., 2017). While each protocol was unique regarding their specific details, each method was based on the concept that, in order to generate accurate microglial surrogates in vitro, one needed to mimic the cues that naturally drive microglia differentiation in vivo. Whereas early attempts at in vitro microglia generation tried to produce microglia by pushing embryonic stem cells through a neuroectodermal lineage (Beutner, Roy, Linnartz, Napoli, & Neumann, 2010; Tsuchiya et al., 2005), this new generation of protocols was highly informed by an elegant set of developmental ontogeny studies that traced the lineage of microglia from mesodermal primitive yolk sac progenitors, to MYB-independent erythromyeloid progenitors (EMPs), yolk sac macrophages, and finally to microglia within the brain (Ginhoux et al., 2010; Kierdorf et al., 2013; Schulz et al., 2012). With this guidance, these new protocols were able to produce cells that transitioned from iPSCs through a range of lineage states resembling primitive hematopoietic precursor cells (HPCs), EMPs, and, ultimately, microglia (Figure 2; Figure 3, iMG), as confirmed in each study by transcriptomic and protein-level analysis of canonical microglia markers.
FIGURE 3.
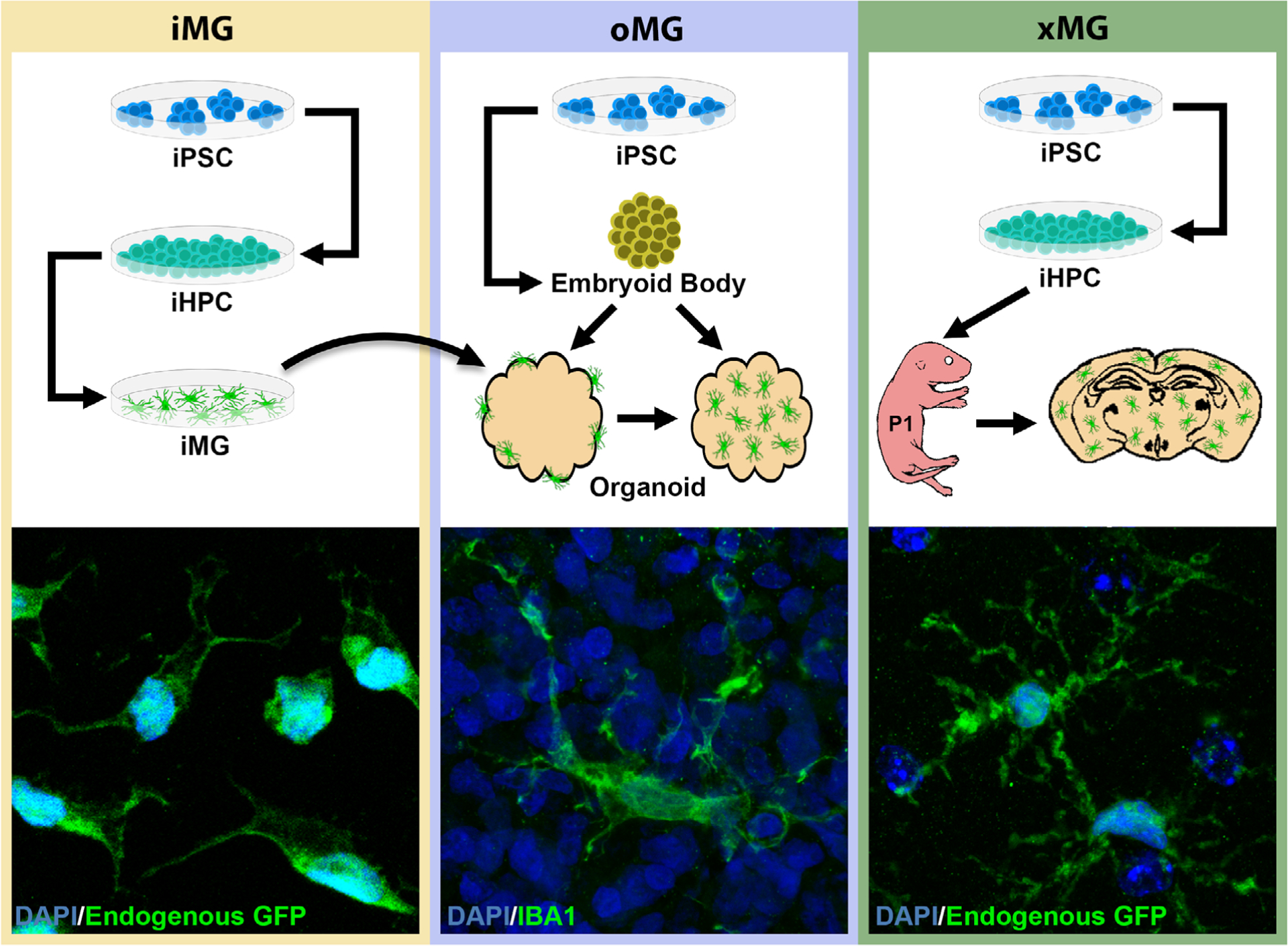
Procedural overview and representative morphology of each iPSC-derived microglia technique. Microglial cells adopt unique, paradigm-dependent morphologies that increase in complexity with more in vivo-like conditions. (iMG) in vitro differentiation of iPSCs into iPSC-derived hematopoietic progenitor cells (iHPCs) and on to iMGs results in cells that typically assume amoeboid, unipolar, or bipolar morphologies. (oMG) Cerebral organoids are generated by first forming embryoid bodies from iPSCs and then further driving differentiation into neuroepithelium and, ultimately, self-assembly into cerebral organoids. Depending on the differentiation approach used, the organoids may contain endogenous microglia or can be cocultured with iMGs which results in iMG infiltration and engraftment. These microglia take on a more ramified morphology than their iMG counterparts, although the branching pattern remains less complex than adult human in vivo microglia. (xMG) iPSCs are differentiated into iHPCs, as in the iMG procedure, followed by intracerebroventricular transplantation into the brains of immune-deficient mice humanized for the CSF1 protein. Transplantation into the murine brain induces a highly ramified morphology, reminiscent of the complex branching patterns observed in in vivo human microglia
In addition to genetic validation, each of these studies sought to functionally validate their iMGs to demonstrate that these in vitro systems could recapitulate a range of traditional microglia behaviors. For example, the characteristic ability of microglia to respond to inflammatory cues was tested via stimulation with lipopolysaccharide, phorbol myristate acetate, IFNγ, or IL1β, and confirmed by measurement of reactive oxygen species or a variety of inflammatory cytokines (Abud et al., 2017; Haenseler, Sansom, et al., 2017; Pandya et al., 2017). The propensity of iMGs to migrate toward sites of damage was also tested by employing a laser induced injury to a 3D culture system, similar to in vivo studies (Davalos et al., 2005), or through the addition of ADP, which is typically released by damaged neurons. In all cases, iMGs exhibited rapid responses by extending process toward the perceived site of damage and/or relocating their cell bodies (Abud et al., 2017; Douvaras et al., 2017; Muffat et al., 2016). Finally, the phagocytic capacity of these cells was determined by utilizing a variety of substrates, ranging from zymosan-coated microbeads, Escherichia coli particles, or more brain-relevant substrates such as synaptosomes, fibrillar beta-amyloid, and Tau oligomers (Abud et al., 2017; Douvaras et al., 2017; Pandya et al., 2017; Takata et al., 2017). While the overall level of uptake varied by assay and substrate, each study demonstrated the genesis of phagocitically-functional cells. As a whole, these results demonstrated a plethora of microglia-related activities that can be modeled in vitro, supporting the use of these cells in a variety of experimental contexts.
Additionally, many of these studies recognized the possibility of transcriptomic deficiencies in their iMGs in the absence of other neuronal cell types and sought out ways to induce a more accurate, brain-specific microglia profile. While each protocol included the critical microglia survival signal CSF1 or IL34 in the culture medium, it was also realized that there were a multitude of signaling molecules and physical interactions missing from these basic cell culture conditions. To address this, most of the protocols attempted to drive these cells closer to a microglia fate by coculturing the iMGs with neurons and/or astrocytes (Abud et al., 2017; Muffat et al., 2018; Pandya et al., 2017; Takata et al., 2017). Additionally, while one study showed that coculture with rat neurons altered the transcriptomic profile of the iMGs, Abud et al. (2017) also attempted to rectify the deficiencies by supplementing the cell culture medium with numerous additional factors aimed at generating more mature microglia. In addition to the CSF1 that was included in the other medium formulations, Abud and colleagues further added in cytokines such as TGFβ, CX3CL1, CD200 which have all been shown to be critical for maintaining microglia homeostasis (Butovsky et al., 2014; Cardona et al., 2006; Hoek et al., 2000; Kierdorf & Prinz, 2013). In all cases, changes to morphology and gene expression were seen following either coculture or medium supplementation, suggesting that these modifications may have further matured the iMGs. However, it must be kept in mind that each of these studies utilized primary cultured microglia as the reference cell type to demonstrate the similarity between iMGs and microglia which raises concerns over how accurate the primary cultured microglia were as a reference point. While each study suggested that coculture or media supplementation further drove the iMGs toward a more accurate microglial fate, additional comparative studies are needed to reveal which deficiencies were corrected and what adjustments can be made to the culture conditions to further improve the accuracy of iMG models.
Due to the rapid popularity of these models, methods have quickly begun to be refined and even commercialized in an effort to reduce variability and broaden access to these techniques. For example, McQuade and colleagues demonstrated that utilizing a commercial kit to generate CD43+ HPCs greatly simplified the initial paradigm presented by Abud et al. (2017), by eliminating the need for hypoxia and cell sorting (McQuade et al., 2018). Additionally, this simplified approach led to the production of 60-fold more HPCs and continued differentiation of these HPCs into iMGs, following the Abud et al. (2017) protocol, resulted in iMGs that were transcriptionally and functionally indistinguishable from cells generated with the original approach. In another example, human iMGs have recently been made available by Cellular Dynamics International, a FujiFilm company that specializes in commercializing iPSC derivatives.
Taken together, these recent advancements clearly demonstrate that the in vitro generation of large amounts of functional human iMGs has greatly diminished the prior bottleneck of acquiring human microglia. Yet, while the possibility of generating a vast quantity of iMGs in vitro is now well within the grasp of most laboratories with basic iPSC-culturing capabilities, the environmental dependencies of microglia and the limited studies comparing iMGs to in vivo reference microglia means the field has yet to develop a validated in vitro system that fully recapitulates the brain environment.
3 |. APPROXIMATING THE BRAIN ENVIRONMENT USING CEREBRAL ORGANOIDS
Researchers looking for ways to maintain the high-throughput nature of in vitro iMG experiments while also attempting to rectify the environment-dependent deficiencies of the iMG transcriptome and phenotype began to examine the potential use of iPSC-derived brain organoids (BORGs) (Lancaster et al., 2013). However, original descriptions of these “mini brains” pointed out that they were lacking microglia due to the ectodermal lineage of the neuroepithelium used to generate them, whereas microglia arise from mesoderm. In an attempt to remedy this, Abud et al. (2017) demonstrated that iMGs migrate to and engraft within BORGs when the two were maintained in a coculture system. Engrafted iMGs in turn adopted a more ramified morphology (Figure 3, oMG) than their purely in vitro counterparts. In addition, engrafted iMGs responded to cerebral damage in the form of a blunt force puncture injury induced by a needle by adopting ame-boid morphologies reminiscent of the “activated” microglia response typically observed in injured brain tissue (Karperien, Ahammer, & Jelinek, 2013). However, the parallel differentiation approach that is required to generate this model is both technically complex and expensive, and a single technique that would innately develop microglia within brain organoids could offer researchers a simpler model.
Interestingly, a recent study demonstrated that the original Lancaster protocol was perhaps capable of doing this all along (Ormel et al., 2018). In this report, organoid-grown microglia (oMGs) were shown to innately develop within BORGs, with only limited alterations to the original protocol published by Lancaster et al. (2013). This finding was predicated on a single-cell RNA sequencing study that demonstrated BORGs contained a cell population defined by markers of mesodermal lineage (Quadrato et al., 2017), strongly suggesting that the presence or absence of this population likely influences the development of microglia within the organoid. Ormel and colleagues were able to demonstrate that their BORGs inherently developed cells that expressed proteins for the myeloid transcription factor PU.1, the lyso-somal marker CD68, and the macrophage/microglia marker IBA1. Furthermore, the transcriptomic signature of oMGs was shown to resemble that of ex vivo human microglia, although the homeostatic microglia gene P2RY12 and the brain-specific gene TMEM119 were expressed at much lower levels in oMGs, suggesting some in vitro environmental effects may not have been fully rectified (Bohlen et al., 2017; Butovsky et al., 2014). Additionally, iPSCs and fibroblasts were included in the bioinformatic comparisons, whereas iMGs and primary cultured microglia were not, suggesting that a higher-resolution comparison is needed to determine whether oMGs exhibit a more “in vivo-like” signature than other in vitro iMG models.
Comparable to the in vitro studies before them, Ormel et al. went on to functionally validate oMGs. As BORGs have been shown to develop functional synapses, complete with neurotransmitter release and recordable electrical activity (Pasca et al., 2015), determining whether oMGs interacted with synapses in a manner consistent with previously described in vivo reports was critical (Schafer et al., 2012). Therefore, super resolution microscopy was employed and showed what appeared to be interactions between IBA1 and PSD95 puncta. While the authors stopped short of clearly demonstrating postsynaptic phagocytosis, which has been the subject of recent debate (Weinhard et al., 2018), the data presented appears consistent with the microglia-mediated pruning of synapses that occurs during murine brain development (Schafer et al., 2012). Additionally, oMGs were isolated from BORGs and subjected to further functional analyses. To test the inflammatory response capabilities of oMGs, isolated cells were exposed to LPS for up to 72 hr and significant increases in IL6 and TNFα were observed. Phagocytic capacity in the isolated cells via the C3-dependent complement pathway was also assesses by incubating the oMGs with iC3b-coated microbeads, demonstrating a rate of complement-dependent phagocytosis similar to that of primary human microglia.
Overall, the ability to generate a surrogate human brain environment in a dish that is populated with human iMGs along with human neurons, astrocytes, and neural progenitors, either endogenously or through coculture, is an exciting prospect. Ongoing studies aimed at producing BORGs that show more consistent ratios of the different cell types across both individual organoids, experiments, and labs, may well prove to be a fertile avenue for discovery.
4 |. TRANSPLANTATION OF IPSC-DERIVED MICROGLIA INTO THE MOUSE BRAIN
While organoids offer an approximation of the brain environment, the logical counterpart to this approach would be a model involving in vivo engraftment of human microglia into a fully developed mam-malian brain. Indeed, this was performed in one of the original iMG studies which delivered iMGs into the cortex and hippocampus of adult “MITRG” mice (Abud et al., 2017). The use of MITRG mice, which are deficient in T cells, B cells, and NK cells, but also humanized for colony stimulating factor (CSF)1, CSF2/IL3, and thrombopoietin, proved to be critical to the success of this application (Rongvaux et al., 2014). Importantly, immune deficiency alone is not sufficient for the engraftment and survival of human microglia, and little to no human microglia survive beyond 1–2 weeks when transplanted into traditional immune deficient mice that lack expression of human versions of either CSF1 or IL34, the two ligands for the CSF1 receptor (Hasselmann et al., 2019; Mathews et al., 2019). This finding is highly consistent with prior studies that show the importance of CSF1 signaling for microglia development and survival and that human monocytes fail to respond appropriately to murine CSF1 (Elmore et al., 2014; Erblich, Zhu, Etgen, Dobrenis, & Pollard, 2011; Rathinam et al., 2011; Sieff, 1987; Y. Wang et al., 2012).
Other recent studies have also used more traditional sources of hematopoietic cells, such as CD34+ cord blood cells, to induce microglial-like differentiation with some success (Capotondo et al., 2017; Mathews et al., 2019). However, cord blood cells include definitive hematopoietic stem cells and their downstream progenitors which differ from the yolk sac-derived primitive progenitors that give rise to microglia during development (Ginhoux et al., 2010). Thus, these cord blood-derived cells likely only partially recapitulate a microglia phenotype, perhaps better mimicking brain-infiltrating blood monocytes. In fact, these differences were demonstrated in another study that transplanted human blood monocytes, human fetal brain macrophages, and postnatal microglia into hCSF1 expressing, immune-deficient mice (Bennett et al., 2018). While the study demonstrated evidence of microglial-like morphology and marker expression following transplantation of each of these cell types, blood-derived human cells expressed the protein, MS4A7 at much higher levels than transplanted microglia. Furthermore, RNA-sequencing analysis of similarly derived murine cell transplants, revealed many additional ontogeny-dependent differences. Taken together, these studies demonstrate that while transplantation of definitive hematopoietic stem cells and peripherally-derived monocytes can result in the expression of several microglial markers, the ontogeny of the transplanted cells likely continues to influence the expression of many key genes, regardless of transplantation into the brain.
While these initial reports opened the door to xenotransplantation as a promising new approach to study human microglia in vivo, key questions remained. For example, does transplantation of human iMGs or their progenitors into the mouse brain induce the expression of an in vivo-like human transcriptome and the adoption of typical in vivo microglial behavior? This question has become particularly important given recent findings from Chris Glass and colleagues demonstrating that culturing of human brain-derived microglia leads to a rapid change in their transcriptome (Gosselin et al., 2017). To try to address this important finding and provide a more comprehensive and robust model of microglia xenotransplantation, Hasselmann et al. (2019) generated human, iPSC-derived primitive hematopoietic progenitor cells (iHPCs) and transplanted them into early postnatal MITRG mice. This combination of primitive microglial progenitors and young mouse pup recipients, in which developmental signaling cascades within the central nervous system (CNS) are still active (Ginhoux et al., 2010; Kierdorf et al., 2013; Schulz et al., 2012), resulted in robust forebrain engraftment of cells that acquired typical microglial surface markers and morphology (Hasselmann et al., 2019) (Figure 3, xMG). More importantly, transcriptomic analyses of the xenotransplanted microglia (xMGs) demonstrated that, by 2 months of age, these cells acquired an expression profile that closely resem-bled human, ex vivo microglia while correcting many of the transcriptomic deficits present in iMGs and cultured human microglia (Gosselin et al., 2017). Importantly, this chimeric approach has recently been validated by two other groups, demonstrating the widespread applicability and reproducibility of this model (Mancuso et al., 2019; Svoboda et al., 2019).
Additional functional studies by Hasselmann et al. (2019) further demonstrated that xMGs responded to peripheral LPS injections, by upregulating inflammatory genes such as CD45, SPP1, and MSR1, rapidly migrated toward the site of a laser ablation injury similarly to the study by Davalos et al. (2005), and reacted to repeated, mild closed-head injury (rmCHI; Gold et al., 2018) by phagocytosing cellular debris within the injury site (Hasselmann et al., 2019). Importantly, this study was also able to demonstrate an application that is unique to in vivo models, the xMG response to endogenously developed beta-amyloid plaque pathology, by employing a transgenic mouse model of AD that had been backcrossed with the MITRG model (5X-MITRG). This functional study revealed that xMGs actively migrated toward amyloid plaques, formed microglial barriers similar to those observed in human patients, and could be observed phagocytosing Aβ. Considering the wide variety of murine disease models already in use, the adoption of this methodology toward other mouse models clearly has potential applications in many active areas of neurological research.
5 |. DISEASE APPLICATIONS
In the short time since iMGs and their derivatives were first developed, they have already been utilized in conjunction with a wide range of disease model systems and a multitude of additional applications can be imagined that have yet to be demonstrated in the literature (Figure 4). Currently, the most prominent published studies have been performed in the context of neurodegenerative diseases and models of viral infections. The following section will focus on the results of these studies while also discussing possible future directions.
FIGURE 4.
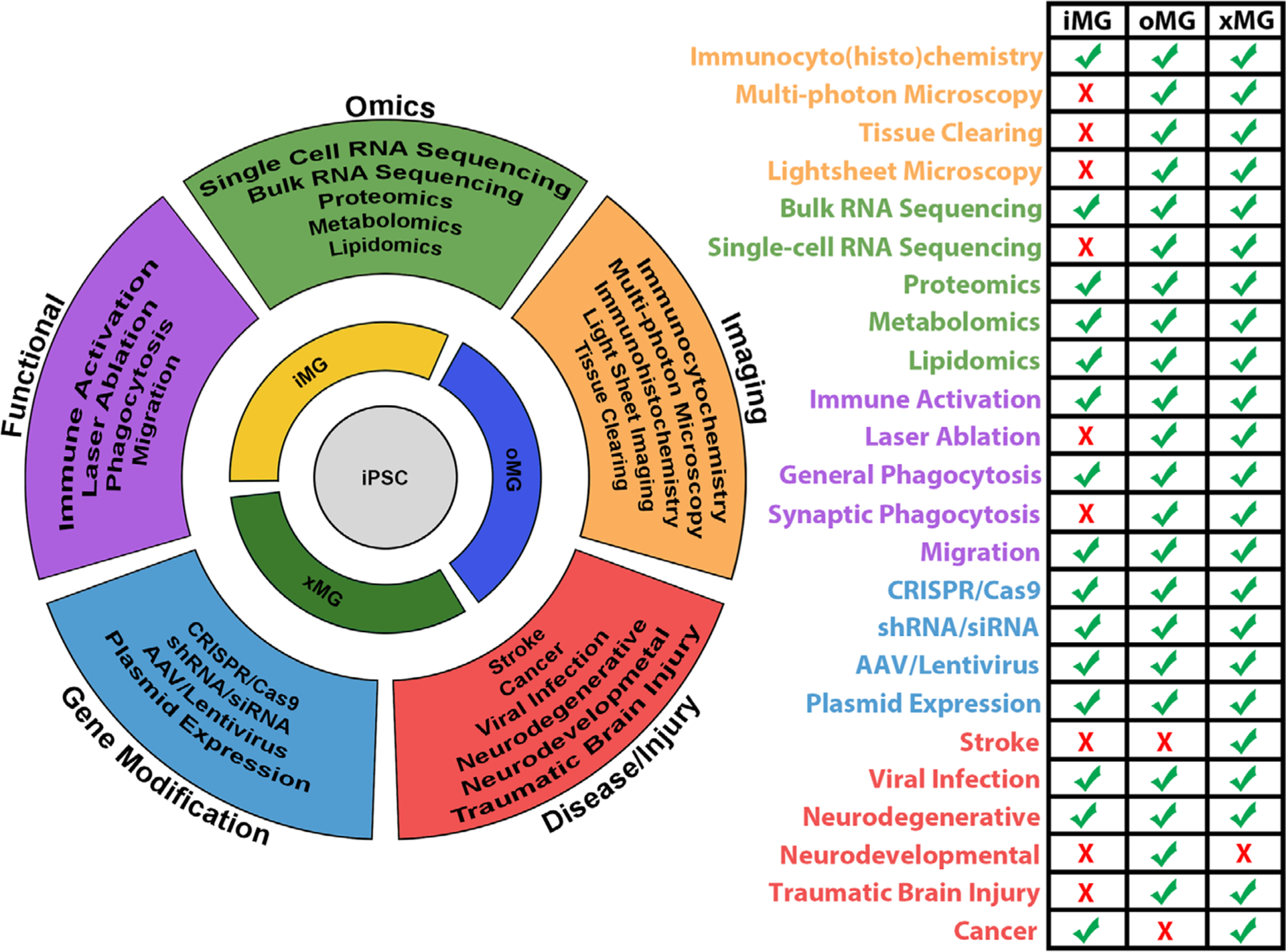
Current and potential applications of the iPSC-derived microglia toolset. It is possible to pair each iPSC-derived microglia technique with an array of research assays used to study various aspects of microglia biology. While some assays may be best suited for use in conjunction with a specific microglial technique, most can be readily applied to all three microglia techniques. This flexibility supports a robust research system in which studies can be conducted and validated across multiple experimental paradigms depending on a laboratory’s expertise and capabilities
5.1 |. Neurodegenerative diseases
5.1.1 |. Parkinson’s disease
One of the first applications of iMG technology to model human disease, examined the effects of early-onset PD mutations on microglia function (Haenseler, Zambon, et al., 2017). In this study, iMGs were generated from iPSC lines derived from familial PD patients that possessed either a triplication of the SNCA gene, which codes for the protein α-synuclein (α-syn), or the SNCA A53T mutation. Phagocytosis assays were then performed, demonstrating that iMGs could phagocytose either monomeric or fibrillar α-syn, but that this activity was significantly impaired following either endogenous overexpression of the protein, in the SCNA triplication mutants, or addition of monomeric α-syn to the culture medium. It was further shown that endocytosis of monomeric and fibrillar α-syn occurred via different mechanisms, with monomeric being taken up by actin-independent pathways and fibrillar being taken up via actin-dependent phagocytosis, providing unique molecular and genetic targets that could potentially be modified to enhance phagocytosis of one form of α-syn over another.
This same study also demonstrated that excess α-syn led to increased levels of the inflammatory cytokines, IL-18, and IL-22, and the chemokine CXCL1, although a more advanced activation state, marked by increased expression of TNFα, was not reported. This is in contrast to studies of microglia isolated from mouse models, in which a more robust activation state in response to α-syn has been described (Su, Federoff, & Maguire-Zeiss, 2009; Thome, Harms, Volpicelli-Daley, & Standaert, 2016), potentially indicating that these activation profiles can be attributed to factors beyond the direct microglial interactions with α-syn. This suggests that examination of iMGs in isolation from the additional inflammatory factors or responsive cells present within α-syn mouse models may be capable of offering a more targeted view of the direct microglial response to α-syn pathology.
These findings also provide an interesting step in furthering our understanding of how microglia respond to and clear excess α-syn. As our knowledge of these phagocytic processes, and the underlying genetics, improves, researchers may be able to use this information to generate targeted approaches that could enhance the clearance of oligomeric α-syn and/or Lewy bodies and potentially slow the progression of neuronal dysfunction and loss in patients suffering from PD and related synucleinopathies. As the cells used in this study have been described as being more reminiscent of macrophages rather than microglia, future studies combining this method with neuronal coculture systems, or even with a more recently developed organoid model of PD (Smits et al., 2019), will likely provide an interesting and informative continuation of this line of research. Additionally, development of xenotransplantation-compatible α-syn mice would provide opportunities to study how xMGs with varying genetic backgrounds respond to α-syn pathology and its downstream consequences over time. Ideally, this will offer a view into the early stages of α-syn clearance and related inflammatory responses which should aid in identifying the most effective targets for therapeutically modulating these activities and potentially slowing disease progression.
5.1.2 |. Alzheimer’s disease
As a multitude of microglial genes have recently been implicated in modulating AD risk (Efthymiou & Goate, 2017; Guerreiro, Wojtas, et al., 2013; Hansen, Hanson, & Sheng, 2018; Huang et al., 2017; Jansen et al., 2019; Jonsson et al., 2013; Kunkle et al., 2019; Lambert et al., 2013; Sims et al., 2017), studies that carefully analyze these risk genes will be critical for enhancing our understanding of AD pathogenesis. Therefore, it is not surprising that some of the first iMG studies examined functions related to AD neuropathology. For example, two studies utilized phagocytosis of AD-related substrates, including beta-amyloid, tau oligomers, and/or synaptosomes, as a means to functionally assess their iMGs and to demonstrate the applicability of this model to the study of this neurodegenerative disease (Abud et al., 2017; Takata et al., 2017). Interestingly, exposure to fibrillar beta amyloid, and to a lesser extent tau oligomers, was further shown to significantly alter the expression of several microglial AD risk genes, suggesting that at least some of these risk genes likely play a role in the microglial response to AD pathology (Abud et al., 2017).
More recent studies have further built upon these initial proof-of-concept experiments to begin to unravel the genetic underpinnings of this disease. In one elegant example, Lin et al., examined the effects of the APOE ε4 allele, the most significant AD risk gene (Farrer et al., 1997; Harold et al., 2009; Lambert et al., 2009), on the transcriptome and function of human microglia (Lin et al., 2018). Using CRISPR/Cas9 genome editing, an APOE3/3 iPSC line was modified to generate an isogenic APOE4/4 line. This alteration was shown to dysregulate over 1,000 genes while inhibiting the in vitro uptake of Aβ1–42 oligomers. These iMGs were then cocultured with BORGs, generated from APP overexpressing iPSCs, which, by 2 months of age, have been shown to exhibit diffuse amyloid pathology (Raja et al., 2016). Following 1 month of engraftment, APOE3/3 oMGs demonstrated an ability to significantly reduce the amount of amyloid within the BORG, while APOE4/4 oMGs were unable to perform the same degree of phagocytosis, mirroring the monoculture deficits. Finally, CRISPR/Cas9 was used to convert an APOE4/4 expressing iPSC line, generated from an AD-affected individual, to the APOE3/3 genotype, which in turn rectified the previously observed phagocytic deficit.
This important study was followed by experiments that examined another critical AD risk gene, triggering receptor expressed on myeloid cells-2 (TREM2) (Claes et al., 2019; Guerreiro, Bilgic, et al., 2013; Jonsson et al., 2013; Xiang et al., 2018). In one study by Xiang et al., mice possessing a heterozygous TREM2 R47H mutation, which, in humans, has been shown to confer almost the same level of risk as a single APOE ε4 allele (Guerreiro, Bilgic, et al., 2013; Jonsson et al., 2013), were shown to display altered splicing of the TREM2 mRNA transcript as well as a significant decreases in TREM2 mRNA and protein expression (Xiang et al., 2018). However, iMGs containing identical mutations to the human form of the TREM2 gene did not display reduced TREM2 expression or altered mRNA splicing patterns, highlighting the critical importance of using human models to study disease risk genes in order to enhance the translatability of results. Adding further support to this finding, Claes et al. (2019) demonstrated that, while iMGs possessing either a heterozygous or homozygous TREM2 deletion displayed phagocytic deficits, heterozygous R47H iMGs did not. This held true for both E. coli fragments, representing a general inflammatory substrate, as well as human amyloid plaques present in an in situ slice culture paradigm. Overall, these data lend further support to the importance of studying not only knockout models, but also human iMG models that faithfully recapitulate disease relevant mutations.
While these studies suggest that the R47H mutation may not induce obvious deficits to iMGs in vitro, a recent in vivo xMG study demonstrated that the TREM2 R47H mutation does indeed result in functional deficits. To demonstrate this, 5X-MITRG were transplanted with homozygous R47H iHPCs at P1 and subsequently aged for 9 months, resulting in robust amyloid pathology within the murine brain. While TREM2 protein expression was not shown to be reduced in these cells, there was a significant deficit in xMG migration toward amyloid plaques, although the overall plaque load did not appear to be altered (Hasselmann et al., 2019). This study went on to also describe the cellular and transcriptomic response of WT xMGs to amyloid pathology via single-cell RNA sequencing. This analysis revealed a transcriptomic expression pattern made up of hundreds of differentially expressed genes that were unique to the human xMGs, when compared to previously reported responses of endogenous mouse microglia (Kamphuis, Kooijman, Schetters, Orre, & Hol, 2016; Keren-Shaul et al., 2017; Krasemann et al., 2017; Yin et al., 2017). Additionally, two of the genes which have been implicated in AD, HLA-DRB1, and LGALS3 (Boza-Serrano et al., 2019; Lambert et al., 2013; Lu et al., 2017), were histologically validated in both xMGs and AD patient tissue, further confirming the validity of this approach. As a whole, these results further demonstrate the utility of the iMG toolset for the examination of AD and suggest that the study of AD-relevant risk genes in the human context is critical for understanding the underlying mechanisms that drive human disease while also being more accessible than ever.
5.1.3 |. Nasu–Hakola disease
While heterozygous TREM2 mutations have been linked to increased risk of AD, homozygous missense mutations in TREM2 or its binding partner TYRO protein tyrosine kinase binding protein (TYROBP), otherwise known as DNAX-activation protein 12 (DAP12), can result in Nasu-Hakola disease (NHD; Paloneva, Autti, Hakola, & Haltia, 1993). This disease initially presents with peripheral symptoms including bone cysts, but significant CNS symptoms are also present and characterized by widespread neurodegeneration and presenile dementia, beginning as early as the fourth decade of life (Paloneva et al., 1993).
Recently, two studies took up the challenge of elucidating the role of human microglia in this devastating disease by comparing iMGs generated from NHD patients that were homozygous for either the T66M or W50C mutations to iMGs generated from unaffected subjects carrying heterozygous nonpenetrant mutations or WT forms of TREM2 (Brownjohn et al., 2018; Garcia-Reitboeck et al., 2018). In both studies, there was a distinct loss of TREM2 localization to the cell membrane in the T66M and W50C mutants, while the heterozygous mutants did not display the same deficit. Furthermore, the c-terminal fragment (CTF) of TREM2, a peptide that is generated upon cleavage of membrane-bound TREM2 into soluble TREM2 (sTREM2), was decreased in a gene dose-dependent manner. This finding suggests that in addition to the loss of membrane expression, these NHD mutations also produce a deficiency in sTREM2 levels, a finding that correlates well with a previous report that examined sTRME2 levels within the CSF of NHD patients (Piccio et al., 2016).
Each of these studies went on to further test the functionality of iMGs by examining their response to a variety of substrates. In both cases, there was no observed differences in the inflammatory cytokine response to LPS, with all genotypes responding in a seemingly appropriate manner. There were also no differences observed in the rate of phagocytosis of E. coli or zymosan particles, suggesting that signaling through WT TREM2 is not required for the maintenance of these specific responses. However, exposure to two substrates that are thought to be more specific to TREM2 binding, acetylated low-density lipoprotein (AcLDL) and phosphatidylserine, which is flipped to the outer leaflet of the membrane in apoptotic cells, demonstrated substrate-specific deficits in phagocytosis. While phagocytosis of AcLDL was only slightly reduced in the homozygous mutants, phagocytosis of apoptotic cells showed significant decreases, in a gene-dose dependent manner, suggesting that these mutations may have a direct effect on proper debris clearance within the CNS. Furthermore, Garcia-Reitboeck and colleagues went on to show that not only does the T66M mutation negatively affect phagocytosis of apoptotic cells, it also induces a clear migratory deficit toward apoptotic cells, suggesting a two-fold deficiency in response to cellular debris. This study also demonstrated that homozygous TREM2 mutants display a significant increase in cell death when M-CSF was removed from the culture medium for 72 hr, which may suggest that mutant cells will have impaired survival under stressful conditions encountered in an inflammatory disease such as NHD.
It is important to note, however, that these two studies did not agree on all points. While Garcia-Reitboeck et al. showed a dose-dependent decrease in both mature and immature TREM2 protein, Brownjohn et al. reported that, while the mature protein decreased, this was balanced by an increase in immature protein. Additionally, the decreased protein levels that were observed by Garcia-Reitboeck were further supported by significant reductions in TREM2 mRNA, whereas this aspect of TREM2 expression was not discussed by Brownjohn. Localization of the TREM2 protein also presented another discrepancy between the two studies. Brownjohn showed that, while homozygous mutants were deficient in membrane localization of TREM2, there was still robust intracellular staining, indicative of a deficit in protein trafficking to the membrane. However, Garcia-Reitboeck showed that not only was membrane localization lost in homozygous mutants, but there was also minimal intracellular staining, suggesting that there was an underlying deficit in protein production, consistent with their observed reduction in TREM2 mRNA.
As such, determining if the observed phenotypes are generated due only to reduced membrane trafficking or due to dysfunctions in transcription will be critical in developing a better understanding or the nature of these mutations. Furthermore, future studies examining how these deficits manifest when mutant iMGs are maintained in a more complex environment such as brain organoids or chimeric models, will be crucial to better understanding the phagocytic and viability deficiencies induced by these mutations within the context of CNS disease and disfunction. Finally, as NHD is a relatively rare disease, and patient cells lines are not as abundant as heterozygous lines, this is a perfect opportunity to leverage the iPSC origin of these cells by utilizing CRISPR/Cas9 gene editing to generate additional experimental cell lines and compare these lines to AD-associated TREM2 mutations in an effort to better understand the similarities and differences between both of these devastating diseases.
5.2 |. CNS viral infection
iMG methods have also recently shown promise for investigating viral infections, especially those that are known to infect human immune cells with subsequent neural symptoms. In particular, researchers have begun to leverage iMGs in experimental applications aimed at studying both human immunodeficiency virus (HIV) and Zika virus (ZIKV).
5.2.1 |. Human immunodeficiency virus
While the processes surrounding HIV-1 infection have been well studied in the context of the peripheral immune system (Swanstrom & Coffin, 2012), invasion and infection in the CNS has been much more difficult to properly model, as the virus specifically infects human, but not murine immune cells. As microglia are the prominent resident immune cell type within the brain, iMG technologies represent a new approach that is beginning to advance research on HIV infection of the CNS. With the advent and widespread application of combined antiretroviral therapy, HIV/AIDS has become a far more treatable, albeit long-lasting chronic disease. However, growing evidence suggests that these effective treatments have in turn led to an increasing prevalence of HIV-associated neurocognitive disorders (HANDs) with as many as 30% of HIV patients exhibiting cognitive impairments (Portilla et al., 2019). Even in patients that do not exhibit cognitive impairment, recent studies have detected significant HIV-associated brain atrophy (Nir et al., 2019). One likely explanation for this finding is that antiretroviral therapies have limited brain penetrance, allowing microglia to serve as a reservoir for HIV infection within the brain (Ko et al., 2019). Interestingly, this line of research also ties into the above discussed role of TREM2 in AD, as HAND patients on antiretroviral therapy exhibit alterations in both TREM2 and beta-amyloid levels (Fields et al., 2018).
Yet until recently, studying the dynamics of HIV infection within the CNS have been extremely challenging because of the species selectivity of the virus. However, one study recently harnessed the promise of chimeric models, demonstrating that injection of CD34+ cord blood cells into the liver of irradiated, immune-deficient, humanized IL34 mice led to differentiation into perivascular macrophages and microglia-like cells (Mathews et al., 2019). This model subsequently allowed for the successful infection of these human cells with HIV following an intraperitoneal injection of the virus (Mathews et al., 2019). Transcriptomic analysis of the infected microglia further demonstrated several upregulated transcriptomic programs related to viral defense and pattern recognition, in the form of interferon response genes and toll-like receptors, while also revealing a significant increase in transcripts aligning to the HIV-1 genes gag, nef, and env, demonstrating robust viral replication within the human microglia. These results indicate that this approach has the potential to serve as a promising new model of neural HIV infection that could be used to research methods of clearing HIV from this understudied CNS viral reservoir.
However, it is important to discuss some caveats to this approach. Notably, the microglial engraftment was derived from intrahepatic transplantation of CD34+ hematopoietic stem progenitor cells (HSPCs), isolated from human cord blood, rather than intracerebroventricular transplantation of iHPCs. As discussed in Section 4 of this review, this approach has been shown to result in engrafted cells that do not completely recapitulate microglia (Capotondo et al., 2017). Furthermore, acquisition of cord blood to isolate the HSPCs requires access to human tissue which is not a simple process for many laboratories. Additionally, while the use of intrahepatic transplantation resulted in the engraftment of many peripheral immune cells, including CD3, CD4, and CD8 T cells, CD19 B cells, and CD14/CD16 monocytes, brain engraftment was dependent on irradiation of the mouse pups prior to transplantation (Mathews et al., 2019). Success of this approach suggests that the CNS-engrafted cells were derived from peripheral monocytes which gained access to the CNS due to damage to the blood brain barrier (BBB) that occurs following irradiation. This damage is concerning as it confounds the researcher’s ability to clearly identify the route of HIV infection in the CNS. Since HIV is known to infect peripheral immune cells (Swanstrom & Coffin, 2012) and the damage to the BBB could allow these cells to enter the CNS, it is possible that the infection of the engrafted “microglia” did not occur via a mechanism that is consistent with the human condition. While this by no means precludes the study of the effects of HIV infection in the CNS, it does present an important confound for studies aimed at treating or preventing the propagation of infection within the CNS. As Mathews et al. did not present any data examining whether additional peripheral immune cells were also present in the CNS, further studies may be needed to determine whether the route of infection resembles that of human patients. However, this could also be addressed with further adaptation of the Mathews model to incorporate transplantation of iHPCs directly into the brain. This would be a useful approach that would eliminate the need for cord blood collections and radiation while also reducing confounding variables related to ontogeny-dependent deficiencies and peripheral cell effects on the xMGs. Yet, this too comes with important caveats such as the lack of peripheral T cells inherent in the use of immune-deficient mice. Nevertheless, it is reasonable to expect that studies will continue to use and improve upon these methods to further investigate the role of microglia in HIV.
5.2.2 |. Zika virus
ZIKV is a member of the Flaviviridae family that also includes Dengue and West Nile viruses and can be transmitted to humans by mosquitos. ZIKV was initially described in 1947 but human cases were rare, with only 13 cases of mild symptoms, including a fever, aches, and rash, being reported between its discovery and 2007. However, from 2007 to 2016 a series of outbreaks that spread through multiple Pacific Island nations, both South and North America, and southeast Asia saw that number grow into the millions (Petersen, Jamieson, Powers, & Honein, 2016). As the number of cases grew, so did our understanding of a set of more serious effects related to ZIKV, including a subset of patients that developed transient paralytic symptoms resembling Guillain-Barré syndrome as well as apparent transmission from mother to fetus, causing fetal brain abnormalities that resulted in microcephaly, preterm birth, and death (Uncini, Shahrizaila, & Kuwabara, 2017). While the number of annual cases has been dramat-ically reduced since the peak of the outbreaks, we still understand little about how this disease is spread throughout the CNS or which cells serve as the viral reservoir within the body, both of which are questions that need to be answered in order to develop an effective treatment for the infection.
However, two studies have recently adapted iMG protocols to study the transmission of ZIKV between microglia and developing neurons in an attempt to address this question (Mesci et al., 2018; Muffat et al., 2016). In these studies, iMGs were found to be susceptible to ZIKV infection, but, while the virus was cytotoxic to iPSC-derived neurons, the iMGs were able to survive the infection. Interestingly, infected iMGs were also found to induce a limited interferon defense in response to ZIKV, resulting in a diminished capacity to clear the infection (Muffat et al., 2016), although this response seems to vary depending on the viral strain (Mesci et al., 2018). Infected iMGs were then cocultured with iPSC-derived neural progenitor cells (NPCs) or BORGs and, in both cases, the iMGs were capable of transferring the ZIKV infection into the NPCs or neurons of the BORG which induced neuronal apoptosis (Mesci et al., 2018) or a reduction in the size of the BORGs, a phenotype resembling microcephaly (Muffat et al., 2016). Interestingly, it was also shown that treatment with Sofosbuvir, an FDA approved drug for Hepatitis C infection, may be capable of reducing ZIKV transmission and cell death in NPCs (Mesci et al., 2018). While studies of ZIKV that utilize human microglia models are in their infancy, these initial reports suggest that iMG, oMG, and perhaps even xMG models will continue to inform and advance our understanding of this virus and its effect on the developing CNS.
6 |. BENEFITS AND LIMITATIONS
While each of the models discussed in this review have proven to be useful in multiple avenues of research, there are, of course, both advantages and limitations to each approach (Table 1) that should be carefully considered. The following section will briefly discuss potential pros and cons of each model, with the purpose of highlighting how the appropriate integration of these techniques into a comprehensive system may help to alleviate the deficiencies present in any single approach.
TABLE 1.
Benefits and limitations of each iPSC-derived microglia technique
| Cell type | Benefits | Limitations |
|---|---|---|
| iMGs | High throughput | Transcriptomic deficiencies related to culture environment |
| Enhanced experimental control | Limited opportunities to study interactions with other CNS cell types | |
| Easily modified via gene editing techniques | Lack of peripheral immune cell interactions | |
| oMGs | Transcriptomic profiles resembling fetal brain tissue | Transcriptomic profiles resembling fetal brain tissue |
| System allows for interactions with other CNS cell types | Lack of structural organization | |
| Matching genetic background between microglia and organoid tissue | High variability between batches of organoids | |
| Easily modified via gene editing techniques | Lack of peripheral immune cell interactions | |
| xMGs | xMGs age within an intact brain environment | Difficult to generate appropriate host models for disease studies |
| Transcriptomic profile resembles that of in vivo human microglia | Genetic background differs from the host organism | |
| xMGs can be transplanted into a variety of disease or injury models in a controlled manner | Presence of host microglia (unless ablated with CSF1R inhibitors or genetic methods) | |
| Easily modified via gene editing techniques | Lack of peripheral immune cell interactions |
6.1 |. In vitro iMGs
One of the key benefits to generating microglia from iPSCs is the exponential increase in cell number that can be achieved when compared to the traditional method of isolating microglia from human brain tissue. In general, over 100 million iMGs can be generated from a starting population of just 1 million iPSCs (McQuade et al., 2018). This improvement allows researchers to perform high throughput experiments that can be well controlled, as the increased cell yield means multiple control groups and technical replicates can be run. Furthermore, the ability to generate iMGs from any iPSC cell line means that increasing the number of biological replicates only requires differentiations of additional iPSC lines rather than acquiring brain tissue from multiple patients. Overall, these benefits underscore the potential of iMG modeling.
However, these important gains are not without drawbacks. Studies focused on the transcriptional deficits induced in primary human or mouse microglia upon maintenance outside of the brain environment have demonstrated rapid and significant changes to microglial gene expression (Bohlen et al., 2017; Gosselin et al., 2017) and many of these deficits are also observed in iMGs, such as limited expression of TMEM119 and SALL1. While many of these transcriptional deficiencies were shown to be corrected upon transplantation into the murine brain (Hasselmann et al., 2019), suggesting that iMGs possess the same transcriptomic potential as primary human microglia, the downstream effects of the altered gene expression profiles are not fully understood. This means that, whenever possible, in vitro results should be validated in a more complex model or human tissue.
Despite the transcriptomic alterations that occur in cultured microglia, we would be remiss not to discuss the fact that this may be a correctable problem and studies have already identified factors that can ameliorate some aspects of this deficiency. For example, some of the “activating” effects of the in vitro environment can be tempered upon the addition of TGFβ to the culture medium (Abud et al., 2017), as signaling through the TGFβ receptor has been shown to be critical for maintaining in vivo microglia in a homeostatic state (Butovsky et al., 2014). Additionally, Ben Barres’ group demonstrated that the removal of bovine serum and the addition of cholesterol to cultures of murine microglia aided in restoring the expression of the brain-specific microglia gene TMEM119 while reducing the “activated” signature (Bohlen et al., 2017). Furthermore, Abud et al. added CD200 and CX3CL1 to the culture medium in order to further mature their iMGs. This was predicated on studies that demonstrated that these markers aid in maintaining microglia in a resting state (Cardona et al., 2006; Hoek et al., 2000; Kierdorf & Prinz, 2013). However, it is possible that complete microglia maturation also requires additional factors or physical interactions with neurons and/or other CNS cell types. Indeed, coculturing of iMGs with neurons and astrocytes was capable of inducing significant alterations in both iMG morphology and transcriptomic profile (Abud et al., 2017; Haenseler, Sansom, et al., 2017; Muffat et al., 2016; Pandya et al., 2017; Takata et al., 2017). As a whole, these findings suggest that by continuing to identify the brain-specific factors that drive the microglia lineage, it may be possible to improve our modeling of in vivo phenotypes in a cell culture environment. Continued improvement in this area has the potential to greatly reduce both the cost and time requirements of microglia research.
In addition to the transcriptomic irregularities that occur in iMG monoculture experiments, the absence of the other CNS cells types is also a problem in and of itself. As endogenous microglia do not exist in isolation, it is difficult to determine whether iMG functionality in a dish is directly applicable to microglia function in the brain. While experiments focused on examining in vitro iMG functions such as migration, phagocytosis, and inflammatory responses represent straightforward and high-throughput paradigms that are easy to interpret, they neglect the reality of the brain environment in which microglia interact with neurons, astrocytes, oligodendrocytes, endothelial cells, and infiltrating peripheral immune cells. Growing evidence has demonstrated the considerable interplay between microglia and astrocyte signaling (Liddelow et al., 2017) and likewise the neuronal regulation of microglia activation state is well-established (Marinelli, Basilico, Marrone, & Ragozzino, 2019; Sheridan & Murphy, 2013). More recently, interactions between vascular endothelial cells and microglia have been implicated in aging, AD, and stroke (Dudvarski Stankovic, Teodorczyk, Ploen, Zipp, & Schmidt, 2016; Merlini et al., 2019; Yousef et al., 2019). Thus, monoculture experiments likely provide only an initial, albeit highly quantitative, assessment of microglial function. The additional application of coculture methods that combine iMGs with iPSC-neurons, astrocytes, endothelial cells, or other cell types will thus likely provide important additional information.
6.2 |. In situ oMGs
As an in vitro model, oMGs afford the experimenter similar benefits to iMGs in that experiments can be designed that can often be completed more rapidly than in vivo experiments while offering a substantial amount of experimental control. However, one of the greatest benefits of oMGs is the fact that one can model the complex interactions between human microglia and human forms of all the core cell types of the CNS within a three-dimensional environment. To add to this, oMGs and the cells making up the rest of the organoid can be generated from the same cell line and thus contain the same genetic background, which is not a benefit that is afforded with xMG applications. Furthermore, one does not need to be concerned with side-by-side engraftment of murine microglia which could provide compensatory or detrimental signaling, especially in the case of studies focused on genetic mutations. Overall, these characteristics of organoid research allows for increased control over some of the extrinsic factors that may affect microglial function.
However, certain considerations need to be taken into account before deciding which experimental paradigms are best addressed using oMGs. First, it has been noted that BORGs, and presumably oMGs within the BORGs, express a transcriptome more closely resembling that of the fetal brain (Camp et al., 2015), which presents both benefits and disadvantages. While this makes the model well suited for the study of neurodevelopmental disorders, such as autism (Mariani et al., 2015; P. Wang et al., 2017), schizophrenia (Ye et al., 2017), or ZIKA transmission (Muffat et al., 2018), it also presents problems when attempting to utilize BORGs and oMGs to study diseases that require a more mature brain environment, such as PD or AD. Ormel et al. (2018) also demonstrated that oMGs display a diminished expression levels of the key microglia genes P2RY12 and TMEM119, further suggesting that these cells have not completely obtained a mature, homeostatic signature. However, this does not mean that oMGs are entirely inappropriate for studying diseases of the developing brain, as it has been shown that the transcriptome may begin to mature as BORGs are aged (Pasca et al., 2015; Quadrato et al., 2017).
There is also a significant concern regarding the quantitative nature of BORG-based experiments. As the generation of BORGs is typically performed in a self-organizing manner, without the aid of external patterning cues, BORGs tend to lack a consistent structure and cellular makeup (Kadoshima et al., 2013; Lancaster et al., 2013; Quadrato et al., 2017). Furthermore, while BORGs from the same bioreactor appear transcriptionally similar, significant batch effects are introduced when comparing BORGs across bioreactors, even if they were generated during the same initial differentiation (Quadrato et al., 2017). Recent modifications to the protocol and application of bioengineering techniques have been shown to ameliorate this to an extent (Velasco et al., 2019), although this requires additional expertise and the presence of endogenous microglia has not yet been investigated in these newer models.
BORGs also lack vascularization and, since nutrient and oxygen distribution are dependent on diffusion through the tissue, the central mass of larger BORGs has a tendency to become necrotic as the tissue is aged (Kelava & Lancaster, 2016). As microglia are known to robustly respond to both hypoxic conditions and cell death (Davalos et al., 2005; Fumagalli, Perego, Pischiutta, Zanier, & De Simoni, 2015; Nimmerjahn et al., 2005; Sierra et al., 2016), the presence of a necrotic core is a serious confound for many oMG experiments. One attempt at rectifying these issues involves the xenotransplantation of BORGs into the immunodeficient mouse brain (Daviaud, Friedel, & Zou, 2018; Mansour et al., 2018). This approach resulted in BORGs that became vascularized, did not develop central necrosis, and proceeded to grow in size following transplantation. While it was shown that host murine microglia invaded the transplanted BORGs, these BORGs were not shown to possess endogenous human microglia prior to transplantation. Further studies examining the transplantation of BORGs containing oMGs and whether this would diminish the infiltration of murine microglia may elucidate a valuable method for combining the oMG and xMG paradigms.
6.3 |. In vivo xMGs
The greatest strength of the xMG approach is the ability to perform in vivo studies of patient-derived microglia in an intact disease model. This allows for the preservation of a disease-relevant polygenic profile paired with the ability of the microglia to age within the context of an intact brain environment. Furthermore, this approach exposes the transplanted microglia to a gradual and chronic disease course, which likely better recapitulates the human condition. As an additional benefit, transplantation has been shown to induce a transcriptomic signature that is highly reminiscent of in vivo human microglia and, perhaps, slightly more homeostatic than microglia isolated from disease-affected brain tissue samples (Hasselmann et al., 2019). This suggests that the results acquired from studies utilizing xMGs have improved odds of remaining relevant upon translation into human treatments.
However, while generating xMGs from a variety of patient iPSC lines represents a fairly straightforward procedure, there are currently very limited murine disease models that are compatible with xMG transplantation. While the use of MITRG, humanized-CSF1 (Jax # 017708), or humanized IL34 mice offers an appropriate environment to study homeostatic function, aging, cancer, viral infections, or acute injuries such as traumatic brain injury or stroke, these models do not provide an environment useful for the study of many neurodegenerative disorders. Indeed, only one transplant-compatible transgenic disease model has thus far been published, the 5X-MITRG, and development of that mouse was a substantial undertaking that required restoration of five genes to homozygosity and the incorporation of two additional disease-related human genes (Hasselmann et al., 2019). Therefore, a significant investment into the development of transplant-compatible murine disease models is required before the full power of this technique can be harnessed.
Even with increased availability of transplant-compatible mouse models, there are still concerns regarding the effects of the murine host cells on xMG functionality. One significant issue is that the host brain tissue possesses a different genome than that of the xMGs. As it has been demonstrated that a number of murine proteins possess limited homology with their human counterparts, the effects that these discrepancies have on cell-cell interactions and the activation or inhibition of microglia signaling cascades are currently unknown. Additionally, the presence of host murine microglia serves as another confounding variable. Although the forebrain regions where engraftment of xMGs was targeted routinely displayed ~80% engraftment of human microglia (Hasselmann et al., 2019), this was not consistent throughout the brain, as the midbrain and hindbrain exhibited far more limited engraftment. This caveat becomes especially relevant in the context of studying genetic mutations, as host murine microglia that do not possess the mutation may confound observations by exerting beneficial effects on pathology or xMGs and altering the overall inflammatory landscape of the brain. Therefore, generation of reliable experimental results may well be dependent on models that utilize chemical ablation of murine microglia prior to transplant (Elmore et al., 2014) or the use of a recently developed mouse model that lacks endogenous microglia (Rojo et al., 2019).
Additionally, the effects of host age on xMG phenotype has yet to be fully examined. Indeed, Hasselmann et al. showed that xMGs isolated from 2 to 6 months old mice appeared to cluster closest with the three youngest ex vivo human microglia samples (mean age 16.5 months), potentially suggesting a transcriptomic signature reminiscent of young microglia. However, subsequent activation of xMGs with LPS seemed drive expression of some of the deficient genes closer to the expression levels associated with microglia isolated from older patients. This raised the possibility that rather than presenting with an immature transcriptomic signature, xMGs isolated from healthy mouse brains presented with a “less-activated” phenotype than microglia that have spent 5–17 years in a diseased human brain. A study directly examining these two possibilities is required to more clearly elucidate the effects of aging on xMG maturation.
6.4 |. Characteristics shared across protocols
It is important to note that there are also drawbacks and benefits that apply to every one of the aforementioned methods. As all three of the discussed techniques involve cells derived from iPSCs, appropriate applications will always need to take into account sources of variation that are intrinsic to iPSC culture such as variation between clonal lines, the potential for karyotypic instability, and batch-dependent variation in differentiation. However, these variables can be greatly diminished through proper quality control and experimental design. Yet, there remain other important limitations shared across iMG, oMG, and xMG models such as the current inability to study the interactions between microglia and peripheral immune cells. While this could be crudely addressed in vitro by obtaining and coculturing human peripheral immune cells from the same patients that donated the iPSC cell lines, it is a complex process that may or may not yield physiologically relevant results. One potential solution to this problem would involve developing a method that allows for the generation of microglia and peripheral immune cells, or their progenitors, from the same cell line followed by engraftment of these cells into the relevant compartments of the same organism. The paradigms presented in Rongvaux et al. and Mathews et al. both reported the presence of peripheral immune cells following engraftment of HSPCs (Mathews et al., 2019; Rongvaux et al., 2014) suggesting that this idea is not so far-fetched. Further development of such a model would provide unprecedented opportunities to investigate the critical and understudied interactions between peripheral and central human immune cells.
A key benefit that relates to all three of these models is the fact that each of these cell types (iMGs, oMGs, and xMGs) are derived from iPSCs, meaning that the application of gene editing technologies, such as CRISPR/Cas9, is relatively straightforward. This capability means that researchers can obtain cell lines from either healthy control patients or disease-affected patients and directly target genes for genetic modification. This allows for the observation of the effects of a single genetic mutation in isolation or in conjunction with a relevant polygenic background. Furthermore, generation of such cell lines con-currently develops isogenic control lines, in which only the targeted gene differs. The incorporation of isogenic controls into experimental designs using iPSC-derived cells has been shown to be critical for the accurate interpretation of complex cellular responses by reducing variation between the experimental and control lines thereby increasing the experimenter ability to claim that the observed response was due to the mutation rather than background genetic variability (Engle, Blaha, & Kleiman, 2018). It is therefore reasonable to hope that continued progress in this direction will further uncover the influence of specific genes on microglial function and disease risk, leading perhaps to eventual applications in the development of individualized disease treatments.
Another important limitation is that there remain gaps in the field’s knowledge regarding precisely how we should define homeostatic human microglia. While current studies have utilized a variety of primary and ex vivo microglia as references to validate the transcriptomic signatures of iPSC-derived cells, there remain some concerns regarding these references. In the case of primary microglia, it has been clearly demonstrated that human and murine microglia that are maintained in vitro rapidly undergo transcriptomic changes when removed from the brain environment, exhibiting a diminished expression of many brain-specific genes while subsequently upregulating a genetic signature resembling an activated state (Bohlen et al., 2017; Butovsky et al., 2014; Gosselin et al., 2017). Furthermore, there is the overarching concern that microglia collected from human patients having originated from resections of diseased or postmortem tissue may not accurately represent a homeostatic human state. It is therefore important to consider these potential limitations when interpreting experiments and further reiterate the importance of validating novel findings in human tissue and other systems whenever possible.
7 |. CONCLUSION
Each technological advancement in the field of microglia research has yielded substantial increases in our understanding of these cells. We are currently in another period of technological growth and the tools at the disposal of today’s researchers are more bountiful than ever. With the recent development of robust methods to study iMGs, oMGs, and xMGs, researchers now have additional techniques that can be utilized to test new hypotheses or further validate and expand findings from murine or immortalized microglial models.
As we have highlighted in this review, there are a multitude of protocols in use and, depending on the study, there may be subtle differences introduced that could provide varying relevance to the human condition. However, we are currently in a state of flux as this relatively young set of protocols are being rapidly applied, independently reproduced, and improved upon. As such, it is important to begin considering not just which protocols to use, but how to combine initial high-throughput in vitro experiments with appropriate in situ or in vivo validation of promising findings in more complex models. Development and use of paradigms that utilize this time-tested approach of advancing promising in vitro findings onto more complex in vivo studies of human microglia, will likely produce more translational results.
ACKNOWLEDGMENTS
This work was supported by NIH grants DA048813, AG055524, AG016573, AG056303 (M.B.J.), and NIA Training Grant AG00096 (J.H.). The authors would like to also thank Dr. Grant McGregor, Dr. Matt Inlay, Amanda McQuade, and Morgan Coburn for contributing images and artwork. Graphics were also obtained from Les Laboratories Servier (smart.servier.com) and used in accordance with the Creative Commons Attribution 3.0 Unported license (https://creativecommons.org/licenses/by/3.0/).
Funding information
Foundation for the National Institutes of Health, Grant/Award Numbers: AG00096, AG016573, AG048099, AG055524, AG056303; Women’s Alzheimer’s Movement, Grant/Award Number: 01-2018
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, … Blurton-Jones M (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron, 94(2), 278–293 e279. 10.1016/j.neuron.2017.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, … Barres BA (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron, 98(6), 1170–1183. 10.1016/j.neuron.2018.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner C, Roy K, Linnartz B, Napoli I, & Neumann H (2010). Generation of microglial cells from mouse embryonic stem cells. Nature Protocols, 5(9), 1481–1494. 10.1038/nprot.2010.90 [DOI] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, & Barres BA (2017). Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron, 94(4), 759–773. 10.1016/j.neuron.2017.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boza-Serrano A, Ruiz R, Sanchez-Varo R, Garcia-Revilla J, Yang Y, Jimenez-Ferrer I, … Deierborg T (2019). Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathologica, 138, 251–273. 10.1007/s00401-019-02013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownjohn PW, Smith J, Solanki R, Lohmann E, Houlden H, Hardy J, … Livesey FJ (2018). Functional studies of missense TREM2 mutations in human stem cell-derived microglia. Stem Cell Reports, 10(4), 1294–1307. 10.1016/j.stemcr.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns TC, Li MD, Mehta S, Awad AJ, & Morgan AA (2015). Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: A systematic bioinformatics-based critique of preclinical models. European Journal of Pharmacology, 759, 101–117. 10.1016/j.ejphar.2015.03.021 [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, … Weiner HL (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature Neuroscience, 17(1), 131–143. 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, … Treutlein B (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proceedings of the National Academy of Sciences of the United States of America, 112(51), 15672–15677. 10.1073/pnas.1520760112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capotondo A, Milazzo R, Garcia-Manteiga JM, Cavalca E, Montepeloso A, Garrison BS, … Biffi A (2017). Intracerebroventricular delivery of hematopoietic progenitors results in rapid and robust engraftment of microglia-like cells. Science Advances, 3(12), e1701211 10.1126/sciadv.1701211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, … Ransohoff RM (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience, 9(7), 917–924. 10.1038/nn1715 [DOI] [PubMed] [Google Scholar]
- Claes C, Van Den Daele J, Boon R, Schouteden S, Colombo A, Monasor LS, … Verfaillie CM (2019). Human stem cell-derived monocytes and microglia-like cells reveal impaired amyloid plaque clearance upon heterozygous or homozygous loss of TREM2. Alzheimers Dement, 15(3), 453–464. 10.1016/j.jalz.2018.09.006 [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, … Gan WB (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience, 8(6), 752–758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- Daviaud N, Friedel RH, & Zou H (2018). Vascularization and engraftment of transplanted human cerebral organoids in mouse cortex. eNeuro, 5(6), ENEURO.0219–18.2018. 10.1523/ENEURO.0219-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Golde TE, & Lagier-Tourenne C (2018). Animal models of neurodegenerative diseases. Nature Neuroscience, 21(10), 1370–1379. 10.1038/s41593-018-0236-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, … Fossati V (2017). Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Reports, 8(6), 1516–1524. 10.1016/j.stemcr.2017.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, & Schmidt MHH (2016). Microglia-blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathologica, 131 (3), 347–363. 10.1007/s00401-015-1524-y [DOI] [PubMed] [Google Scholar]
- Efthymiou AG, & Goate AM (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Molecular Neurodegeneration, 12(1), 43 10.1186/s13024-017-0184-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, … Green KN (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82(2), 380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SJ, Blaha L, & Kleiman RJ (2018). Best practices for translational disease Modeling using human iPSC-derived neurons. Neuron, 100(4), 783–797. 10.1016/j.neuron.2018.10.033 [DOI] [PubMed] [Google Scholar]
- Erblich B, Zhu L, Etgen AM, Dobrenis K, & Pollard JW (2011). Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One, 6(10), e26317 10.1371/journal.pone.0026317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, … van Duijn CM (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta Analysis Consortium. JAMA, 278(16), 1349–1356 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9343467 [PubMed] [Google Scholar]
- Fields JA, Spencer B, Swinton M, Qvale EM, Marquine MJ, Alexeeva A, … Desplats P (2018). Alterations in brain TREM2 and amyloid-beta levels are associated with neurocognitive impairment in HIV-infected persons on antiretroviral therapy. Journal of Neurochemistry, 147(6), 784–802. 10.1111/jnc.14582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, … Hansen DV (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Reports, 22(3), 832–847. 10.1016/j.celrep.2017.12.066 [DOI] [PubMed] [Google Scholar]
- Fumagalli S, Perego C, Pischiutta F, Zanier ER, & De Simoni MG (2015). The ischemic environment drives microglia and macrophage function. Frontiers in Neurology, 6, 81 10.3389/fneur.2015.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, … Eggen BJL (2017). Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nature Neuroscience, 20(8), 1162–1171. 10.1038/nn.4597 [DOI] [PubMed] [Google Scholar]
- Garcia-Reitboeck P, Phillips A, Piers TM, Villegas-Llerena C, Butler M, Mallach A, … Pocock JM (2018). Human induced pluripotent stem cell-derived microglia-like cells Harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Reports, 24(9), 2300–2311. 10.1016/j.celrep.2018.07.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons HM, Smith AM, Teoh HH, Bergin PM, Mee EW, Faull RL, & Dragunow M (2011). Valproic acid induces microglial dysfunction, not apoptosis, in human glial cultures. Neurobiology of Disease, 41(1), 96–103. 10.1016/j.nbd.2010.08.024 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, … Merad M (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845. 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold EM, Vasilevko V, Hasselmann J, Tiefenthaler C, Hoa D, Ranawaka K, … Cummings BJ (2018). Repeated mild closed head injuries induce long-term White matter pathology and neuronal loss that are correlated with Behavioral deficits. ASN Neuro, 10, 1759091418781921 10.1177/1759091418781921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, … Glass CK (2017). An environment-dependent transcriptional network specifies human microglia identity. Science, 356(6344), eaal3222 10.1126/science.aal3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, … Emre M (2013). Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiology of Aging, 34(12), 2890 10.1016/j.neurobiolaging.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, … Alzheimer Genetic Analysis G (2013). TREM2 variants in Alzheimer’s disease. The New England Journal of Medicine, 368(2), 117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenseler W, & Rajendran L (2019). Concise review: Modeling neurodegenerative diseases with human pluripotent stem cell-derived microglia. Stem Cells, 37(6), 724–730. 10.1002/stem.2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, … Cowley SA (2017). A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Reports, 8(6), 1727–1742. 10.1016/j.stemcr.2017.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenseler W, Zambon F, Lee H, Vowles J, Rinaldi F, Duggal G, … Cowley SA (2017). Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Scientific Reports, 7(1), 9003 10.1038/s41598-017-09362-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, & Sheng M (2018). Microglia in Alzheimer’s disease. The Journal of Cell Biology, 217(2), 459–472. 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, … Williams J (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics, 41(10), 1088–1093. 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, … Blurton-Jones M (2019). Development of a chimeric model to study and manipulate human microglia in vivo. Neuron, 103, 1016–1033. 10.1016/j.neuron.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy LM, Perron G, Won SY, Rao VTS, Guiot MC, Moore C, … Antel JP (2018). Differential transcriptional response profiles in human myeloid cell populations. Clinical Immunology, 189, 63–74. 10.1016/j.clim.2016.04.006 [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, … Sedgwick JD (2000). Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science, 290(5497), 1768–1771. 10.1126/science.290.5497.1768 [DOI] [PubMed] [Google Scholar]
- Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, … Goate AM (2017). A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nature Neuroscience, 20(8), 1052–1061. 10.1038/nn.4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janabi N (2002). Selective inhibition of cyclooxygenase-2 expression by 15-deoxy-Delta(12,14)(12,14)-prostaglandin J(2) in activated human astrocytes, but not in human brain macrophages. Journal of Immunology, 168(9), 4747–4755. 10.4049/jimmunol.168.9.4747 [DOI] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, … Posthuma D (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nature Genetics, 51(3), 404–413. 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, … Lamb BT (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. The Journal of Experimental Medicine, 212(3), 287–295. 10.1084/jem.20142322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, … Stefansson K (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England Journal of Medicine, 368(2), 107–116. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, & Sasai Y (2013). Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proceedings of the National Academy of Sciences of the United States of America, 110(50), 20284–20289. 10.1073/pnas.1315710110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Schetters S, Orre M, & Hol EM (2016). Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochimica et Biophysica Acta, 1862(10), 1847–1860. 10.1016/j.bbadis.2016.07.007 [DOI] [PubMed] [Google Scholar]
- Karperien A, Ahammer H, & Jelinek HF (2013). Quantitating the sub-tleties of microglial morphology with fractal analysis. Frontiers in Cellular Neuroscience, 7, 3 10.3389/fncel.2013.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, & Lancaster MA (2016). Dishing out mini-brains: Current progress and future prospects in brain organoid research. Developmental Biology, 420(2), 199–209. 10.1016/j.ydbio.2016.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, … Amit I (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell, 169(7), 1276–1290 e1217. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, … Prinz M (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nature Neuroscience, 16(3), 273–280. 10.1038/nn.3318 [DOI] [PubMed] [Google Scholar]
- Kierdorf K, & Prinz M (2013). Factors regulating microglia activation. Frontiers in Cellular Neuroscience, 7, 44 10.3389/fncel.2013.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, & Kim WK (2019). Macrophages but not Astrocytes Harbor HIV DNA in the brains of HIV-1-infected Aviremic individuals on suppressive antiretroviral therapy. Journal of Neuroimmune Pharmacology, 14(1), 110–119. 10.1007/s11481-018-9809-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, … Butovsky O (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity, 47(3), 566–581 e569. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, … Pericak-Vance MA (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nature Genetics, 51(3), 414–430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, … Amouyel P (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics, 41(10), 1094–1099. 10.1038/ng.439 [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, … Amouyel P (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics, 45(12), 1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, … Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature, 501(7467), 373–379. 10.1038/nature12517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry RP, Jacobs VL, Romero-Sandoval EA, & DeLeo JA (2012). Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Experimental Neurology, 234(2), 340–350. 10.1016/j.expneurol.2011.11.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, … Barres BA (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, … Tsai LH (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron, 98(6), 1141–1154 e1147. 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu RC, Yang W, Tan L, Sun FR, Tan MS, Zhang W, … Tan L (2017). Association of HLA-DRB1 polymorphism with Alzheimer’s disease: A replication and meta-analysis. Oncotarget, 8(54), 93219–93226. 10.18632/oncotarget.21479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, … De Strooper B (2019). Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nature Neuroscience, 22(12), 2111–2116. 10.1038/s41593-019-0525-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour AA, Goncalves JT, Bloyd CW, Li H, Fernandes S, Quang D, … Gage FH (2018). An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology, 36(5), 432–441. 10.1038/nbt.4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, … Vaccarino FM (2015). FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism Spectrum disorders. Cell, 162(2), 375–390. 10.1016/j.cell.2015.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Basilico B, Marrone MC, & Ragozzino D (2019). Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Seminars in Cell & Developmental Biology, 94, 138–151. 10.1016/j.semcdb.2019.05.017 [DOI] [PubMed] [Google Scholar]
- Mathews S, Branch Woods A, Katano I, Makarov E, Thomas MB, Gendelman HE, … Gorantla S (2019). Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Molecular Neurodegeneration, 14(1), 12 10.1186/s13024-019-0311-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, & Blurton-Jones M (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Molecular Neurodegeneration, 13(1), 67 10.1186/s13024-018-0297-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, … Akassoglou K (2019). Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron, 101(6), 1099–1108 e1096. 10.1016/j.neuron.2019.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesci P, Macia A, LaRock CN, Tejwani L, Fernandes IR, Suarez NA, … Muotri AR (2018). Modeling neuro-immune interactions during Zika virus infection. Human Molecular Genetics, 27(1), 41–52. 10.1093/hmg/ddx382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Li Y, Omer A, Durbin A, Bosch I, Bakiasi G, … Jaenisch R (2018). Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to Zika and dengue infections. Proceedings of the National Academy of Sciences of the United States of America, 115(27), 7117–7122. 10.1073/pnas.1719266115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, … Jaenisch R (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nature Medicine, 22(11), 1358–1367. 10.1038/nm.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, & Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308(5726), 1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Nir TM, Jahanshad N, Ching CRK, Cohen RA, Harezlak J, Schifitto G, … Consortium HIVN (2019). Progressive brain atrophy in chronically infected and treated HIV+ individuals. Journal of Neurovirology, 25, 342–353. 10.1007/s13365-019-00723-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormel PR, Vieira de Sa R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer MAM, … Pasterkamp RJ (2018). Microglia innately develop within cerebral organoids. Nature Communications, 9(1), 4167 10.1038/s41467-018-06684-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Hakola P, & Haltia MJ (1993). Polycystic Lipomembranous Osteodysplasia with Sclerosing leu-koencephalopathy (PLOSL) In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews([R]). Seattle, WA: University of Washington. [Google Scholar]
- Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, … Park JK (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nature Neuroscience, 20(5), 753–759. 10.1038/nn.4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, … Pasca SP (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nature Methods, 12(7), 671–678. 10.1038/nmeth.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen LR, Jamieson DJ, Powers AM, & Honein MA (2016). Zika Virus. The New England Journal of Medicine, 374(16), 1552–1563. 10.1056/NEJMra1602113 [DOI] [PubMed] [Google Scholar]
- Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, … Cruchaga C (2016). Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathologica, 131(6), 925–933. 10.1007/s00401-016-1533-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, & Piers TM (2018). Modelling microglial function with induced pluripotent stem cells: An update. Nature Reviews. Neuroscience, 19(8), 445–452. 10.1038/s41583-018-0030-3 [DOI] [PubMed] [Google Scholar]
- Portilla I, Reus S, Leon R, van-der Hofstadt C, Sanchez J, Lopez N, … Portilla J (2019). Neurocognitive Impairment in Well-Controlled HIV-Infected Patients. A Cross-Sectional Study. AIDS Res Hum Retroviruses, 35(7), 634–641. 10.1089/AID.2018.0279 [DOI] [PubMed] [Google Scholar]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, … Arlotta P (2017). Cell diversity and network dynamics in photosensitive human brain organoids. Nature, 545(7652), 48–53. 10.1038/nature22047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, & Tsai LH (2016). Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One, 11(9), e0161969 10.1371/journal.pone.0161969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, … Flavell RA (2011). Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood, 118(11), 3119–3128. 10.1182/blood-2010-12-326926 [DOI] [PubMed] [Google Scholar]
- Río-Hortega P (1919a). El “tercer elemento” de los centros nerviosos: La microglia en estado normal. Bol Soc Esp Biol VIII: 67–82. [Google Scholar]
- Río-Hortega P (1919b). El “tercer elemento de los centros nerviosos”. II. Intervención de la microglía en los procesos patológicos (células en bastoncito y cuerpos gránuloadiposos). Bol Soc Esp Biol VIII: 91–103. [Google Scholar]
- Río-Hortega P (1919c). El “tercer elemento” de los centros nerviosos. III. Naturaleza probable de la microglía. Bol Soc Esp Biol VIII: 108–115. [Google Scholar]
- Río-Hortega P (1919d). El “tercer elemento de los centros nerviosos”. IV. Poder fagocitario y movilidad de la microglía. Bol Soc Esp Biol VIII: 154–166. [Google Scholar]
- Río-Hortega P (1939). The microglia. The Lancet, 233(6036), 1023–1026. 10.1016/S0140-6736(00)60571-8 [DOI] [Google Scholar]
- Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, … Pridans C (2019). Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nature Communications, 10(1), 3215 10.1038/s41467-019-11053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, … Flavell RA (2014). Development and function of human innate immune cells in a humanized mouse model. Nature Biotechnology, 32(4), 364–372. 10.1038/nbt.2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, … Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74 (4), 691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, … Geissmann F (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science, 336 (6077), 86–90. 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- Sheridan GK, & Murphy KJ (2013). Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take Centre stage. Open Biology, 3 (12), 130181 10.1098/rsob.130181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieff CA (1987). Hematopoietic growth factors. The Journal of Clinical Investigation, 79(6), 1549–1557. 10.1172/JCI112988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, de Castro F, Del Rio-Hortega J, Rafael Iglesias-Rozas J, Garrosa M, & Kettenmann H (2016). The “big-bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia, 64(11), 1801–1840. 10.1002/glia.23046 [DOI] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, … Schellenberg GD (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nature Genetics, 49(9), 1373–1384. 10.1038/ng.3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, & Dragunow M (2014). The human side of microglia. Trends in Neurosciences, 37(3), 125–135. 10.1016/j.tins.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Smits LM, Reinhardt L, Reinhardt P, Glatza M, Monzel AS, Stanslowsky N, … Schwamborn JC (2019). Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinson’s Disease, 5, 5 10.1038/s41531-019-0078-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Federoff HJ, & Maguire-Zeiss KA (2009). Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotoxicity Research, 16(3), 238–254. 10.1007/s12640-009-9053-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda DS, Barrasa MI, Shu J, Rietjens R, Zhang S, Mitalipova M, … Jaenisch R (2019). Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proceedings of the National Academy of Sciences of the United States of America, 116(50), 25293–25303. 10.1073/pnas.1913541116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R, & Coffin J (2012). HIV-1 pathogenesis: The virus. Cold Spring Harbor Perspectives in Medicine, 2(12), a007443 10.1101/cshperspect.a007443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, … Ginhoux F (2017). Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for Modeling tissue-resident macrophage differentiation and function. Immunity, 47(1), 183–198. 10.1016/j.immuni.2017.06.017 [DOI] [PubMed] [Google Scholar]
- Thome AD, Harms AS, Volpicelli-Daley LA, & Standaert DG (2016). microRNA-155 regulates alpha-Synuclein-induced inflammatory responses in models of Parkinson disease. The Journal of Neuroscience, 36(8), 2383–2390. 10.1523/JNEUROSCI.3900-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman R, Burm SM, & Bajramovic JJ (2018). An overview of in vitro methods to study microglia. Frontiers in Cellular Neuroscience, 12, 242 10.3389/fncel.2018.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Lecours C, Samson L, Sanchez-Zafra V, & Sierra A (2015). From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Frontiers in Neuroanatomy, 9, 45 10.3389/fnana.2015.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya T, Park KC, Toyonaga S, Yamada SM, Nakabayashi H, Nakai E, … Shimizu K (2005). Characterization of microglia induced from mouse embryonic stem cells and their migration into the brain parenchyma. Journal of Neuroimmunology, 160(1–2), 210–218. 10.1016/j.jneuroim.2004.10.025 [DOI] [PubMed] [Google Scholar]
- Ueda Y, Gullipalli D, & Song WC (2016). Modeling complement-driven diseases in transgenic mice: Values and limitations. Immunobiology, 221(10), 1080–1090. 10.1016/j.imbio.2016.06.007 [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, … Holtzman DM (2014). Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Molecular Neurodegeneration, 9, 20 10.1186/1750-1326-9-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uncini A, Shahrizaila N, & Kuwabara S (2017). Zika virus infection and Guillain-Barre syndrome: A review focused on clinical and electrophys-iological subtypes. Journal of Neurology, Neurosurgery, and Psychiatry, 88(3), 266–271. 10.1136/jnnp-2016-314310 [DOI] [PubMed] [Google Scholar]
- Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, … Arlotta P (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature, 570, 523–527. 10.1038/s41586-019-1289-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, & Lachman HM (2017). CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Molecular Autism, 8, 11 10.1186/s13229-017-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, … Colonna M (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell, 160(6), 1061–1071. 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, … Colonna M (2012). IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nature Immunology, 13(8), 753–760. 10.1038/ni.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, … Gross CT (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9(1), 1228 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, & Kettenmann H (2017). Microglia in physiology and disease. Annual Review of Physiology, 79, 619–643. 10.1146/annurev-physiol-022516-034406 [DOI] [PubMed] [Google Scholar]
- Xiang X, Piers TM, Wefers B, Zhu K, Mallach A, Brunner B, … Haass C (2018). The Trem2 R47H Alzheimer’s risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Molecular Neurodegeneration, 13(1), 49 10.1186/s13024-018-0280-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Kang E, Yu C, Qian X, Jacob F, Yu C, … Zhang M (2017). DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron, 96(5), 1041–1054 e1045. 10.1016/j.neuron.2017.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, … Boddeke E (2017). Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer’s disease. Neurobiology of Aging, 55, 115–122. 10.1016/j.neurobiolaging.2017.03.021 [DOI] [PubMed] [Google Scholar]
- Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, … Wyss-Coray T (2019). Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nature Medicine, 25(6), 988–1000. 10.1038/s41591-019-0440-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, … Grutzendler J (2016). TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron, 90(4), 724–739. 10.1016/j.neuron.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]