

Summary
Many bacteria resist invasive DNA by incorporating sequences into CRISPR loci, which enable sequence-specific degradation. CRISPR systems have been well-studied from isolate genomes, but culture-independent metagenomics provides a new window into their diversity. We profiled CRISPR loci and cas genes in the body-wide human microbiome using 2,355 metagenomes, yielding functional and taxonomic profiles for 2.9 million spacers by aligning the spacer content to each sample’s metagenome and corresponding gene families. Spacer and repeat profiles agree qualitatively with those from isolate genomes but expand their diversity by approximately 13-fold, with the highest spacer load present in the oral microbiome. The taxonomy of spacer sequences parallels that of their source community, with functional targets enriched for viral elements. When coupled with cas gene systems, CRISPR–Cas subtypes are highly site- and taxon-specific. Our analysis provides a comprehensive collection of natural CRISPR–cas loci and targets in the human microbiome.
Keywords: metagenomics, CRISPR-Cas, CRISPR system, vial defense, bacteriophages
eTOC
In this study, Münch et al. carried out a taxonomic and functional characterisation of CRISPR systems in 2,355 human microbiomes. Together with quantification of cas gene abundance, this informs the potential roles of CRISPR/Cas systems and their targets, as well as evolutionary properties and principles of bacteria-virus relationships.
Graphical Abstract
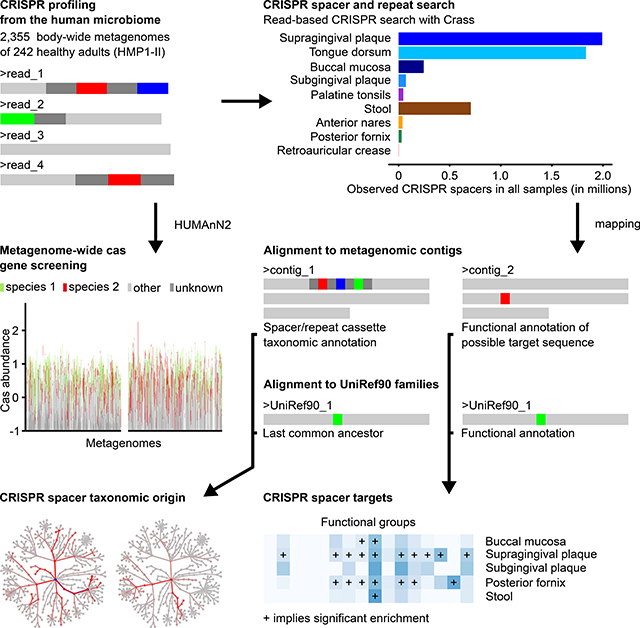
Introduction
Bacteriophages are one of the most abundant entities in our biosphere. To prevent infection by bacteriophages, 40% (Godde and Bickerton, 2006; Kunin et al., 2007) of sequenced bacterial species and most archaea possess Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), which together with CRISPR-associated (cas) genes form a defense system against foreign DNA (van der Oost et al., 2009). Such CRISPR loci are mosaics of a short repeat unit and multiple unique spacer sequences, which they acquire continuously as part of their defense strategy. Upon phage exposure, genomic fragments of 24–48 nucleotides in length are incorporated proximal to the leader end of the CRISPR array as spacers (Kunin et al., 2007). These spacer regions are transcribed and processed into CRISPR RNAs (crRNAs), which together with cas gene products such as the DNA exonuclease Cas9 recognise and subsequently cleave complementary nucleic acid sequences, called protospacers (Hatoum-Aslan and Marraffini, 2014), thus providing microbes with a molecular “immune” system (Horvath and Barrangou, 2010).
While this general mechanism of CRISPR inference has been extensively studied(Brouns et al., 2008; Karginov and Hannon, 2010), both the spacers and repeats captured by diverse microbes, and the diversity of associated cas genes suggest a largely unexplored range of CRISPR systems (Crawley et al., 2018). Independently of the spacer/repeat systems, associated genes comprise up to 65 different proteins, which can be classified into dozens of families (Makarova et al., 2011a). Two of these, Cas1 and Cas2, are highly conserved, while others vary greatly between organisms (Deveau et al., 2010; Horvath and Barrangou, 2010). All known active CRISPR–Cas systems contain Cas1 and Cas2, which coordinate spacer integration into the repeat cassette (Makarova et al., 2011a, 2020), while the other proteins cluster into three system types. The type I system includes Cas3 and the RAMP superfamily (encompasses Cas5 and Cas6), while the bacterial type II system includes Cas9 and the type III Cas10, which occurs in bacteria and archaea (Makarova et al., 2011a). Subsystems are classified based on these proteins, e.g. Cas12 for type V and Cas13 for type VI (Makarova et al., 2020). Thus, one potential driver of CRISPR subtype differentiation is in the architecture of captured sequences’ associated protein machinery, which might differ both in its phylogeny and in its potential ecological associations (e.g. among human body habitats).
In addition to the functions carried out by cas gene products, the adaptive memory itself is stored in the form of spacer sequences (surrounded by repeats), and has been studied as a record of microbes’ encounters with foreign DNA and RNA (Gogleva et al., 2014; Horvath et al., 2009; Shmakov et al., 2017; Stern et al., 2012; Vatanen et al., 2019). Such studies have confirmed, for example, that a large fraction of protospacers were found on phage and prophage genomes, which is in line with a main CRISPR function directed at defense against viral and mobile genetic elements (MGE). However, self-targeting spacers were found in ~18% of CRISPR-encoding organisms, which implies that the CRISPR–Cas system may also have a regulatory role (Stern et al., 2010) or cause detrimental “autoimmune” reactions, due to accidental incorporation of self sequences. Furthermore, anti-CRISPR systems could be chromosomally encoded to limit self-targeting effects (Rauch et al., 2017; Wimmer and Beisel, 2019). However, no potential targets can be identified for a large fraction of spacer sequences using current databases, leaving the question of the function and origin of the CRISPR “dark matter” (Shmakov et al., 2017).
Previous studies of microbial cas gene diversity and their accompanying CRISPR arrays and spacer sequences are mostly based on genomic isolates (Grissa et al., 2007; Makarova et al., 2015). These may not provide an accurate view of this important regulatory system’s distribution in microbial communities, particularly those of the human microbiome, due to their restriction to cultivable organisms. Furthermore, spacers may be lost during cultivation (Lopez-Sanchez et al., 2012) or assembly (Skennerton et al., 2013), thus potentially altering the observed CRISPR locus configuration. Cultivation-independent approaches, such as metagenomics (Quince et al., 2017), allow for a more comprehensive characterisation of microbial CRISPR systems across the range of taxa from a particular ecosystem as well as their phage counterparts, although this presents its own methodological challenges (Burstein et al., 2016; Sun et al., 2016).
Previous metagenomic studies of CRISPR locus distribution in the human microbiome focused mostly on streptococci in the oral environment (Naidu et al., 2014; Pride et al., 2011, 2012), finding that CRISPR spacers, like viruses, are even more subject-specific than the bacterial composition of the microbiome. Another study recovered 3,545 unique CRISPR spacers from gut metagenomic assemblies (Gogleva et al., 2014); here, spacers with matches in their paired metagenomes tended to occur at a proximal location in the CRISPR cassettes, indicating relatively recent acquisition. Since this spacer collection relied on metagenomic assemblies, it can easily miss many CRISPR arrays, since their repetitive structure is difficult to assemble de novo (Skennerton et al., 2013). In part to overcome this limitation, another study used targeted assembly instead to uncover 7,815 total CRISPR spacers. This collection again confirmed the site- and subject-specificity of spacer sequences, but was conversely limited to the set of 150 pre-selected CRISPR loci (Rho et al., 2012) and thus not intended to survey total CRISPR diversity. In addition to these challenges in characterising community spacer diversity, it can also be difficult to identify the targets of these spacers, since most studies rely on external databases to search for putative protospacers. Shotgun metagenomics in principle provides the opportunity to identify CRISPR elements comprehensively, while also searching the same samples for potential target sequences from community intrinsic viruses or mobile genetic elements.
To address these gaps, we present here a comprehensive taxonomic and functional characterisation of natural CRISPR–Cas systems in the human microbiome including spacers, repeats, cas genes, and their putative targets. We identified over 2.9 million unique CRISPR spacers, of which 98.63% are not present in data repositories (corresponding to a 13-fold increase compared to CRISPRCasdb) (Pourcel et al., 2020), with virus-associated proteins as one of the most targeted functional groups. We further quantified over 9,038 cas gene variants distributed across all 13 members (cas1-13) of the family, carried by a variety of subject- and site-specific taxa. Our CRISPR–Cas system and protospacer collection thus provides a map of potential microbe-phage interactions in the human microbiome and represents a useful resource for further in-depth studies of the functions and taxa associated with this intricate biological system.
Results
Quality of HMP-derived CRISPR cassettes and length-dependent sequence regularities
To first focus on CRISPR spacer and repeat sequences in the human microbiome, we screened all 2,355 metagenomes from the HMP1-II using Crass (Skennerton et al., 2013) (Supplementary Datasets S1-S2). We also determined the presence and abundances of Cas proteins for all samples using the HMP Unified Metabolic Analysis Network (HUMAnN2) (Franzosa et al., 2018) (Table S3) and examined their taxonomic provenance and co-occurrence across samples and body sites, detailed later.
We confirmed the validity of the spacer and repeat sets by four kinds of quality controls: (i) using comparisons of the nucleotide composition to CRISPR loci published in CRISPRCasdb, (ii) by kmer-based analysis of the repeat stability within and between samples, (iii) by quantification of singleton spacers, and (iv) direct sequence comparison using global alignments (STAR Methods). Interestingly, analysis of the nucleotide distribution per relative position in the spacer sequence uncovered several general trends. These included a symmetric pattern at the spacer centre, with a peak of C/G at the first quarter and third quarter of sequence length, and an overrepresentation of thymine at the beginning and guanine at the spacer end (Fig. 1B). The mean GC content of spacer sequence was highly similar between HMP and CRISPRCasdb spacers (47% GC and 48% GC, respectively). On repeat sequences, a consistent inverse-symmetric pattern to the relative sequence center was found on both datasets (Fig. S1B).
Figure 1: High consistency and agreement of spacer sequences from HMP to public databases and presence of a length-specific GC bias.

A) Sequence lengths of spacers were largely consistent between the minimum of 28 nucleotides and a tail permitted up to 43 nucleotides over different body areas. B) HMP spacers were highly similar to CRISPRCasdb spacers in position-wise nucleotide composition normalised by spacer length and showed a palindromic pattern in both datasets. C) Nucleotide composition stratified by spacer length showed a consistent pattern for HMP- and CRISPRCasdb-derived spacer sequences D) Stability of repeat sequences (as measured by Bray–Curtis dissimilarity of k-mer counts of repeat sequences) across (i) technical replicates, (ii) samples taken from the same individuals over time and (iii) between individuals randomly selected individuals, respectively. Samples containing fewer than 25 repeats are not shown. E) HMP samples generally contain few CRISPR repeats that are sample-specific (singleton repeats). Histogram shows the proportion of singleton repeats among all repeats per sample for all samples.
To compare HMP and CRISPRCasdb-derived spacer composition, we binned the relative nucleotide abundance per relative position and compared the mean values of each bin; these were generally well-correlated (Pearson’s ρ: A = 0.67, C = 0.67, G = 0.74, T = 0.74, Fig. S3A). To more deeply assess the similarity of the two data sets, we stratified the compositional profiles by spacer length and calculated mean position-wise relative nucleotide frequencies binned by spacer length (Fig. 2C). This showed that especially longer spacers (> 34 nt) were differentially enriched for A/T nucleotides, and that this overall reduction of G/C content was dependent on spacer length. Again, overall nucleotide frequencies of HMP and CRISPRCasdb spacers were strongly correlated (Pearson’s ρ: A = 0.66; C = 0.68; G = 0.70, T = 0.70, Fig. S3B).
Figure 2: High body-site dependent differences in spacer loads (regardless of host or target) on the HMP1-II dataset.

A) Three oral associated body sites, supra- and subgingival plaque and tongue, have significantly increased CRISPR spacer counts (Wilcoxon rank sum test on spacer counts, P < 10−6) relative to other body sites, such as the urogenital and skin microbiota. Mean values (points for spacers and triangles for repeats) and SD (lines) of the read-depth normalised load per body site are shown for observed reads and repeat and for cluster representatives to account for repetitive sequences. B) The lengths of observed CRISPR spacer and repeat sequences are consistent between most body sites, especially between gut and oral samples, but different from the spacers and repeats present in CRISPRCasdb. Mean (points and triangles) and SD (line) sizes of the spacer and repeat sequences across body sites and within CRISPRCasdb (grey). C) Correlation of species richness (number of species exceeding 1% RA) and spacer load (cluster representatives, defined as the longest sequence within a cluster of > 80% of sequence identity) of selected samples.
We quantified similarity in repeat structure among pairs of HMP samples as the Bray-Curtis distance over their respective repeat 5-mer profiles. Individuals’ longitudinal samples tended to be stable over time, and spacers found in technical replicates were highly similar (both relative to samples drawn from different individuals; Fig. 2D). More specifically, stability was highest (BC distances were smaller) for technical replicates (mean BC = 0.24 ± 0.14) and significantly greater than longitudinal stability within individuals (mean BC = 0.28 ± 0.10, P < 10−6, two-sided Wilcoxon rank sum test). Stability was lowest for randomly chosen sample pairs and markedly lower than samples taken from the same individual (mean BC = 0.41 ± 0.18, P < 10−13).
Similarly, the co-occurrence of most repeats across samples further supported the validity of the CRISPR collection. Overall, the number of singleton repeats, defined here as repeat clusters of size one across the whole HMP1-II repeat set, was reasonably low (3.8% of all repeats) with an average 6% of singleton repeats per sample (Fig. 1E). This is expected, as valid CRISPR repeats should co-occur in multiple samples with similar taxonomic profiles, since they tend to be species-specific. Most singleton clusters were found in the less deeply sampled and less diverse urogenital and skin body sites (where 18% and 17% of all clusters were singletons, respectively), while singleton clusters in the gut and oral body sites were comparatively less common (7% and 4% of clusters, respectively).
CRISPR spacer loads and sequences differ across human body habitats
On average, we identified 43.1 ± 44.1 (mean ± s.d.) spacer sequences per million reads (CPM) (2,361 ± 2,808 spacer sequences per metagenomic sample) and 4.7 ± 5.0 CPM (in total 225 ± 222) repeat sequences across all body areas (Fig. 2A). These exclude outliers with unusual lengths compared to a collection of CRISPR loci from genome sequenced isolates (Grissa et al., 2007), which exceeded the inner fence of the CRISPRdb spacer length distribution (STAR Methods). The average length of all spacers (regardless of cassette host or spacer target) after this filtering was 32.7 ± 2.9 nt, slightly lower and significantly different from that of spacer from CRISPRCasdb (35.7 ± 4.7, P < 10−10, Wilcoxon rank sum test, Fig. 2B). The average length of repeat sequences (cluster representatives) was 34.1 ± 4.9, significantly longer than repeats from CRISPRCasdb with 31.8 ± 5.1 (P < 10−15, Wilcoxon rank sum test). Assuming that the spacer length distribution is unimodal, 31 nt spacers were underrepresented in the HMP1-II (Fig. 1A, Fig. S1A). However, this was also true for CRISPRCasdb content, where spacers of length 30 (n = 16,992) and 32 (n = 54,038) are much more frequent than 31 nt spacers (n = 2,314).
To account for possible redundancy of the spacer and repeat sets, we clustered predicted spacers using CD-HIT (Li and Godzik, 2006) and used the representative sequences reported by CD-HIT for downstream analysis. Of 965,495 spacer clusters with 80% identity, 33% (316,572) were only observed in one HMP sample, and no cluster was found across all HMP samples (Fig. S2). The mean (± s.d.) number of clusters per sample was 1,879 ± 2,185, with most found in oral body sites (2,881 ± 2,433 clusters). Each HMP sample included on average 33 highly prevalent clusters defined as appearing in at least 100 samples. The most prevalent cluster had 563 spacer instances, distributed across 300 samples, with the cluster centroid spacer sequence of GCACTTGTTGAAGCTGATGTACTTGCTGACGTGCTTGCACTT. The prevalence of these clusters was driven mainly by their distribution in the oral microbiome, which was highly sampled by the HMP; of the ten most prevalent clusters, half of cluster centroid sequences mapped uniquely to Streptococcus pneumoniae, itself highly prevalent in the oral cavity and occasionally other body sites. The remaining prevalent sequences had no blastn matches to nr/nt (default parameters, October 2019), thus likely driven by other microbes in the oral sites.
In addition to their sequence compositions, spacer loads also differed significantly across body areas and sites (P < 10−15, Kruskal-Wallis test, Table S2). Several oral sites - supragingival plaque (93.0 ± 49.8, n = 360 samples), subgingival plaque (79.0 ± 59.7, n = 19), and tongue dorsum (68.0 ± 32.7, n = 389) - had two- to three-fold higher spacer load than other oral sites, such as buccal mucosa (29.7 ± 30; n = 340), hard palate (33.8, n = 1) and keratinised gingiva (12.3 ± 9.7; n = 14, Fig. 2A). Gut, skin, and urogenital body areas had significantly fewer CRISPR spacers than oral sites (Dunn’s test FDR corrected q < 10−62, 10−90, 10−8, respectively, Table S2). Since these site-dependent differences in spacer load were not correlated with differences in species diversity or sequencing depth, factors such as the difference in prevalence of biofilm-forming microbes or exposure to viruses might explain the observed spacer load differences.
The high spacer load of the three oral body sites from the two plaque and tongue microbiotas correlates with Archaeal and Bacterial species richness (number of species with relative abundance exceeding 1%) normalised by the sample’s sequencing depth (Spearman’s ρ = 0.45, P < 10−104), and load was even higher for samples from the subgingival plaque (Spearman’s ρ = 0.89, Fig. 2C). This suggests that the CRISPR system is more active on these oral communities (i.e. due to longer repeat cassettes) and that most spacers are species-specific, and particular CRISPR-containing microbes are exclusively present in these three body sites. To investigate this further, we searched for uniquely enriched taxa in the three oral sites. One species enriched in supragingival plaque (relative abundance = 14%) and subgingival plaque (relative abundance = 10%) is Corynebacterium matruchotii, but the relative abundance of this species did not correlate with spacer load. The abundance of further taxa such as Veillonellaceae, Rothia and Rothia dentocariosa, which were enriched in the three body sites, also had no correlation and are therefore not explanatory for the high spacer load in these communities, maybe due to the absence of CRISPR–Cas system in these strains.
Diverse taxonomic origins of CRISPR spacers
To characterise the taxonomic affiliations, we mapped the HMP spacers and repeat sequences to sample-specific assemblies (Human Microbiome Project Consortium, 2012). Spacers without matches were aligned to the UniRef90 database using DIAMOND (Buchfink et al., 2015) (STAR Methods) to identify putative, unassembled targets. Since spacers acquired in the past can be retained for variable lengths of time, complementary protospacer sequences need not necessarily be present in a particular present-day sample. A taxonomic annotation of the global spacer collection was assigned using the provided last common ancestor (LCA) of all species that contributed a sequence to the UniRef cluster (Suzek et al., 2007).
To characterize the extent to which CRISPR utilization (i.e. spacer carriage) agrees or deviates from the ecological background taxonomic profile, we first focused on the whole spacer set, which is dominated by the taxonomic annotation of the spacer hosts (i.e. CRISPR cassettes), followed by an analysis of spacer targets outside cassettes, for which we use the distance of matches to direct repeats for filtering. Alpha diversities (Shannon Index) of LCA-derived taxonomic annotation of all observed spacers (i.e. all hosts and targets) to those from assemblies’ annotations were significantly correlated (Spearman’s ρ = 0.67 and 0.78 at the order and genus level, respectively).
Subsequent analysis also identified a correlation of the spacer and general microbiota taxonomic compositions, based on MetaPhlAn estimates for the latter and direct spacer assignments for the former (Spearman’s ρ = 0.53 and 0.58, at the order and genus level, Fig. S4), with diversity again consistently higher (mean ± s.d.) on the spacer profile (2.23 ± 0.91) on order level and 2.75 ± 1.12 on genus level (versus 1.28 ± 0.67 and 1.70 ± 0.74, respectively). As with assemblies above, the taxonomic profiles of CRISPR spacers and microbiota were very similar, though more diverse for spacers, especially in the gut microbiota (Fig. 3A). This trend was also observed at other taxonomic levels (Fig. S5), suggesting that overall spacers sample microbiome taxa randomly and no specific taxa dominate spacers in the studied body sites. The strongest exceptions from this trend of high similarity were seen on gut samples, where e.g. the relative proportion of spacer sequences associated to the Bacteroidia class 38% ± 22% (mean ± s.d) lower than the relative abundance of Bacteroidia in the general microbiota (74% ± 19%). The same trend was also observed for samples taken from the oral sites, where the mean abundance of Bacteroidia was higher in the general microbiota (10% ± 9%) than on spacers (5% ± 6%). Bacteroidia load in the general microbiota was below 1% on all other (skin and vaginal) body sites (Fig. 3A).
Figure 3: Body site dependence and a high overall high taxonomic agreement between all observed HMP1-II spacers and the general community.

A) Overview of the relative abundances of the seven most enriched taxa for the overall HMP microbiota (bottom) and for taxonomic assignments to HMP CRISPR spacers (top). B) PCoA of spacers per body area (based on BC dissimilarities on order level) show most variation to be driven by distinct stool communities and variation among oral samples. Point size indicates the number of spacer cluster (at 80% identity) per sample.
Relatedly, the gut and oral body areas tended to carry taxonomically distinct spacer sets from other body sites even at the order level (Fig. 3B, Fig. S6A for other ranks), which was less evident for overall microbiome taxonomic profiles (Fig. S6B, in which e.g. skin samples are not particularly similar). A PERMANOVA analysis indicated that 21% of the taxonomic spacer variance at the genus level was explained by body site (classes: 29%; orders: 27%; families: 23%), which was lower than the variance explained by body site among the microbiota as a whole (genera: 46%; classes: 50%; orders: 50% families: 42%). This is likely due to the particularly diverse CRISPR sampling of the well-sequenced oral and gut communities in this population, in contrast to the more balanced sampling of all taxa in the body-wide microbiome overall.
The previous sections all describe taxonomic annotation of the total observed spacer set (regardless of cassette host or spacer target). We next sought to identify potential targets using the mapping information of the CRISPR repeats (STAR Methods). In detail, we quantified spacer density per taxon by filtering out spacers with less than 500 nt distance to the next repeat for well-assembled taxa of each sample’s metagenome. This allowed us to focus on putative spacer targets (i.e. matches to viral content, or protospacers present in bacterial chromosomes, e.g. due to regulatory functions of the CRISPR system) by filtering out matches of spacers to the CRISPR cassette itself (i.e. the spacer host). Overall, mean protospacer density was 1.46 spacers per Mb. The body sites with the highest spacer density were mid vagina (3.94 Mb−1), hard palate (3.56 Mb−1), and supragingival plaque (2.37Mb−1), while the lowest spacer densities were seen on body sites such as stool (0.53 Mb−1), saliva (0.75 Mb−1), and throat (0.77 Mb−1). Genera with high spacer density were body site specific, the genus with the highest spacer load being Fusobacterium for tongue (87.3 Mb−1) and supra- and subgingival plaque (19.1 Mb−1, 8.62 Mb−1, respectively) while Prevotella (5.95 Mb−1) showed the highest spacer density in stool.
Spacers targets encode proteins involved in methylation processes and membrane activity and phage proteins
To further characterize the functions of the CRISPR system within the human microbiota, we used the same mapping approach as for taxonomic analyses, but now focusing on spacers with homology to UniRef90 annotations within each sample’s assembly or the UniRef90 database itself (Franzosa et al., 2018), resulting in 1,816,735 spacers with an associated UniRef90 term (best hit). Since spacer matches to CRISPR cassettes are on the non-coding regions of the genome, we used the full spacer set for subsequent analysis. We identified Gene Ontology (GO) terms (Ashburner et al., 2000) that were specifically enriched among spacer annotations relative to the gene families present in each corresponding sample. Specifically, we calculated the ratio of spacer annotation abundance for each UniRef90 family, per sample, to that of the sample’s HUMAnN2 estimate of that family. We then ranked these ratios and tested for gene set enrichment (Fisher’s exact test). We found that enriched GO terms using both the assembly-based (Table S3, Fig. 4A) and direct mapping approach were generally consistent (Table S4, Fig. S7). As anticipated, assembly-based mapping demonstrated more recent events (Fig. 4A, Fig. S8), which we focused on subsequently.
Figure 4: Functional enrichments within predicted spacer targets.

A) Log fold enrichment of Gene Ontology (GO) terms for all spacer targets within sample assemblies per body site. Terms shown here achieved at least one FDR corrected q value < 0.05 based on a Fisher test of enriched UniRef90 terms with respect to the overall contig annotation of the site (STAR Methods). Corresponding spacer targets by the global UniRef90 approach of remaining spacers are shown in Fig. S7) B) GO terms of spacers matching contigs outside of CRISPR-cassettes without any phage-related term on whole contigs, thus potentially within bacterial chromosomes. In both panels, a plus sign denotes an q value < 0.05 and GO groups cellular component (CC) and biological processes (BP) are shown (full version shown in Fig. S8.
As expected, many such significantly enriched GO terms (FDR corrected q < 0.05) for genes targeted by spacers were virus-related, such as viral capsid (- assembly GO:0019069), virion assembly (GO:0019068) (Fig. 4A, Table S3), or had phage-related functions, such as N-acetylmuramoyl-L-alanine amidase (GO:0008745) (Regamey and Karamata, 1998). To estimate the fraction of spacers involved in phage-related activity, we categorised UniRef90 groupings based on phage and virus search terms (“virus”, “phage”, or “viral”, see STAR Methods). These estimates are likely conservative, as viral processes are often unannotated or use less specific terms, such as DNA maintenance, DNA integration, or DNA transfer, which are challenging to distinguish from non-viral forms of DNA integration, such as conjugation. 7.3% of annotated spacers matched to 982 viral-term associated UniRef90 groups (Table S5). These groups accumulated slightly more spacer hits per gene than non-viral-associated UniRef groups (0.07 vs. 0.06 spacer hits per annotated gene). Spacer density varied by anatomic location, and was high for some oral sites, e.g. 0.48 for throat, and lower within stool (0.03). A large portion of spacers without assembly matches and matches to the UniRef90 database (32.8% of spacer matches with annotation) could be mapped to 2,283 unique, virus-associated UniRef90 groups, further demonstrating the prevalence of viral targeting for the CRISPR systems in the human microbiome. This difference may be attributable to spacer-related resistance against the matching phage, which could result in a low proportion of assembly-based phage matches due to low abundance of these viruses.
Several other bacterial processes are known to be specifically involved in phage invasion or replication, and these were often also enriched in our results. We identified significantly enriched GO terms associated with methyltransferase activity (GO:0009007, Table S3) and methylation (GO:0006306, Fig. 4A, Fig. S8). DNA methylation sites within bacteria are associated with restriction-modification systems (RMSs) (Rocha et al., 2001), a widely distributed defense mechanism that provides protection against incominging DNA such as phages (Vasu and Nagaraja, 2013). Phages may acquire protection against RMSs by phage-encoded self-methylation (Shapiro, 2012; Warren, 1980), but methylation is also involved in regulatory functions by modulating or interfering with DNA-binding proteins (Reisenauer et al., 1999; Sánchez-Romero et al., 2015) or influencing the expression of virulence genes (Heithoff et al., 1999). This enrichment may indicate that either CRISPR acts to target phages that adapted to the RMS, or that the system interacts directly with epigenetic regulation. We found a similar result for conjugation-related functions, such as unidirectional conjugation and DNA integration (Fig. 4A): 66 individual conjugation-associated UniRef90s were assigned 0.3% of all annotated spacers matched to the assembly (Table S6). These conjugation and horizontal gene transfer (HGT) related functions are also known mechanisms of action of the CRISPR–Cas system (Marraffini and Sontheimer, 2008).
UniRef90 groups with descriptions containing “transferase” accounted for 11.4% of all functionally annotated spacers, with an average load of 0.08 spacers per annotated open reading frame (ORF, Table S7). One such transferase is represented by the UniRef90 group “Prenyltransferase/squalene oxidase,” which has a high spacer load on tongue samples (3,653 spacers on 61 ORFs) and supragingival plaque (9,037 spacers on 156 ORFs). Genes for membrane-associated proteins appeared to be a common target of CRISPR, with 2% of all annotated spacers directed against such ORFs, especially in supragingival plaque, tongue, and stool sites (Fig. 4). We also found that the “LPXTG-motif cell wall anchor domain” was targeted by 0.9% of all spacers and highly prevalent: found in 14 of 16 anatomical locations. The “KxYKxGKxW signal domain” accounted for 0.3% of all spacers, with a high spacer load in keratinised gingiva and hard palate samples. A high spacer load (28 spacers per ORF) was similarly found for the “histone regulatory homologue binding (HIRA B) motif family” at all major oral sites.
With growing appreciation for the CRISPR system in non-defense-related functions (Westra et al., 2014), we focused subsequently on matches to spacer sequences outside of CRISPR arrays (based on the next CRISPR repeat match to the assembly) and removed ORFs with viral-associated sequences on the same contig, to identify putative spacer targets on chromosomally encoded genes. We then searched for enriched functions based on Fisher’s exact test of spacer counts and annotated ORFs at the contig level (Fig. 4B). This revealed that ORFs targeted by CRISPR associated with methylation activity such as the R-M system (q < 10−4) and DNA methylation (q < 0.003) appear on bacterial chromosomes rather than on viral contigs, highlighting its potential role in regulatory function. Further chromosomally enriched GO terms included membrane and cell-wall related functions, cell wall (GO:0005618, q < 10−89), cell adhesion (GO:0007155, q < 10−24) and extracellular region (GO:0005576, q < 10−33), more general functions such as pathogenesis (GO:0009405, q < 10−6), as well as general cell cycle-related functions such as DNA replication (GO:0006260, q < 10−14), recombination (GO:0006310, q = 0.009), and integration (GO:0015074, q < 10−4). However, since the annotation of phage and prophages remains challenging and they can easily be interspersed with putative bacterial chromosomal sequences, both in reference genomes and in metagenomic assemblies, these results should be interpreted with caution.
Variation in cas gene dominance across body sites
We next compared the CRISPR system repeats and spacers identified above to the Cas systems carried in corresponding communities and taxa. Three distinct subtypes are known among CRISPR–Cas systems on the basis of co-occurrence of cas genes (Haft et al., 2005), while ecological drivers and associations with these subsystems remain largely unexplored (CRISPR still needs microbiologists, 2018). To screen for cas genes associated with CRISPR subtypes and their prevalence and abundance in the human microbiome, we profiled the abundance of the ten main cas genes in HMP1-II samples using HUMAnN2 (Franzosa et al., 2018). From this, we generated a collection of 9038 cas gene family UniRef90 entries (Supplementary Dataset 3). Gene abundances were determined as gene length and sequencing depth normalised copies per million (CPM) reads, and the species-resolved functional profiling of HUMAnN 2 was used to assign taxa to cas genes.
Cas genes occurred widely across HMP samples. In 2,365 out of 2,388 samples, we detected at least one of the cas1-13 genes (Fig. S9A). Samples without cas genes mostly came from anterior nares (n = 12), posterior fornix (n = 5), and stool (n = 3), the former likely due to their relatively low sequencing coverage. 24% (583) samples included all ten Cas proteins. On average, 7.3 cas genes were found per sample, associated with a wide range of microbes. The most prevalent cas genes were cas2 and cas1, which were found in 98% of all samples, while UniRef90 groups associated to cas11-13 are not found in any sample. Taken together all samples and all taxons, cas1 (137 ± 91 CPM; mean ± s.d.) cas2 (145 ± 97 CPM) is the most abundant cas gene followed by the CRISPR associated genes cas3 (29 ± 32 CPM), cas4 (26 ± 28 CPM), and cas5 (25 ± 29 CPM). cas10, a signature gene for the CRISPR subtype III (Makarova et al., 2015), was the least prevalent (prevalence of 28%) and least abundant (0.24 ± 1 CPM) (Fig. S9A); it was predominantly found in some oral sites and in some gut and skin samples. cas10, cas7 and cas8 were notably also represented by the fewest available UniRef90 reference sequences (41, 96, and 96 respectively of 9,038 total), potentially contributing to the former’s lower prevalence (if additional variants within the family remain to be annotated).
The overall relative abundance and gene load of cas genes differed between body sites, with the highest levels occurring in several vaginal sites, mainly due to increased cas9 abundance (as carried by ecologically dominant lactobacilli within these communities), while skin samples had the lowest cas abundances (Fig. S9A, Fig. S9B). These trends were not a straightforward function of community diversity, however, since vaginal and skin communities are less diverse than oral or gut communities. The overall mean cas gene load among oral body sites was 3.4 times higher than for the remaining body sites, most profoundly for cas7 (15-fold increase to 4 CPM on oral sites), cas10 (11-fold increase to 0.4 CPM), and cas8 (7-fold increase to 4 CPM). Three body sites, namely anterior nares, stool, and left retroauricular crease, showed comparably low cas loads dominated by cas2 and an absence of cas9 and cas10 (Fig. S9B).
The taxa contributing to cas abundances were body-site specific, sometimes corresponding to the site’s abundant taxa, in other cases showing unique enrichments (Fig. 5). This high body-site specificity of the former is driven by a joint association between taxa and CRISPR systems. This could be indicative of ecologically driven mechanisms - like any other taxon - or ecology-specific molecular function - since the body sites are associated with different environmental factors such as aerobicity, nutrient availability, and viral load, among others. In the oral cavity, for example, Corynebacterium matruchotii is common. It is not the most abundant organism per se, but the top contributor of cas1-3. Conversely, this was true of Neisseria subflava in saliva samples. Overall, cas1 and cas2 occurred in similar taxa in oral and vaginal sites, and most cas-containing taxa had both proteins, such as B. vulgatus (cas1, cas2, cas4) and Cutibacterium (formerly Propionibacterium) acnes, a Gram-positive skin bacterium mostly carrying cas1, cas2, and cas5. One of the main contributing taxa for cas3 within the oral area (across all sites) was Haemophilus haemolyticus, a gram-negative bacterium found as a commensal in the respiratory tract (Pickering et al., 2016). This had a high load of cas3, but also cas1–5 at lower levels. Within the urogenital body area, Lactobacillus species were the major contributors, where the samples divided into two main clades based on Cas1/2 gene abundance dominated by L. crispatus or L. iners. In addition to these two Cas genes, some vaginal samples show high cas9 abundances contributed by L. jensenii. This can be explained since this species belonging to the type II-C (Makarova et al., 2015) which includes cas1–2 and cas9, which we confirmed by screening of the cas annotation of the genome. The cas6 system was mainly found in Streptococcus infantis for oral samples and Fusobacterium periodonticum in tongue dorsum samples. Rothia aeria contributed nearly all cas7 genes on non-saliva oral samples, while cas8 was present nearly solely in Neisseria sicca genomes.
Figure 5: Difference and similarities of cas gene abundances across body sites stratified by contributing species.

The height of each set of stacked bars (y axis) indicates the total cas abundance within a single sample, normalised for gene length and sequencing depth on a log10 scale. The taxonomic stratifications are done using a linear linearly (proportionally) scale. Species, “other,” and “unclassified” stratifications are linearly (proportionally) scaled within the total bar height. Highlighted taxa account for at least 35% of overall species abundance for each cas gene. Order of samples (bars) is according to the global Bray Curtis dissimilarity of the full microbiota within the body areas. Body areas with less than 30 samples are not shown. The y axis scale can be negative to facilitate the visualization of small abundances.
The pronounced co-occurrences of cas genes indicate that, as expected, multiple cas genes are generally present to provide a functional CRISPR–Cas system (Fig. S9B). 37 of the non-identical cas-cas pairs (87%) exhibited significant correlations (Fig. S10). The lowest co-occurrence to other cas genes was found for cas9, which only correlates with cas1 and cas2. As expected, cas1 and cas2 abundances were highly correlated across the population (Pearson’s ρ = 0.87, P < 10−16, Fig. S9A, Fig. S10), since they are considered as essential for a functional CRISPR system. However, this correlation was weaker for skin samples (ρ = 0.47), which show a reduced Cas2 load. We also saw a substantial, body site specific co-occurrence of Cas4 and Cas5 on the oral samples (ρ = 0.88), which is not present in the gut (ρ = 0.07) and weaker for skin and vaginal samples (ρ = 0.43 and 0.20, respectively). This might be explained by the oral predominance of CRISPR subtypes that requires both cas4 and cas5, which is the case for the subtypes I-A, I-B and I-C (Makarova et al., 2015). Cas4 is a nuclease present in the majority of CRISPR–Cas systems and is involved in the spacer acquisition. In some systems, cas4 is fused to cas1, suggesting a common function, however, other functions of cas4 such as involvement in programmed cell death have been proposed (Makarova et al., 2011b). While many of these variants may be due to technical limitations in the detection of species-specific cas genes within metagenomes, others may suggest additional Cas system architectures employed by members of the human microbiome.
Discussion
This study provides a large scale assessment of CRISPR spacers and repeats from across the human microbiome, incorporating 2,355 metagenomes from 17 different body sites in the HMP1-II population. By identifying CRISPR cassettes in communities and taxa for which no published reference genome exists, this extended the set of CRISPR spacers identified within the human microbiome by an order of magnitude. Using this resource, we (i) estimated the CRISPR activity on different body sites, identifying the oral plaque and tongue as ecologies with high activity compared to gut and urogenital sites; (ii) characterised the nucleotide-sequence properties of spacer and repeat sequences, identifying a hitherto-unknown palindromic nucleotide distribution pattern and an association of GC content to spacer length; (iii) uncovered the functional potential of the human-associated CRISPR spacers, including proteins by which they target bacteriophages. Beyond CRISPR targets likely to be phage-derived, we highlighted potential CRISPR interference to genes involved in bacterial methylation activity, suggesting an as-yet-unknown connection of the CRISPR and R-M defense systems.
A variety of biochemical and ecological factors appear to influence CRISPR distributions across the human body. Host-microbe interactions are especially prevalent on mucosal surfaces, where commensal microbial communities are maintained via nutrient absorption and controlled by the host through a variety of immune strategies (Aymeric and Sansonetti, 2015; Turner, 2009). The spacer load on mucosal surfaces (buccal mucosa, hard palate, keratinised gingiva, palatine tonsils, saliva, throat, tongue dorsum, gut, vaginal) is nearly one magnitude higher than on non-mucosal surfaces (skin) but highest on mucosal-adjacent surfaces (supragingival and subgingival plaque). Though environmental factors such as aerobicity correlate with CRISPR-incidence (Weissman et al., 2019) across taxa, HMP-1-II derived spacer load was not directly associated to the oxygen exposure of the body site (median spacer load for high-O2 sites = 2.71 CPM; 46.96 CPM for mid-O2 sites and 18.19 CPM for low-O2 sites). Instead, functions of the CRISPR system beyond phage targeting, such as control of genes involved in commensalism and virulence (Sampson and Weiss, 2013) and regulation of inter-microbial interactions within the host (Sampson et al., 2013), could explain the difference in spacer load between these surfaces. However, genetic experiments (likely in vitro) would be needed to control for potential confounders such as nutrient and oxygen availability and spatial differences such as biofilm formation and viral load.
Overall, the highest spacer load itself was in the oral cavity, particularly in dental plaque (Fig. 2A). This environment is a common entry point for microorganisms to the human host (Edlund et al., 2015) and is densely populated by viruses including bacteriophages (Naidu et al., 2014; Wang et al., 2016) that can persist over time (Abeles et al., 2014). Some of these properties are also true in the gut, but host physiology and immunity exert a much greater control over the live viruses that reach the colon, unlike the oral cavity. We hypothesised increased viral abundance coupled with longer exposure durations would lead to selection for spacer maintenance over many bacterial generations (Weinberger et al., 2012). The differences of spacer load are partially attributable to increasing species richness (Fig. 2C), also true in the oral cavity relative to the gut, suggesting that sites with a high spacer load harbour more distinct species with a CRISPR system. A link between biofilm formation and CRISPR activity (Cady and O’Toole, 2011; Zegans et al., 2009) also seems plausible, since samples of the supra and subgingival dental plaque originate from perhaps the most structurally-organised biofilms (Marsh, 2006) and show the highest spacer densities. These biofilms are known to facilitate the action of bacteriophages due high density of bacteria (Harper et al., 2014), which in turn would cause evolutionary pressure on bacteria to survive these phage attacks via adaptation and upregulation of the CRISPR system (Patterson et al., 2016). Additionally, since HGT is facilitated in these biofilms (Madsen et al., 2012), inter-bacterial spread of the CRISPR system might contribute to the high spacer loads observed, or might provide an additional evolutionary pressure to tightly regulate other forms of potentially invasive DNA.
In terms of sequence structure, the lengths of the HMP1-II derived CRISPR elements were generally similar to previously reported sequence sizes (21–72 nt for spacers and ~23 to ~48 for repeats) (Horvath and Barrangou, 2010; Kunin et al., 2007). Averages differed slightly across body sites, with larger repeat sizes present in stool, palatine tonsils, and throat, potentially reflecting differences in phage load or genome maintenance strategies among the dominant bacteria in these environments (Fig. 2B). Three distinct repeat length classes of small (~24 nt), medium (~29 nt), and large (~36 nt) sizes have been previously reported (Grissa et al., 2007), where longer repeat sequences are found in the genomes of e.g. Bacteroides fragilis and smaller repeat group is present in archaea (Grissa et al., 2007). Our results mimic these findings, including B. fragilis and other Bacteroides spp. abundant in gut (Huang et al., 2011) and archaeal species rare in human-associated communities (Horz, 2015). To ensure that this difference in spacers and repeat sizes was not due to the read-based CRISPR detection method, we recovered spacers using an alternative method based on assemblies (MinCED, https://github.com/ctSkennerton/minced) and confirmed that the resulting spacer length and size distributions were not significantly different from those recovered by Crass (Fig. S12B). We identified some interactions between sequence and composition, with smaller spacers having higher C/G content, suggesting different DNA targets (or, again, genome maintenance strategies) by size class. Viruses are often rich in A/T base pairs, and increasing length would result in more selectivity towards specific viral strains, while shorter G/C rich classes could target non-viral sequences for other types of regulation. Interestingly, the unexpectedly low number of 31 nt spacer sequences observed here (Fig. S1), which was also the case in published genomes, has neither a clear biochemical nor evolutionary driver to date.
The beginning and ends of CRISPR spacers were also found to be A/T-enriched here, which we again confirmed with spacers derived from sequenced genomes (Fig. 1B). This could arise from multiple synergistic sources. Structurally, such palindromic sequences arise when repeats contribute to the stability of RNA secondary structures (Mojica et al., 2000). In previous RNA stability studies of CRISPR repeats and spacers, only the former (repeats) showed elevated folding stability, but not the latter (spacers) (Kunin et al., 2007). Another property associated with these end sequences might be cleavage or integration efficiency, which has been found previously for AT-motifs at the end of spacer sequences in E. coli (Yosef et al., 2013). The AT enrichment that we observe throughout spacer sequences, with additional enrichments at both beginnings and ends, could thus be due to a combination of effects driven by both RNA secondary structural stability and efficiency of spacer acquisition.
Functionally, spacer-targeted GO terms were most associated with viral processes, bacterial transduction, and conjugation, as expected (Fig. 3). However, overall HGT rates by body site (Liu et al., 2012) were not correlated with the observed spacer loads in the HMP1-II results. This could be due to a variety of reasons, particularly the extremely different measurement strategies for the two effects, or possibly biological confounding from the large number of targeted non-viral sequences with diverse functions outside of transduction and conjugation. Purely natural competence from extracellular DNA was also not a complete explanation, since we compared the number of CRISPR spacers in a collection of 13,337 complete genomes with the presence of competence-conferring protein family (PF03772) and did not find a significant association (Fig. S11). Among other functional enrichments, a third group of pathways were associated with methylation processes and the R-M system, indicating that CRISPR could act as a second-stage defence against phages that acquire R-M functions, or that CRISPR regulates R-M or interacts with its downstream regulation. Intriguingly, this is in line with the finding that R-M and CRISPR activity might be functionally coupled (Dupuis et al., 2013; Makarova et al., 2013).
Finally, in addition to CRISPR repeat and spacer sequences themselves, the ecology of CRISPR systems overall is driven in large part by their associated cas genes, which have been grouped into five subtypes (I-V) with differential function and phylogeny that is not yet fully clear (Makarova et al., 2015, 2020). These differ in which combination of cas genes are carried by their associated host microbes, e.g. type I often including all of cas1-8 vs. type II including only cas1-2, cas4, and cas9. In part, subtypes differ in their intended targets, where the type II and III systems are associated with self-regulation in addition to phage defense. In particular, the type III system, which is thought to facilitate transcriptional regulation (Ledford, 2017), is the least prevalent system found in the HMP1-II set. The type II system, enriched in host-associated bacteria and potentially associated with virulence regulation (Sampson et al., 2013), was highly abundant in the vaginal microbiome and present to a lesser degree in the oral cavity. In our dataset, the canonical type I system was most prevalent and present on all body sites. Overall, though, we did not tend to see strong differentiation among cas subtypes in vivo, and it would be of interest to more closely study cas gene co-segregation within different microbes in a high-throughput, culture-independent manner.
As evidenced by this study, certain properties of CRISPR cassettes are more or less difficult to examine from metagenomic sequencing. While individual Cas proteins are relatively easy to detect and differentiate, the repetitive nature of CRISPR spacers and repeats is challenging to handle in metagenomes. The read-based detection approach employed in this study is not affected by the need to de novo assemble repetitive elements. However, as a consequence, recovered CRISPR cassettes cannot be seen as discrete CRISPR loci and can originate from multiple organisms that share the same repeat. This could in turn inflate the number of spacers due to isolated sequencing errors, and the exact spacer sequence can be erroneous if many spacers start or end with a shared subsequence. For a CRISPR locus to even be detected by read-based analysis requires the presence of at least some sequences that share sufficient homology across the length of a read and of a repeat, and thus such approaches fail to detect small CRISPR cassettes and cassettes from low abundance organisms. Conversely, other short repeats with sequence characteristics similar to CRISPR cassettes could inflate the overall spacer set by contributing false positives. Since many of these drawbacks are intrinsic to the interaction between short read sequencing and any type of underlying repetitive elements, they might best be reduced in the future through the use of culturomics, or by long read or linked read sequencing techniques.
Despite potential limitations, the spacer and repeat set described in this study is, to our knowledge, the largest and most comprehensive assessment of CRISPR carriage and ecology in the human microbiome. Together with our quantification of cas gene abundances, this informs both the potential functional roles of CRISPR–Cas systems and their targets in the human microbiome, as well as evolutionary properties and general principles of bacteria-virus relationships in and on human hosts. These data could also aid in the identification of viral sequences associated with human microbiome, which remains technically challenging for additional reasons (Edwards and Rohwer, 2005), and for the development of bioinformatic CRISPR detection methodology. Ultimately, these resources may be further translational for optimisation of phage-based treatments (Fischetti et al., 2006; Nobrega et al., 2015) or plasmid vectors, which must avoid similarity to prevalent natural spacer sequences within the microbiome in order to be effective for therapy.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Curtis Huttenhower chuttenh@hsph.harvard.edu.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The Human Microbiome Project (HMP) metagenomes analyzed in this work are available via http://hmpdacc.org. The CRISPR reads and spacer datasets (Supplementary Datasets S1 - S3) and the UniRef90 profiles (Supplementary Datasets S4 - S6) are available via http://huttenhower.sph.harvard.edu/crispr2020.
METHOD DETAILS
CRISPR identification from metagenomes
We processed all 2,355 shotgun metagenomic samples of the expanded Human Microbiome Project (HMP1-II), which has been described in depth (Human Microbiome Project Consortium, 2012; Lloyd-Price et al., 2017). Briefly, the cohort comprises 2,103 unique metagenomes and 252 technical replicates from 256 individuals obtained from 15 to 18 distinct body sites. From reads, we extracted spacer and repeats as well as CRISPR associated reads using Crass version 0.3.12 (Skennerton et al., 2013) with the parameters, which yielded 5,613,734 spacers and 479,632 repeats sequences (Supplementary Dataset 1–2) associated to 78,523,306 individual reads. These Crass parameters use a higher length threshold of 100 nt, since longer spacers above 50 nt (the default settings) have been reported (Pourcel et al., 2020). We further increased the kmerCount parameter to 23, which controls how similar direct repeats must be to define a cluster, to improve sensitivity in the highly complex human microbiome samples. We have chosen Crass over other tools such as MetaCRAST (Moller and Liang, 2017) since the latter require the presence of a database containing known repeat clusters to search for. We expect that current databases such as the database provided by MetaCRAST (6456 DR repeat clusters) is not covering the true diversity in complex communities and would therefore oversample for known repeats. On the other hand, tools such as Minced are optimized for longer sequences and find less spacers compared to Crass on HMP metagenomes, probably due to the fact that repeats are challenging to assemble de novo.
We compared the number of recovered spacers and spacer lengths of this read-based method (Crass) to MinCED (https://github.com/ctSkennerton/minced), a method optimized for longer sequences (such assembled contigs), using ten randomly selected samples of different body sites (IDs SRS063621, SRS146746, SRS893278, SRS043239, SRS044366, SRS016575, SRS148979, SRS077751, SRS143290, SRS019120). Crass, the method used in our study, showed a higher number of spacers detected when comparing with MinCED (Fig. S12B). The set of spacers recovered by MinCED showed similar length-distributions as we have reported in the manuscript using Crass, including the absence of 31-nt long spacer sequences. The non-normality of spacer length distributions was thus not explained by a Crass specific length bias.
Unusually long or short spacer sequences (potential false positives) were filtered out using a fixed distance from the interquartile range (Q3 – Q1) (inter-fence) from a spacer length distribution of public available CRISPR dataset. For the spacer dataset, the lower limit was calculated to be 27 nt and the upper limit to be 43 nt in length. Any observation outside these fences was considered a potential outlier and was removed from the analysis. The lower limit is defined as Q1 – (1.5 * IQR) while the upper limit is defined as Q3 + (1.5 * IQR). In total, 10.3% of spacer sequences were removed using this method (5,033,299 sequences after filtering). These outliers were especially prevalent in samples taken from the anterior nares and left and right retroauricular creases.
We created clusters as 95%, 90% and 80% similarity using CD-HIT v. 4.7 (Li and Godzik, 2006) with default settings resulting in sets of 1,859,558, 1,656,661, and 732,293 cluster representatives, respectively. For most sequence comparisons (such as in Fig. 2) we use the 80% identify cutoff based on an evaluation of different cutoff thresholds (Figure S12A). We downloaded all spacer and repeat sequences marked as ‘convincing’ from CRISPRCasdb (Pourcel et al., 2020) (Accessed August 2020). Spacer and repeats were clustered at 90% and 80% similarity using the same method, to create a bowtie2 database using these sequences. To quantify similarity of our dataset to CRISPRCasdb, we mapped all 479,632 HMP1-II derived repeat sequences to the bowtie database of CRISPRCasdb repeat sequences (allowing 1 mismatch in SEED alignment) using the --local parameter. Kruskal-Wallis tests were used to compare spacer and repeat counts with Dunn’s post-hoc test and the Benjamini-Hochberg multiple hypothesis test correction procedure using the R package “FSA”. Heatmaps were created using the R Complex heatmaps package (Gu et al., 2016). We quantified the mean GC content using the GC and s2c functions of the seqinr R library on filtered spacers sequences (the same mean GC content seems not be affected by the filtering).
We quantified the correlation between the raw (non-sequencing-depth corrected) species count based on MetaPhlAn2 (Truong et al., 2015) estimates of species exceeding 1% RA, and the number of spacers per million reads stratified by body site (Fig. 2C). The reason to choose non-sequencing-depth corrected species counts was that most samples in HMP1-II are sequenced to sufficient depth to detect most species and re-normalizing would introduce more bias than it removes. Anterior nares is potentially the only boddy site with both high diversity and (often) insufficient high sequencing depth to saturate detection.
We quantified the median spacer load as related to aerobicity by grouping samples based on the aerobicity of their body sites into three classes based on the O2 exposure (Segata et al., 2011). Skin samples were in the high-O2 exposure class, oral and vaginal samples in the mid-O2 exposure class, and gut on the low-O2 exposure class. We similarly quantified differences in spacer load between mucosal and non-mucosal body sites by grouping body sites, where the oral cavity, gut, and vaginal sites were classified as sources of mucosal communities and skin as non-mucosal (Segata et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
Validation of spacer and repeat sequences
K-mer profiles were calculated from observed HMP repeats using a publicly available tool developed in-house (https://github.com/algbioi/kmer_counting). Bray-Curtis (BC) similarity between all sample pairs was calculated using 5-mer data. We calculated mean stabilities (1-BC) for samples marked as technical replicates, taken from the same individual at two different time points and on randomly chosen pairs of two different individuals. We filtered out samples with only a small number of repeats (< 25 repeats per sample) to prevent a high k-mer similarity caused by undersampling rather than a biological effect. To quantify the cluster co-occurrence patterns across HMP samples, we used CD-HIT at 80% identity (see Fig. S12A for a comparison of the influence of the cluster threshold) and translated the CD-HIT output using the clstr2txt.pl tool, which was then analysed within R. We analysed the nucleotide frequency using the consensusMatrix function of the Biostrings R package. To account for different spacer length, we calculated the relative length by dividing each position by the total spacer length and taking all spacers into account that remained after filtering using the interfence criterion of a public CRISPR database (length of 28 nt to 42 nt). Confidence intervals were calculated by the geom_smooth function of ggplot2 using Local Polynomial Regression Fitting (loess) with standard parameters. Binnings were generated using the stats.bin function fields package with number of bins (N parameter) set to 100. Pearson’s correlation coefficients were calculated using the cor.test function in R.
Taxonomic analysis
We remapped the spacer content to the samples’ individual assemblies which contains both CRISPR cassettes and spacer targets (e.g. on contigs of vial origin) since current public available phage and viral databases are not covering the true viral diversity. Mapping was performed using the bowtie2-build command to create an index of the samples’ assembly, followed by bowtie2 for local alignment (--local option) with the parameters “-N 1 -a –very-sensitive”. This mapped 48% of all spacer sequences to sample-specific assemblies, similar to the overall fraction of mapped HMP1-II reads (36–42%) (Lloyd-Price et al., 2017). This produces a BAM file for each mapping, which we processed in R using the scanBam function of the R library Rsamtools. We saved the unmapped fraction using the bowtie2 “--un” parameter for later mapping to a more global database. Taxonomic information for the metagenomic samples were generated using MetaPhlAn2 (Truong et al., 2015) (available via http://hmpdacc.org). Using this mapping approach, we mapped 2,468,324 spacers to the assembly, for which we identified taxonomic information for 1,630,590. Since the samples’ individual assemblies provide UniRef90 annotations for ORFs, we determined the LCA of UniRef90 annotations if the spacer match overlapped with this ORF and parsed the LCA taxonomy for spacers with UniRef90 annotations using the R library taxonomizr by mapping the taxon UID to the phylogenetic tree using names.dmp and nodes.dmp annotation files. Since the taxonomy of both approaches agreed largely, we used the MetaPhlAn2 annotation for further analysis (Supplementary Dataset 3). FDR corrected p-values are denoted as q-values throughout the manuscript.
We mapped the remaining spacers without a match to the samples’ assemblies to the human-microbiota relevant subset of the UniRef90 database (Franzosa et al., 2018) database we used DIAMOND blastx using the uniref90_annotated.1.1 database with the parameters “-e 5000000 --more-sensitive --threads 30 --max-target-seqs 1 --query-cover 80 --compress 1 --id 8”. We filtered the mapping and retained matches with more than 80% percentage identity. We assigned taxonomic information to the UniRef90 groups using the LCA approach described before, resulting in 768,068 taxonomically annotated spacer sequences. We merged the set of annotated spacer sequences with the set generated using the bowtie2 approach, resulting in overall 2,398,658 taxonomically annotated spacers.
We calculated the Shannon entropy using the diversity function of the vegan package. Phylogenetic trees (Fig. S5) were generated using the metacoder and taxa R package based on the taxonomy of the 2,398,658 spacer sequences. PCoA plots were generated in R using the ape package on Bray Curtis dissimilarity calculated using the vegan package. PERMANOVA tests were performed using the anova.cca function of the vegan package with default parameters. Alpha diversity were consistently higher for LCA-based spacer assignment (3.40 ± 1.18 for mean ± s.d. on order level and on 4.03 ± 1.48 genus level versus 0.97 ± 0.60 and 1.31 ± 0.72), likely due to the substantially greater coverage possible by including unassembled targets. The two types of annotations together provided potential taxonomic assignments for 2,398,658 spacers (48%).
To quantify putative protospacer density, we matched the repeat set to the samples’ individual assemblies using bowtie2 similar to the spacer mapping. We filtered the bowtie2-based spacer mapping based on the mapping locations of the repeats and excludes spacers within 500nt of a repeat match, since these are putative CRISPR regions, after which 294,550 putative protospacers remained, of which 142,446 are taxonomically annotated based on the MetaPhlAn2 profile of the contigs. On a per-sample level, we filtered out genera with less than 10,000 genes to focus our analysis on well-assembled genera. We aggregated the number of spacer matches by contigs and stratified these by the genus annotation and sample ID. To calculate the mean density, we divided the sum of all spacer matches to contigs by the sum of the contig lengths. Mean densities per body site were calculated by averaging over the spacer density values for the individual samples.
Functional analysis
We mapped spacer and repeat sequences to the samples’ individual assemblies (contigs) generated by the HMP1 (Human Microbiome Project Consortium, 2012; Lloyd-Price et al., 2017) using their IDBA-UD assembly protocol (Peng et al., 2012) on a per-sample basis. Annotations of the assemblies (position and putative function of ORFs) were generated in HMP1 (Lloyd-Price et al., 2017) using MetaGeneMark based on several sequence-based searches leading to functional annotation of 35–45% of genes. We aggregated the assembly annotations, leading to 1,071,685 unique associations of genes to UniRef90 tems, functioning as a background for our statistical test and stratified these background occurrences by the body site (Fig. 4A) or body area (Fig. 4B). From the spacers matched to the samples’ individual assemblies, we annotated 1,003,429 spacers with one Uniref90 term (best hit), resulting in 16,462 UniRef90 terms based on overlaps of the spacer match to the contig annotation (Supplementary Dataset 3). Based on the number of spacer hits per UniRef90 term and the UniRef90 occurrence on the assemblies, we create for each body site a ranked list of Uniref90 terms based on the number of spacer matches per ORF found in the background were used to identify significantly enriched gene ontology (GO) terms using Fisher’s exact test for gene rank enrichment (Table S3).
Spacers without a match to the samples’ individual assemblies were mapped to a human-microbiota relevant subset of the UniRef90 database (Franzosa et al., 2018), resulting in 813,306 spacers with a UniRef90 annotation (in total 335,739 different UniRef90 terms). As a background, we use HUMMaN2 estimates of the assembly to these UniRef90 groups and generate ranked lists of UniRef90 spacer matches relative to the background for the GO enrichment analysis (Table S4).
We quantified conjugation-associated UniRef90 groups based on a non-case-sensitive text search on the terms “conjugation” or “integration” or “horizontal” or “conjugative”). Viral-associated groups were defined by a non-case-sensitive grep match of “virus” or “phage” or “viral” and transferase-associated groups were identified by a grep match to “transferase”. Conjugation related functions were searched via a grep match of “conjugation” or “integration” or ”horizontal” or “conjugative”, embrane related functions via “cytoplasmic” or “cytoplasma” or “membrane”.
To identify autoimmune-related functions, we mapped the repeat set to the samples’ assemblies using the same methods as for the spacer set and annotated the spacer dataset based on the distance to the next repeat on the same contig, and filtered out spacers that occured within +−500 nucleotides near a repeat match. We further filtered out spacers that occured on a contig with ORFs annotated as “Phage” or “Bacteriophage” or “phage” or “virus” or “tail” or “head”. We quantified the spacer density based on the sum of spacers found on ORFs associated with the UniRef90 annotation and divided this by the overall number of ORFs associated with the UniRef90 group.
Analysis of complete genomic isolates
We screened all open reading frames (ORFs) of a collection of 13,337 quality-controlled complete genomes for protein family matches using HMMSEARCH of the HMMER 3.1b2 software (Eddy, 1998) against the PF03772 (“Competence protein”) from the PFAM database v. 31.0, using a E-value cutoff of 1e-4. CRISPR loci and repeats were identified using CRISPR Recognition Tool (CRT) version 1.1 (Bland et al., 2007) with standard parameter setting. We calculated the Pearson correlation coefficient and the Spearman’s rank correlation coefficient using R 3.6.3.
Quantification of cas genes
The presence or absence of CRISPR associated genes (cas) was quantified using HUMAnN2 0.9.9 (Franzosa et al., 2018) based on the quantification of a set of UniRef90 terms within the metagenomic samples (Supplementary Dataset 4, Table S1). We searched for UniRef90 groups that are annotated with Cas1–10 (www.uniprot.org, accessed February 2019) and Cas11–13 (accessed March, 2020) (in total 80 UniRef90 terms, with no term associated to Cas11) due to an recent update on the CRISPR subtype classification scheme (Makarova et al., 2020). Based on this we quantified gene-length and community-wise normalised abundance (in reads per kilobase, default settings of humann2_renorm_table script of HUMAnN2) of each cas gene. We created unstratified and stratified output using taxonomic information for the UniRef90 matches (Supplementary Dataset 5, 6). Plots were created using the HUMAnN2 humann2_barplot script and in-house developed functionality present in the R library https://github.com/philippmuench/PMtools with the humann2Barplot and makeHumann2Barplot scripts and num.bugs.explained.fraction parameter set to 0.35. Kruskal-Wallis tests were carried out to compare each feature’s community total to the associated body site, and per-site means were created using humann2_associate script with default settings. Body site and overall mean Cas CPM values were computed using the sum over the taxonomic stratified HUMAnN2 table.
Supplementary Material
Document S1. Figures S1–S12
Table S1: Gene length and sequencing depth normalized Cas1-13 gene abundance based on HUMAnN2 prediction; Related Figure 5 and STAR methods.
Table S2: Post-hoc comparisons of number of observed CRISPR spacer and body sites; Related to Figure 2 and STAR methods. Differences in CRISPR spacer counts across human body areas and sites via two-sided Dunn’s test of multiple comparisons. FDR correction using the Benjamini and Hochberg procedure
Table S3: List of GO terms and associated q values of spacer matches to the assembly; Related to Figure 4 and STAR methods.
Table S4: List of GO terms and associated q values of spacer matches to the global UniRef90 database; Related to Figure 4 and STAR methods.
Table S5: Viral associated uniref groups based on mapping to assembly; Related to Figure 4 and STAR methods.
Table S6: Conjugation associated UniRef90 groups based on mapping to assembly; Related to Figure 4 and STAR methods.
Table S7: Transferase associated UniRef90 groups based on mapping to assembly; Related to Figure 4 and STAR methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Crass 0.3.12 | Skennerton et al., 2013 | https://github.com/ctSkennerton/crass |
| R version 3.6.3 | The R Project for Statistical Computing | https://www.r-project.org/ |
| MetaPhlAn 2 | Truong et al., 2015 | https://huttenhower.sph.harvard.edu/metaphlan |
| HMMER 3.1b2 | Eddy, 1998 | http://hmmer.org/ |
| Bowtie 2 | Langmeadet al., 2013 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| CRISPR Recognition Tool 1.1 | Bland et al., 2007 | http://www.room220.com/crt/ |
| GNU parallel | O. Tange, 2011 | http://www.gnu.org/s/parallel |
| HUMAnN 2 | Franzosa et al., 2018 | https://huttenhower.sph.harvard.edu/humann |
| CD-HIT v. 4.7 | Li and Godzik et al., 2006 | http://cd-hit.org |
| Other | ||
| HMP1-II metagenomes | available from the HMP DACC (http://hmpdacc.org) and from SRA BioProjects PRJNA48479 and PRJNA275349 | http://hmpdacc.org |
| CRISPRCasdb | Pourcel et al., 2020 | https://crisprcas.i2bc.paris-saclay.fr/ |
| CRISPRdb | Grissa et al., 2007 | http://crisprdbcrispr.i2bc.paris-saclay.fr/ |
| Accompanying dataset to the study “Natural CRISPR systems and targets in the human microbiome” | This study | http://huttenhower.sph.harvard.edu/crispr2020 and https://data.mendeley.com/datasets/bsmmy8pwrt/1 |
| UniProt Reference Clusters (UniRef) | Baris E. Suzek | https://www.uniprot.org/uniref/ |
Highlights.
2.9 million CRISPR spacers from 2,355 body-wide human metagenomes are profiled
Oral habitats show high CRISPR load compared to gut/urogenital sites
Functional potential of CRISPR spacers suggests link to restriction-modification system
cas gene profiles accompany CRISPR subtype differentiation by body site
Acknowledgements
P.C.M. received funding from the German Research Foundation (DFG, Grant number 315980449 and 405892038). The work was funded in part by NIH grants R24DK110499 and U54DK102557 to C.H.. We thank Peter Turnbaugh for helpful discussions.
Footnotes
Declaration of Interests
The authors declare no competing interests.
ADDITIONAL RESOURCES
Datasets S1-S6, Related to STAR methods: http://huttenhower.sph.harvard.edu/crispr2020
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeles SR, Robles-Sikisaka R, Ly M, Lum AG, Salzman J, Boehm TK, and Pride DT (2014). Human oral viruses are personal, persistent and gender-consistent. ISME J 8, 1753–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymeric L, and Sansonetti P (2015). Chapter 50 - Discriminating Pathogens from Commensals at Mucosal Surfaces In Mucosal Immunology (Fourth Edition), Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroutre H, and Lambrecht BN, eds. (Boston: Academic Press; ), pp. 975–984. [Google Scholar]
- Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, and Hugenholtz P (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Burstein D, Sun CL, Brown CT, Sharon I, Anantharaman K, Probst AJ, Thomas BC, and Banfield JF (2016). Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun 7, 10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady KC, and O’Toole GA (2011). Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol 193, 3433–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley AB, Henriksen ED, Stout E, Brandt K, and Barrangou R (2018). Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci. Rep 8, 11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRISPR still needs microbiologists (2018). CRISPR still needs microbiologists. Nat Microbiol 3, 641. [DOI] [PubMed] [Google Scholar]
- Deveau H, Garneau JE, and Moineau S (2010). CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol 64, 475–493. [DOI] [PubMed] [Google Scholar]
- Dupuis M-È, Villion M, Magadán AH, and Moineau S (2013). CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat. Commun 4, 2087. [DOI] [PubMed] [Google Scholar]
- Eddy SR (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. [DOI] [PubMed] [Google Scholar]
- Edlund A, Santiago-Rodriguez TM, Boehm TK, and Pride DT (2015). Bacteriophage and their potential roles in the human oral cavity. J. Oral Microbiol 7, 27423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, and Rohwer F (2005). Viral metagenomics. Nat. Rev. Microbiol 3, 504–510. [DOI] [PubMed] [Google Scholar]
- Fischetti VA, Nelson D, and Schuch R (2006). Reinventing phage therapy: are the parts greater than the sum? Nat. Biotechnol 24, 1508–1511. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godde JS, and Bickerton A (2006). The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol 62, 718–729. [DOI] [PubMed] [Google Scholar]
- Gogleva AA, Gelfand MS, and Artamonova II (2014). Comparative analysis of CRISPR cassettes from the human gut metagenomic contigs. BMC Genomics 15, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, and Pourcel C (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, and Nelson KE (2005). A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol 1, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DR, Parracho HMRT, Walker J, Sharp R, Hughes G, Werthén M, Lehman S, and Morales S (2014). Bacteriophages and Biofilms. Antibiotics 3, 270–284. [Google Scholar]
- Hatoum-Aslan A, and Marraffini LA (2014). Impact of CRISPR immunity on the emergence and virulence of bacterial pathogens. Curr. Opin. Microbiol 17, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithoff DM, Sinsheimer RL, Low DA, and Mahan MJ (1999). An essential role for DNA adenine methylation in bacterial virulence. Science 284, 967–970. [DOI] [PubMed] [Google Scholar]
- Horvath P, and Barrangou R (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. [DOI] [PubMed] [Google Scholar]
- Horvath P, Coûté-Monvoisin A-C, Romero DA, Boyaval P, Fremaux C, and Barrangou R (2009). Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol 131, 62–70. [DOI] [PubMed] [Google Scholar]
- Horz H-P (2015). Archaeal Lineages within the Human Microbiome: Absent, Rare or Elusive? Life 5, 1333–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Lee SM, and Mazmanian SK (2011). The human commensal Bacteroides fragilis binds intestinal mucin. Anaerobe 17, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). A framework for human microbiome research. Nature 486, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, and Hannon GJ (2010). The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol. Cell 37, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Sorek R, and Hugenholtz P (2007). Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 8, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H (2017). Five big mysteries about CRISPR’s origins. Nature 541, 280–282. [DOI] [PubMed] [Google Scholar]
- Li W, and Godzik A (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Liu L, Chen X, Skogerbø G, Zhang P, Chen R, He S, and Huang D-W (2012). The human microbiome: a hot spot of microbial horizontal gene transfer. Genomics 100, 265–270. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez M-J, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, and Glaser P (2012). The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol. Microbiol 85, 1057–1071. [DOI] [PubMed] [Google Scholar]
- Madsen JS, Burmølle M, Hansen LH, and Sørensen SJ (2012). The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol 65, 183–195. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, et al. (2011a). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol 9, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, and Koonin EV (2011b). Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, and Koonin EV (2013). Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res 41, 4360–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh PD (2006). Dental plaque as a biofilm and a microbial community - implications for health and disease. BMC Oral Health 6 Suppl 1, S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Díez-Villaseñor C, Soria E, and Juez G (2000). Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol 36, 244–246. [DOI] [PubMed] [Google Scholar]
- Moller AG, and Liang C (2017). MetaCRAST: reference-guided extraction of CRISPR spacers from unassembled metagenomes. PeerJ 5, e3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu M, Robles-Sikisaka R, Abeles SR, Boehm TK, and Pride DT (2014). Characterization of bacteriophage communities and CRISPR profiles from dental plaque. BMC Microbiol 14, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega FL, Costa AR, Kluskens LD, and Azeredo J (2015). Revisiting phage therapy: new applications for old resources. Trends Microbiol 23, 185–191. [DOI] [PubMed] [Google Scholar]
- van der Oost J, Jore MM, Westra ER, Lundgren M, and Brouns SJJ (2009). CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci 34, 401–407. [DOI] [PubMed] [Google Scholar]
- Patterson AG, Jackson SA, Taylor C, Evans GB, Salmond GPC, Przybilski R, Staals RHJ, and Fineran PC (2016). Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol. Cell 64, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, and Chin FYL (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Pickering JL, Prosser A, Corscadden KJ, de Gier C, Richmond PC, Zhang G, Thornton RB, and Kirkham L-AS (2016). Haemophilus haemolyticus Interaction with Host Cells Is Different to Nontypeable Haemophilus influenzae and Prevents NTHi Association with Epithelial Cells. Front. Cell. Infect. Microbiol 6, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Touchon M, Villeriot N, Vernadet J-P, Couvin D, Toffano-Nioche C, and Vergnaud G (2020). CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res 48, D535–D544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pride DT, Sun CL, Salzman J, Rao N, Loomer P, Armitage GC, Banfield JF, and Relman DA (2011). Analysis of streptococcal CRISPRs from human saliva reveals substantial sequence diversity within and between subjects over time. Genome Res 21, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pride DT, Salzman J, and Relman DA (2012). Comparisons of clustered regularly interspaced short palindromic repeats and viromes in human saliva reveal bacterial adaptations to salivary viruses. Environ. Microbiol 14, 2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Walker AW, Simpson JT, Loman NJ, and Segata N (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol 35, 833–844. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regamey A, and Karamata D (1998). The N-acetylmuramoyl-L-alanine amidase encoded by the Bacillus subtilis 168 prophage SP beta. Microbiology 144 ( Pt 4), 885–893. [DOI] [PubMed] [Google Scholar]
- Reisenauer A, Kahng LS, McCollum S, and Shapiro L (1999). Bacterial DNA methylation: a cell cycle regulator? J. Bacteriol. 181, 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M, Wu Y-W, Tang H, Doak TG, and Ye Y (2012). Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 8, e1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A, and Viari A (2001). Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res 11, 946–958. [DOI] [PubMed] [Google Scholar]
- Sampson TR, and Weiss DS (2013). Alternative roles for CRISPR/Cas systems in bacterial pathogenesis. PLoS Pathog 9, e1003621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Saroj SD, Llewellyn AC, Tzeng Y-L, and Weiss DS (2013). A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Romero MA, Cota I, and Casadesús J (2015). DNA methylation in bacteria: from the methyl group to the methylome. Curr. Opin. Microbiol 25, 9–16. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J (2012). Mobile Genetic Elements (Elsevier; ). [Google Scholar]
- Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, and Koonin EV (2017). The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skennerton CT, Imelfort M, and Tyson GW (2013). Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res 41, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Keren L, Wurtzel O, Amitai G, and Sorek R (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet 26, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Mick E, Tirosh I, Sagy O, and Sorek R (2012). CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CL, Thomas BC, Barrangou R, and Banfield JF (2016). Metagenomic reconstructions of bacterial CRISPR loci constrain population histories. ISME J 10, 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, and Wu CH (2007). UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Turner JR (2009). Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol 9, 799–809. [DOI] [PubMed] [Google Scholar]
- Vasu K, and Nagaraja V (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev 77, 53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatanen T, Plichta DR, Somani J, Münch PC, Arthur TD, Hall AB, Rudolf S, Oakeley EJ, Ke X, Young RA, et al. (2019). Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat Microbiol 4, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gao Y, and Zhao F (2016). Phage-bacteria interaction network in human oral microbiome. Environ. Microbiol 18, 2143–2158. [DOI] [PubMed] [Google Scholar]
- Warren RA (1980). Modified bases in bacteriophage DNAs. Annu. Rev. Microbiol 34, 137–158. [DOI] [PubMed] [Google Scholar]
- Weinberger AD, Sun CL, Pluciński MM, Denef VJ, Thomas BC, Horvath P, Barrangou R, Gilmore MS, Getz WM, and Banfield JF (2012). Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol 8, e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JL, Laljani RMR, Fagan WF, and Johnson PLF (2019). Visualization and prediction of CRISPR incidence in microbial trait-space to identify drivers of antiviral immune strategy. ISME J 13, 2589–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, Buckling A, and Fineran PC (2014). CRISPR-Cas systems: beyond adaptive immunity. Nat. Rev. Microbiol 12, 317–326. [DOI] [PubMed] [Google Scholar]
- Wimmer F, and Beisel CL (2019). CRISPR-Cas Systems and the Paradox of Self-Targeting Spacers. Front. Microbiol 10, 3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef I, Shitrit D, Goren MG, Burstein D, Pupko T, and Qimron U (2013). DNA motifs determining the efficiency of adaptation into the Escherichia coli CRISPR array. Proc. Natl. Acad. Sci. U. S. A 110, 14396–14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, and O’Toole GA (2009). Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol 191, 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S12
Table S1: Gene length and sequencing depth normalized Cas1-13 gene abundance based on HUMAnN2 prediction; Related Figure 5 and STAR methods.
Table S2: Post-hoc comparisons of number of observed CRISPR spacer and body sites; Related to Figure 2 and STAR methods. Differences in CRISPR spacer counts across human body areas and sites via two-sided Dunn’s test of multiple comparisons. FDR correction using the Benjamini and Hochberg procedure
Table S3: List of GO terms and associated q values of spacer matches to the assembly; Related to Figure 4 and STAR methods.
Table S4: List of GO terms and associated q values of spacer matches to the global UniRef90 database; Related to Figure 4 and STAR methods.
Table S5: Viral associated uniref groups based on mapping to assembly; Related to Figure 4 and STAR methods.
Table S6: Conjugation associated UniRef90 groups based on mapping to assembly; Related to Figure 4 and STAR methods.
Table S7: Transferase associated UniRef90 groups based on mapping to assembly; Related to Figure 4 and STAR methods.
Data Availability Statement
The Human Microbiome Project (HMP) metagenomes analyzed in this work are available via http://hmpdacc.org. The CRISPR reads and spacer datasets (Supplementary Datasets S1 - S3) and the UniRef90 profiles (Supplementary Datasets S4 - S6) are available via http://huttenhower.sph.harvard.edu/crispr2020.