

Abstract
Introduction
Macrophages and neutrophils have been separately implicated in cerebral aneurysm formation. The interactions between different myeloid subsets and the contributions of macrophage phenotypes in these lesions over time are not known. The purpose of the study was to examine macrophage phenotypic changes in cerebral aneurysms.
Methods
We induced aneurysm formation in C57BL/6 mice and quantified contributions of M1 and M2 macrophages in aneurysm specimens with or without neutrophil blockade. In our aneurysm model, the left common carotid and right renal arteries were ligated, and mice were placed on a hypertensive high fat diet. One week later, stereotactic injection with elastase solution into the basal cisterns was performed. An angiotensin II secreting osmotic pump was implanted. The mice were then treated with anti-CXCL1 antibody or IgG control antibody. Animals were euthanized at 3 days, or 1 or 2 weeks. The circle of Willis was analyzed using immunohistochemistry for M1 and M2 macrophage phenotype contributions.
Results
Proinflammatory M1/M2 ratio increased in cerebral aneurysm formation over time, from 0.56 at 3 days to 1.75 at 2 weeks (p<0.0001). In contrast, anti-CXCL1 antibody blockade led to polarization towards an anti-inflammatory phenotype with an M1/M2 ratio of 0.95 at 2 weeks compared with IgG treated mice (p=0.0007).
Conclusions
CXCL1 dependent neutrophil inflammation appears to have an important role in macrophage polarization to M1 phenotype in cerebral aneurysm development.
INTRODUCTION
Cerebral aneurysm (CA) development is driven by inflammation and hemodynamic stress.1 It is characterized by dysfunctional endothelium, secretion of matrix metalloproteinases, and activity of innate immune cells.1,2 Previous studies of myeloid cells in CA progression showed that depleting macrophages alone prevents lesion formation.1–4 Conversely, we reported that blockade of CXCL1 decreased murine CA formation by significantly decreasing neutrophil, but not macrophage, infiltration.5 To resolve this conflict, we examined the phenotypic changes in macrophages within aneurysms with and without neutrophils. We reasoned that it is the macrophage phenotype rather than the total burden that is responsible for CA formation.
METHODS
Animals
All animal experimentation was performed under the Institutional Animal Care and Use Committee (IACUC) approved protocol No 201303748 and the guidelines ‘Animal Research: Reporting in Vivo Experiments’. The animals were monitored daily for default IACUC endpoints.
Murine intracranial aneurysm model
Intracranial aneurysms were created in female 8–12-week-old C57BL/6 mice (Charles River Laboratories, Wilmington, Massachusetts, USA) using a previously described method with ligations of the left common carotid and right renal arteries; angiotensin II (Bachem AG, Switzerland) at 1000 ng/kg/min (via a micro-osmotic pump implanted subcutaneously); intracranial injection of 10 μL of 0.8% porcine elastase (Worthington Biochemical Corp, Lakewood, New Jersey, USA); and hypertensive diet with 8% NaCl and 0.12% β-aminopropionitrile (Harlan Laboratories, Indianapolis, Indiana, USA) (figure 1).5,6 This model replicates all features of human CAs: inflammatory infiltration, angiogenesis, robust aneurysm formation, and rupture.6,7
Figure 1.
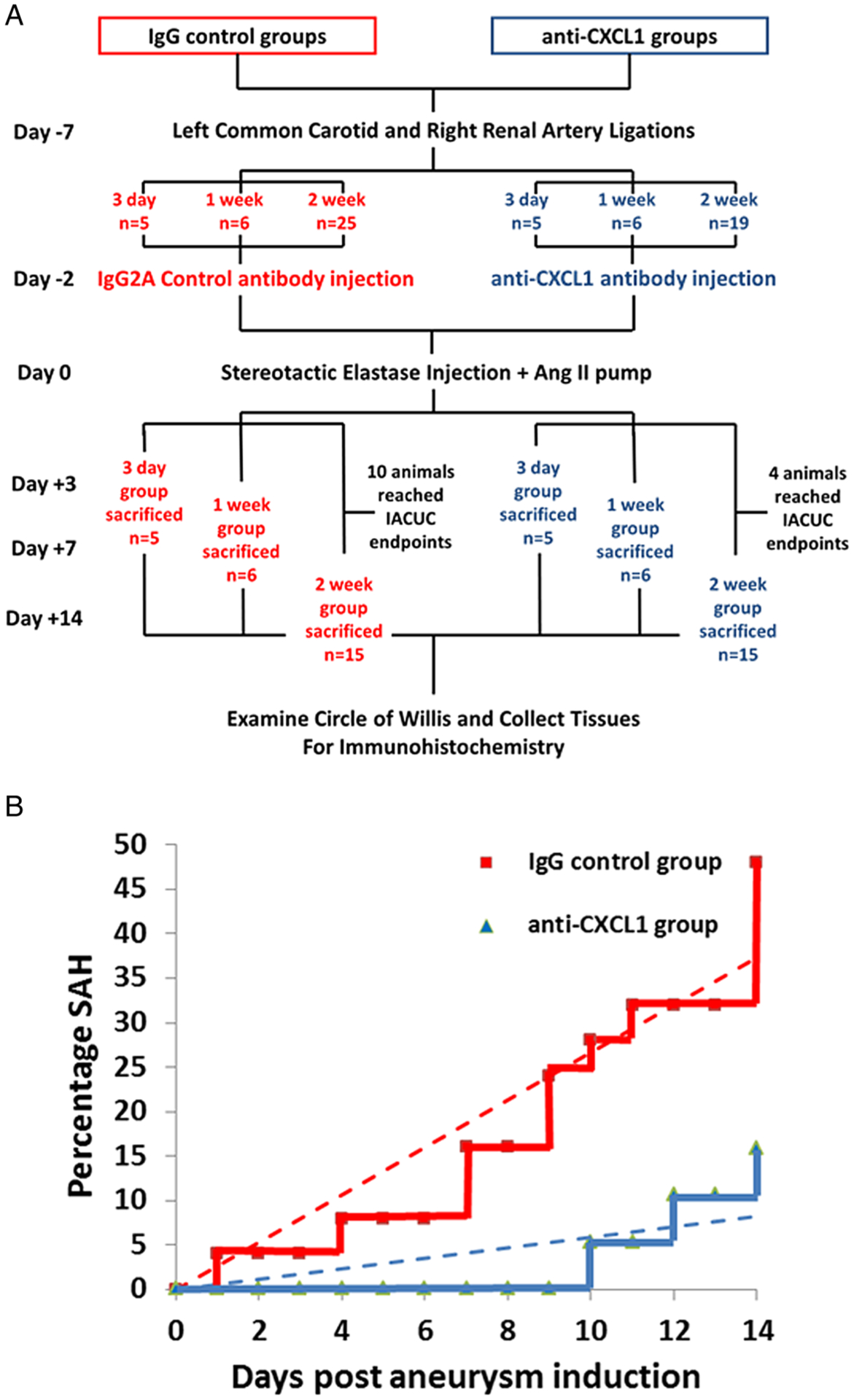
Experimental scheme. (A) Protocol flowchart and (B) associated mortality due to subarachnoid hemorrhage (trendlines superimposed). IACUC, Institutional Animal Care and Use Committee.
The animals were housed in an SPF facility with a 12/12 hour light/dark cycle, and were randomly assigned to groups. In the anti-CXCL1 treatment group, 0.2 mL of 100 μg/mL anti-CXCL1/GRO-α/KC/CINC-1 antibody (MAB453; R&D Systems, Minneapolis, Minnesota, USA) were injected retro-orbitally 2 days before, on the day of, and every 2 days after aneurysm induction. Retro-orbital venous plexus injection is routinely used to gain access to the intravascular compartment.8 In IgG controls, 0.2 mL of 100 μg/mL rat IgG2A isotype control antibody (MAB006; R&D Systems) was injected every 2 days. According to the manufacturer, the neutralization dose (ND50) is 0.05–0.25 μg/mL. For both antibodies, the final delivered dose was 1 μg/1 g animal body weight.
Mice were euthanized 3 days, or 1 or 2 weeks after aneurysm induction (5, 6, and 15 animals per time point for each group, respectively). To account for decreased survival due to default IACUC endpoints, 25 animals were initially included in the 2 week IgG group with 10 animals not reaching the experimental time point (figure 1A). In the 2 week anti-CXCL1 group, 19 animals were initially included, with 4 animals not surviving until the endpoint. One animal was excluded due to anesthetic overdose during surgery. In the 3 day and 1 week experimental groups, all animals reached the experimental time points. The number of animals per each time point was chosen based on prior expected variability in histopathological findings in the model.6
Aneurysm grading scale
Following animal euthanasia, each brain was collected and the circle of Willis carefully examined under a light microscope for vascular abnormalities. Grade 0 was assigned to a circle of Willis with normal vessels found in control animals, 1 to irregular morphology but without identifiable aneurysm, 2 to a single saccular aneurysm, 3 to multiple saccular aneurysms, and 4 to saccular aneurysm with rupture.6
Immunohistochemistry of mouse aneurysm specimens
Murine specimens were fixed in 4% paraformaldehyde for 24 hours, dehydrated in 18% sucrose solution, mounted in Tissue-Tek OCT compound (Sakura Finetek USA, Torrance, California, USA), and sectioned at 5 μm. Heat mediated retrieval in Dako Target Retrieval Solution (Dako, Carpinteria, California, USA) was performed. The primary antibodies used were anti-F4/80 (MCA497R; AbD Serotec, Düsseldorf, Germany), anti-inducible nitric oxide synthase (iNOS) (ab15323–500; Abcam, Cambridge, Massachusetts, USA), and anti-arginase antibody (ab60176; Abcam). Secondary antibodies used were Alexa Fluor 488 (A-21208; Life Technologies, Grand Island, New York, USA) and Alexa Fluor 594 (A-21207; Life Technologies) or Alexa Fluor 594 (A-11058; Life Technologies). The nuclei were counterstained with DAPI (H-1200; Vector Labs). M1 macrophages were defined as F4/80+ iNOS+, while M2 macrophages were defined as F4/80+ Arg I+ cells.
Statistical analysis
All analyses were performed by a biostatistician (DWN). Sample sizes were chosen based on previous power calculations and published data (figures 2 and 3). The Mann–Whitney test was used to test for differences in CA grade (figure 2B, C). Linear trend across time and phenotype ratio between time points in the treatment groups was tested using linear regression and ANOVA (figure 3A, B). To meet linear model assumptions, the natural log of ratio was taken as the outcome. Bonferroni correction was used to test for differences between individual groups. For the anti-CXCL1 group, the Kruskal–Wallis test (non-parametric ANOVA) was used as the log transformation was not possible due to one observation ratio having a value of zero (figure 3A). For the final analysis of phenotype ratio at 2 weeks between IgG and anti-CXCL1 treated mice, the Mann–Whitney test was used (figure 3C).
Figure 2.
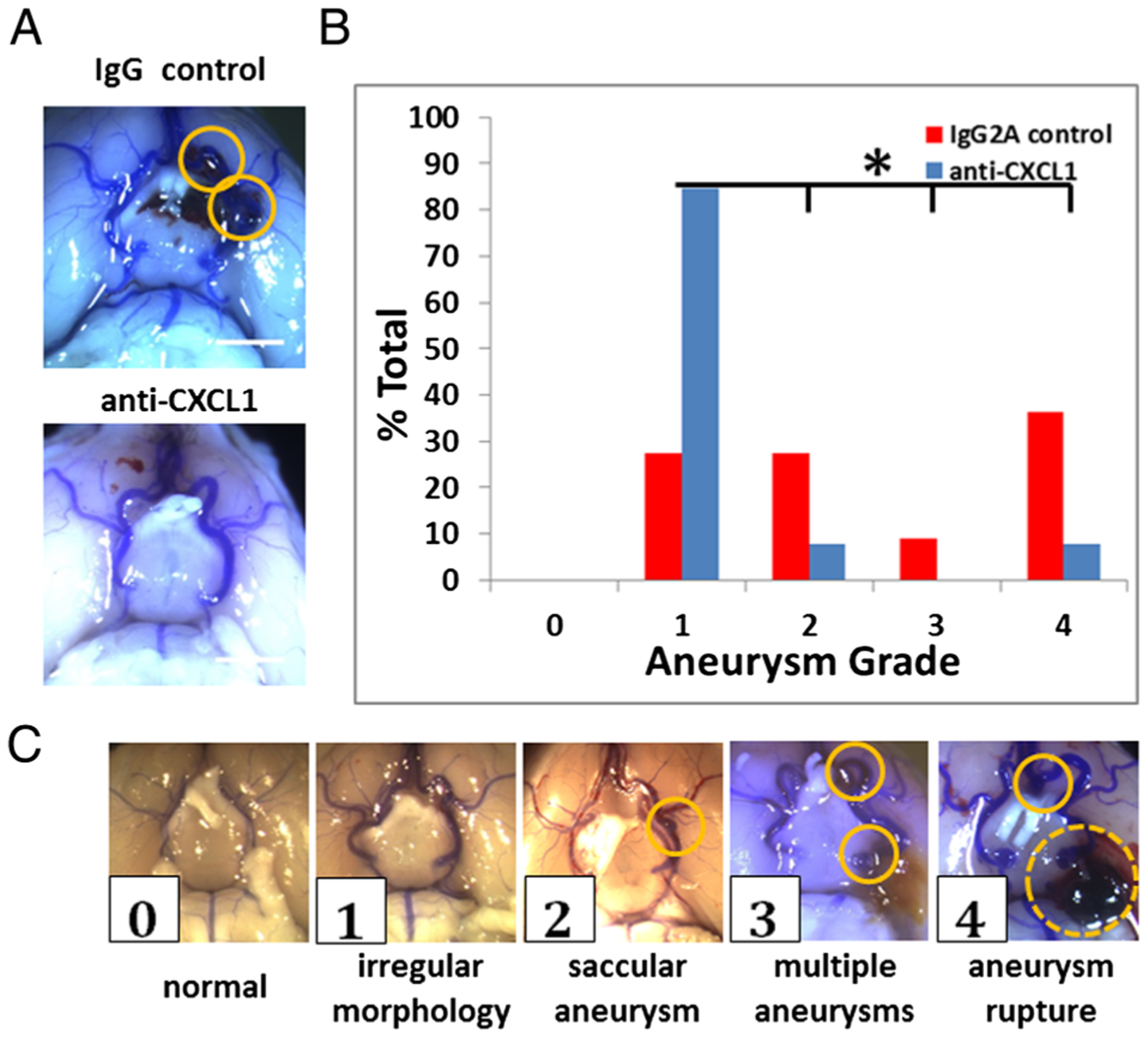
Severity of aneurysm development in mouse cerebral aneurysm model. (A) Anti-CXCL1 treated mice did not develop aneurysms at 2 weeks compared with IgG treated mice. Scale bar=2 mm. (B) Aneurysm development in the IgG control group displayed varying levels of severity that were significantly different from anti-CXCL1 treated mice when scored using (C) the 5 point grading scale (circle=aneurysm, dotted circle=ruptured aneurysm) (p=0.0046 by Mann–Whitney test, *p<0.05).
Figure 3.
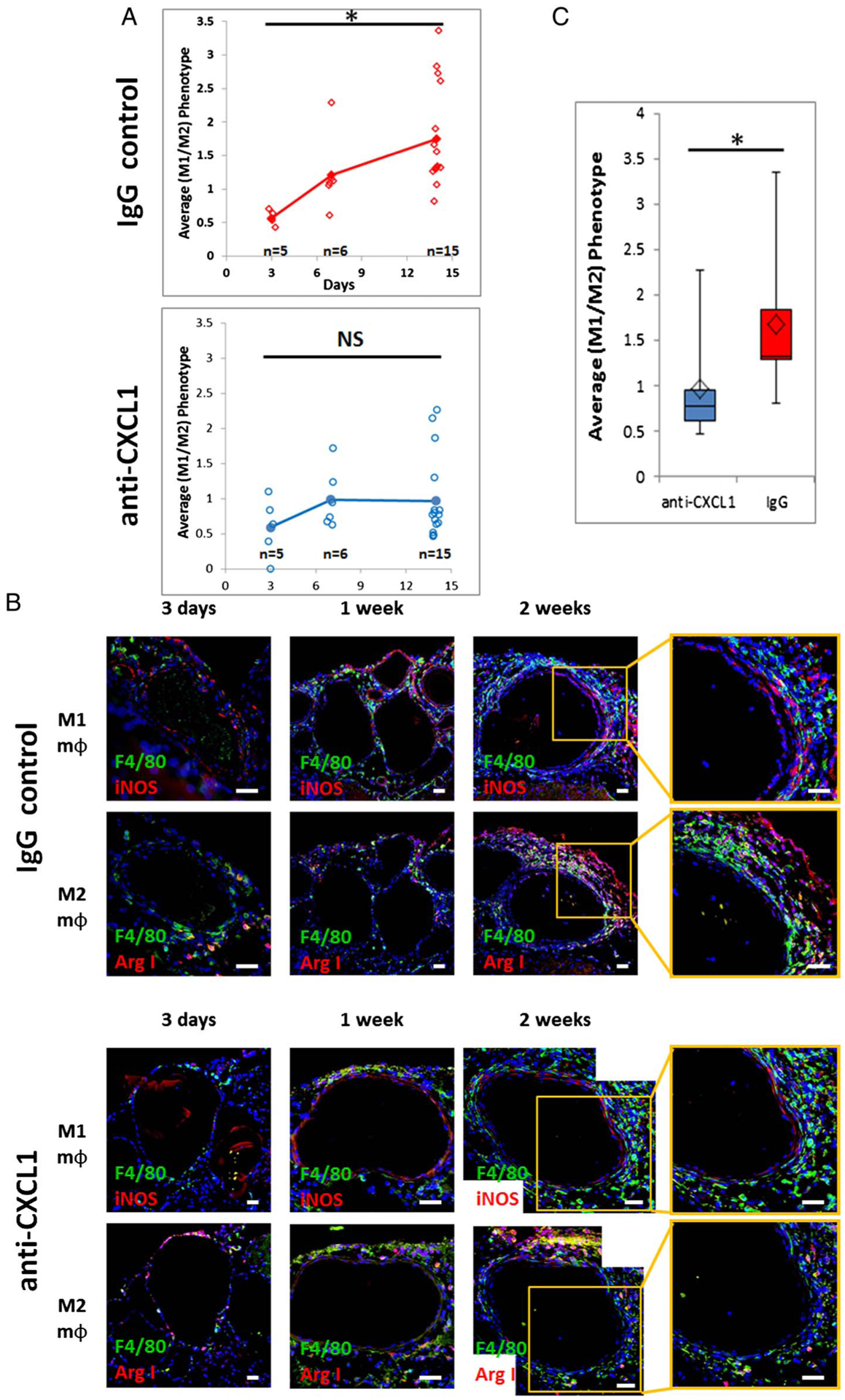
Macrophage phenotype dynamics over time in the mouse cerebral aneurysm model. (A) Macrophage M1 to M2 phenotype ratio dynamics over 2 weeks in the IgG control and anti-CXCL1 groups (IgG: linear model and ANOVA with Bonferroni pairwise comparisons; anti-CXCL1: Kruskal-Wallis; *p<0.05 for both). (B) Representative immunohistochemistry images of macrophage phenotype in mouse aneurysms (blue: DAPI; green: F4/80; red: inducible nitric oxide synthase (iNOS) or Arg I. Scale bar=20 μm). (C) Neutrophil blockade with anti-CXCL1 antibody prevents M1 macrophage polarization (Mann–Whitney test, *p<0.05).
RESULTS
CXCL1 mediated neutrophil blockade prevents development of higher grade cerebral aneurysm lesions
CXCL1 blockade decreased aneurysm formation without significant change in local macrophage burden.5 Associated mortality/morbidity due to subarachnoid hemorrhage in the 2 week experimental time period was 48% in the IgG control group and 15.8% in the anti-CXCL1 treatment group (figure 1B). For this study, we analyzed macrophage phenotype in harvested tissues to ascertain why the majority did not develop aneurysms (figure 2A). Aneurysm grade (figure 2B) using a gross morphological 5 point scale (figure 2C) showed that the difference between developing aneurysms in IgG and anti-CXCL1 treated mice was highly significant. Mice in the IgG control group had a higher aneurysm scale score than those in the anti-CXCL1 group (2.4 vs 1.3, p=0.0046 by Mann–Whitney test) at 2 weeks.
Cerebral aneurysm development is characterized by increasing polarization towards the M1 macrophage phenotype
In our study, the M1 (F4/80+iNOS+) to M2 (F4/80+ArgI+) macrophage phenotype ratio increased during the 2 week period as aneurysms developed in IgG treated mice. The average M1/M2 ratio was 0.56 at 3 days (n=5), 1.21 at 1 week (n=6), and 1.75 at 2 weeks (n=15) (figure 3A). It was estimated that the log(ratio) increased by 0.09/day (95% CI 0.051 to 0.121; p<0.0001). Linear model showed the difference in M1/M2 ratio over time was highly significant (p<0.0001). The M1/M2 ratio was significantly higher at 2 weeks than at 3 days (1.56 vs 0.56, p<0.0001) (figure 3A). No significant differences were found between other pairs (figure 3B).
Anti-CXCL1 neutrophil blockade prevents increased M1 macrophage phenotype polarization
Clearance or deactivation of M1 macrophages is a critical step in lowering inflammation. Neutrophil blockade using anti-CXCL1/GRO-α antibody attenuated polarization towards the M1 phenotype during the 2 weeks post-aneurysm induction (n=15). The average M1/M2 ratio was found to be 0.59 at 3 days (n=5), 0.99 at 1 week (n=6), and 0.95 at 2 weeks (n=15) (figure 3A). The Kruskal–Wallis test showed that the M1/M2 ratio was not significantly different between the three groups (p=0.442) (figure 3B). Comparison of M1/M2 ratio between IgG control antibody treated mice and anti-CXCL1 antibody treated mice at 2 weeks revealed that the phenotype ratio was significantly lower in the anti-CXCL1 group (1.75 vs 0.95, p=0.0007 by Mann–Whitney test) (figure 3C).
DISCUSSION
CXCL1 is a functional murine homolog of human CXCL8/IL-8 with similar functions, such as neutrophil chemotaxis.9 Neutrophils exhibit specific phenotypes with opposing effects on macrophage polarization.10,11 Once activated by CXCL1, neutrophils release proinflammatory cytokines.10 Proinflammatory neutrophils (PMN-I) express interleukin 12 and macrophage inflammatory protein-1α (MIP-1α/CCL3), while anti-inflammatory neutrophils (PMN-II) are thought to secrete interleukin 10 and MCP-1.11 PMN-I polarize macrophages into the M1 proinflammatory phenotype while PMN-II shift the balance towards M2.11
Macrophages are monocyte derived cells capable of remarkable plasticity. In the mouse, Gr1+/Ly6ChighCCR2+CX3CR1low monocytes (CD14highCD16low in humans) are the source of M1 proinflammatory macrophages.12,13 Gr1−/Ly6ClowCCR2 −CX3CR1high mouse monocytes (CD14lowCD16high in humans) predominate physiologically, promote wound healing, and give rise to M2 anti-inflammatory macrophages.12,13 Several surface markers have been used to describe macrophage phenotypes. M1 macrophages have been associated with CD68 (MHC II), iNOS, Arg II, STAT1, and STAT2. M2 Macrophages have been associated with Arg I, CD163, STAT3, and STAT6.14
Frosen et al15 were the first to report asymmetry in the CD68 + classical M1 and alternative CD163+ macrophage distribution in human aneurysm specimens. In ruptured aneurysm walls, there was a trend towards predominance of CD68+ classical macrophages in areas other than those involving myointimal hyperplasia. In the areas of hyperplasia, this trend was reversed.15 Ollikainen et al16 showed that CD68 macrophages increased in human CA specimens with a higher density of mast cells and neo-vessels, which was attributed to disease progression. CD68 is not restricted to macrophages, however, and can be expressed in non-hematopoietic cells, such as fibroblasts.17,18 Similarly, CD163 can be co-expressed with phosphorylated STAT1, a transcription factor, in response to interferons in Th1 response driven M1 macrophages.14,19 In solid tumors, a phenotype known as tumor associated macrophages, sharing features of M1 and M2 subtypes, has been described.14 Hasan et al20 reported that macrophages in ruptured human aneurysms are primarily of the HLA DR+ M1 proinflammatory phenotype compared with unruptured aneurysms. It is not known whether the proinflammatory macrophage burden leads to aneurysm rupture or whether it is the result of rupture.
Macrophage phenotype identification requires careful marker selection as protein expression and cell phenotype exist on a spectrum of function.12,19,21 F4/80 is a member of the TM-7 family of proteins with functions in signaling and adhesion, and is a potent murine macrophage marker.22 It is expressed at high levels by various macrophages, including microglia, and also by monocytes although at lower levels. Macrophage phenotype can be distinguished using L-arginine utilization. M1 macrophages upregulate arginase II (Arg II) and iNOS to produce nitric oxide, while M2 macrophages shunt arginine to arginase I (Arg I) to produce ornithine and L-proline.21 This set of markers can be used to classify macrophages based on whether L-arginine is shifted towards local microenvironment destruction or repair. In our study, the M1(F4/80+iNOS+) macrophage predominance over M2 (F4/80+ArgI+) phenotype increased during the 2 week period as the aneurysms developed in IgG mice. In contrast, in anti-CXCL1 treated mice, the macrophage phenotype population was more balanced and did not shift towards the M1 phenotype.
Previous experiments have shown that macrophage depletion prevents murine CA formation.4 However, such methods are not viable clinical avenues in lesions with an established macrophage burden.23 Specifically, even if influx of additional inflammatory cells is stopped, established macrophage burden in advanced lesions may lead to biomechanical failure and rupture. Future therapies should focus on leveraging the immune cells within the lesion to induce repair. Peroxisomal proliferator activated receptor γ agonists, which modulate macrophage polarization, can be used to prevent CA rupture.24 Another promising approach involves shifting the balance towards the M2 macrophages using micro-RNAs, such as miR-181a.25 Macrophage egress could also be promoted.26 Statins have been found to promote macrophage egress from atherosclerotic lesions,27 but could inadvertently increase aneurysmal rupture.28 We hypothesize that early on during CA development, M1 proinflammatory macrophages are detrimental; consequently, M2 macrophage presence could be protective in advanced lesions by favorably affecting local wall biomechanics.
CONCLUSIONS
High M1 macrophage activity is the primary driver behind murine CA formation. We have also shown that CXCL1 dependent neutrophil infiltration is critical in modulating the macrophage phenotype in this process.
Acknowledgements
We would like to thank Dan W Neal for performing the statistical analyses.
Funding This work was supported by a Shirley Dudek Demmer Chair of Research grant from the Brain Aneurysm Foundation to KWN and NIH grant K08 NS067058-01 to BLH.
Footnotes
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data available on request.
REFERENCES
- 1.Tulamo R, Frösen J, Hernesniemi J, et al. Inflammatory changes in the aneurysm wall: a review. J Neurointerv Surg 2010;2:120–30. [DOI] [PubMed] [Google Scholar]
- 2.Aoki T, Kataoka H, Ishibashi R, et al. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40:942–51. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MB, van Rooijen N, Izui S, et al. Liposomal clodronate as a novel agent for treating autoimmune hemolytic anemia in a mouse model. Blood 2003;101:594–601. [DOI] [PubMed] [Google Scholar]
- 4.Kanematsu Y, Kanematsu M, Kurihara C, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011;42:173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nowicki KW, Hosaka K, He Y, et al. Novel high-throughput in vitro model for identifying hemodynamic-induced inflammatory mediators of cerebral aneurysm formation. Hypertension 2014;64:1306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hosaka K, Downes DP, Nowicki KW, et al. Modified murine intracranial aneurysm model: aneurysm formation and rupture by elastase and hypertension. J Neurointerv Surg 2014;6:474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Emeto TI, Lee J, et al. Mouse models of intracranial aneurysm. Brain Pathol 2015;25:237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yardeni T, Eckhaus M, Morris HD, et al. Retro-orbital injections in mice. Lab Anim (NY) 2011;40:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hol J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J Leukoc Biol 2010;87:501–8. [DOI] [PubMed] [Google Scholar]
- 10.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13:159–75. [DOI] [PubMed] [Google Scholar]
- 11.Tsuda Y, Takahashi H, Kobayashi M, et al. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004;21:215–26. [DOI] [PubMed] [Google Scholar]
- 12.Daley JM, Brancato SK, Thomay AA, et al. The phenotype of murine wound macrophages. J Leukoc Biol 2010;87:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007;204:3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavez-Galan L, Olleros ML, Vesin D, et al. Much more than M1 and M2 macrophages, There are also CD169(+) and TCR(+) macrophages. Front Immunol 2015;6:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frosen J, Piippo A, Paetau A, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke 2004;35:2287–93. [DOI] [PubMed] [Google Scholar]
- 16.Ollikainen E, Tulamo R, Frosen J, et al. Mast cells, neovascularization, and microhemorrhages are associated with saccular intracranial artery aneurysm wall remodeling. J Neuropathol Exp Neurol 2014;73:855–64. [DOI] [PubMed] [Google Scholar]
- 17.Kunz-Schughart LA, Weber A, Rehli M, et al. [The “classical” macrophage marker CD68 is strongly expressed in primary human fibroblasts]. Verh Dtsch Ges Pathol 2003;87:215–23. [PubMed] [Google Scholar]
- 18.Gottfried E, Kunz-Schughart LA, Weber A, et al. Expression of CD68 in non-myeloid cell types. Scand J Immunol 2008;67:453–63. [DOI] [PubMed] [Google Scholar]
- 19.Barros MH, Hauck F, Dreyer JH, et al. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013;8:e80908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasan D, Chalouhi N, Jabbour P, et al. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflammation 2012;9:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisser SB, McLarren KW, Kuroda E, et al. Generation and characterization of murine alternatively activated macrophages. Methods Mol Biol 2013;946: 225–39. [DOI] [PubMed] [Google Scholar]
- 22.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 1981;11:805–15. [DOI] [PubMed] [Google Scholar]
- 23.Hasan DM, Mahaney KB, Magnotta VA, et al. Macrophage imaging within human cerebral aneurysms wall using ferumoxytol-enhanced MRI: a pilot study. Arterioscler Thromb Vasc Biol 2012;32:1032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada K, Furukawa H, Wada K, et al. Protective role of peroxisome proliferator-activated receptor-γ in the development of intracranial aneurysm rupture. Stroke 2015;46:1664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bi J, Zeng X, Zhao L, et al. miR-181a induces macrophage polarized to M2 phenotype and promotes M2 macrophage-mediated tumor cell metastasis by targeting KLF6 and C/EBPα. Mol Ther Nucleic Acids 2016;5:e368. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res 2014;114:1757–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tada Y, Kitazato KT, Yagi K, et al. Statins promote the growth of experimentally induced cerebral aneurysms in estrogen-deficient rats. Stroke 2011;42:2286–93. [DOI] [PubMed] [Google Scholar]