

Abstract
Emerging evidence indicates the deubiquitinase USP22 regulates transcriptional activation and modification of target substrates to promote pro-oncogenic phenotypes. Here, in vivo characterization of tumor-associated USP22 upregulation and unbiased interrogation of USP22-regulated functions in vitro demonstrated critical roles for USP22 in prostate cancer (PCa). Specifically, clinical datasets validated that USP22 expression is elevated in PCa, and a novel murine model demonstrated a hyperproliferative phenotype with prostate-specific USP22 overexpression. Accordingly, upon overexpression or depletion of USP22, enrichment of cell cycle and DNA repair pathways was observed in the USP22-sensitive transcriptome and ubiquitylome using PCa models of clinical relevance. Depletion of USP22 sensitized cells to genotoxic insult, and the role of USP22 in response to genotoxic insult was further confirmed using mouse adult fibroblasts from the novel murine model of USP22 expression. As it was hypothesized that USP22 deubiquitylates target substrates to promote pro-tumorigenic phenotypes, analysis of the USP22-sensitive ubiquitylome identified the nucleotide excision repair protein, XPC, as a critical mediator of the USP22-mediated response to genotoxic insult. Thus, XPC undergoes deubiquitylation as a result of USP22 function and promotes USP22-mediated survival to DNA damage. Combined, these findings reveal unexpected functions of USP22 as a driver of pro-tumorigenic phenotypes and have significant implications on the role of USP22 in therapeutic outcomes.
Introduction:
Elevated expression of the ubiquitin specific peptidase 22 (USP22) has been associated with poor prognosis in numerous tumor types[1–5]. To date, increased USP22 mRNA or protein levels have been reported in colorectal[1,3,5], breast[6], pancreatic[7], and prostate[8] cancers, correlating with unfavorable outcomes in all tumor types examined. Furthermore, elevated USP22 was identified as a key component of an eleven-gene “death from cancer” signature that predicts for therapeutic failure, with a particularly robust prognostic value in prostate cancer (PCa)[9]. PCa remains the second leading cause of cancer-related death in US men[10], and USP22 has been shown to increase with disease progression[8]. Initial reports identified USP22 as a potent mediator of PCa progression, manifesting in part through USP22-dependent modulation of c-Myc and hormone receptor activity[8]. Despite these advances, the overall mechanisms by which USP22 contributes to cancer progression remain incompletely defined.
Functionally, USP22 was initially described as a deubiquitinase that removes ubiquitin moieties from histones H2A and H2B as well as a member of the human Spt-Ada-Gcn5-acetyl transferase (SAGA) deubiquitinase (DUB) module that promotes activator-driven transcription[11]. A multitude of transcription factors are influenced by the SAGA complex in cancer, including the androgen receptor (AR), the oncogene c-Myc, and the tumor suppressor p53[8,11,12]. In addition to histones H2A and H2B, USP22 directly deubiquitinates other substrates, including SIRT1 in a c-Myc dependent manner, consequently inhibiting p53 transcriptional and pro-apoptotic functions, further implicating USP22 in critical cancer-related signaling axes[12]. Moreover, USP22 has been linked to processes outside transcriptional regulation, including telomere maintenance[13], maintenance of genomic integrity[13], and cell cycle regulation[14]. Additionally, USP22 has been associated with the DNA damage response (DDR) through regulation of class switch recombination and double strand break (DSB) repair in B cells[15,16]. Despite these advances, it remains uncertain which mechanisms of USP22 action are required to promote cancer phenotypes, and what substrates support USP22 pro-tumorigenic functions.
In this study, unbiased assessment of USP22 activity was achieved in clinically relevant models to discern the oncogenic capacity of the deubiquitinase USP22. Using a series of in vitro models, a novel genetically engineered mouse model (GEMM) developed to mimic tumor-associated USP22 induction, and assessment of previously published human tumor datasets, critical roles for USP22 in promoting cell cycle progression and DNA repair were identified. Complementing these findings, delineation of the USP22-dependent ubiquitylome revealed novel putative substrates associated with both cell cycle progression and DNA repair, and identified USP22 as a major modulator of XPC, a DNA repair factor recently shown to be associated with PCa risk[17]. Functional assessment revealed that USP22 significantly modulates XPC function through deubiquitylation, and that this pathway is critical for USP22-mediated DNA repair processes. In sum, these studies identify USP22 as a regulator of the response to DNA damage, and ultimately define a major node by which USP22 promotes cancer phenotypes.
Materials and Methods
Cell lines, cell culture, and maintenance:
All LNCaP and C4-2 derived cell lines were maintained in minimum essential media (IMEM) supplemented with 5% FBS (heat inactivated fetal bovine serum). Parental LNCaP and C4-2 cells were obtained and authenticated by ATCC. All media were supplemented with 2 mmol/l of L-glutamine and 100 units/ml penicillin-streptomycin. Cell lines, LNCaP and C4-2, were authenticated by ATCC and checked for mycoplasma upon thawing and termination of maintenance (<20 passages). For Ubiscan analysis, the cell lines (LNCaP, shUSP22, and USP22) were used as previously indicated after transfection with indicated plasmids[8,14]. For generation of stable cell lines used throughout the study, LNCaP and C4-2 cells were transduced with lentivirus and underwent at least three rounds of selection with the appropriate antibiotic. SMARTvector Human Inducible non-targeting mCMV-TurboGFP control shRNA or USP22 shRNA (Dharmacon) were used for inducible knockdown of USP22. USP22 shRNA include: USP22 shRNA V3SH11252- 225591928 (shUSP22-9578), 227392804 (shUSP22-0454), 230910043 (shUSP22-4693). The USP22 shRNA cell lines used for RNA-seq analysis and subsequent molecular studies expressed shUSP22-4693. Stable USP22 overexpression was achieved by cloning pcDNA3.1 FLAG-USP22 into the pLenti6.3/V5/TO-DEST vector using the Virapower T-Rex system (ThermoFisher)[14]. Cells were either transduced with pLenti6.3/V5/TO-DEST-FLAG-USP22 or control vector pLenti4/TO/V5-DEST-pLPLUC.
Generation of MAFs:
The peritoneal tissue from an 8-week old heterozygous, male mouse with the hUSP22 transgene was isolated and digested with collagenase and DNase. Cells were immortalized using the NIH 3T3 guidelines. Cells were maintained in DMEM supplemented with 10% heat inactivated FBS, 2 mmol/l of L-glutamine, and 100 units/ml penicillin-streptomycin.
Proliferation Assays:
LN- or C42- shCon and shUSP22–4693 cells were plated at equal densities, treated as indicated, and cell number was determined by either trypan blue exclusion with a hemocytometer or the Quanti-iT Pico Green dsDNA assay kit (Thermo Fisher). Doxycycline was refreshed every 48 hours. Mouse adult fibroblasts (GFP and hUSP22) were plated at equal densities, treated as indicated, and cell number was quantified using the Quanti-iT Pico Green dsDNA assay kit (Thermo Fisher).
Chromatin Tethering Assays:
Cells were harvested from a 10 cm plate (~70% confluent) and immediately resuspended in 100–200 μL CSK buffer (10 nM PIPES, pH 6.8, 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF) with 0.2% Triton X and protease inhibitor (PI). After removing 50 μL of suspension (Total Protein Fraction), the remaining suspension was brought up to a volume of 1 mL in CSK buffer with 0.2% Triton X+PI, then incubated on ice for 20 minutes. The chromatin-tethered fraction was pelleted, and subsequently re-extracted in 1 mL of CSK buffer for 10 minutes. The chromatin-tethered fraction was then re-pelleted, and sample was aspirated to 50 μL. Sample buffer was added, and the samples were boiled for 5 minutes. Immunoblot analysis ensued using indicated antisera.
Immunofluorescence:
Cells were plated at equal densities on poly-L-lysine coverslips in a 6 well plate and treated as indicated. The cells were then fixed with 3.7% formaldehyde in PBS for 20 minutes at room temperature (RT), and washed with PBS. 0.3% Triton-X in PBS was added to wells for 15 minutes at RT. Cells were blocked with 2% goat serum (Atlanta Biologicals S13110) in PBS for 30 minutes at RT. Antibody (XPC at 1:250 PA3–956 Thermo Fisher; Rad23B at 1:250 C-4 sc-166507) was then added to each coverslip, incubated for 2 hours at 37°C. Coverslips were washed with PBSx3 and incubated with FITC conjugated goat anti-rabbit or mouse IgG (1:1000 in 2% goat serum) for 45 minutes at 37°C. Coverslips were washed with PBSx3, stained with DAPI (100 μL per coverslip, 1:1000 dilution in PBS) for 10 minutes in dark. Slides were mounted using Prolong (without DAPI) and allowed to dry overnight in the dark.
Western Blotting:
As previously described[8,18,19], using antisera described. Antibodies: XPC (D-10) sc-74410; Rad23B (C-4) sc-166507; USP22 Novus Biologicals NBP1–49644; GAPDH sc-25778; H4 EMD Millipore 07–108; Vinculin Sigma (V9264); Lamin B (M-20) sc-6217. Cyclohexamide used as described (Sigma Aldrich). All antibodies used at 1:1000.
Gene Expression:
Cells were treated as indicated, and RNA was isolated using TRIzol (Life Technologies). RT-PCR was performed using Superscript VILO cDNA Synthesis Kit (Thermo Fisher), following the manufacturer’s instructions.
Primers:
The following primers were used for qRT-PCR: 18S Forward 5’ GCAATTATTCCCCATGAACG 3’; 18S Reverse 5’ GGCCTCACTAAACCATCCAA 3’; USP22 Forward 5’ CTCCTGTCTGGTCTGTGAGATG 3’; USP22 Reverse 5’ CAGCAACTTATACGGGATGTGA 3’; mouse S16 Forward 5’ AGGAGCGATTTGCTGGTGTGGA 3’; mouse S16 Reverse 5’ GCTACCAGGGCCTTTGAGATGGA 3’; mouse USP22 forward 5’ TTGCATAGTGTCACCCGCTT 3’; mouse USP22 reverse 5’ GCTACCAGGGCCTTTGAGATGGA 3’.
Generation of hUSP22 KI mice:
A cDNA insert of human USP22 with a 3X FLAG tag was knocked into the Rosa26 locus of C57BL/6 mice (See Figure 3A). Generation and cloning of targeting constructs, embryonic injection and selection, clone verification, and generation of chimeric mice were conducted by InGenious Laboratories LLC. Verification of hUSP22 allele was achieved using primers (ROSA SQ1: 5’ AGCACTTGCTCTCCCAAAGTC 3’; ROSA SQ2: 5’ TGCTTACATAGTCTAACTCGCGAC 3’; LAN1: 5’ CCAGAGGCCACTTGTGTAGC 3’). ROSA SQ1 +ROSA SQ2 = wt, 565bp and ROSA SQ1 + LAN1 = KI, 493bp.
Figure 3. Human USP22 expression confers a hyperproliferative phenotype in vivo and a survival advantage to genotoxic insult.
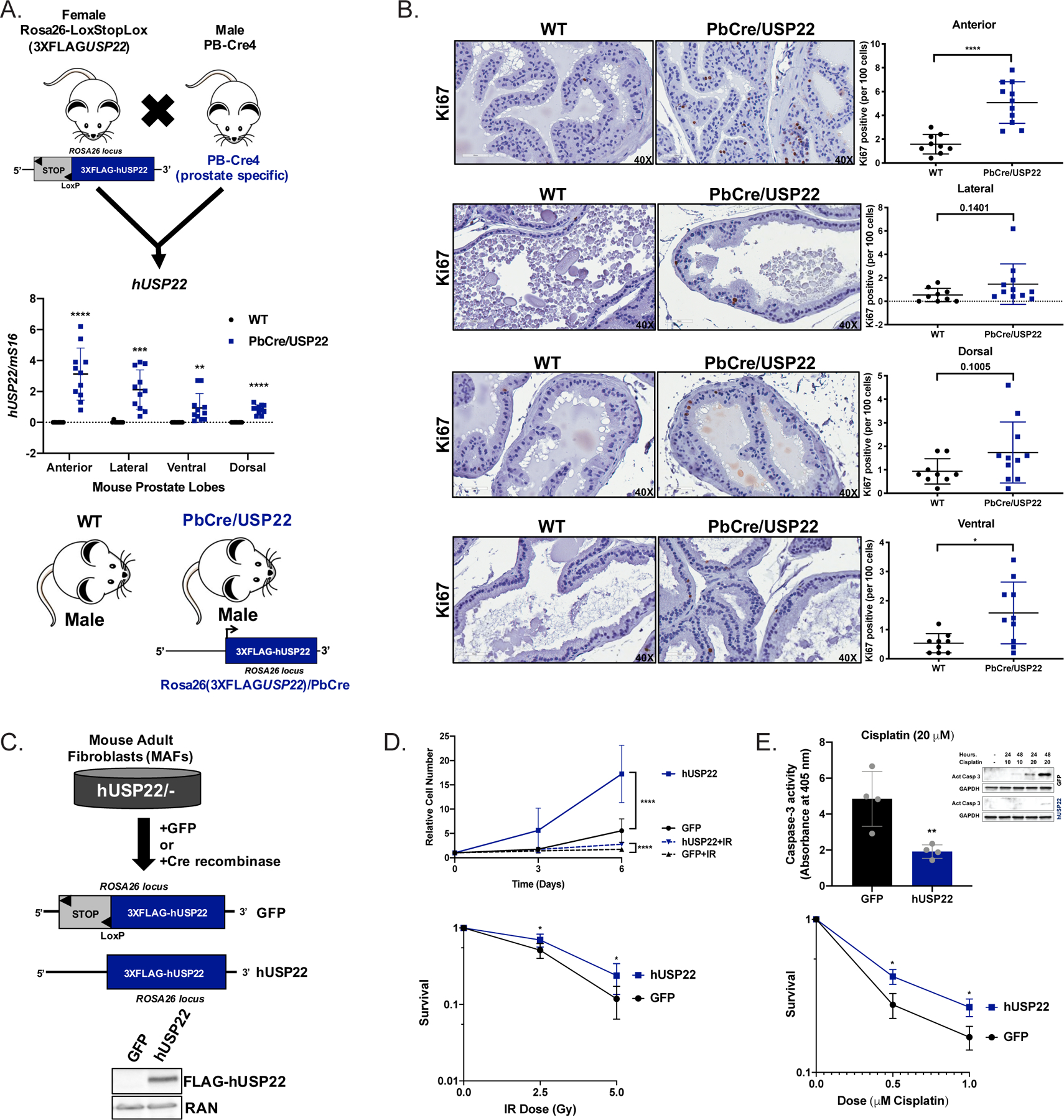
(A) Female heterozygous mice with human USP22 in the Rosa26 locus (Rosa26-Lox-Stop-Lox(3XFLAGUSP22)) were crossed with male prostate-specific probasin cre recombinase mice (PB-Cre4) to produce offspring with no expression of hUSP22 (WT) or with expression of hUSP22 (PbCre/USP22). Mice were aged at least 12 months and their prostate lobes were micro-dissected to detect expression of hUSP22 RNA (WT N=9; Pb-Cre/USP22 N=11). Student’s t-test. (B) The prostate lobes of WT and PbCre/USP22 mice aged at least 12 months underwent IHC analysis for Ki67. Representative images are shown at 40X and quantitative analysis are shown to the right (WT N=9; Pb-Cre/USP22 N=10). Student’s t-test. (C) Mouse adult fibroblasts (MAFs) from a male heterozygous mouse with human USP22 in the Rosa26 locus (Rosa26-Lox-Stop-Lox(3XFLAGUSP22)) were immortalized and then transduced with GFP control (GFP) or cre recombinase (hUSP22) to induce expression of hUSP22. (D) GFP or hUSP22 MAFs were plated for proliferation or survival assays and were treated with vehicle (N=5) or 5Gy irradiation (N=4) and measured using pico green fluorescence on Day 0, 3, and 6. Survival assays were plated and then treated on Day 0 with the designated dose of IR (N=6). (E) GFP or hUSP22 MAFs were plated for survival (N=3) or caspase-3 activity assays/western blot and treated with the indicated dose of cisplatin on Day 0. Student’s t-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Mouse Lines and Handling:
Probasin-Cre (PB-Cre4) mice Tg(Pbsn-cre)4Prb/J (Stock#026662) were acquired from Jackson Laboratories. Verification of Pb-Cre allele was assessed with the following primers: P021 (Pb/Cre F 5’ CTGAAGAATGGGACAGGCATT 3’) and C031 (Pb-Cre R 5’ CATCACTCGTTGCATCGACC 3’). Mice were housed in animal facilities within the Sidney Kimmel Cancer Center at Thomas Jefferson University, and all protocols used for this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Thomas Jefferson University. All mice used for the study were male.
Immunohistochemistry (IHC):
IHC procedures were performed by the SKCC Shared Resource Translational Pathology Core. 4 μm paraffin slides were deparaffinized in Shandon Varistain Gemini ES Autostainer. Antigen retrieval was performed with DAKO PT-Link using Citrate Buffer (pH 6.0) at 98˚C for a total time of 20 min. The major IHC procedures were performed in DAKO autostainer Plus platform. Primary immunostaining was performed using antibodies against Ki67 (abcam, Cat#: ab16667, 1:200). Antibodies were incubated at room temperature for 30 minutes. Biotinylated anti-Rabbit or anti-Rat (Vector Laboratories, cat#: BA-1000 and BA-4001) secondary antibodies and ABC-HRP complexes (Vector Laboratories, Cat#: PK6100) were applied following the primary antibodies with 30 minutes incubation each reagent at room temperature. Three 1XTBST (pH7.5) washes were performed between each step above. The signals were visualized using DAB substrate (DAKO, Cat#:K3468). Slides were then washed with DI water and proceeding with Hematoxylin counter stain, dehydration and clearing in Shandon Varistain Gemini ES Autostainer and coverslipped with Permount Mounting Medium.
Ubiscan Analysis:
Cell models of LNCaP parental, LN-shUSP22, and LN-USP22 were used as previously described[8]. Samples were analyzed using the Cell Signaling PTMScan method as previously described[20–22] (Cell Signaling). Cell lysates were protease-digested with trypsin and purified through C18 solid phase extraction columns to produce lyophilized peptides. Ubiquitinated peptide enrichment using the Ubiquitin Branch Motif Antibody (K-ε-GG #3925) was followed by LC-MS/MS analysis using LTQ-Orbitrap-Velos, ESI-CID and sequences were assigned to MS/MS spectra with the Sorcerer-2 platform[23] as duplicate injections. Peptides were eluted using a 72-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. MS parameter settings were the following: MS Run Time 96 min, MS1 Scan Range (300.0 – 1500.00), Top 20 MS/MS (Min Signal 500, Isolation Width 2.0, Normalized Coll. Energy 35.0, Activation-Q 0.250, Activation Time 20.0, Lock Mass 371.101237, Charge State Rejection Enabled, Charge State 1+ Rejected, Dynamic Exclusion Enabled, Repeat Count 1, Repeat Duration 35.0, Exclusion List Size 500, Exclusion Duration 40.0, Exclusion Mass Width Relative to Mass, Exclusion Mass Width 10ppm). Searches were performed against the NCBI Homo sapiens database with mass accuracy of +/− 50 ppm for precursor ions and 1 Da for product ions. Results were filtered with mass accuracy of +/− 5 ppm on precursor ions and presence of the intended motif (K-ε-GG). A 5% default false discovery rate was used to filter the SORCERER results. Differential ubiquitylation was characterized using a 1.5-fold cut-off compared to control, less than 35 Max % CV, and a max intensity of >1×106 compared to control.
RNA-sequencing analysis:
LN-pLPLUC, LN-USP22hi, LN-shCon, and LNshUSP22–4693 cells were treated as indicated in Figure 2A in biological triplicate. RNA was extracted and purified using the miRNeasy kit (QIAGEN) according to manufacturer’s instructions. RNA-seq libraries were constructed using the TruSeq Stranded Total RNA Library Prep Gold kit (protocol # 15031048 Rev E) and sequenced on Illumina’s NextSeq 500 sequencer at the Sidney Kimmel Cancer Sequencing core facility using paired-end 75bp reads. RNA-Seq was aligned against the hg19 human genome using STAR v2.5.2a[24]. Differential gene expression was generated using DESeq2 v1.12.4[25]. Gene set enrichment analysis (GSEA) was performed using gene sets from the Molecular Signature Database[26]. RNA-seq files have been deposited to the GEO repository under accession GSE140164.
Figure 2. USP22 modulates DNA repair factor expression and survival after DNA damage.
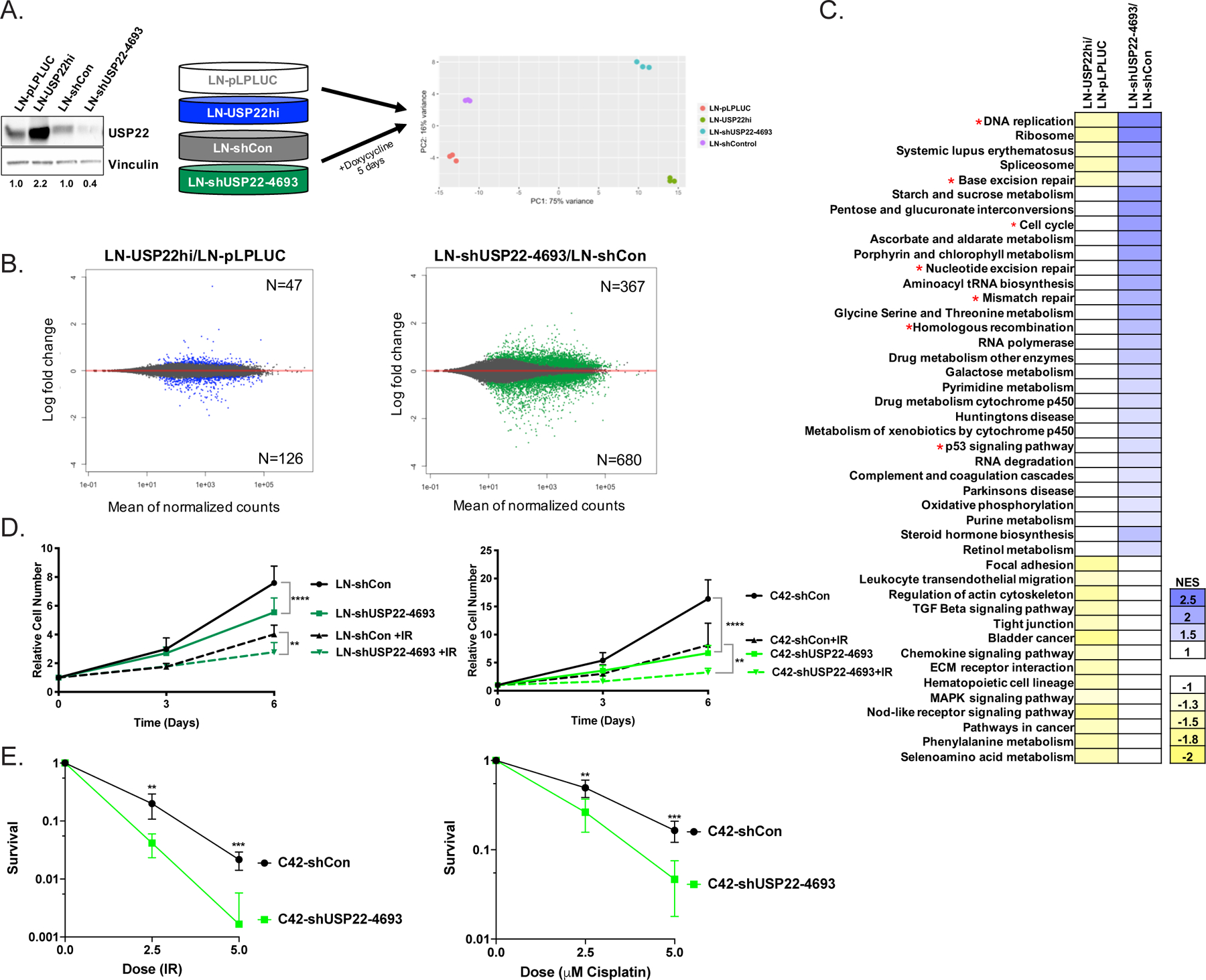
(A) RNA-seq analysis was performed in LN-pLPLUC and LN-USP22hi cells, as well as LN-shCon and LN-shUSP22–4693 cells after 5 days of 1 μg/mL doxycycline treatment in biological triplicate, as shown in the PCA analysis. Cells were treated as described and immunoblotted with the indicated antisera. (B) MA plots show differential gene expression in LN-USP22hi cells compared to LN-pLPLUC (Left; Blue) and LN-shUSP22–4693 cells compared to LN-shCon (Right; Green). (C) Using normalized RNA-seq count data, GSEA was used to identify MSigDB pathways that were enriched upon USP22 depletion and de-enriched upon USP22 overexpression. A p value of <0.05 was used to determine significant enrichment. *represents cell cycle or DNA repair-related pathways. (D) Cells were plated for proliferation assays and treated with vehicle (UT) or irradiation (IR; LN-shCon and LN-shUSP22–4693 +2Gy IR (N=5); C42-shCon and C42-shUSP22–4693 +5Gy IR (N=3)). Cell numbers were measured using pico green fluorescence on Day 0, 3, and 6. Cells were treated with 1 μg/mL doxycycline every 48 hours. Relative cell number was achieved by normalizing to Day 0. 2-way ANOVA. (E) C42-shCon or C42-shUSP22–4693 cells were plated for clonogenic survival assays and treated with IR or cisplatin at the designated doses. Cells were treated with 1 μg/mL doxycycline every 2–3 days. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Statistical analysis:
In vitro and In vivo data are presented as mean +/− standard deviation, unless otherwise indicated. Statistical analyses were performed using GraphPad Prism 7.
Ubitest:
Cells were plated on 10 cm plates and after 48 to 72 hours, cells were lysed using the M-PER Mammalian Protein Extraction Reagent with Halt protease inhibitor (Thermo Fisher), Halt phosphatase inhibitor (Thermo Fisher), and 5 μM PR-619 followed by 3×30sec sonication in Bioruptor Pico (Diagenode). The magnetic TUBE Elution Kit (Lifesensors) was then used per manufacturer’s instructions.
Immunoprecipitation:
Cells were plated on 10 cm plates and after 48 to 72 hours, cells were lysed using the M-PER Mammalian Protein Extraction Reagent with Halt protease and phosphatase inhibitors (Thermo Fisher) followed by 3×30sec sonication in Bioruptor Pico (Diagenode). Protein A Dynabeads (Thermo Fisher) were incubated with USP22 antibody (Novus Biologicals). The antibody (Ab) was crosslinked to the beads using 10 mM DiMethylPimelimidate in 0.2 M TriEthanolAmine, pH 8.2 for 30 minutes at RT. The Ab-Bead resin was washed with 50 mM Tris (pH 7.5) for 10 minutes, and then PBST three times. The resin was blank eluted twice with 100 mM TriEthylAmine (pH 11.5) and elute was neutralized with 2M Tris pH 6.0. Resin was washed with PBST twice and 500 μg of cell lysate was added to resin. Lysate and bead mixture rotated overnight, and resin was washed with PBST three times then 100 mM ammonium bicarbonate (AB). Immunoprecipitate was eluted in 100 mM AB/8M Urea.
Transfection with siRNA:
Cell lines were seeded on poly-L-lysine coated plates in culture media for 24 hours. Cells were then transfected for 8 hours in serum-free media conditions with either control (siCon) or XPC (siXPC) siRNA pools (Dharmacon) according to manufacturer’s protocol as previously described[18]. Cells were then maintained in complete media for 72 hours post transfection and processed for either protein or growth assays.
Analysis of clinical datasets (cBioportal):
All information used from the designated cBioportal studies was from February 2019, unless otherwise indicated in the text.
Caspase Assays:
Caspase 3 activation was measured using the Colorimetric Caspase-3 assay kit (ab39401) according to the manufacturer’s procedure. Briefly: 0.75 −1×106 cells (MAFs - GFP and USP22) were seeded on 10 cm plates and next day treated with 20 μM of cisplatin for 24 hours. Freshly isolated protein extracts were incubated in 96-well plate with caspase 3 substrate, DEVD-pNa, for 2 hours. Absorbance was read at 405 nm and adjusted to a protein content after subtraction of background signal (wells with no protein extract). Caspase activation is presented as fold-increase compared to untreated control, graph represents four independent experiments in duplicate. For additional confirmation of apoptosis, cells were treated as above, protein extracts were isolated and subjected to immunoblot analysis using anti-active caspase-3 antibody (BD Pharmingen, 559585)
Clonogenic Assays:
Clonogenic assays were performed as described previously[27]. Briefly, each cell line was counted and plated at three dilutions in a 6 well plate. Cells were treated ~24 hours after plating and harvested approximately 14 days post-treatment. Cells were fixed in 0.5% crystal violet with 37% formaldehyde in PBS. Aggregates of more than 50 cells were counted as clones. Plating efficiency (PE) was calculated from untreated cells ((No. colonies/No. cells seeded)x100). Survival was calculated by ((No. colonies formed after treatment)/(No. cells seeded)xPE).
CPD ELISA:
NER was assessed by measurement of CPD after UV radiation using OxiSelect UV-induced DNA damage ELISA kit (CPD quantitation, Cell Biolabs, STA-322) according to manufacturer’s recommendations. Briefly, 1×106 MAF-GFP and MAF-USP22 were seeded on 10 cm plates, and after 24 hours subjected to 20J/m2 of UV radiation in Stratalinker 1800. LN-shCon and LN-shUSP22 were plated at the density of 2.5 – 3×105 /per well onto 6 well plates, and subjected to UV (10 J/m2) 36–48 hours later. Cells were collected at the indicated post-treatment time and DNA was isolated with DNeasy Blood and Tissue kit (Quagen 69504). DNA was probed in ELISAs with anti-CPD primary and HRP-conjugated secondary antibodies, followed by the measurement of absorbance at 450 nm. The absorbance at 0 hours (cells collected immediately after UV treatment) was designated as 100%, and the extent of NER was determined by comparing these values with the absorbance in the rest of the samples. Graphs represent 4 independent experiments in duplicates for MAF-GFP/MAF-USP22 and three independent experiments in triplicates for LN-shCon/LN-shUSP22 cell lines.
Results:
USP22 alterations occur across prostate cancer stages and are associated with disease progression
Expression of USP22 increases as a function of disease progression in numerous tumor types and has been linked with poor outcome[1–5]. However, the underlying mechanisms of USP22 function in cancer remain incompletely defined. In the case of PCa, USP22 protein expression increases as tumors progress from hormone therapy (HT)-sensitive to castration-resistant disease (CRPC)[8]. Analysis of recent genomic and transcriptomic clinical specimen studies revealed that USP22 alterations, defined as amplification, homozygous deletion, mutation, mRNA upregulation/downregulation, or fusion events, were observed in both primary and advanced PCa (Figure 1A), consistent with a known role of USP22 in modulating AR stability and activity[8]. Importantly, PCa cells rely upon the AR for proliferation and survival, and AR cooperates with c-Myc to drive disease progression[28,29]. USP22 modulates both AR and MYC activity[8], and USP22 mRNA expression was positively correlated with both AR and MYC mRNA expression (p=7.76e-14 and p=3.36e-7, respectively; Figure 1B), further supporting a pro-oncogenic role for USP22 in tumors with elevated AR and MYC expression. In addition, alterations in USP22 co-occurred with alterations in both AR and MYC (p<0.001 and p=0.036, respectively; Figure 1B inset), unlike with several other oncogenic factors associated with disease progression (Supplemental Figure 1A–C), further suggesting a function for USP22 in PCa progression. Consistent with previous observations showing that USP22 expression is critical for CRPC maintenance both in vitro and in vivo[8], USP22 amplification and/or mRNA upregulation was associated with decreased progression-free survival in primary (p=0.0459; Figure 1C Left) and advanced disease (p=0.0342; Figure 1C Right). Moreover, elevated USP22 expression occurred with increasing Gleason Grade (Figure 1D), associating USP22 with aggressive phenotypes. Finally, in clinical specimens, analyses revealed that USP22 was most frequently amplified or upregulated at the mRNA level (Figure 1E). Combined these data extend previous findings implicating USP22 as a pro-oncogenic factor in PCa, further underscoring the need to discern its functions.
Figure 1. USP22 alterations occur across prostate cancer stages and are associated with disease progression.

(A) Alterations, defined as amplification, homozygous deletion, mutation, mRNA upregulation, mRNA downregulation, or fusion, in USP22 occur in both primary and metastatic prostate adenocarcinoma from studies available in the cBioportal (Excluded are those studies with <1% alterations in USP22: Broad-Cornell 2013; CPC-GENE 2017; MSKCC-DFCI 2018); Studies with * represent those studies that include only genomic alterations (amplification, homozygous deletion, or mutation). (B) USP22 gene expression is positively correlated with AR or MYC gene expression using the cBioportal TCGA provisional prostate adenocarcinoma study (N=498). Inset represents co-occurrence (p value, Fisher Exact Test) of alterations in USP22 with MYC or AR. (C) Amplification and/or mRNA upregulation of USP22 are associated with decreased progression free survival in the TCGA provisional dataset (p=0.0459) using all complete tumors and the MSKCC Cancer Cell dataset (p=0.0342) using all complete tumors. Log rank test. (D) Using the MSKCC Cancer Cell 2010 dataset (cBioportal), USP22 expression was measured as a function of Gleason Grade. (E) USP22 alterations are most commonly amplification events or mRNA upregulation events. Left, those studies with only genomic alterations available (See * in Figure 1A). Right, those studies with both genomic and RNA alterations available. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
USP22 modulates DNA repair factor expression and survival after DNA damage
While initial studies identified USP22 as a modulator of AR and MYC, the overall mechanisms by which USP22 promotes disease progression remain unclear. To model tumor-associated USP22 upregulation, isogenic models of constitutive USP22 induction and doxycycline-inducible USP22 downregulation were generated and used to identify the USP22-sensitive transcriptome by RNA-Seq analysis (Figure 2A; Supplemental Figure 2A). As expected, differential gene expression was observed after USP22 induction (n=173) or suppression (n=1047) as compared to controls (Figure 2B; Supplemental Figure 2B). Gene set enrichment analysis (GSEA) identified cell cycle and DDR-related pathways as significantly and differentially enriched, including DNA replication, base excision repair, cell cycle, nucleotide excision repair, mismatch repair, homologous recombination, and p53 signaling (Figure 2C; Supplemental Figure 2C–D). These findings are consistent with previous studies linking USP22 with cell cycle regulation[14] and unmask unexpected impact on DNA repair factor expression.
Given these findings, the cellular response to USP22 downregulation and upregulation was investigated in the isogenic models. Doxycycline-inducible USP22 knockdown in both HT-sensitive (LN-shUSP22) and CRPC (C42-shUSP22) cell lines decreased cell growth by 1.4-fold and 2.4-fold, respectively, compared to controls (Figure 2D). Moreover, a concomitant decrease in cell growth occurred post-irradiation in both LN-shUSP22 (1.4-fold) and C42-shUSP22 (2.5-fold) compared to doxycycline treated controls (Figure 2D). Decreased cellular survival upon USP22 depletion in response to 2.5 Gy (4.8-fold; p=0.002) and 5 Gy (13-fold; p=0.0001) was also apparent in CRPC cells (Figure 2E left) and HT-sensitive cells at 2.5 Gy (p<0.0001; Supplemental Figure 2E). Moreover, a 1.7-fold increase (p=0.017) in survival upon USP22 overexpression in response to irradiation occurred (Supplemental Figure 2F), further indicating a role for USP22 in the response to DNA damaging agents. As multiple DNA repair pathways were altered in the USP22-sensitive transcriptome, the response of USP22 knockdown was also investigated using the chemotherapeutic agent cisplatin, known to induce inter- and intra-strand crosslinks, ultimately resulting in both double-stranded and single-stranded breaks[30]. As shown, USP22 knockdown in combination with cisplatin treatment decreased cell growth in both LN-shUSP22 and C42-shUSP22 models by 1.2-fold and 1.7-fold, respectively, compared to controls (Supplemental Figure 2G). Moreover, USP22 depletion decreased cellular survival in response to cisplatin at 2.5 μM (1.9-fold; p=0.004) and 5 μM (3.5-fold; p=0.0002) in CRPC cells (Figure 2E, right) as well as HT-sensitive cells (p=0.001; Supplemental Figure 2E, right), indicating that USP22 is required for cellular survival after DNA damage. Thus, the USP22-sensitive transcriptome uncovers a role for USP22 in DNA repair factor expression and cellular survival after genotoxic insult.
USP22 confers a hyperproliferative phenotype in vivo and a survival advantage to genotoxic insult
USP22 upregulation was observed to be the most frequent type of USP22 alteration in both primary and advanced PCa (Figure 1). Thus, to further delineate the role of USP22 in tumor development, a novel GEMM was designed to mimic the clinical observation of USP22 upregulation in vivo. As such, Lox-Stop-Lox human 3XFLAG-USP22 cDNA was cloned into the Rosa26 locus, resulting in the GEMM, Rosa26-Lox-Stop-Lox(3XFLAGUSP22). Upon breeding the Rosa26-Lox-Stop-Lox(3XFLAGUSP22) GEMM with prostate-specific Probasin-cre (PB-Cre4) mice, Rosa26(3XFLAGUSP22) mice express 3XFLAG-USP22 in the mouse prostate lobes (PbCre/USP22), while no expression was observed in wildtype (WT) mice (Figure 3A). No changes were observed in mouse USP22 expression (Supplemental Figure 3A). While expression of human USP22 did not overtly alter the histologic appearances of the hematoxylin and eosin (H&E) stained mouse prostate lobes (Supplemental Figure 3B), a hyperproliferative phenotype, as determined by immunostaining for Ki67, was observed in the anterior (3.2-fold increase in Ki67 positive prostatic epithelial cells) and ventral (3 fold increase in Ki67+ prostatic epithelial cells) prostate lobes of PbCre/USP22 mice compared to controls (Figure 3B; Supplemental Figure 3C), further supporting a role for USP22 in cell cycle modulation and consistent with pathways uncovered in human PCa models (Figure 2). As such, the prostate-specific expression of human USP22 in vivo supports the role of USP22 in driving aberrant cell proliferation.
While increased proliferative indices were observed in vivo, assessment of the role of human USP22 expression on the response to DNA damage was accomplished by using heterozygous, immortalized mouse adult fibroblasts (MAFs) generated from the Rosa26-Lox-Stop-Lox(3XFLAGUSP22) GEMM (hUSP22/-). Transduction of a GFP vector control or Cre recombinase to immortalized MAFs derived from the heterozygous Rosa26-Lox-Stop-Lox(3XFLAGUSP22) GEMM (hUSP22/-) resulted in GFP expression (GFP) or human USP22 expression (hUSP22) (Figure 3C). In concordance with the in vivo hyperproliferative phenotype, human USP22 expression increased relative MAF cell number in hUSP22 MAFs compared to GFP controls by 3.1-fold (Figure 3D). Complementary to human PCa cell models, hUSP22 MAFs demonstrated 1.6-fold increased cell number after irradiation as well as a 1.4-fold increase in survival (p=0.01) after 2.5 Gy and a 2.0-fold increase in survival after 5 Gy irradiation (p=0.007), compared to GFP MAFs (Figure 3D), further implicating USP22 in modulation of the response to DNA damaging agents. As PCa models demonstrated increased sensitivity to cisplatin upon USP22 depletion, the hUSP22 MAFs, a model of tumor-associated USP22 overexpression, demonstrated increased survival after 0.5 μM (1.5-fold; p=0.02) or 1 μM (1.6-fold; p=0.02) cisplatin treatment as well as a 2.5-fold decrease in caspase-3 activity upon cisplatin treatment (Figure 3E). Thus, mimicking the clinical alterations in USP22 drive hyperproliferative phenotypes in vivo as well as cellular survival after genotoxic insult.
The USP22-sensitive ubiquitylome reveals altered modification of DNA repair-related proteins
The USP22-sensitive transcriptome implicated USP22 in affecting DDR-related pathways (Figure 2), and in vitro and in vivo studies demonstrated that USP22 modulates proliferative phenotypes as well as cellular survival (Figures 2–3). USP22 acts as a deubiquitinase, removing ubiquitin moieties from substrate proteins, to affect downstream signaling pathways[12,14]. Thus, strategies were employed to define and identify putative substrates of USP22 activity, nominating those proteins that may have an impact on the response of USP22-altered cells to genotoxic insult. Using previously published models of USP22 knockdown or overexpression[8], LNCaP cells with USP22 knockdown (shUSP22) or USP22 overexpression (USP22) underwent Ubiscan analysis to define the USP22-sensitive ubiquitylome (Figure 4A). In short, cell lysates were immunoprecipitated for the KεGG motif present on ubiquitylated proteins, and quantitative mass spectrometry identified those peptides differentially ubiquitylated compared to LNCaP controls (Figure 4A). USP22 was initially described to deubiquitinate histones H2A and H2B to promote activator driven transcription[11], and both H2A and H2B were confirmed as differentially ubiquitinated upon USP22 modulation (Figure 4B). Based on the function of USP22 as a deubiquitinase, putative direct targets of USP22 function would be expected to demonstrate increased ubiquitylation upon USP22 knockdown and decreased ubiquitylation with USP22 upregulation compared to control. As predicted, histones H2A and H2B demonstrate this pattern of differential ubiquitylation, confirming H2A and H2B as targets of USP22 function (Figure 4B), and serving as a control for subsequent interpretation of the unbiased USP22-sensitive ubiquitylome. Upon depletion of USP22, many DNA repair-related peptides demonstrated increased ubiquitylation, compared to controls, including the non-homologous end joining factor, DNA-PK, as well as nucleotide excision repair factors, XPC and RAD23B (Figure 4C; Supplemental Figure 4A–B).
Figure 4. The USP22-sensitive ubiquitylome reveals altered modification of DNA repair-related proteins.

(A) Ubiscan workflow and models used for analyses. LNCaP parental cells were transfected with shUSP22 or USP22. Three samples (LNCaP: parental, shUSP22, and USP22) were run as duplicate injections for a total of 6 LC-MS/MS experiments. (B) Differential ubiquitylation of histones H2A and H2B upon USP22 knockdown and overexpression compared to LNCaP parental control. (C) Ubiscan analysis demonstrates differentially ubiquitylated peptides upon USP22 overexpression and USP22 knockdown. Putative targets of USP22 differential ubiquitination designated as >1.5-fold decreased peptide ubiquitylation in LN-USP22 compared to control and >1.5-fold increased peptide ubiquitylation in LN-shUSP22 compared to parental control (Peptides denoted in purple). (D) Putative direct targets of USP22 function as determined by (C) with DNA repair-related peptides designated by inclusion in gene set enrichment analysis pathways (KEGG or Hallmark GSEA).
To identify those changes likely to be directly regulated by USP22, the data was stringently analyzed to include only those proteins which showed both increased ubiquitylation upon knockdown of USP22 and decreased ubiquitylation upon overexpression of USP22 (Figure 4C, purple). Among these differentially ubiquitylated USP22 targets numerous DNA repair proteins, including XPC and RAD23B, were identified. As USP22 depletion sensitized cells to cisplatin and USP22 overexpression resulted in augmented cisplatin resistance (Figures 2,3), the nucleotide excision repair sensor XPC was nominated as a direct target of USP22 enzymatic function potentially mediating USP22-dependent DNA damage responses. Thus, analysis of the USP22-sensitive ubiquitylome identified potential USP22 substrates responsible for the USP22-mediated response to genotoxic insult, and alterations in the NER pathway were identified as a putative basis of these phenotypes.
USP22 deubiquitylates the nucleotide excision repair protein XPC, modulating foci formation
The NER protein XPC was identified as a putative direct target of the deubiquitinase USP22 in the USP22-sensitive ubiquitylome (Figure 4D). XPC is responsible for sensing and recognition of DNA damage with its binding partners RAD23B and Centrin-2 in global genome NER, where damaged DNA is removed as part of an oligonucleotide fragment after helix-distorting injuries, like cisplatin treatment[31]. During effective NER, enhanced DNA binding of XPC occurs via polyubiquitylation, and this is reversed by de-ubiquitylation to promote efficient repair at damaged foci[32,33]. Thus, active XPC foci promote efficient DNA repair. In addition to its role in NER, increased sensitivity to both etoposide and gamma irradiation have been observed upon depletion of XPC, suggesting a role for XPC in efficient DSB repair[34,35]. Importantly, germline mutations in XPC have also recently been associated with aggressive disease and early onset PCa in a large sequencing study[17], thus further raising interest in understanding the role of XPC in this tumor type.
As prioritized through unbiased assessment of the USP22-sensitive ubiquitylome, XPC was found to interact with USP22 in both the HT-sensitive LN-USP22hi and the CRPC C42-USP22hi models (Figure 5A), which mimic tumor-associated USP22 upregulation in PCa models. An interaction between USP22 and XPC also occurred in the GEMM-derived MAFs, GFP and hUSP22 cells (Figure 5A). This interaction occurred in control cells as well, indicating that XPC and USP22 interact regardless of USP22 overexpression (Figure 5A). Notably, while XPC protein levels remained unchanged upon USP22 modulation (Figure 5B), XPC was confirmed as a downstream target of USP22 de-polyubiquitylation because decreased polyubiquitylated XPC was observed in PCa cells overexpressing USP22, using a polyubiquitin pulldown assay (Ubitest by LifeSensors; Figure 5C)[14,36–38]. USP22 induced deubiquitylation of XPC without an increase of the protein half-life (Supplemental Figure 5A), suggesting the alternative hypothesis that USP22 may modulate XPC activity through de-polyubiquitylation, as XPC is known to be polyubiquitylated to promote efficient DNA repair[32,33,39]. To explore this hypothesis, the biological impact of XPC knockdown in USP22 overexpression cells was explored. As shown, there was no measurable effect on cell number with XPC depletion in models tested (Figure 5D). However, XPC knockdown decreased cell number after irradiation by 1.3-fold, which was alleviated upon USP22 overexpression (Figure 5D; Supplemental Figure 5B), indicating XPC plays a role in the cellular response after irradiation that can be rescued by USP22 overexpression.
Figure 5. USP22 deubiquitylates the nucleotide excision repair protein XPC, modulating foci formation.
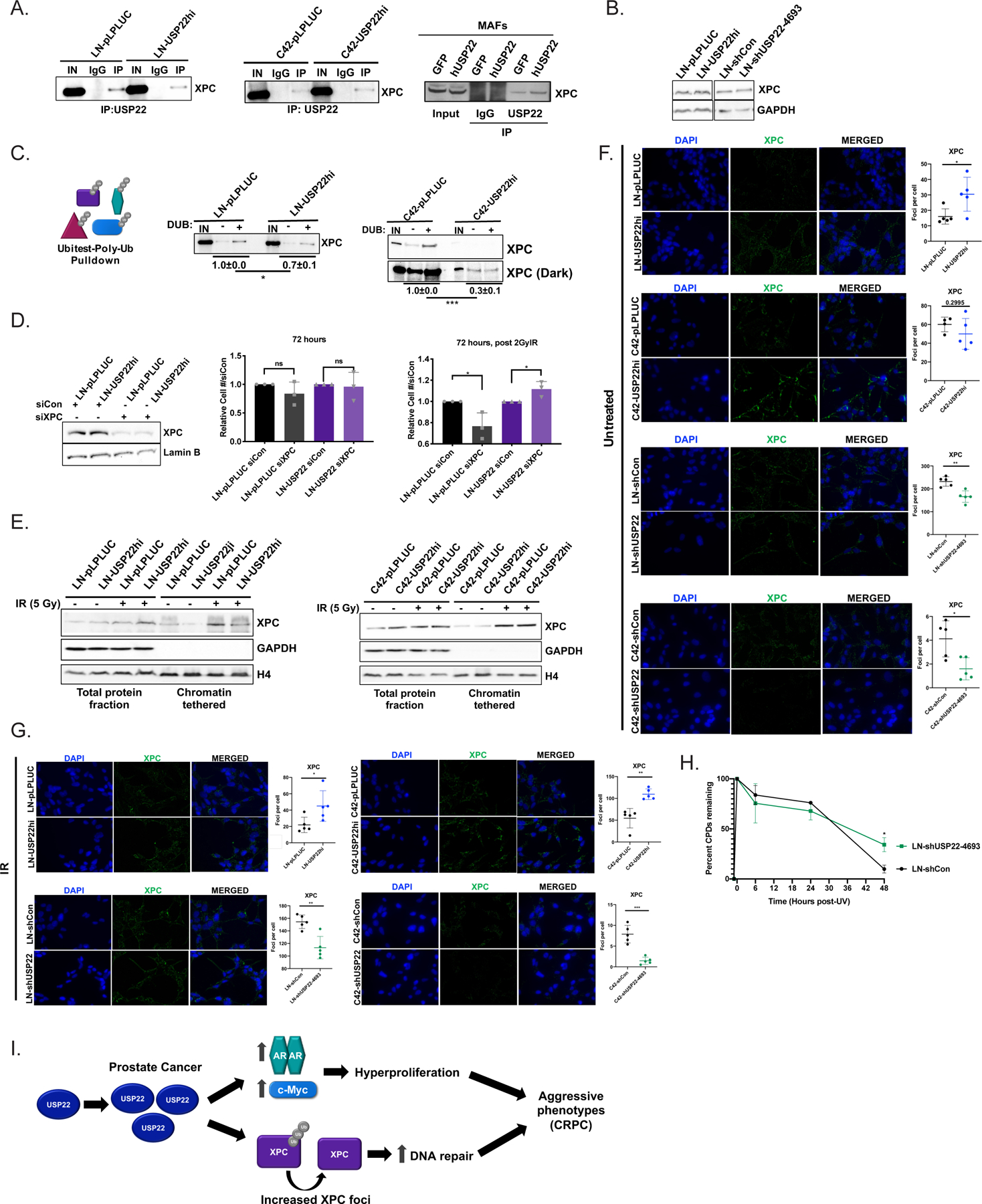
(A) Cell lysates underwent immunoprecipitation of USP22 and immunoblot analysis of XPC in LN-pLPLUC and LN-USP22hi (left), C42-pLPLUC and C42-USP22hi (middle), and GFP and hUSP22 MAFs (right). (B) Indicated cells grown in full serum were isolated for immunoblot analysis with the designated antisera. (C) Cell lysates underwent Ubitest analysis (Life Sensors) with pulldown of poly-ubiquitylated proteins, then +/− deubiquitylase (DUB) treatment, and subsequent immunoblot analysis of XPC in LN-pLPLUC and LN-USP22hi (left) and C42-pLPLUC and C42-USP22hi (right). The change in XPC levels was compared +/− DUB relative to input. The blots are a representative image from three independent experiments. (Student t-test; N=3). (D) LN-pLPLUC or LN-USP22hi cells underwent immunoblot analysis or proliferation assays 72 hours post-transfection of siControl or siXPC +/− 2Gy irradiation (N=3). Student’s t-test. (E) LN-pLPLUC and LN-USP22hi (left) and C42-pLPLUC and C42-USP22hi (right) cells were used for immunoblot analysis of chromatin tethering experiments. (F) LN-pLPLUC or LN-USP22hi, C42-pLPLUC or C42-USP22hi, LN-shCon or LN-shUSP22–4693, and C42-shCon or C42-shUSP22–4693 cells were plated for immunofluorescence analysis of XPC. A representative experiment is shown and at least three independent experiments were performed. Student’s t-test. (G) LN-pLPLUC or LN-USP22hi, C42-pLPLUC or C42-USP22hi, LN-shCon or LN-shUSP22–4693, and C42-shCon or C42-shUSP22–4693 cells were plated and immunofluorescence of XPC was analyzed 24 hours post-5Gy irradiation. A representative experiment is shown and at least three independent experiments were performed. Student’s t-test. (H) LN-shCon or shUSP22–4693 cells were treated with 10 J UV irradiation (N=3 independent experiments in technical triplicate) and cyclobutane pyrimidine dimers (CPDs) were measured by ELISA. (I) Model of study findings. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
XPC activity promotes DNA repair through recognition of DNA damage and efficient binding at damaged foci, and increased XPC was observed on chromatin after irradiation, further suggesting increased XPC activity after DNA damage (Figure 5E). Accordingly, XPC foci formation was also observed to be increased in LN-USP22hi cells compared to control (Figure 5F). Upon USP22 knockdown, decreased XPC foci formation was observed in both LN-shUSP22 and C42-shUSP22 cells (Figure 5F), suggesting XPC foci formation requires sufficient USP22 expression. After treatment with irradiation, XPC foci were significantly increased in LN-USP22hi and C42-USP22hi cells as well as decreased in LN-shUSP22 and C42-shUSP22 compared to corresponding controls (Figure 5G), suggesting that USP22 modulates XPC activity basally and in response to genotoxic insult. Importantly, XPC levels or stability were not changed after irradiation upon USP22 modulation (Supplemental Figure 5C–E), suggesting foci formation was an accurate readout of XPC activity. Moreover, as Rad23B partners with XPC to sense and initiate NER, Rad23B foci formation was also increased basally and after irradiation in LN-USP22hi and C42-USP22hi cells (Supplemental Figure 5F), further suggesting USP22 upregulation augments XPC activity. Additionally, depletion of USP22 resulted in incomplete repair of cyclobutane pyrimidine dimers (CPDs; Figure 5H), an established NER substrate[33], with 3.5-fold more CPDs at 48 hours post-insult (p=0.019). Moreover, hUSP22 MAFs demonstrated increased CPD repair with 1.4-fold less CPDs at 24 hours after insult (p=0.04; Supplemental Figure 5G), supporting a role for USP22 in modulating XPC function. In summary, USP22 removes ubiquitin moieties from poly-ubiquitylated XPC, thus promoting XPC foci formation and subsequently XPC activity, implicating USP22 in proper NER.
In sum (as depicted in schematic Figure 5I), these analyses demonstrated USP22 increases as a function of PCa progression, and that the USP22-sensitive transcriptome included key cell cycle and DDR-related pathways. Thus, in combination with findings correlating increased AR and MYC expression with USP22 overexpression, a hyperproliferative phenotype was observed in the novel Rosa26(3XFLAGUSP22) GEMM model of prostate-specific human USP22 expression. Additionally, upon genotoxic insult, both MAF and PCa models revealed that USP22 modulated the response to DNA damaging agents. Furthermore, identification of the USP22-sensitive ubiquitylome demonstrated that proteins in DDR-related pathways were enriched as putative substrates of USP22-directed deubiquitylation. Specifically, the NER protein XPC was found to be a target of de-polyubiquitylation by USP22, modulating the response to DNA damage. Thus, the novel findings herein indicate that USP22 modulates the response to genotoxic insult by regulation of NER through XPC.
Discussion:
The deubiquitinase USP22 has been previously linked to cancer progression and poor prognosis, but the underlying mechanisms are not fully understood. Studies herein unveil novel mechanisms by which the deubiquitinase USP22 exerts pro-oncogenic phenotypes, supported by the following key findings: (i) USP22 is upregulated in both primary and advanced PCa, and is associated with poor outcome; (ii) transcriptome and ubiquitylome analyses reveal unexpected roles for USP22 in cell cycle and DNA repair; (iii) in vitro modeling and a novel genetically engineered mouse model demonstrate the impact of USP22 in promoting cell proliferation and DNA repair competency; (iv) USP22-mediated DNA repair manifests through regulation of the DNA repair NER factor, XPC, promoting efficient response to genotoxic insult. These studies further confirm the oncogenic nature of USP22 and uncover novel USP22 functions that contribute to tumor progression.
The data reported in Figure 1 demonstrate that the most frequent USP22 alteration in PCa is mRNA upregulation, consistent with previous observations that USP22 increases during progression from early stage PCa to CRPC[8]. USP22 alterations are associated with pro-proliferative oncogenes, including AR and MYC[8], and accordingly, data demonstrated that suppression of USP22 expression decreased cell proliferation in PCa models (Figure 2D). Downregulation of USP22 in various in vitro and in vivo tumor models has been previously reported to decrease cell proliferation through G1 arrest as well as increase pro-apoptotic phenotypes[11,14,40,41], but the mechanisms by which USP22 drives these phenotypes remain incompletely defined. While genetic loss or depletion of USP22 in vivo has been observed to be embryonic lethal or result in growth impairment, respectively[12,42], the in vivo effects of tumor-associated USP22 upregulation had not been assessed until this study. As a result, the data herein were the first to demonstrate USP22 independently promotes proliferation in normal tissue. The present study reports the generation of a novel in vivo model that mimics tumor-associated USP22 deregulation, demonstrating that USP22 is sufficient to drive a hyperproliferative phenotype in the normal murine prostate. While neoplastic lesions were not apparent in the context of human USP22 expression in the murine prostate lobes examined, this is not without precedent as single oncogenes rarely are sufficient to promote tumorigenesis, with the exception of c-Myc[43,44]. Taken together, these findings are the first to demonstrate that USP22 alone induces pro-tumorigenic phenotypes in normal tissue, and further implicates USP22 as an oncogenic driver in PCa.
Ongoing studies will address which cooperative alterations act in concert with USP22 to complete the transition to tumorigenesis. The availability of PCa models that express high MYC as well as models that express the AR would provide the means to assess the requirement of USP22 on MYC or AR-mediated tumor development and progression in the mouse prostate[28,45–47]. To complement in vivo studies interrogating the role of AR and MYC in the context of USP22 expression, the study herein identified XPC as a substrate of USP22 action. Thus, the requirements of XPC and USP22 on cancer development can be identified through interrogation of USP22 overexpression in XPC null mice[48–50]. Analysis of oncogenic drivers that were significantly and positively correlated with USP22 expression in the TCGA provisional dataset revealed TRIM25 as a potential target with high USP22 expression (Supplemental Figure 1). The E3 ubiquitin ligase, TRIM25, has been shown to promote progression of hormone-driven cancers[51,52], and the relationship and requirements of USP22 and TRIM25 in PCa models will be interrogated. Additionally, hnRNP K was identified in the USP22-sensitive ubiquitylome as a putative substrate of USP22 function. The RNA processing protein, hnRNP K, is a known substrate of ATM action[53,54], a critical modulator of the DNA damage response, as well as a regulator of the androgen receptor[48]. Thus, the role of USP22 and hnRNPK action will be thoroughly addressed in the context of AR and DNA repair. Finally, the involvement of two critical DNA damage response phosphatidylinositol 3-kinase-related kinases (PIKKs), DNA-PK and ATM (via hnRNP K), warrants investigation into USP22 function in double strand break repair in addition to XPC-mediated nucleotide excision repair.
In addition to the role of USP22 in proliferation, data herein implicate USP22 in DNA repair competency. USP22 has previously been associated with maintenance of genomic stability through deubiquitylation of target substrates, including the telomere-associated shelterin component, TRF1[13], and the p53 tumor suppressor, which has established roles in DNA repair checkpoint integrity[13,55]. In this study, p53 signaling was confirmed as altered upon USP22 modulation, but ubiquitylome data do not support the concept that p53 serves as a putative USP22 substrate. By contrast, the USP22-sensitive ubiquitylome demonstrated robust enrichment for DNA repair pathways. Interestingly, USP22 has been previously implicated in efficient class switch recombination (CSR) and DSB repair in B cells with potential roles in homologous recombination (HR) and non-homologous end joining (NHEJ)[15,16]. The studies herein identified the NHEJ factor DNA-PK as putatively regulated by USP22 based on ubiquitylome mapping, which could contribute to the role of USP22 in NHEJ. Accordingly, USP22 promotes cellular survival after double strand break-inducing irradiation (Figure 2; Supplemental Figure 2; Figure 3). Future studies will address this concept, as DNA-PK has been previously defined as an effector of PCa progression[18,19]. However, the most striking observations were associated with NER factors, as the NER pathway was the most significantly enriched DNA repair pathway in the USP22-sensitive ubiquitylome. Intriguingly, protein-truncating variants of XPC have been significantly associated with aggressive disease in PCa[56], furthering the need to understand the role of XPC in PCa progression. As such, the combination of recent evidence identifying the NER protein XPC as a risk factor for aggressive PCa and candidate-driven interrogation of the factors identified in USP22-sensitive unbiased analyses, nominated XPC as a critical effector of USP22 function.
In the NER pathway, XPC senses DNA damage as part of a complex that includes RAD23B and Centrin-2[31]. After DNA damage, XPC is post-translationally modified through both SUMOylation and ubiquitylation[57–59]. Damage-induced XPC ubiquitylation occurs via the cullin-RING ubiquitin ligase (CRL) complex, consisting of damage-specific DNA binding protein 1 (DDB1), DDB2, regulator of cullins 1 (ROC1), and cullin 4A (CUL4A) in addition to RING finger protein 111 (RNF111)[60]. XPC polyubiquitylation promotes the DNA binding of XPC, while removal of ubiquitin moieties triggers the release of XPC from damaged DNA, protecting XPC from proteasomal degradation[32,60]. Thus, regulation of XPC polyubiquitylation is a key requirement for an efficient response to DNA damage. The studies herein demonstrated increased XPC chromatin residency after genotoxic insult, consistent with previous observations that XPC is a critical DNA damage sensor[57–59]. Moreover, USP22 was determined to positively regulate XPC activity to promote efficient DNA repair after DNA damage through regulation of XPC polyubiquitylation. Interestingly, USP7 has also been identified to deubiquitinate XPC, critical for the UV-induced, XPC-mediated NER reaction[61], and USP11 has been shown to promote radiation-induced DNA damage repair through USP11-mediated XPC deubiquitylation on chromatin after UV damage. Interestingly, USP11-depleted cells still demonstrated XPC deubiquitylation after UV damage, possibly in a USP11-independent mechanism that could be associated with USP22 function. Similarly, USP11 does not impact degradation of XPC after UV damage, as was also observed in this study upon the USP22-mediated response to irradiation[62]. Finally, degradation of XPC is ubiquitylation-independent[39], which is consistent with the data demonstrating no change in XPC levels or stability upon modulation of USP22. Thus, efficient regulation of XPC polyubiquitylation is required for an effective response to DNA damage, and USP22 modulates XPC ubiquitylation to promote DNA repair.
The NER pathway is generally associated with repair of DNA damage caused by UV radiation, and the studies herein identify USP22 as a critical regulator of XPC, a DNA damage sensor in the NER pathway. Importantly, the data here implicate XPC in the response to irradiation and cisplatin, which induce both single and double strand breaks. The concept that XPC modulates DNA repair pathways has strong precedent as XPC or RAD23B depletion sensitizes cells to γ-radiation or etoposide, and this correlates with decreased NHEJ[34,35], further confirming the observations in this study that decreased XPC increased sensitivity to irradiation in a USP22-dependent manner. XPC has also been observed to prevent cisplatin-induced apoptosis through activation of the ATM pathway, as XPC is required for the association of ATM with genomic DNA[63]. Through activation of the ATM pathway, cell cycle arrest would allow for efficient repair of DNA damage[63]. Thus, USP22 could be potentiating efficient ATM-mediated DNA repair through modulation of XPC to promote DSB repair. Moreover, as the USP22-mediated response to DNA damage through XPC promotes therapeutic resistance mechanisms, XPC could contribute to the hyperproliferative phenotype observed in the absence of insult in vivo as well as the survival advantage observed upon tumor-associated USP22 upregulation in MAFs. Thus, the observations here that USP22-mediated regulation impacts response to single and double strand breaks repair likely involves XPC function in multiple DDR pathways.
In sum, these studies uncover novel insights into the molecular functions of USP22 in malignancy. The data presented demonstrate that USP22 is deregulated in human PCa, and that USP22 potentially regulates cell proliferation and DNA repair through regulation of XPC. These findings identify critical mechanisms by which USP22 drives pro-oncogenic functions and nominate USP22 as a putative therapeutic target.
Supplementary Material
Statement of Significance:
The studies herein present a novel mouse model of tumor-associated USP22 over-expression and implicate USP22 in modulation of cellular survival and DNA repair, in part through regulation of XPC.
Acknowledgments:
We gratefully thank all members of the Knudsen laboratory for their invaluable input and support. We also thank the institutions that supported this work: The National Cancer Institute (F99CA212225 JJM, R01 CA176401, R01 CA182569, and R01 CA217329 KEK), the Sidney Kimmel Cancer Center (Support Grant 5P30CA056036-17; Laboratory Animals, MetaOmics, Translational Pathology), and awards from the Prostate Cancer Foundation.
Footnotes
The authors declare no potential conflicts of interest
References:
- 1.Liu YL, Yang YM, Xu H, Dong XS. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J Gastroenterol Hepatol 2010;25:1800–1805. [DOI] [PubMed] [Google Scholar]
- 2.Tang B, Tang F, Li B, Yuan S, Xu Q, Tomlinson S, et al. High USP22 expression indicates poor prognosis in hepatocellular carcinoma. Oncotarget 2015;6:12654–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhai R, Tang F, Gong J, Zhang J, Lei B, Li B, et al. The relationship between the expression of USP22, BMI1, and EZH2 in hepatocellular carcinoma and their impacts on prognosis. Onco Targets Ther 2016;Volume 9:6987–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ning J, Zhang J, Liu W, Lang Y, Xue Y, Xu S. Overexpression of ubiquitin-specific protease 22 predicts poor survival in patients with early-stage non-small cell lung cancer. Eur J Histochem 2012;56:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y-L, Yang Y-M, Xu H, Dong X-S. Aberrant expression of USP22 is associated with liver metastasis and poor prognosis of colorectal cancer. J Surg Oncol 2011;103:283–289. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, et al. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol 2011;137:1245–1253. [DOI] [PubMed] [Google Scholar]
- 7.Liang J-X, Ning Z, Gao W, Ling J, Wang A-M, Luo H-F, et al. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol Rep 2014:2726–2734. [DOI] [PubMed]
- 8.Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res 2014;74:272–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005;115:1503–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 11.Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, et al. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell 2008;29:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 Antagonizes p53 Transcriptional Activation by Deubiquitinating Sirt1 to Suppress Cell Apoptosis and Is Required for Mouse Embryonic Development. Mol Cell 2012;46:484–494. [DOI] [PubMed] [Google Scholar]
- 13.Atanassov BS, Evrard Y a., Multani AS, Zhang Z, Tora L, Devys D, et al. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell 2009;35:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gennaro VJ, Stanek TJ, Peck AR, Sun Y, Wang F, Qie S, et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc Natl Acad Sci 2018;115:E9298–E9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran S, Haddad D, Li C, Le MX, Ling AK, So CC, et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep 2016;15:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Irrazabal T, So CC, Berru M, Du L, Lam E, et al. The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non-homologous end joining. Nat Commun 2018;9:1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leongamornlert DA, Saunders EJ, Wakerell S, Whitmore I, Dadaev T, Cieza-Borrella C, et al. Germline DNA Repair Gene Mutations in Young-onset Prostate Cancer Cases in the UK: Evidence for a More Extensive Genetic Panel. Eur Urol 2019;76:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015;28:97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A Hormone-DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov 2013;3:1254–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 2005;23:94–101. [DOI] [PubMed] [Google Scholar]
- 21.Stokes MP, Farnsworth CL, Moritz A, Silva JC, Jia X, Lee KA, et al. PTMScan direct: Identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol. Cell. Proteomics, vol. 11, 2012:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stokes MP, Farnsworth CL, Gu H, Jia X, Worsfold CR, Yang V, et al. Complementary PTM Profiling of Drug Response in Human Gastric Carcinoma by Immunoaffinity and IMAC Methods with Total Proteome Analysis. Proteomes 2015;3:159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lundgren DH, Martinez H, Wright ME, Han DK. Protein identification using sorcerer 2 and SEQUEST. Curr Protoc Bioinforma 2009;28:13.3.1–13.3.21. [DOI] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1:2315–2319. [DOI] [PubMed] [Google Scholar]
- 28.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003;4:223–238. [DOI] [PubMed] [Google Scholar]
- 29.Nadiminty N, Tummala R, Lou W, Zhu Y, Zhang J, Chen X, et al. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J Biol Chem 2012;287:1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vare D, Groth P, Carlsson R, Johansson F, Erixon K, Jenssen D. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA Repair (Amst) 2012;11:976–985. [DOI] [PubMed] [Google Scholar]
- 31.Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer 2001;1:22–33. [DOI] [PubMed] [Google Scholar]
- 32.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005;121:387–400. [DOI] [PubMed] [Google Scholar]
- 33.Hannah J, Zhou P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair (Amst) 2009;8:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biard DSF. Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res 2007;35:3535–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Despras E, Pfeiffer P, Salles B, Calsou P, Kuhfittig-Kulle S, Angulo JF, et al. Long-term XPC Silencing Reduces DNA Double-Strand Break Repair. Cancer Res 2007;67:2526–2560. [DOI] [PubMed] [Google Scholar]
- 36.Wang F, Wang L, Wu J, Sokirniy I, Nguyen P, Bregnard T, et al. Active site-targeted covalent irreversible inhibitors of USP7 impair the functions of Foxp3+ T-regulatory cells by promoting ubiquitination of Tip60. PLoS One 2017;12:e0189744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dybas JM, O’Leary CE, Ding H, Spruce LA, Seeholzer SH, Oliver PM. Integrative proteomics reveals an increase in non-degradative ubiquitylation in activated CD4+ T cells. Nat Immunol 2019;20:747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali MAM, Strickfaden H, Lee BL, Spyracopoulos L, Hendzel MJ. RYBP Is a K63-Ubiquitin-Chain-Binding Protein that Inhibits Homologous Recombination Repair. Cell Rep 2018;22:383–395. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q-E, Prætorius-Ibba M, Zhu Q, El-Mahdy MA, Wani G, Zhao Q, et al. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res 2007;35:5338–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell 2012;46:484–494. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D, Jiang F, Wang X, Li G. Downregulation of Ubiquitin-Specific Protease 22 Inhibits Proliferation, Invasion, and Epithelial-Mesenchymal Transition in Osteosarcoma Cells. Oncol Res Featur Preclin Clin Cancer Ther 2017;25:743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kosinsky RL, Wegwitz F, Hellbach N, Dobbelstein M, Mansouri A, Vogel T, et al. Usp22 deficiency impairs intestinal epithelial lineage specification in vivo. Oncotarget 2015;6. [DOI] [PMC free article] [PubMed]
- 43.Ward JM, Herbert R, Rozengurt N, Sundberg JP, Shappell SB, Humphrey PA, et al. Prostate Pathology of Genetically Engineered Mice: Definitions and Classification. The Consensus Report from the Bar Harbor Meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res 2005;64:2270–2305. [DOI] [PubMed] [Google Scholar]
- 44.Valkenburg KC, Williams BO, Valkenburg KC, Williams BO. Mouse Models of Prostate Cancer. Prostate Cancer 2011;2011:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albanese C, Rodriguez O, Johnson MD, Fricke S. Models of prostate cancer. Drug Discov Today Dis Model 2005;2:7–13. [Google Scholar]
- 46.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes Cancer 2010;1:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borowsky AD, Chen Z, Matusik RJ, Jin RJ, Yu X, Grabowska MM, et al. Mouse models of prostate cancer: picking the best model for the question. Cancer Metastasis Rev 2014;33:377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukhopadhyay NK, Kim J, Cinar B, Ramachandran A, Hager MH, Di Vizio D, et al. Heterogeneous Nuclear Ribonucleoprotein K Is a Novel Regulator of Androgen Receptor Translation. Cancer Res 2009;69:2210–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheo DL, Ruven HJ, Meira LB, Hammer RE, Burns DK, Tappe NJ, et al. Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res Mol Mech Mutagen 1997;374:1–9. [DOI] [PubMed] [Google Scholar]
- 50.Sands AT, Abuin A, Sanchez A, Conti CJ, Bradley A. High susceptibility to ultraviolet-induced carcinogenesis in mice lacking XPC. Nature 1995;377:162–165. [DOI] [PubMed] [Google Scholar]
- 51.Hatakeyama S TRIM proteins and cancer. Nat Rev Cancer 2011;11:792–804. [DOI] [PubMed] [Google Scholar]
- 52.Takayama KI, Suzuki T, Tanaka T, Fujimura T, Takahashi S, Urano T, et al. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018;37:2165–2180. [DOI] [PubMed] [Google Scholar]
- 53.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013;14:197–210. [PubMed] [Google Scholar]
- 54.Moumen A, Magill C, Dry K, Jackson SP. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 2013;12:698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atanassov BS, Mohan RD, Lan X, Kuang X, Lu Y, Lin K, et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol Cell 2015:1–14. [DOI] [PMC free article] [PubMed]
- 56.Leongamornlert DA, Saunders EJ, Wakerell S, Whitmore I, Dadaev T, Cieza-Borrella C, et al. Germline DNA Repair Gene Mutations in Young-onset Prostate Cancer Cases in the UK: Evidence for a More Extensive Genetic Panel. Eur Urol 2019;76:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Q-E. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res 2005;33:4023–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akita M, Tak Y-S, Shimura T, Matsumoto S, Okuda-Shimizu Y, Shimizu Y, et al. SUMOylation of xeroderma pigmentosum group C protein regulates DNA damage recognition during nucleotide excision repair. Sci Rep 2015;5:10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, Poulsen JW, et al. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat Cell Biol 2012;14:1089–1098. [DOI] [PubMed] [Google Scholar]
- 60.Van Cuijk L, Van Belle GJ, Turkyilmaz Y, Poulsen SL, Janssens RC, Theil AF, et al. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat Commun 2015;6:7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, et al. Ubiquitin-specific Protease 7 Regulates Nucleotide Excision Repair through Deubiquitinating XPC Protein and Preventing XPC Protein from Undergoing Ultraviolet Light-induced and VCP/p97 Protein-regulated Proteolysis. J Biol Chem 2014;289:27278–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shah P, Qiang L, Yang S, Soltani K, He Y. Regulation of XPC deubiquitination by USP11 in repair of UV- induced DNA damage 2017;8:96522–96535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colton SL, Xu XS, Wang YA, Wang G. The Involvement of Ataxia-telangiectasia Mutated Protein Activation in Nucleotide Excision Repair-facilitated Cell Survival with Cisplatin Treatment. J Biol Chem 2006;281:27117–27125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.