

Abstract
Fifty five members of the nineteen Receptor Tyrosine Kinase subfamilies in humans are all single-span transmembrane receptors in which relatively conserved intracellular kinase domains are coupled to divergent extracellular modules. The extracellular domains initiate receptor signaling upon binding to either soluble or membrane-embedded ligands. The diversity of extracellular domain structures allows for coupling of many unique signaling inputs to intracellular tyrosine phosphorylation but, importantly, this diversity is further increased by the fact that multiple ligands can typically interact with the same receptor. Despite engaging with the same receptor, such ligands elicit specific and unique control over receptor signaling. Mechanisms behind such biased agonism are largely unknown and have been shown to include direct regulation of the kinase activation mechanisms, of the composition of recruited signaling regulators and sometimes of trafficking of the activated receptor complexes. Using recent progress in understanding the structures of active RTK signaling units, we discuss selected mechanisms by which ligands couple receptor activation to distinct signaling outputs.
RTK ligands may act as biased agonists.
Receptor tyrosine kinases (RTKs) enable communication between cells and with their extracellular environment, contributing to fundamental steps of tissue patterning and organogenesis in development and to the maintenance of adult organismal homeostasis [1]. Nineteen different RTK sub-families specialize in different functions but all are built similarly and contain an N-terminal extracellular domain (ECD), a single transmembrane domain (TMD) and an intracellular kinase domain followed by a largely unstructured C-terminal tail region. The kinase domains share a conserved architecture among most RTKs. In contrast, ECDs are highly divergent between the receptor subfamilies, varying in size and composition of individual subdomains. This diversity allows receptors to recognize structurally distinct protein ligands which then exert unique control over the activation of the kinase domain.
Most human RTKs can be activated by more than one ligand. For instance, three of four EGFR/HER receptors respond to a family of ligands expressed by 15 genes and a number of their splice variants ([2,3], Figure 1). Nine EphA receptors are each activated by five ephrin A ligands and five EphB receptors are each activated by five ephrin B ligands [4,5]. In case of the four human FGF receptors, their ligands are encoded by 22 genes and many have multiple isoforms [6–8]. The remarkable combinatorial power of the FGF signaling system is further diversified by engagement with additional binding partners such as heparin, heparan sulfate proteoglycans, and α- and β-Klotho co-receptors [6,7,9]. The expression of some of the RTK ligands is tissue-specific, however many are broadly expressed yet modulate the extent of receptor activation in a distinct way. Frequently, several ligands bind to the same receptor exerting qualitatively different signaling outputs, for instance by favoring signaling via the ERK pathway over AKT activation [10,11]. This phenomenon, called “biased agonism”, defined also as functional selectivity, has been extensively described for G protein coupled receptors (GPCRs). The mechanisms by which GPCR ligands exert biased agonism are complex and involve the kinetic and thermodynamic properties of the receptor ligand complex, kinetics of receptor endocytosis and its subsequent degradation, association with unique downstream effectors, triggering distinct negative/positive feedbacks and even induction of altered signaling properties within intracellular compartments after receptor internalization [12,13]. A lot of effort has been invested in trying to understand how receptors can modulate these variables differently depending on the bound ligand. One widely accepted view is that GPCRs exist in a dynamic equilibrium between multiple active conformations and that biased agonists shift this equilibrium stabilizing conformationally distinct active states [12–14]. These states then uniquely couple to downstream G proteins, β-arrestins and GPCR kinases (GRKs) resulting in distinct cellular responses [15]. Several such structures of GPCRs have been solved and demonstrated differential a range of conformational states that activating ligands can stabilize [14].
Figure 1: Diversity of the human RTK ligands.
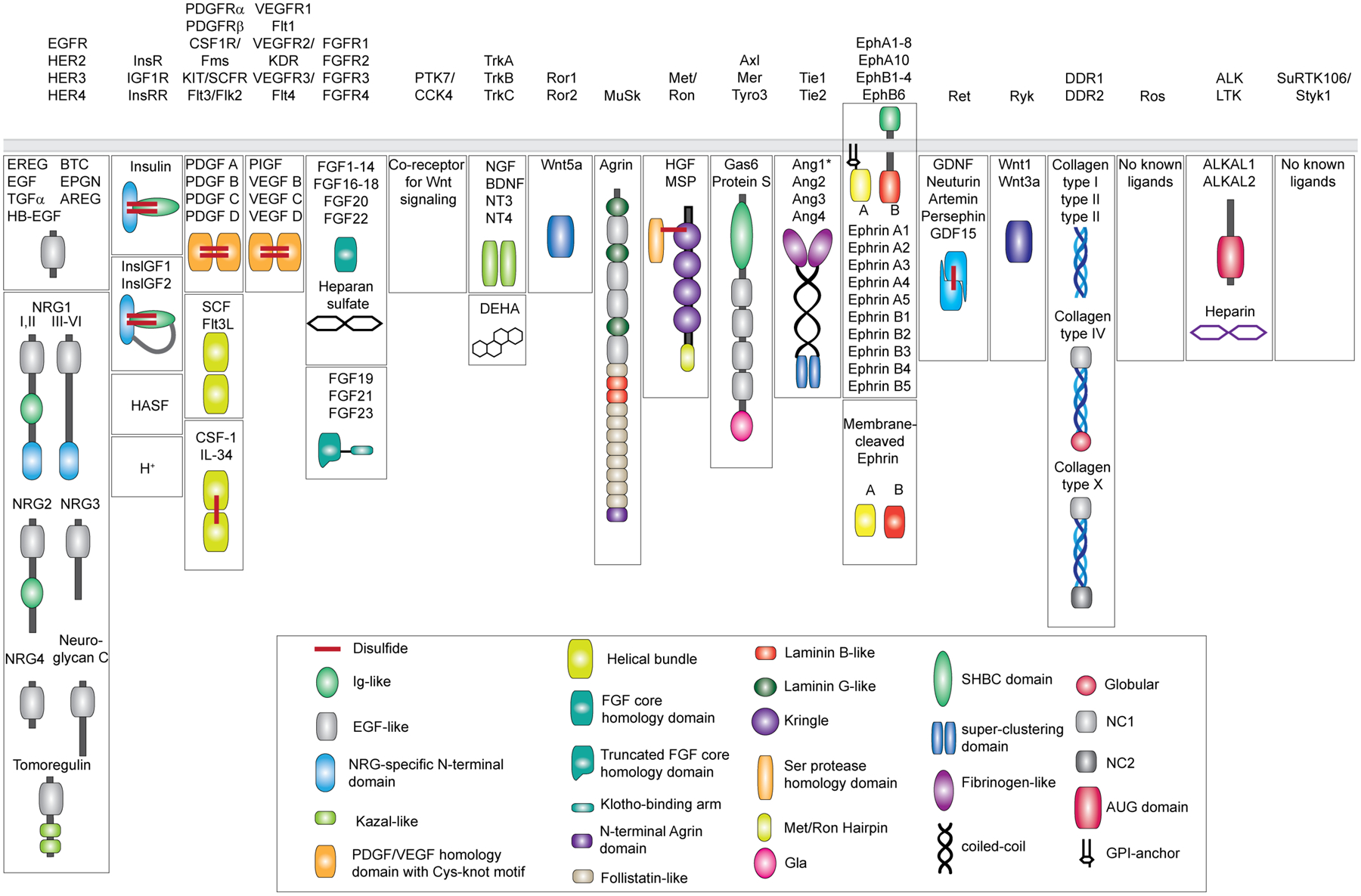
RTKs are shown on the top categorized into 19 different families as originally described [1] and recently revised to remove the LMR1–3 family due to its re-classification as Ser/Thr receptor kinases [127]. Ligands for each RTK family are shown underneath in mature, secreted form. All membrane-tethered ligands are cleaved off the membrane except for ephrins, which activate their cognate receptors in a juxtacrine fashion. The ligands are drawn with their N-terminus pointing away from the membrane. Main structural domains are depicted in a cartoon form in their known oligomeric state except for Angiotensins*, which may form higher-order oligomers in addition to dimers. If applicable, domain labels are included as captions. Sizes of individual domains are not drawn up to scale.
In analogy to the GPCR field, a concept of biased agonism in RTK signaling was proposed a decade ago to explain functionally discrete outcomes of activation of the same receptor by different ligands [10]. Similarly to GPCRs, many factors determine unique signaling properties of an RTK ligand, including altered kinetics of receptor binding, induction of distinct receptor oligomeric states, association with signaling co-receptors, differential regulation of receptor endocytosis, trafficking, degradation and finally signaling from intracellular compartments. However, from a receptor perspective, in contrast to GPCRs, a much sparser repertoire of RTK structures, in particular of those spanning the ligand-bound extracellular domains, and lack of any high-resolution structures of full-length receptors limit the extent of mechanistic insights into whether ligands actually stabilize structurally distinct receptor states. Moreover, in the absence of direct insights into whether ligand binding to the ECDs is conformationally coupled to the activation of the kinase domains, it is difficult to know if structural changes associated with binding of different ligands can be robustly interpreted by the intracellular receptor module on the other side of the membrane. Here we discuss recent work in the EGFR/HER, Ephrin, RET, SCF and FGF receptor families to illustrate the current understanding of these mechanisms for RTKs.
Direct effect of ligands on receptor conformation and dimerization
In the canonical model of HER receptor activation, ligand binding to the ECDs results in receptor dimerization by promoting an extended conformation of the ECD [16–19]. Through a still unclear mechanism, the ECD dimer communicates with the intracellular fragments of the receptor. It is well established that in the active complex, the intracellular kinase domains form an asymmetric dimer in which one kinase allosterically activates the other [20]. This conformation is stabilized by the interactions made by the intracellular JM domains [21]. The activated kinase then phosphorylates C-terminal tails of the receptors providing docking sites for numerous downstream signaling molecules (Figure 2)[20–22]. Three out of the four different EGFR/HER receptors, EGFR, HER3 and HER4, recognize multiple ligands, while HER2 is an orphan receptor. EGFR itself binds EGF, TGFα, epigen, epiregulin, amphiregulin, HB-EGF and betacellulin (BTC), which are believed to be monomeric in solution and engage the receptor through their conserved albeit slightly divergent EGF-like domains (Figure 1 and 2) [23]. The seven cognate ligands activate EGFR signaling in qualitatively different ways, resulting in responses ranging from cell differentiation, proliferation, migration to plethora of other phenotypes [10,24–34]. For example, TGFα and HB-EGF are more potent stimulators of DNA synthesis in isolated rat hepatocytes than EGF despite similar receptor binding affinities [35], and amphiregulin causes greater mobility and invasiveness of MCF10A human mammary gland epithelial cells than EGF [36]. Whether different ligands cause distinct structural changes in the intracellular receptor modules is not known, but they certainly promote unique phosphorylation states of the receptor tails and modulating patterns of recruited downstream signaling proteins such as AKT, ERK, PLCγ and STAT3 [25,34,37]. Signaling downstream from HER4 is also differentially modulated by saturating levels of its cognate ligands that include four family members of the neuregulin family, in addition to BTC, epiregulin and HG-EGF [38].
Figure 2: Ligand-specific signaling through EGFR homodimers.
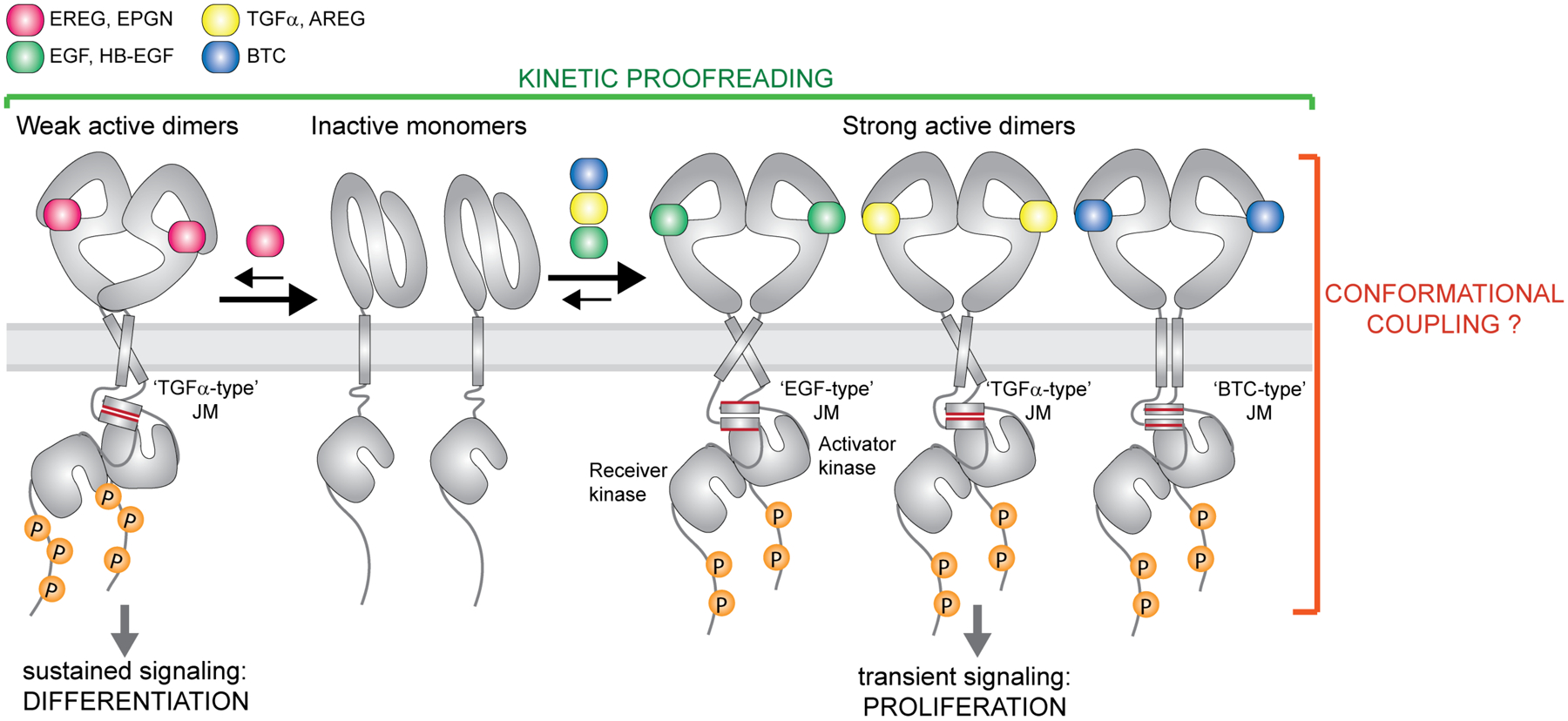
EGFR forms high-affinity complexes with its cognate ligands Transforming Growth Factor α (TGFα), Epidermal Growth Factor (EGF), Amphiregulin (AREG), Betacellulin (BTC) and Heparin-Binding-EGF (HB-EGF) but lower-affinity complexes with Epiregulin (EREG) and Epigen (EPGN). Available structures of EGF, TGFα±EPGN and EREG bound to EGFR ECDs are so far consistent with the EGFR ECDs adopting symmetric dimers with high affinity ligands and asymmetric structures with lower affinity ligands. Weaker EGFR dimers lead to sustained signaling and cell differentiation while formation of strong complexes causes transient receptor signaling and cell proliferation. Thus, the dynamics of ligand-dependent receptor association and dissociation has an important impact on the recruitment of downstream effectors and the ligand-specific cellular responses (kinetic proofreading). Biochemical studies on the EGFR juxtamembrane (JM) segment and studies of full-length EGFR in cells are consistent with the presence of the JM coiled-coil dimer in active receptor complex. Conformation of the JM dimer appears to change depending on a bound ligand. At least three specific JM dimer modes have been described for EGFR, denoted here as ‘TGFα-type’, the ‘EGF-type’ and the ‘BTC-type’.
Some of the reported ligand-unique responses might be a result of the cross-reactivity between ligands and multiple HER receptors. However, recent structural insights into the interaction modes between EGFR ECD homodimers and two of its ligands, epiregulin and epigen, offer mechanistic insights how the ligands could act as biased structural agonists. A crystal structure of the EGFR ECD with epiregulin revealed that this ligand promotes a less symmetric ECD dimer than seen in previously solved structures of EGFR ECD in complex with EGF and TGFα [18,19,26]. Although epigen crystalized in a monomeric complex with the EGFR ECD, it induced ECD dimers in solution, and in the structure stabilized an EGFR ECD conformation that resembled HER2. A HER2-like conformation is predicted to weaken the canonical ECD dimerization interface [26,39]. While it is unclear whether these different conformations of ECDs are directly involved in modulation of the active kinase dimer module, itself asymmetric [20], it appears that non-canonical, asymmetric complexes of EGFR ECDs are less stable than the symmetric ones [26]. Epigen and epiregulin consequently promote the formation of weaker EGFR dimers than EGF and TGFα. These altered dimerization affinities lead to ligand-specific differences in the average lifetime of active signaling complexes. Counterintuitively, weaker EGFR dimers promoted more sustained signaling resulting in cellular differentiation in comparison to EGF, which activated transient signaling responses and cell proliferation [26].
Regulation of the signaling outcomes via stability of active receptor complex formation is a signature of kinetic proofreading - a mechanism first described in the receptor field for the T cell receptor [40]. This model assumes a time lag between initial receptor-ligand interactions and the formation of active, phosphorylated complexes, and consequently further recruitment of downstream signal transducers. Such delay can be caused by quick dissociation of unstable complexes and/or their effective dephosphorylation by phosphatases, ensuring that unproductive receptor complexes fail to signal. If a pool of downstream proteins varies in recruitment kinetics to the receptor then the assembly of signaling complexes will be determined by the lifetime of active receptor complexes. An interesting example is provided by the FGFR1 receptor in which autophosphorylation of tyrosine residues in the kinase domain, which serve as docking sites for downstream signaling proteins, occurs sequentially in time [41]. Therefore, the extent of FGFR1 phosphorylation and its ability to recruit signaling effectors depends on the lifetime of the active receptor complexes [41]. On the other hand, the extent of autophosphorylation of HER receptors seems to depend more on the recruitment of phospho-tyrosine phosphatases (PTPs) to active receptor complexes [42,43].
Kinetic proofreading can play an important role in tuning RTK signaling in response to different agonists, and this property allowed for its harnessing for a number of therapeutic strategies [44]. When a native ligand for the c-Kit receptor, stem cell factor (SCF), binds to the receptor as a non-covalent homodimer it stimulates both the maintenance and survival of bone marrow hematopoietic stem and progenitor cells (HSPCs) as well as the development and activation of mast cells [45]. Selective activation of HSPCs without mast cell activation is often desired in the clinic but cannot be achieved using native SCF dimers. However, an engineered dimerization-deficient SCF variant reduced the dimerization propensity ligand-bound receptors and achieves biased activation of HSPCs without simultaneous activation of mast cells in vitro and in vivo [44]. Similarly, activation of several isoforms of FGFR by FGF1 can induce different biological pathways and for instance regulates both cell proliferation and glucose metabolism in various cell lines [46]. A mutant FGF1 ligand that induces weaker FGFR dimers selectively abrogates FGFR’s mitogenic potential while preserving its full metabolic activities [47], thereby providing another example for how the stability of receptor ligand complexes may regulate distinct signaling responses.
Allosteric coupling across the membrane
It remains poorly understood how much unique signaling responses induced by RTK ligands are a result of conformational coupling between the ECDs and the intracellular domains, in addition to kinetic proofreading. Lack of high resolution structures of full-length receptors precludes direct insights into such coupling, but several less direct approaches support the notion that at least in a number of receptors the conformational changes in different receptor domains are coordinated, and that coordination can be further tuned by ligand binding. An increasing number of structural and biochemical studies of the interactions between the transmembrane (TM) helices of RTKs suggest that these single spanning modules adopt different orientations depending on a ligand that engages the receptor, which in some cases affects the orientation of the adjacent intracellular JM domains [48–52]. In EGFR, this has been investigated using bipartite tetracysteine display, which relies on the presence of four cysteine residues that when brought into close proximity for long enough can form a fluorescent complex by coordinating bis-arsenical fluorophore (ReASH). In these experiments, the relative orientation of the JM domains containing engineered cysteines was used as a sensor of conformational states of ECDs upon binding to different ligands [53,54]. The JM domains have been previously shown to dimerize in the active receptor complex but the structure of this dimer in the context of the full-length receptor has not been solved [21,55]. The ReASH measurements showed that binding of seven cognate EGFR ligands to ECDs generates at least three distinct cytosolic juxtamembrane domain conformations propagated through unique configurations of the transmembrane domains [53,54,56]. Interestingly, while EGF and TGFα both stabilize symmetric ECD dimer structures and consequently stable dimers, their corresponding juxtamembrane domain arrangements within a dimer seem to differ (denoted as ‘EGF-type’ and ‘TGFα-type’ in Figure 2). Thus the intracellular JM and kinase conformations seem sensitive to the identity of a bound ligand.
Recent biochemical studies in cells provide evidence that different FGF ligands might also stabilize structurally unique conformations of full-length FGF receptors (FGFRs), providing an explanation for observed biased agonism. Most FGF ligands serve as autocrine and paracrine growth factors and activate their cognate receptors in a heparin or heparan sulfate proteoglycan-dependent manner [6]. When engineered constructs of FGFR1, FGFR2 and FGFR3, in which the ICDs are replaced by fluorescent proteins as FRET pairs, are stimulated with saturating levels of FGF1 or FGF2, FRET signals are different for each ligand suggesting different conformational states [50]. These states then are coupled to unique receptor activation profiles, as evidenced in case of FGFR1 and FGFR3, where saturating FGF2 levels induced stronger receptor phosphorylation than treatment with FGF1. While these studies do not provide direct structural evidence for the existence of multiple, conformationally distinct active FGFR complexes at the plasma membrane, they are in agreement with such structures existing and being functionally unique.
Are high resolution structures of full-length RTKs attainable? As of now, numerous full-length RTKs have been characterized by lower resolution negative-stain electron microscopy, including the EGF, PDGFRβ, Insulin and Kit receptors [57–61]. While these studies do not yet offer sufficient detail to discriminate between conformational changes resulting from binding of different ligands, they do show promise in capturing distinct conformations of full-length receptor dimers [57–60]. First cryo-EM attempts have been recently published for the Insulin and type-I Insulin-like growth factor receptors [62,63]. These receptors form disulfide-linked constitutive dimers even in the absence of a ligand and are activated by conformational rearrangements within the ECD dimer upon ligand binding [57,62,64–70]. It is still unclear how kinase domains of the Insulin and Insulin-like growth factor receptor behave before and after ligand binding in the full-length receptor complex. Structural studies on intracellular modules of this receptor alone have identified distinct inactive and active dimer complexes [71–75]. Regretfully, the cryo-EM analysis, which was conducted on detergent-solubilized receptors, failed to resolve intracellular kinase domains raising a concern that full-length receptor might be too flexible to enable high resolution structure determination. However, it is likely that this flexibility is ameliorated by the presence of the membrane bilayer with which receptors, such as EGFR have been speculated to extensively interact [48,76–78]. Such interactions would not be adequately modelled experimentally in detergent micelles, but rather by utilization of membrane mimetics, such as nanodiscs [79].
Ligands can induce distinct changes in receptor oligomerization
In addition to stabilizing structurally distinct RTK homodimers with different half-lives, a number of growth factors promotes association of RTKs into higher-order oligomers. Such is the case for ephrins, which are membrane tethered growth factors that engage Eph receptors on neighboring cells in a juxtacrine fashion [5]. This leads to activation of many distinct signaling pathways with often paradoxical cellular responses [5,80]. For instance, receptor activation can either lead to strong cell adhesion or facilitate cell separation, all within the same cellular context [81–83]. Activation of Eph receptors by ephrins proceeds through formation of higher-order receptor oligomers that condensate in a time- and concentration-dependent manner into aggregates that eventually become inactive [84]. It is known that different Eph receptors can form structurally distinct clusters [83,85,86]. Recent FRET studies provided evidence that different EphA2 agonists can induce conformationally distinct dimeric units of the same receptor, resulting in formation of structurally distinct oligomers [87]. The extent to which other Eph receptors can flexibly adopt different oligomeric structures depending on a bound ephrin, as well as the functional consequences of these distinct structures for Eph receptor signaling, are yet to be comprehensively analyzed.
An intricate interplay between structural differences of ligand-bound receptors and their distinct propensities for higher-order oligomerization over time appears to also play an important role in determination of the signaling outcome from the EGFR/HER receptors. Upon EGF binding, EGFR is often observed to form large clusters on the plasma membrane that appear to have functional consequences on downstream signaling [88–96]. The extent of oligomerization under different conditions and how such different oligomeric EGFR/HER states affect signaling still remains to be fully understood, but they clearly matter. For example - like the native ligand, EGF, synthetic EGFR ligands and some therapeutic antibodies can constitutively dimerize the receptor and potently stimulate its phosphorylation, however they fail to activate the Ras/MAPK pathway (Figure 3A) [97,98]. In case of the synthetic ligands, this effect is correlated with lack of receptor clustering [99]. Inability of phosphorylated EGFR to form clusters and signal in response to these ligands suggests that they might fail to induce a specific EGFR conformation that is required to form functional higher order oligomers. What is this conformation is unclear because high-resolution structures of higher-order oligomeric EGFR states do not exist, and thus far only their molecular models emerged from molecular dynamics simulations [94,96,100]. Ligands could possibly additionally tune these oligomeric receptor states by stabilizing distinct ECD structures, as observed in crystal structures of EGFR ECDs with different ligands [18,19,101]. While EGFR clusters have been frequently observed, it is important to note that receptor dimers seem to be the main population of EGFR on the cell surface [102]. Ligand binding is also not absolutely necessary for EGFR to signal as indicated by the ability of certain oncogenic mutations in EGFR to promote EGFR dimerization and activation via the Ras/MAPK pathway in a ligand-independent manner [103,104]. Thus, the exact differences in structural identity of different EGFR oligomeric states and their contribution to signaling remain an open question.
Figure 3: Ligands can diversify RTK signaling via promotion of higher-order receptor oligomers.
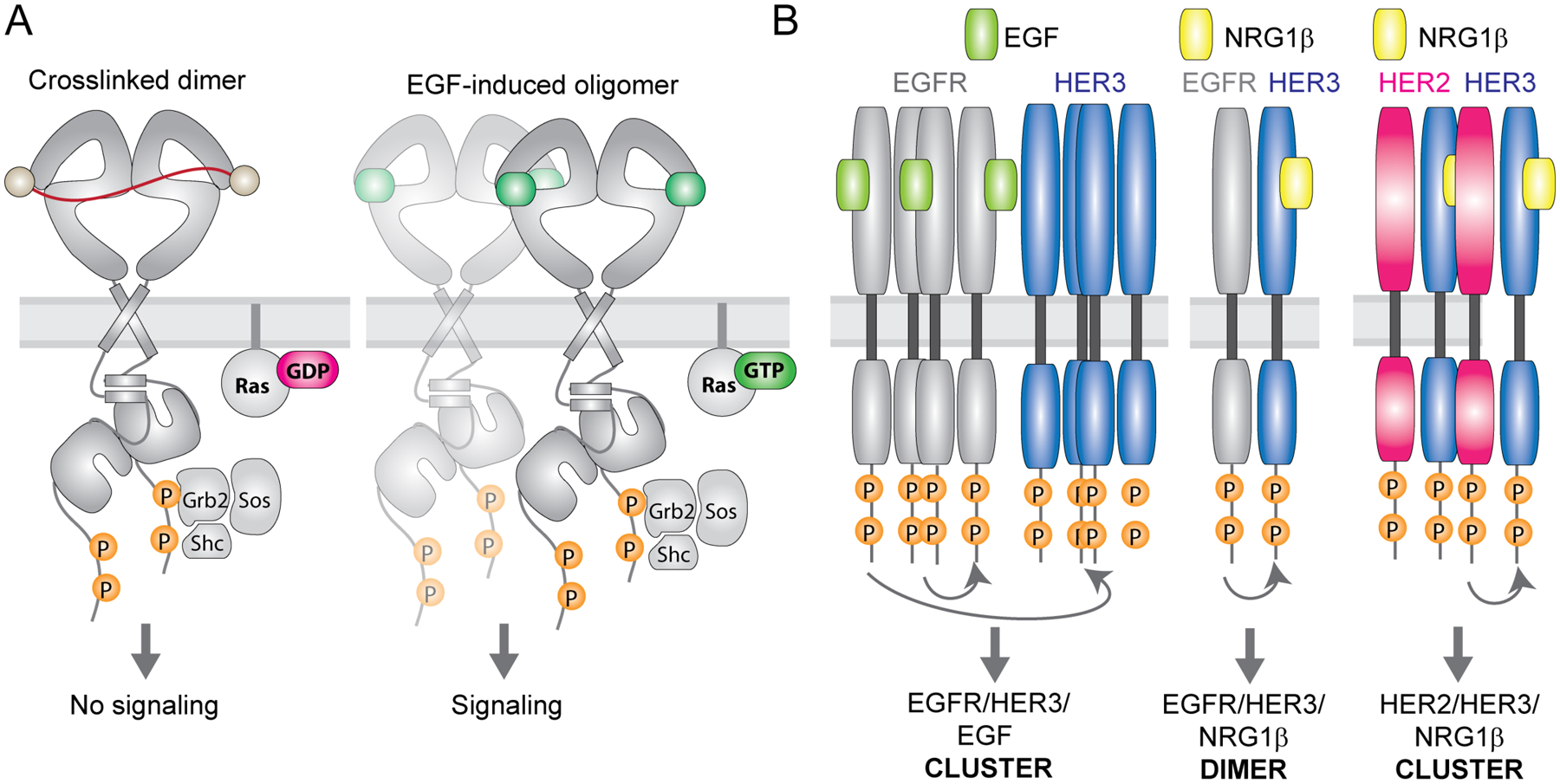
(A) Cartoon representation of active EGFR dimers in the plasma membrane dimerized via DNA-bases crosslinkers (left) or upon EGF binding (right). Both receptors undergo autophosphorylation but only EGF-bound receptors further progress into clusters and induce signaling via Ras. (B) Simplified illustration of EGFR, HER2 and HER3 oligomerization patterns upon EGF or NRG1β stimulation in cells. EGF treatment of cells co-expressing EGFR and HER3 causes phosphorylation and clustering of both receptors with HER3 phosphorylation occuring via a non-canonical mechanism that does not rely on asymmetric dimerization of the kinase domains. In contrast, NRG1β treatment induces HER3 phosphorylation by EGFR via canonical asymmetric kinase dimerization, without promoting clustering of the receptors. In yet another scenario, NRG1β stimulation of HER2/HER3 expressing cells induces clusters of both receptors in which HER3 phosphorylation is dependent on formation of asymmetric kinase dimers.
HER receptor ligands also act as biased agonists by preferentially stabilizing unique hetero-oligomeric receptor complexes [29,37,92]. Due to cross-reactivity of these ligands with different receptors, studies of such effects need to be conducted under conditions when HER receptor expression is known and controlled. We have shown that the catalytically inactive member of the EGFR family, HER3, organizes into larger complexes at the plasma membrane with its dimerization partners EGFR and HER2 but does so differently depending on the bound ligand (Figure 3B) [92]. In response to EGF, HER3 clusters with EGFR, which subsequently phosphorylates HER3 without engaging it in a canonical allosteric kinase dimer. In contrast, in response to NRG1β, HER3 and EGFR form the allosteric kinase dimer which does not evolve with time into higher-order clusters. The same ligand, NRG1β, leads to formation of HER3 clusters when HER2 is its dimerization partner. In this case signaling depends on the allosteric kinase dimer interface. Another study found that the EGFR ligands EGF and BTC have different effects on EGFR interaction with HER3. BTC induced weaker EGFR but stronger HER3 phosphorylation than EGF, leading to increased cell migration [29]. This suggests that EGF might preferentially stabilize EGFR homodimers while BTC promotes EGFR/HER3 heterodimerization. Given that each HER receptor has distinct tyrosine phosphorylation sites that serve as docking sites for downstream signaling proteins, such ligand-specific, biased formation of signaling complexes will result in distinct signaling outcomes. Thus, promotion of different heterodimers and/or oligomers emerges as important ways by which ligands establish the astounding combinatorial potential of the HER family of receptors to signal.
Ligand-distinct signaling can be mediated by co-receptors
A number of RTKs have low intrinsic affinity for their cognate ligands and either partially or fully rely on non-catalytic co-receptors for ligand binding and activation. Examples include the MuSK, FGF and Ret receptor families [7,9,105–107]. The RET receptor offers a particularly captivating case of such regulation as illustrated recently by elegant structural analysis of its ECD complexes with different co-receptors. Upon activation, RET interacts with one of four glial cell-derived growth factor (GDNF) receptor α family members (GFRα 1–4) or with GFRα-like protein (GFRAL) which act as non-catalytic co-receptors. These interactions are mediated by specific ligands: GDNF, Neurturin (NRTN), Artemin (ARTN), Persephin or GDF15, respectively, and result in unique cellular responses (Figure 4A) [106,108–112]. Cryo-EM structures of four different RET ECD/co-receptor/ligand complexes outline common features of the receptor activation mechanism by the four ligands that involves the formation of V-shaped complexes with 2:2:2 stoichiometry (Figure 4B) [113]. This unifying principle is then customized by each ligand which stabilizes a unique angle of protomers within the V-shaped structure, suggesting a mechanism for how they lead to distinct signaling outputs. One of these ECD complexes, NRTN/RET/GFRa2, further oligomerizes into tetramers, which show a reduced rate of endocytosis, prolonged signaling and unique signaling profiles [113,114]. Exactly how these structural differences in RET ECD complexes translate into activation of the kinase domain modules and further to distinct signaling remains to be shown.
Figure 4: Co-receptors assure ligand specificity and control of downstream signaling in the FGF and RET signaling systems.
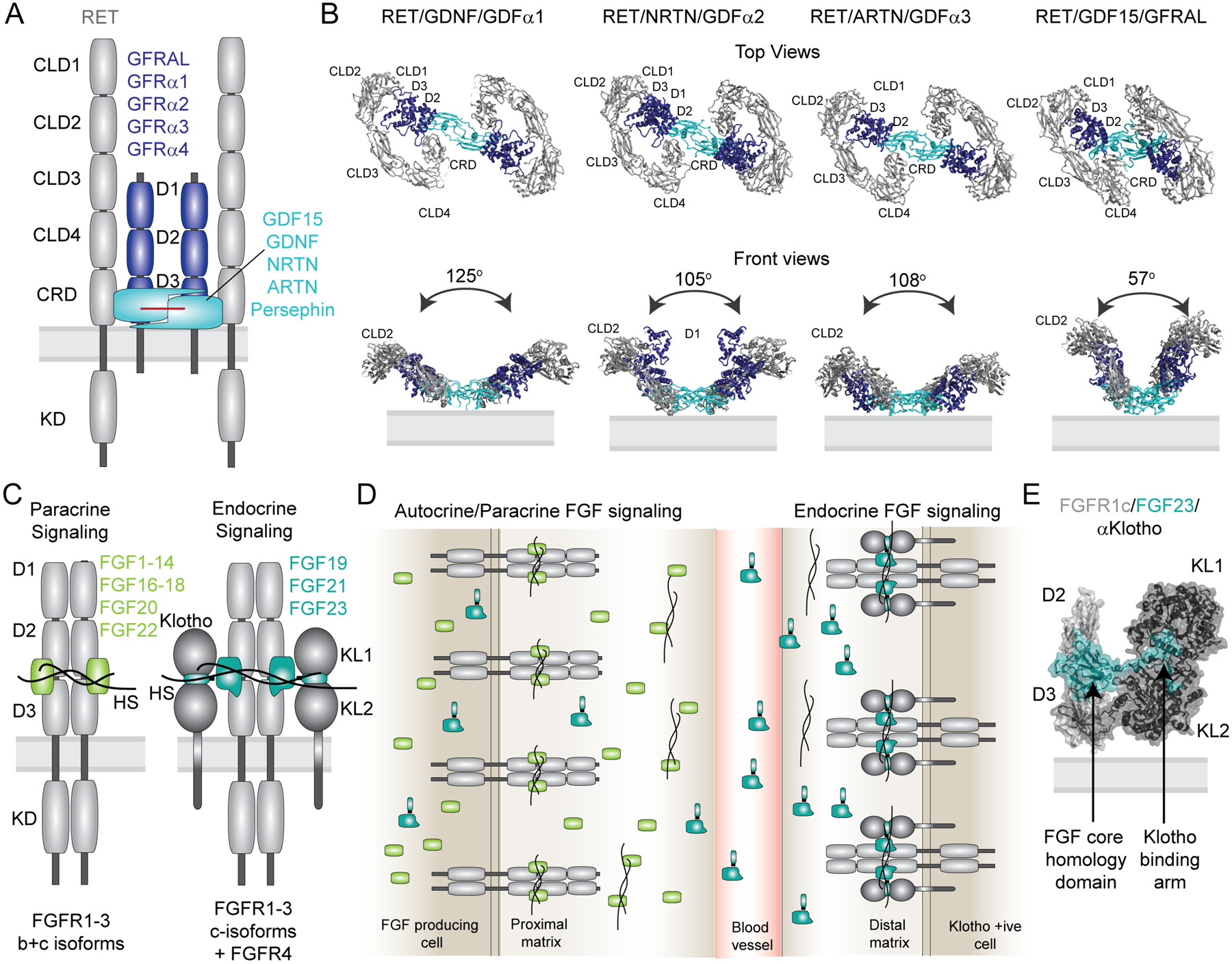
(A) Domain architecture of the 2/2/2 RET receptor/co-receptor/ligand complex. The receptor with its structural domains is shown in grey. The co-receptors GFRAL and GFRα1–4 are shown in blue and the ligands GDF15, GDNF, NRTN, ARTN, Persephin are shown in cyan. (B) Cryo-EM structures of the 2:2:2 RET ECD receptor/co-receptor/ligand complexes are shown as top and front views (PDB codes from left to right: 6Q2N, 6Q2O, 6Q2S, 6Q2J). Adapted from [113]. (C) Domain architecture of the autocrine/paracrine and endocrine active 2:2 and 2:2:2 complexes of receptor/ligand and receptor/ligand/Klotho complexes, respectively. Both representations illustrate heparan sulfate (HS) bound to the complexes. (D) Simplified overview of receptor specificity for autocrine/paracrine and endocrine ligands. Paracrine/autocrine ligands bind to HS with high affinity, which traps the ligands close to the site of secretion and enables them to bind the cognate receptors. Autocrine/paracrine ligands can be either specific towards FGFR1–3b or c isoforms or bind both isoforms promiscuously. The presence of HS is mandatory for activation by the paracrine/autocrine ligands. Endocrine ligands FGF19, FGF21 and FGF23 have low affinity for both FGFR isoforms and HS and require the presence of either α-Klotho or β-Klotho co-receptors for binding and activation of their cognate FGF receptors. The low affinity of endocrine ligands to HS facilitates secretion from the tissue of origin to distal organs that express the respective Klotho/FGFRc complexes. Despite low affinity in the absence of Klotho, HS appears to be required for receptor dimerization upon ligand binding. (E) Crystal structure of the FGFR1c/α-Klotho/FGF23 ECD complex (PDB code: 5W21). The structure illustrates tight interactions between FGF23 and the α-Klotho ECD via the Klotho binding arm, and with the receptor ECD via a truncated FGF core homology domain.
Similar to RET receptors, all four human FGFR family members rely on the presence of co-factors for activation by FGF ligands. Depending on their distribution within the body, FGF ligands are categorized as paracrine/autocrine or endocrine FGFs. Paracrine/Autocrine ligands (FGF 1–14, FGF16–18, FGF20, FGF22) generally have high affinity for heparan sulfate, which concentrates the ligands at the site of secretion and is obligatory for receptor binding and activation (Figure 4C+D) [115,116]. Endocrine FGF ligands (FGF19, FGF21, FGF23) have lower affinity for heparan sulfate, allowing them to enter the blood stream and act as functionally distinct hormones. Due to their low affinity for both heparan sulfate and receptors, endocrine FGFs require the co-receptors α- or β−Klotho, which preassemble with FGFRc receptor isoforms creating a high affinity ligand-binding site (Figure 4C+D) [9,107]. This specific, bipartite recognition of two receptors by the ligand has recently been unraveled by several crystal structures [117–119]. The ECD complex of the FGFR1c receptor bound to α−Klotho and FGF23 revealed that Klotho engages the C-terminal tail region of the ligands, which is unique to endocrine FGFs (Figure 4C) [119]. Interestingly, the complex could only be crystalized in the presence of heparan sulfate, which, despite its reduced affinity to endocrine ligands, was still required for receptor dimerization. Thus, numerous FGF ligands explore a broad array of mechanisms to stabilize unique ligand/receptor complexes in different tissues, linking them to distinct signaling responses.
Regulation by endocytosis
RTK activation generally leads to rapid internalization of activated receptor complexes. As illustrated by the example of the RET receptor [113], different ligands can alter signaling outcomes by altering the kinetics of RTK endocytosis. In case of EGFR, stimulation via TGFα and EGF leads to comparable rates of endocytosis, but EGF/EGFR-containing early endosomes more frequently fuse with lysosomes than those carrying TGFα/EGFR complexes, possibly due to a more stable interaction between EGF and EGFR under acidifying conditions [120,121]. In result, EGF is more efficient in promoting EGFR degradation terminating signaling. In contrast, a significant portion of TGFα/EGFR-containing endosomes is recycled back to the plasma membrane, keeping the cell more responsive to TGFα over time and changing the overall cellular response to the two ligands.
In addition to signaling from the plasma membrane, many RTKs continue to signal from intracellular compartments generating qualitatively different outputs [122–125]. Thus differential regulation of subcellular distribution of RTKs by different ligands could be another mechanism behind biased agonism. In support of this notion comes a recent study showing that the ability of TGFα and EGF to induce distinct dynamic changes in the EGFR interactome, ubiquitome, phosphoproteome and late proteome is dependent on the trafficking route of EGFR [126].
Conclusions
RTK ligands employ a wide array of mechanisms to achieve unique RTK signaling outcomes. But how cells differentiate between different ligand/receptor complexes to orchestrate these unique responses is still a big mystery. We are only starting to discover the detailed molecular mechanisms governing the formation of ligand/receptor assemblies and how their different structure, composition and/or stability are interpreted by intracellular signaling machinery. Recent advancements in high resolution structural studies by cryo-EM of these “difficult” targets bring promise of many exciting future discoveries that will help to uncover the architecture of such complexes and to provide new clues to their regulation.
Acknowledgments
We sincerely apologize to all colleagues whose work was omitted in this review due to space constraints. N.J. would like to acknowledge support from the National Institute of General Medical Sciences (R01 GM109176), National Cancer Institute (R01 CA230263) and the UCSF Program in Breakthrough Biomedical Research (PBBR).
References
* of special interest
** of outstanding interest
- 1.Lemmon MA, Schlessinger J: Cell signaling by receptor tyrosine kinases. Cell 2010, 141:1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falls D: Neuregulins: functions, forms, and signaling strategies. Experimental Cell Research 2003, 284:14–30. [DOI] [PubMed] [Google Scholar]
- 3.Wieduwilt MJ, Moasser MM: The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cellular and Molecular Life Sciences 2008, 65:1566–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flanagan JG, Gale NW, Hunter T, Pasquale EB, TessierLavigne M: Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 1997, 90:403–404. [DOI] [PubMed] [Google Scholar]
- 5.Pasquale EB: Eph receptor signalling casts a wide net on cell behaviour. Nature Reviews Molecular Cell Biology 2005, 6:462–475. [DOI] [PubMed] [Google Scholar]
- 6.Itoh N, Ornitz DM: Evolution of the Fgf and Fgfr gene families. Trends in Genetics 2004, 20:563–569. [DOI] [PubMed] [Google Scholar]
- 7.Ornitz DM, Itoh N: Fibroblast growth factors. Genome Biology 2001, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen SK: Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes & Development 2006, 20:185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu M-C, Moe OW, et al. : Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. Journal of Biological Chemistry 2006, 281:6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ: Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacology & Therapeutics 2009, 122:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bareja A, Patel S, Hodgkinson CP, Payne A, Dzau VJ: Understanding the mechanism of bias signaling of the insulin-like growth factor 1 receptor: Effects of LL37 and HASF. Cellular Signalling 2018, 46:113–119. [DOI] [PubMed] [Google Scholar]
- 12.Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM: Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nature Reviews Molecular Cell Biology 2018, 19:638–653. [DOI] [PubMed] [Google Scholar]
- 13.Smith JS, Lefkowitz RJ, Rajagopal S: Biased signalling: from simple switches to allosteric microprocessors. Nature Reviews Drug Discovery 2018, 17:243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilger D, Masureel M, Kobilka BK: Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology 2018, 25:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ: Functional antagonism of different G protein-coupled receptor kinases for -arrestin-mediated angiotensin II receptor signaling. Proceedings of the National Academy of Sciences 2005, 102:1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson K, Berger M, Mendrola J, Cho H-S, Leahy DJ, Lemmon MA: EGF Activates Its Receptor by Removing Interactions with Autoinhibit Ectodomain Dimerization. Molecular Cell 2003, 11:507–517. [DOI] [PubMed] [Google Scholar]
- 17.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM: Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Molecular and Cellular Biology 2005, 25:7734–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garrett TPJ, McKern NM, Lou MZ, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et al. : Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110:763–773. [DOI] [PubMed] [Google Scholar]
- 19.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim J-H, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. : Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110:775–787. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J: An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125:1137–1149. [DOI] [PubMed] [Google Scholar]
- 21.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J: Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009, 137:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan J: A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem 2015, 84:739–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh B, Carpenter G, Coffey RJ: EGF receptor ligands: recent advances. F1000Research 2016, 5:2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hotchin NA, Krall JA, Beyer EM, MacBeath G: High- and Low-Affinity Epidermal Growth Factor Receptor-Ligand Interactions Activate Distinct Signaling Pathways. PLoS ONE 2011, 6:e15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronan T, Macdonald-Obermann JL, Huelsmann L, Bessman NJ, Naegle KM, Pike LJ: Different Epidermal Growth Factor Receptor (EGFR) Agonists Produce Unique Signatures for the Recruitment of Downstream Signaling Proteins. Journal of Biological Chemistry 2016, 291:5528–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study links binding of seven EGFR ligands to different recruitment kinetics of downstream signaling effectors to the receptor.
- 26.Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, Valley CC, Ferguson KM, Leahy DJ, Lidke DS, et al. : EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171:683–+. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study presents crystal structures of the EGFR extracellular domain (ECD) bound to its two low affinity ligands, epigen and epiregulin, and shows that these ligands stabilize distinct dimeric ECD conformations from the previously seen for ECD complexes with EGF and TGFα. These ECD dimers are less stable and weakened EGFR dimerization via epigen and epiregulin is shown to induce more sustained downstream receptor signaling in comparison to EGF and TGFα.
- 27.Kochupurakkal BS, Harari D, Di-Segni A, Maik-Rachline G, Lyass L, Gur G, Kerber G, Citri A, Lavi S, Eilam R, et al. : Epigen, the Last Ligand of ErbB Receptors, Reveals Intricate Relationships between Affinity and Mitogenicity. Journal of Biological Chemistry 2005, 280:8503–8512. [DOI] [PubMed] [Google Scholar]
- 28.Rizzi M, Pittarella P, Sabbatini M, Renò F: Epiregulin induces human SK-N-BE cell differentiation through ERK1/2 signaling pathway. Growth Factors 2013, 31:90–97. [DOI] [PubMed] [Google Scholar]
- 29.Rush JS, Peterson JL, Ceresa BP: Betacellulin (BTC) Biases the EGFR To Dimerize with ErbB3. Molecular Pharmacology 2018, 94:1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito T, Okada S, Ohshima K, Yamada E, Sato M, Uehara Y, Shimizu H, Pessin JE, Mori M: Differential Activation of Epidermal Growth Factor (EGF) Receptor Downstream Signaling Pathways by Betacellulin and EGF. Endocrinology 2004, 145:4232–4243. [DOI] [PubMed] [Google Scholar]
- 31.Shin HS, Lee HJ, Nishida M, Lee M-S, Tamura R, Yamashita S, Matsuzawa Y, Lee I-K, Koh GY: Betacellulin and Amphiregulin Induce Upregulation of Cyclin D1 and DNA Synthesis Activity Through Differential Signaling Pathways in Vascular Smooth Muscle Cells. Circulation Research 2003, 93:302–310. [DOI] [PubMed] [Google Scholar]
- 32.Streicher KL, Willmarth NE, Garcia J, Boerner JL, Dewey TG, Ethier SP: Activation of a nuclear factor kappa B/interieukin-1 positive feedback loop by amphiregulin in human breast cancer cells. Molecular Cancer Research 2007, 5:847–861. [DOI] [PubMed] [Google Scholar]
- 33.Wilson KJ, Mill C, Lambert S, Buchman J, Wilson TR, Hernandez-Gordillo V, Gallo RM, Ades LMC, Settleman J, Riese DJ: EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 2012, 30:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeppe Knudsen SL, Wai Mac AS, Henriksen L, Deurs Bv, Grøvdal LM: EGFR signaling patterns are regulated by its different ligands. Growth Factors 2014, 32:155–163. [DOI] [PubMed] [Google Scholar]
- 35.Ødegård J, Sondresen JE, Aasrum M, Tveteraas IH, Guren TK, Christoffersen T, Thoresen GH: Differential effects of epidermal growth factor (EGF) receptor ligands on receptor binding, downstream signalling pathways and DNA synthesis in hepatocytes. Growth Factors 2018, 35:239–248. [DOI] [PubMed] [Google Scholar]
- 36.Willmarth NE, Ethier SP: Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human. Journal of Biological Chemistry 2006, 281:37728–37737. [DOI] [PubMed] [Google Scholar]
- 37.Macdonald-Obermann JL, Pike LJ: Different Epidermal Growth Factor (EGF) Receptor Ligands Show Distinct Kinetics and Biased or Partial Agonism for Homodimer and Heterodimer Formation. Journal of Biological Chemistry 2014, 289:26178–26188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sweeney C, Carraway KL: Ligand discrimination by ErbB receptors: differential signaling through differential phosphorylation site usage. Oncogene 2000, 19:5568–5573. [DOI] [PubMed] [Google Scholar]
- 39.Ferguson KM: Structure-based view of epidermal growth factor receptor regulation. Annual Review of Biophysics 2008, 37:353–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mckeithan TW: Kinetic Proofreading in T-Cell Receptor Signal-Transduction. Proceedings of the National Academy of Sciences of the United States of America 1995, 92:5042–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lew ED, Furdui CM, Anderson KS, Schlessinger J: The Precise Sequence of FGF Receptor Autophosphorylation Is Kinetically Driven and Is Disrupted by Oncogenic Mutations. Science Signaling 2009, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleiman LB, Maiwald T, Conzelmann H, Lauffenburger DA, Sorger PK: Rapid Phospho-Turnover by Receptor Tyrosine Kinases Impacts Downstream Signaling and Drug Binding. Molecular Cell 2011, 43:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanoev A, Mhamane A, Schuermann KC, Grecco HE, Stallaert W, Baumdick M, Bruggemann Y, Joshi MS, Roda-Navarro PR, Fengler S, et al. : Interdependence between EGFR and Phosphatases Spatially Established by Vesicular Dynamics Generates a Growth Factor Sensing and Responding Network. Cell Systems 2018, 7:295–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho CCM, Chhabra A, Starkl P, Schnorr P-J, Wilmes S, Moraga I, Kwon H-S, Gaudenzio N, Sibilano R, Wehrman TS, et al. : Decoupling the Functional Pleiotropy of Stem Cell Factor by Tuning c-Kit Signaling. Cell 2017, 168:1041–1052.e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study describes how stem cell factor was engineered to be a partial, biased agonist for its cognate receptor.
- 45.Zhang ZT, Zhang RG, Joachimiak A, Schlessinger J, Kong XP: Crystal structure of human stem cell factor: Implication for stem cell factor receptor dimerization and activation. Proceedings of the National Academy of Sciences of the United States of America 2000, 97:7732–7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Z, Tan Y, Gu J, Liu Y, Song L, Niu J, Zhao L, Srinivasan L, Lin Q, Deng J, et al. : Uncoupling the Mitogenic and Metabolic Functions of FGF1 by Tuning FGF1-FGF Receptor Dimer Stability. Cell Reports 2017, 20:1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang ZF, Tan Y, Gu JL, Liu Y, Song LT, Niu JL, Zhao LW, Srinivasan L, Lin Q, Deng JJ, et al. : Uncoupling the Mitogenic and Metabolic Functions of FGF1 by Tuning FGF1-FGF Receptor Dimer Stability. Cell Reports 2017, 20:1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, et al. : Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 2013, 152:543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franco ML, Nadezhdin KD, Goncharuk SA, Mineev KS, Arseniev AS, Vilar M: Structural basis of the transmembrane domain dimerization and rotation in the activation mechanism of the TRKA receptor by nerve growth factor. Journal of Biological Chemistry 2020, 295:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarabipour S, Hristova K: Mechanism of FGF receptor dimerization and activation. Nature Communications 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; **These investigations of the effect of binding of different ligands to FGFR receptors in cells on relative changes in conformations of the transmembrane and intracellular domains, demonstrates unique structures for each ligand, hence providing a mechanistic explanation for biased agonism.
- 51.Trenker R, Call MJ, Call ME: Progress and prospects for structural studies of transmembrane interactions in single-spanning receptors. Current Opinion in Structural Biology 2016, 39:115–123. [DOI] [PubMed] [Google Scholar]
- 52.Bocharov EV, Mineev KS, Pavlov KV, Akimov SA, Kuznetsov AS, Efremov RG, Arseniev AS: Helix-helix interactions in membrane domains of bitopic proteins: Specificity and role of lipid environment. Biochimica Et Biophysica Acta-Biomembranes 2017, 1859:561–576. [DOI] [PubMed] [Google Scholar]
- 53.Doerner A, Scheck R, Schepartz A: Growth Factor Identity Is Encoded by Discrete Coiled-Coil Rotamers in the EGFR Juxtamembrane Region. Chemistry & Biology 2015, 22:776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheck RA, Lowder MA, Appelbaum JS, Schepartz A: Bipartite tetracysteine display reveals allosteric control of ligand-specific EGFR activation. ACS Chem Biol 2012, 7:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G: The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 2009, 34:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinclair JKL, Walker AS, Doerner AE, Schepartz A: Mechanism of Allosteric Coupling into and through the Plasma Membrane by EGFR. Cell Chemical Biology 2018, 25:857–870.e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gutmann T, Kim KH, Grzybek M, Walz T, Coskun U: Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol 2018, 217:1643–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study describes reconstitution of a full-length insulin receptor in nanodiscs and its analysis by negative-stain EM. Ligand-bound receptor is shown to adopt large conformational changes relative to its ligand-free state. This structural study is one of the few conducted on a full-length RTK.
- 58.Mi LZ, Lu C, Li Z, Nishida N, Walz T, Springer TA: Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol 2011, 18:984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen PH, Unger V, He X: Structure of Full-Length Human PDGFRbeta Bound to Its Activating Ligand PDGF-B as Determined by Negative-Stain Electron Microscopy. J Mol Biol 2015, 427:3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Opatowsky Y, Lax I, Tome F, Bleichert F, Unger VM, Schlessinger J: Structure, domain organization, and different conformational states of stem cell factor-induced intact KIT dimers. Proc Natl Acad Sci U S A 2014, 111:1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diwanji D, Thaker T, Jura N: More than the sum of the parts: Toward full-length receptor tyrosine kinase structures. Iubmb Life 2019, 71:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uchikawa E, Choi E, Shang GJ, Yu HT, Bai XC: Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Choi E, Yu HT, Bai XC: Structural basis of the activation of type 1 insulin-like growth factor receptor. Nature Communications 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Several cryo-EM structures of the extracellular domain of the RET receptor bound to different co-receptors and ligands show that while the overall architecture of the different active complexes is the same, the relative orientation of the receptor ECDs differs between them. This study provides structural evidence that a single receptor can be engaged in conformationally distinct complexes with its co-receptors and ligands.
- 64.Ward CW, Lawrence MC: Similar but different: ligand-induced activation of the insulin and epidermal growth factor receptor families. Curr Opin Struct Biol 2012, 22:360–366. [DOI] [PubMed] [Google Scholar]
- 65.Ward CW, Menting JG, Lawrence MC: The insulin receptor changes conformation in unforeseen ways on ligand binding: sharpening the picture of insulin receptor activation. Bioessays 2013, 35:945–954, doi/9101002/bies 201370111. [DOI] [PubMed] [Google Scholar]
- 66.McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, Elleman TC, Richards KM, Bentley JD, Pilling PA, et al. : Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443:218–221. [DOI] [PubMed] [Google Scholar]
- 67.Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, Watson CJ, Zakova L, Kletvikova E, Jiracek J, et al. : How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scapin G, Dandey VP, Zhang Z, Prosise W, Hruza A, Kelly T, Mayhood T, Strickland C, Potter CS, Carragher B: Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature 2018, 556:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weis F, Menting JG, Margetts MB, Chan SJ, Xu Y, Tennagels N, Wohlfart P, Langer T, Muller CW, Dreyer MK, et al. : The signalling conformation of the insulin receptor ectodomain. Nat Commun 2018, 9:4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, Ramek A, Sarabipour S, Shan Y, Shaw DE, Hristova K, et al. : How IGF-1 activates its receptor. Elife 2014, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hubbard SR, Wei L, Ellis L, Hendrickson W: Crystal Structure of the Tyrosine Kinase Domain of the Human Insulin Receptor. Nature 1994, 372. [DOI] [PubMed] [Google Scholar]
- 72.Hubbard SR: Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. The EMBO Journal 1997, 16:5573–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Favelyukis S, Till JH, Hubbard SR, Miller WT: Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 2001, 8:1058–1063. [DOI] [PubMed] [Google Scholar]
- 74.Li S, Covino ND, Stein EG, Till JH, Hubbard SR: Structural and biochemical evidence for an autoinhibitory role for tyrosine 984 in the juxtamembrane region of the insulin receptor. J Biol Chem 2003, 278:26007–26014. [DOI] [PubMed] [Google Scholar]
- 75.Cabail MZ, Li S, Lemmon E, Bowen ME, Hubbard SR, Miller WT: The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat Commun 2015, 6:6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McLaughlin S, Smith SO, Hayman MJ, Murray D: An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. Journal of General Physiology 2005, 126:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sato T, Pallavi P, Golebiewska U, McLaughlin S, Smith SO: Structure of the membrane reconstituted transmembrane-juxtamembrane peptide EGFR(622–660) and its interaction with Ca2+/calmodulin. Biochemistry 2006, 45:12704–12714. [DOI] [PubMed] [Google Scholar]
- 78.Arkhipov A, Shan Y, Kim ET, Shaw DE: Membrane interaction of bound ligands contributes to the negative binding cooperativity of the EGF receptor. PLoS Comput Biol 2014, 10:e1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Denisov IG, Sligari SG: Nanodiscs in Membrane Biochemistry and Biophysics. Chemical Reviews 2017, 117:4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pasquale EB: Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133:38–52. [DOI] [PubMed] [Google Scholar]
- 81.Santiago A, Erickson CA: Ephrin-B ligands play a dual role in the control of neural crest cell migration. Development 2002, 129:3621–3632. [DOI] [PubMed] [Google Scholar]
- 82.Holmberg J, Clarke DL, Frisen J: Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature 2000, 408:203–206. [DOI] [PubMed] [Google Scholar]
- 83.Seiradake E, Schaupp A, del Toro Ruiz D, Kaufmann R, Mitakidis N, Harlos K, Aricescu AR, Klein R, Jones EY: Structurally encoded intraclass differences in EphA clusters drive distinct cell responses. Nature Structural & Molecular Biology 2013, 20:958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ojosnegros S, Cutrale F, Rodriguez D, Otterstrom JJ, Chiu CL, Hortiguela V, Tarantino C, Seriola A, Mieruszynski S, Martinez E, et al. : Eph-ephrin signaling modulated by polymerization and condensation of receptors. Proceedings of the National Academy of Sciences of the United States of America 2017, 114:13188–13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, et al. : Architecture of Eph receptor clusters. Proceedings of the National Academy of Sciences of the United States of America 2010, 107:10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY: An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nature Structural & Molecular Biology 2010, 17:398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singh DR, Kanvinde P, King C, Pasquale EB, Hristova K: The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Communications Biology 2018, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ariotti N, Liang H, Xu Y, Zhang Y, Yonekubo Y, Inder K, Du G, Parton RG, Hancock JF, Plowman SJ: Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Mol Cell Biol 2010, 30:3795–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clayton AH, Orchard SG, Nice EC, Posner RG, Burgess AW: Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors 2008, 26:316–324. [DOI] [PubMed] [Google Scholar]
- 90.Clayton AH, Walker F, Orchard SG, Henderson C, Fuchs D, Rothacker J, Nice EC, Burgess AW: Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J Biol Chem 2005, 280:30392–30399. [DOI] [PubMed] [Google Scholar]
- 91.Saffarian S, Li Y, Elson EL, Pike LJ: Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys J 2007, 93:1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Lengerich B, Agnew C, Puchner EM, Huang B, Jura N: EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proceedings of the National Academy of Sciences 2017, 114:E2836–E2845. [DOI] [PMC free article] [PubMed] [Google Scholar]; *HER3 organizes into higher-order oligomers at the plasma membrane when co-expressed with its dimerization partners HER2 and EGFR, but does it differently depending on which dimerization partner and which ligands are present. This study suggests that different ligands can induce distinct signaling responses by exploring unique HER receptor hetero-dimerization and oligomerization behavior.
- 93.Ichinose J, Murata M, Yanagida T, Sako Y: EGF signalling amplification induced by dynamic clustering of EGFR. Biochemical and Biophysical Research Communications 2004, 324:1143–1149. [DOI] [PubMed] [Google Scholar]
- 94.Huang Y, Bharill S, Karandur D, Peterson SM, Marita M, Shi X, Kaliszewski MJ, Smith AW, Isacoff EY, Kuriyan J: Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozer N, Barua D, Orchard S, Nice EC, Burgess AW, Hlavacek WS, Clayton AH: Exploring higher-order EGFR oligomerisation and phosphorylation--a combined experimental and theoretical approach. Mol Biosyst 2013, 9:1849–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Needham SR, Roberts SK, Arkhipov A, Mysore VP, Tynan CJ, Zanetti-Domingues LC, Kim ET, Losasso V, Korovesis D, Hirsch M, et al. : EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat Commun 2016, 7:13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang SI, van Lengerich B, Eichel K, Cha M, Patterson DM, Yoon TY, von Zastrow M, Jura N, Gartner ZJ: Phosphorylated EGFR Dimers Are Not Sufficient to Activate Ras. Cell Rep 2018, 22:2593–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study shows that formation of phosphorylated EGFR dimers by covalent cross-linking with engineered DNA ligands is not sufficient for downstream signaling via the Ras/MAPK pathway in contrast to stimulation with EGF. These findings underscore an importance of the native physiological structure of the active receptor complex for signaling and suggest that different ligands may differentially exploit an EGFR conformational landscape for generation of unique signaling outputs.
- 98.Yoshida T, Okamoto I, Okabe T, Iwasa T, Satoh T, Nishio K, Fukuoka M, Nakagawa K: Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. International Journal of Cancer 2008, 122:1530–1538. [DOI] [PubMed] [Google Scholar]
- 99.Liang SI, van Lengerich B, Eichel K, Cha M, Patterson DM, Yoon T-Y, von Zastrow M, Jura N, Gartner ZJ: Phosphorylated EGFR Dimers Are Not Sufficient to Activate Ras. Cell Reports 2018, 22:2593–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zanetti-Domingues LC, Korovesis D, Needham SR, Tynan CJ, Sagawa S, Roberts SK, Kuzmanic A, Ortiz-Zapater E, Jain P, Roovers RC, et al. : The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nature Communications 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, Valley CC, Ferguson KM, Leahy DJ, Lidke DS, et al. : EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171:683–695.e618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I: Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464:783–787. [DOI] [PubMed] [Google Scholar]
- 103.Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao DW, Habib AA: Ligand-Independent EGFR Signaling. Cancer Research 2015, 75:3436–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valley CC, Arndt-Jovin DJ, Jovin TM, Steinkamp MP, Chizhik AI, Karedla N, Hlavacek WS, Wilson BS, Lidke KA, Lidke DS: Inside-Out Signaling of Oncogenic EGFR Mutants Promotes Ligand-Independent Dimerization. Biophysical Journal 2015, 108:351a–351a. [Google Scholar]
- 105.Zong Y, Jin R: Structural mechanisms of the agrin–LRP4–MuSK signaling pathway in neuromuscular junction differentiation. Cellular and Molecular Life Sciences 2012, 70:3077–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ibanez CF: Structure and Physiology of the RET Receptor Tyrosine Kinase. Cold Spring Harbor Perspectives in Biology 2013, 5:a009134–a009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuro-o M: The Klotho proteins in health and disease. Nature Reviews Nephrology 2018, 15:27–44. [DOI] [PubMed] [Google Scholar]
- 108.Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Milbrandt J: Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFR alpha 3-RET receptor complex. Neuron 1998, 21:1291–1302. [DOI] [PubMed] [Google Scholar]
- 109.Durbec P, MarcosGutierrez CV, Kilkenny C, Grigoriou M, Wartiovaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, et al. : GDNF signalling through the Ret receptor tyrosine kinase. Nature 1996, 381:789–793. [DOI] [PubMed] [Google Scholar]
- 110.Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Milbrandt J: Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 1996, 384:467–470. [DOI] [PubMed] [Google Scholar]
- 111.Milbrandt J, de Sauvage FJ, Fahrner TJ, Baloh RH, Leitner ML, Tansey MG, Lampe PA, Heuckeroth RO, Kotzbauer PT, Simburger KS, et al. : Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron 1998, 20:245–253. [DOI] [PubMed] [Google Scholar]
- 112.Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, SalazarGrueso E, Pachnis V, Arumae U, et al. : Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature 1996, 381:785–789. [DOI] [PubMed] [Google Scholar]
- 113.Li J, Shang GJ, Chen YJ, Brautigam CA, Liou J, Zhang XW, Bai XC: Cryo-EM analyses reveal the common mechanism and diversification in the activation of RET by different ligands. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Richardson DS, Lai AZ, Mulligan LM: RET ligand-induced internalization and its consequences for downstream signaling. Oncogene 2006, 25:3206–3211. [DOI] [PubMed] [Google Scholar]
- 115.Hidai C: FGF-1 enhanced cardiogenesis in differentiating embryonal carcinoma cell cultures, which was opposite to the effect of FGF-2. Journal of Molecular and Cellular Cardiology 2003, 35:421–425. [DOI] [PubMed] [Google Scholar]
- 116.Belov AA, Mohammadi M: Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harbor Perspectives in Biology 2013, 5:a015958–a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kuzina ES, Ung PM-U, Mohanty J, Tome F, Choi J, Pardon E, Steyaert J, Lax I, Schlessinger A, Schlessinger J, et al. : Structures of ligand-occupied β-Klotho complexes reveal a molecular mechanism underlying endocrine FGF specificity and activity. Proceedings of the National Academy of Sciences 2019, 116:7819–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee S, Choi J, Mohanty J, Sousa LP, Tome F, Pardon E, Steyaert J, Lemmon MA, Lax I, Schlessinger J: Structures of β-klotho reveal a ‘zip code’-like mechanism for endocrine FGF signalling. Nature 2018, 553:501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu M-C, Moe OW, Liang G, Li X, Mohammadi M: α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study reports a crystal structure of the ECD module of FGF23 bound to FGFR1c and the co-receptor αKlotho and describes a mechanism for FGF receptor activation by low affinite how endocrine ligands.
- 120.Ebner R, Derynck R: Epidermal Growth-Factor and Transforming Growth Factor-Alpha - Differential Intracellular Routing and Processing of Ligand-Receptor Complexes. Cell Regulation 1991, 2:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.French AR, Tadaki DK, Niyogi SK, Lauffenburger DA: Intracellular Trafficking of Epidermal Growth-Factor Family Ligands Is Directly Influenced by the Ph Sensitivity of the Receptor-Ligand Interaction. Journal of Biological Chemistry 1995, 270:4334–4340. [DOI] [PubMed] [Google Scholar]
- 122.Bakker J, Spits M, Neefjes J, Berlin I: The EGFR odyssey - from activation to destruction in space and time. Journal of Cell Science 2017, 130:4087–4096. [DOI] [PubMed] [Google Scholar]
- 123.Villasenor R, Nonaka H, Del Conte-Zerial P, Kalaidzidis Y, Zerial M: Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miaczynska M: Effects of Membrane Trafficking on Signaling by Receptor Tyrosine Kinases. Cold Spring Harbor Perspectives in Biology 2013, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bergeron JJM, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI: Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annual Review of Biochemistry, Vol 85 2016, 85:573–597. [DOI] [PubMed] [Google Scholar]
- 126.Francavilla C, Papetti M, Rigbolt KTG, Pedersen A-K, Sigurdsson JO, Cazzamali G, Karemore G, Blagoev B, Olsen JV: Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nature Structural & Molecular Biology 2016, 23:608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wendler F: The LMTK-family of kinases: Emerging important players in cell physiology and disease pathogenesis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2018. [DOI] [PubMed] [Google Scholar]