

SUMMARY
Precise discrimination of tumor from normal tissues remains a major roadblock for therapeutic efficacy of chimeric antigen receptor (CAR) T cells. Here, we perform a comprehensive in silico screen to identify multi-antigen signatures that improve tumor discrimination by CAR T cells engineered to integrate multiple antigen inputs via Boolean logic, e.g., AND and NOT. We screen >2.5 million dual antigens and ~60 million triple antigens across 33 tumor types and 34 normal tissues. We find that dual antigens significantly outperform the best single clinically investigated CAR targets and confirm key predictions experimentally. Further, we identify antigen triplets that are predicted to show close to ideal tumor-versus-normal tissue discrimination for several tumor types. This work demonstrates the potential of 2- to 3-antigen Boolean logic gates for improving tumor discrimination by CAR T cell therapies. Our predictions are available on an interactive web server resource (antigen.princeton.edu).
Graphical Abstract
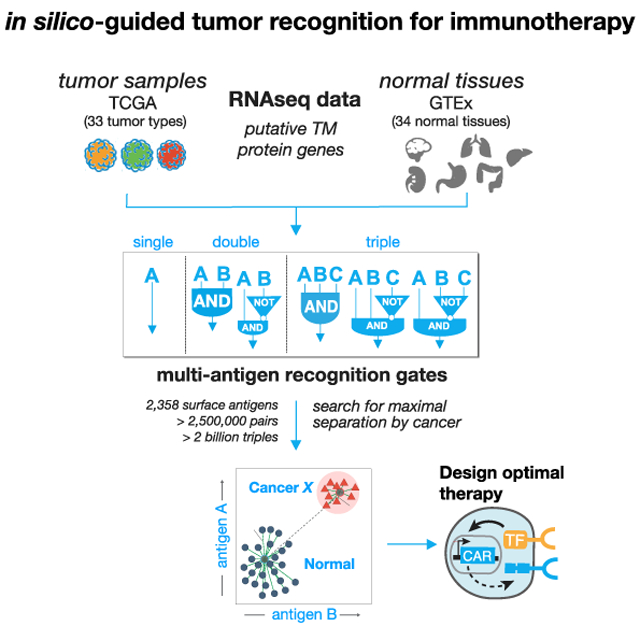
In Brief
The application of CAR T cells to solid tumors is limited by the difficulty in identifying single target antigens that adequately discriminate between tumor and normal tissue to avoid toxicity. We leverage large-scale RNA-seq databases from tumor and normal tissues to evaluate the discriminatory power of single antigens and antigen combinations. Most single antigens, including those currently under investigation as CAR targets in solid tumors, perform poorly. The addition of a second or third antigen using AND or NOT gating can significantly improve CAR T cell performance. We construct and test a pair of potential AND-gated T cells for renal cell carcinoma. A full database of all predicted high-performing antigen pairs and triplets is made available in an associated web server (antigen.princeton.edu).
INTRODUCTION
Despite recent clinical success in using engineered T cells to treat hematologic cancers (Maude et al., 2018; Neelapu et al., 2017), a major barrier in expanding their use to solid tumors is the challenge of specific tumor recognition. Although it is possible to engineer chimeric antigen receptors (CARs) directed toward tumor associated antigens, many of those antigens, especially in the case of solid tumors, are also expressed, often at lower levels, in other normal tissues, leading to cases of toxic cross-reactivity (Lamers et al., 2013; Morgan et al., 2010; Parkhurst et al., 2011; Thistlethwaite et al., 2017). While toxicity can in some cases be ameliorated by reducing CAR T dosage, the small therapeutic window caused by poor discrimination leads to a trade-off between efficacy and toxicity. The difficulty of finding absolutely tumor unique surface antigens that can be distinctly recognized by CARs has led some to question the capability of such engineered T cells to ultimately achieve success in safely treating solid tumors (Rosenberg and Restifo, 2015).
Current approaches for engineering CAR T cells, however, focus only on recognition of a single target antigen. If we consider that solid tumors express an array of antigens, it is possible that improved specificity could be achieved through recognition of combinatorial antigen signatures (Figure 1A). Such considerations, however, have only recently become actionable with advances in synthetic biology approaches to engineering T cell therapies. Engineered cells are unique among therapeutic modalities in that they can in principle be engineered with multi-antigen recognition circuits. For example, recent advances have shown that it is possible to engineer CAR T cells that recognize target cells with combinatorial Boolean logic: one can engineer T cells with multi-receptor circuits that function as AND gates (requiring two antigens to be present) (Kloss et al., 2013; Roybal et al., 2016a, 2016b; Srivastava et al., 2019; Wilkie et al., 2012), NOT gates (Fedorov et al., 2013), and OR gates (requiring the presence of one of two possible antigens) (Grada et al., 2013; Hegde et al., 2013). AND gates (high expression of two antigens) and NOT gates (high expression of one antigen, low expression of another) could, in principle, significantly increase tumor selectivity by limiting cross-reactivity with healthy tissues that also express the CAR/TCR target antigen (Figure 1A). It may also be possible to engineer T cells with more complex recognition circuits, based on more than two antigens. A critical question that remains is how significantly such combinatorial antigen recognition circuits could improve targeting of cancers and limit cross-reaction with normal tissues.
Figure 1. Computationally Enumerating Combinatorial Antigen Sets Predicted to Improve T Cell Discrimination of Cancer versus Normal Cells.

(A) Single antigen targets for CAR T cells often show cross reactivity with subset of normal tissues. Combinatorial recognition circuits (AND, NOT, etc.) could improve discrimination.
(B) Single antigen targets theoretically hit samples that have high expression of antigen A or B. Using Boolean T cells we can target specific patterns of antigen expression reducing off-target toxicity.
(C) Computational pipeline for identifying antigen pairs with improved tumor discrimination. For each cancer type (N = 33), normalized RNA-seq expression data are combined with RNA-seq data for 34 normal tissues. All potential transmembrane antigen pairs are then evaluated for their potential to separate samples of a given tumor type from all normal samples in expression space. Shaded boxes highlight specific steps of the pipeline starting with a representation of the expression data, followed by the scoring method, and toy examples highlighting how evaluation metrics are calculated.
Here, we performed a comprehensive computational search of all possible pairs of predicted surface antigens in the human genome (2,358 total predicted surface genes with >2.5 million total possible surface-presented antigen combinations) to explore the strategy of tumor cell targeting by CAR T cells engineered to express multi-receptor circuits that function as Boolean logic gates. We score all AND and NOT gates by how well the putative combination separates tumor and normal tissue samples for 33 distinct tumor types and 34 major healthy tissues and then add a third surface antigen to explore more than 60 million additional unique AND and NOT gates for triplets. For these logic gates, we define both how much off-target toxicity can be avoided (precision) and the potential number of tumor samples we can target (recall).
We find that cellular recognition programs which incorporate information from multiple (2 or 3) antigens, outperform standard single antigen recognition circuits. As the number of antigens used to discriminate tumor-versus-normal tissue is increased, the precision of tumor detection increases at the cost of decreased recall of all tumor specimens. For most cancer types, there are numerous dual antigen combinations that significantly improve the precision and recall of the best single antigen, including currently clinically investigated CAR targets. For several tumor types, antigen triplets are predicted to show close to ideal tumor-versus-normal tissue discrimination. We also experimentally validate improved detection of renal cell carcinoma (RCC) using computationally identified antigen pairs for proof of principle. In total, our work illustrates an overall strategy for merging computational analysis with increasing synthetic biology capabilities to identify and target sectors of antigen recognition space that precisely identify and discriminate particular tumor types.
RESULTS
Pipeline for Identifying Antigen Combinations that Improve Tumor Discrimination
Candidate antigens must be recognizable from the cell surface. Toward that end we first curated a list of more than 5,000 genes expected to have cell surface expression. Using the COMPARTMENTS database (Binder et al., 2014), we further pruned our curated list to only include predicted transmembrane proteins that are annotated to be expressed on the plasma membrane. Of these, the genes predicted to encode transmembrane proteins, we classified potential target antigens as either: “clinical”—involved as a target of a CAR T cell therapy in a currently registered clinical trial (29 genes; see Table 1); or “novel” — not currently targeted in a known therapeutic T cell clinical trial. In total, this yielded approximately 2,400 surface-expressed genes across 33 tumor types and 34 normal tissue samples (Supplemental Information).
Table 1.
“Clinical” Antigens Currently under Investigation as CAR Targets in Clinical Trials as Identified from Clinicaltrials.gov
| Antigen | Clinical Trial |
|---|---|
| AXL | NCT03393936 |
| CAIX-2 | DDHK97-29 |
| CD133 | NCT02541370 |
| CD137 | NCT02862704 |
| CD147 | NCT04045847 |
| CD70 | NCT02830724 |
| CD80 | NCT03198052 |
| CEA | NCT01373047 |
| CLDN18 | NCT03159819 |
| EGFR | NCT02331693 |
| EpCAM | NCT02915445 |
| EPHA2 | NCT03423992 |
| FAP | NCT01722149 |
| FOLR1 | NCT00019136 |
| GD2 | NCT00085930 |
| GPC3 | NCT02959151 |
| IL13RA2 | NCT00730613 |
| HER2 | NCT02442297 |
| L1CAM | NCT02311621 |
| MSLN | NCT02930993 |
| MET | NCT01837602 |
| MUC1 | NCT02617134 |
| MUC16 | NCT02498912 |
| PD-L1 | NCT0330834 |
| PSCA | NCT02744287 |
| PSMA | NCT01929239 |
| ROR1 | NCT02706392 |
| ROR2 | NCT03393936 |
| VEGFR2 | NCT01218867 |
We then use RNA sequencing (RNA-seq) expression data across 9,084 samples taken from the Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga) and 12,402 samples from Genotype Tissue Expression project (GTEx) (GTEx Consortium et al., 2017) to measure the level of potential target antigen gene expression. To reduce expression differences due to technical variation, we batch corrected all samples and used log transformed TPM (transcript per million) normalized read counts. Samples were partitioned using geometric sketching (Hie et al., 2019) to get an equal representation of all tissue types and the tumor samples in both partitions, with 20% of the data taken for training and the remaining 80% set aside for evaluation.
Using the gene expression values of potential target antigens, we calculated a clustering-based score to quantify the separation between samples of a single tumor type versus all normal tissue samples (Figure 1C). Specifically, we chose the Davies-Bouldin metric, which measures the ratio of within cluster spread to between cluster distance, as the key component of our cluster-based scores. Before settling on Davies-Bouldin we investigated other cluster evaluation metrics that could be applied to the cluster separation problem including: Silhouette, Dunn’s index, and the Xie-Bene validity measure. While all methods yield to similar results, some drawbacks with other metrics made Davies-Bouldin our preferred choice. Namely, that: Silhouette gives too much weight to compactness and did not have enough variation to differentiate between top antigens; Dunn’s index did not produce enough variation in scores; and Xie-Bene generated too many missing values in practice.
Final clustering-based scores for a given antigen combination, utilize the Davies-Bouldin index with a modification to give extra weight for the distance between cluster centers. Together, clustering scores take into account the average distance between the two types of samples (tumor and normal) and the overall distribution of samples in expression space. Clustering-based scores are scaled from 0 to 1, for ease of ordering, with scores close to 1 indicating the best performing combinations with larger distance and less scatter between the classes of samples (see STAR Methods for additional details).
On the training set, we used clustering-based scores to rank all putative target antigens for each tumor type by their potential to separate samples of one tumor type from all normal tissue samples (Figure 1C). We calculated clustering scores for all surface antigens as a single (n = 2,358) and as a pair (n = 2,778,903) for each tumor type. Only singles with high antigen expression in the target cancer samples, and pairs of antigens (doubles) that are either both highly expressed in the target (AND-gate), or one with high expression and the other with low expression in the target (AND-NOT-gate) are useful as viable CAR/TCR targets. Given the large number of surface antigens (2,358), the space of potential triplets for our set of antigens is >2.2 billion (2,358 choose 3), for efficiency we restricted the search of triple antigen gates to single antigens that have at least some discrimination potential as assessed in the single antigen search (see STAR Methods). We then calculated clustering scores over this restricted set per tumor type for triple AND, AND-AND-NOT, and AND-NOT-NOT gates.
The clustering-based scores prioritize antigens that have a large distance between tumor and normal samples, but we are also interested in a metric that can more directly capture how much off-target toxicity can be avoided (precision) and the potential number of tumor samples we can target (recall) if Boolean logic gates are used. Decision tree classifiers can find boundaries that divide data into groups, while optimizing for the purity of the division. New samples can then be labeled with a group depending on which side of the boundary they fall on. In our case, decision trees can be used to find an expression value for each antigen where samples of a given tumor type are the most separated from normal tissue samples, then use the boundary to classify a new sample point as tumor or normal. Since clustering scores prioritize antigens that spread the two sample types, we should be able to find clear boundaries. To train the decision tree models we used the same training data we used for the clustering-based scores and evaluate the resulting models on our held out test set of samples (Figure S1). Applying this to the top antigen combinations found via clustering, we can then assess how well each of the top performing single, double, and triple gates separate tumor samples from normal tissue samples using the resulting F1 score (harmonic mean of precision and recall) from classification.
Recognition of All Cancer Types Can Be Improved by Adding Secondary Antigens to Current Clinical CAR T Targets
We first calculated clustering-based scores for the current clinically targeted antigens described in (Table 1). We compared these single antigen scores with those obtained for antigen pairs in which two clinically targeted antigens (clinical antigens) are combined, a clinical antigen is paired with a novel putative surface antigen, or two novel surface antigens are paired. A spreadsheet listing the top 10 antigen pairs from each type of combination (e.g., clinical-clinical, clinical-novel, etc.) per cancer type is provided in the Supplemental Information, Table S1 and is ranked by clustering-based scores.
To give us more insight into how well antigens combinations separate normal versus tumor samples, we used decision tree models for each single antigen and each antigen pair in the top-ranked antigens, as identified by their clustering scores. Decision trees find expression level cutoffs for each antigen that separate the classes of samples (tumor and normal). Applying them on held out sample data yielded additional metrics describing how well potential antigen combinations separate tumor and normal samples when using distinct expression level boundaries (Figure S1).
More specifically for each gate type, we took the top 10 antigen singles or pairs for each tumor (330 data points per gate type, from 33 tumors × 10 top combos) and quantified their tumor-versus-normal discrimination potential using F1. As shown in Figure 2A, current clinical antigens on average lack sensitivity and specificity when used as the sole recognition antigen (μF1 = 0.09). However, combining two clinical antigens with AND or NOT logic for tumor recognition leads to significant improvement in both precision and recall as seen by the jump in F1 (μtop10 F1 = 0.25; Wilcoxon rank sum p = 5.96 × 10−32; n = 669). This suggests that simple combinations of already well-verified CAR targets can greatly improve the discriminatory ability of CAR T cells. Using the larger pool of novel antigens (that is, those identified by our pipeline that are not currently being investigated in clinical trials) allows for even more improvement in discrimination both alone, as single antigens (μ1 = 0.37), and when paired with clinical antigens (μtop10 F1 = 0.5; Wilcoxon ranksum p = 1.33 × 10−46; n = 676). Novel-novel pairings show even more potential (μtop10 F1 = 0.57). Taken together these results suggest that discrimination achievable by current clinical antigens can be dramatically improved by incorporating them into antigen pairs recognized by Boolean gated T cells.
Figure 2. Dual Antigen Use Greatly Improves the Precision of Cancer Detection.

Antigen combinations were ranked by their clustering scores for each tumor and each gate type (e.g., clinical, novel, clinical-clinical, clinical-novel, or novelnovel). In this figure different subsets of the top antigens (e.g., the top scoring singlet/pair or the top 10 combinations) are taken and their F1 scores are used to describe their potential discriminatory power.
(A) Distribution of tumor-versus-normal discrimination scores (F1) for the top clinical antigens or top 10 novel antigens for each cancer type, and for the top 10 antigen pairs (clinical-clinical, clinical-novel, or novel-novel) for each cancer type. F1 scores range between 0 (no sensitivity and specificity) and 1 (perfect precision and recall). Here, we see significant gains in discrimination power going from a clinical antigen to a single novel antigen (p = 8.41 × 10−69; n = 646) and from a clinical-clinical antigen pair to a clinical-novel pair (p = 1,38 × 10−11; n = 660).
(B) Improvement in tumor-versus-normal discrimination with dual antigen recognition by cancer type. F1 scores are shown for the highest clustering score single clinical antigen and the highest clustering score dual antigen pair. All antigen pairs improve over the highest performing single clinical antigen.
(C) Pie chart showing the composition of different gate types of pairs in the top 10 per tumor type. A AND B gates have high expression of both antigens, A AND NOT B have high expression of one antigen and low expression of the second antigen. The majority of pairs are AND NOT gates.
(D) Novel antigens (hubs, blue) identified that form high-performing pairs with numerous current clinically targeted CAR antigens (spokes, orange). Edge weights and color correspond to the number of applicable cancer types.
We also looked at the highest cluster-based scoring clinical and the highest antigen pair for each of the 33 individual cancer types (Figure 2B). Thirty-one of the cancers examined showed marked improvement from the best clinical antigen to the best double antigen (μΔF1 = 0.58). Among these best pairs per cancer type we saw reductions in overall cross-reactivity (μΔprec = 0.76; n = 31) and an increase in sensitivity (μΔrecall = 0.12; n = 31), with clinical-novel and novel-novel antigen pairs showing the best discrimination performance. Comparing the abundance of AND gates with AND-NOT gates reveals that AND-NOT gating is more common among the identified high-performing antigen pairs (Figure 2C).
Within the top 10 clinical-novel antigens for each of the 33 tumor types (330 pairs), we found a subset of novel antigens that repeatedly form high-ranking antigen pairs with current clinical CAR targets across multiple cancers. We show the most frequently occurring three novel antigens in Figure 2D, along with their highest-ranking clinical pairings and prevalence across tumor types. These novel antigens encode genes that have been noted by prior groups to be upregulated in individual tumors and play a role in tumorigenesis. This includes KREMEN2, a paralog of KREMEN1 that has recently been found to promote cell survival by blocking KREMEN1 homo-dimerization and induction of cell death (Sumia et al., 2019). GRIN2D a glutamate-dependent NMDA receptor has previously been found to be upregulated in cancer by IHC (Ferguson etal., 2016), confirming RNA-based results of this work, and is believed to play a role tumor vascularization. The Cadherin EGF LAG seven-pass G-type receptor 3 (CELSR3) has also been identified to be upregulated in malignancies and is believed to play a role in cell-cycle regulation (Xie et al., 2020). Taken together, these results highlight the power of our approach to systematically identify potentially useful novel antigens that can pair with current clinical antigens, across many different tumor types, which otherwise might have been “lost in the shuffle.”
Examples of Antigen Pairs Predicted to Improve Tumor Recognition
Our top possible antigen pairs as ranked by clustering score and their ability to discriminate a given cancer type is available through an interactive webserver (http://antigen.princeton.edu). The webserver allows users to browse top single and doubles per tumor type and generate an interactive scatterplot for any possible transmembrane pair. In Figure 3, we highlight a few examples of high-performing antigen pairs (high clustering scores). These 2D scatterplots show the RNA-seq expression level (as log transformed TPM counts) of both antigens where each sample is represented by a point—red signifying cancer samples and light gray signifying normal tissue samples. Dark circles highlight the centroids for each normal tissue type, as labeled. In these plots, a high degree of separation occurs when a cluster of cancer (red) samples are segregated away from the bulk of normal tissue samples. This segregation can occur in the upper right quadrant (high:high representing AND gate); in the upper left quadrant (low:high representing AND-NOT gate), or lower right quadrant (high:low, AND-NOT gate).
Figure 3. Numerous Potential Antigen Pairs Show Significant Improvement in the Precision of Tumor Recognition.

(A) Examples of antigen pairs with improved tumor-versus-normal discrimination by switching from single to dual antigen recognition. 2D plots show expression level of both antigens in normal tissue samples (gray) versus specific cancer-type samples (red). Navy circles show centroids for each of the normal tissue types (labeled when close to red cancer cluster). Pairs were scored by clustering as well as by F1 score. Density function of single antigen expression in tumor (red) and normal (gray) tissue are plotted on respective axis, including an optimal point of discrimination showing the best potential tumor-versus-normal discrimination using a single antigen.
(B) Example 2D plots as in (A) highlighting potential AND gates that combine known CAR target pairs (clinical-clinical), known CAR targets paired with new potential antigens (clinical-novel), and pairs of new potential targets (novel-novel).
(C) Example 2D plots as in (B) highlighting potential NOT gates.
The RNA-seq expression data show significant overlap between tumor and normal tissue for single clinical antigens currently being tested as CAR targets in clinical trials (Figure 3A), suggesting that true discrimination between tumor and normal tissue using single antigens may be quite difficult. This overlap is greatly reduced by combining information from both antigens, with a concomitant improvement in the calculated F! score. The plots shown in Figure 3 represent only a small fraction of possible high-performing combinations. Other examples are also shown in Figure S3, with all other examples accessible via the webserver. We discuss some of these specific antigen pairs in the following sections.
Experimental Validation: Secondary Antigens that Improve CAR T Recognition of Renal Cell Carcinoma
This analysis provides a very large dataset of potential antigen pairs (on the order of hundreds of thousands; Figure S2B) for clinical translation as AND gate CAR T cells, many of which are actionable using currently available antigen recognition domains. To outline how antigen pairs can be translated to cellular design and to validate our bioinformatic predictions, we have constructed a pair of engineered cell designs capable of specifically recognizing RCC. Two predicted examples of combinatorial antigens for RCC recognition are shown in Figure 4.
Figure 4. Computationally Predicted Antigen Pairs Can Be Constructed as AND-Gated CAR T Cells in a Laboratory Setting, with Precise In Vitro Discrimination.

(A) RCC recognition circuit: CD70 and AXL. Segregation of RCC samples (red points) versus normal tissue samples (gray points) in antigen expression space, highlighting overlap of CD70 expression with normal blood samples (green points). We constructed an anti-AXL synNotch receptor and validated that human T cells expressing the receptor can detect 769-P renal cell cancer cell line (CD70+AXL+), but not Raji B cell line (CD70+AXL−)via FAC detection of GFP reporter induction. In cell killing assays, we compared human primary CD8+ T cells constitutively expressing the anti-CD70 CAR with the same cells transfected with the anti-AXL synNotch driving anti–CD70 CAR AND-gate circuit. The single antigen targeting anti-CD70 CAR T cells killed both RCC and B cell lines, while the circuit T cells selectively killed RCC cells (n = 3, p value from unpaired two sample student’s t test).
(B) RCC recognition circuit: AXL and CDH6. Segregation of RCC samples (red points) versus normal tissue samples (gray points) in antigen expression space, highlighting overlap of AXL expression with normal lung samples (green points). We constructed an anti-CDH6 synNotch receptor and validated that human T cells expressing the receptor can detect 769-P renal cell cancer cell line (AXL+CDH6+), but not the Beas2B lung epithelial cell line (AXL+CDH6−)via FAC detection of GFP reporter induction. In cell killing assays, we compared human primary CD8+ T cells constitutively expressing the anti-AXL CAR with the same cells transfected with the anti-CDH6 synNotch driving anti–AXL CAR AND-gate circuit. The single antigen targeting anti-AXL CAR T cells killed both RCC and lung cell lines, while the circuit T cells selectively killed RCC cells (n = 3, p value from unpaired two sample student’s t test).
RCC is known to overexpress the tumor associated antigens CD70 and AXL (Jilaveanu et al., 2012; Yu et al., 2015), which we experimentally confirmed in an RCC cell line (769-P) (Figure S4A). Both of these antigens are currently involved in CAR T trials. However, as single CAR targets they are imperfect. CD70 is also expressed on a number of blood cells, including activated T cells, germinal center B cells, and dendritic cells in lymph nodes (Hintzen et al., 1994; Tesselaar et al., 2003). AXL is also expressed in many normal tissues including the lung (Qu et al., 2016). However, we find that the cross-reactive normal tissues for these two antigen targets are non-overlapping and, thus, the combination of these two complementary clinical antigen targets is predicted to greatly improve discrimination of tumor-versus-normal tissue (Figure 4A).
To take advantage of this complementary pair of AND antigens for RCC, we engineered a CAR that recognizes CD70 using its cognate binding partner CD27 as the recognition domain (Wang et al., 2017). In vitro cytotoxicity assays showed that this CAR T cell was able to clear a RCC line (769-P) but also showed significant cytotoxicity against a B cell line (Raji cells). To create a T cell that recognizes AXL AND CD70, we first engineered a synNotch receptor (Morsut et al., 2016) using an α-AXL scFv recognition domain fused to the Notch transmembrane domain and an orthogonal transcription factor (GAL4-VP64). We found that T cells expressing an α-AXL synNotch that are co-cultured with RCC cells activate a synNotch GFP reporter; in contrast, the same T cells co-cultured with Raji B cells, which do not express AXL, do not activate the AXL synNotch receptor. We then engineered AND gate T cells in which an α-AXL synNotch drives expression of a CD70 CAR. We find that this AND circuit caused the specific lysis of RCC cells, but not of Raji B cells (Figure 4A). Thus, the combinatorial recognition of AXL AND CD70 improves upon the CD70 single target CAR, allowing discrimination between RCC cells and B cells.
Similarly, the single target α-AXL CAR is by itself a potential treatment for RCC (Zhu et al., 2019). However, as above targeting AXL is predicted to have toxic cross-reactivity with lung tissue. We constructed an AXL CAR, and when expressed in human primary CD8+ T cells it was found to have cytotoxic activity against both an RCC cell line and an immortalized lung epithelial cell line (Beas2B) (Figure 4B). Based on the current bioinformatics analysis of combinatorial antigens, we predicted that the novel antigen CDH6 (cadherin 6), which has not previously been used as cellular therapy target, would improve the precision of an AXL CAR (Figure 4B). CDH6 is a protein that mediates calcium-dependent cell-cell adhesion with PAX8 lineage-linked expression (de Cristofaro et al., 2016) in the fetal kidney (Mbalaviele et al., 1998) as well as proximal tubule epithelium and is overexpressed in renal and ovarian cancer (Paul et al., 1997). A synNotch receptor targeting CDH6 was generated by screening four potential CDH6 scFv’s fused to the synthetic Notch core receptor. We found that α-CDH6 synNotch receptors expressed in human primary T cells would specifically drive GFP reporter activity when co-cultured with an RCC cell line, but not with CDH6 negative lung epithelium cells (Beas2B). When we constructed an AND-gate T cell with α-CDH6 synNotch driving expression of an α-AXL CAR, we found that specific lysis was only seen for the RCC cell line, and not the lung epithelial cell line. Thus, the combinatorial recognition of CDH6 and AXL improves upon the AXL single target CAR in that it allows discrimination between RCC cells and lung epithelial cells.
These two examples show that there are multiple ways to improve recognition of a specific cancer like RCC by harnessing combinatorial antigen recognition. In total, our analysis predicts 25 antigen pairs that discriminate RCC from normal tissues with a clustering score of >0.85 (Figure S2B). This set of experiments illustrates a pipeline by which improved combinatorial CAR T circuits can be computationally identified and validated.
Triple Antigen Combinations Increase Precision of Cancer Recognition but with Trade-Off of Reduced Recall
Adding a third antigen helps improve overall discrimination performance across tumor types. We observed an overall increase in classification performance with each additional antigen, averaged over the top 10 combos for all tumor and gate types (Figure 5A). With significant increases in the mean F1 from 1 to 2 antigens (μF1 single = 0.15; μtop10 F1 double = 0.37; Wilcoxon ranksum p = 7.86 × 10−68; n = 2,979) and for 2 to 3 antigens (μtop10 F1 triple = 0.58; Wilcoxon ranksum p = 2.83 × 10−48; n = 1,578). Likewise, we see a significant increase in discrimination potential, moving from a clinical to a clinical-novel pair (μF1 C = 0.09; μtop10 F1 C:N = 0.5; Wilcoxon ranksum p = 6.06 × 10−81; n = 676) and to a clinical-clinical-novel triplet (μtop10 F1 C:C:N = 0.66, Wilcoxon ranksum p = 5.00 × 10−8; n = 438), suggesting that novels still have additional value when combined with two clinical antigens.
Figure 5. Antigen Triplets Can Significantly Improve Recognition of Challenging Cancers with Some Reduction in Sensitivity.

(A) (Left) Distribution of tumor-versus-normal discrimination scores (F1) for top 10 antigen singlets, doublets, and triplets. We see significant performance improvements going from 1 to 2 antigens (p = 7.68 × 10−68; n = 2,979) and 2 to 3 antigens (p = 2.83 × 10−48; n = 1,578). The same plot is shown on the right for top 10 clinical antigen singlets, clinical-novel antigen doublets, and clinical-clinical-novel antigen triplets. Again, we see significant increases in performance going from clinical to clinical-novel pairs (p = 6.06 × 10−81; n = 676) and from clinical-novel pairs to clinical-clinical-novel triplets (p = 5.00 × 10−8; n = 438). F1 scores range between 0 (no sensitivity and specificity) and 1 (perfect precision and recall).
(B) Improvement in tumor-versus-normal discrimination with triplet antigen recognition by cancer type. F1 score ranges from best single clinical antigen (gray circle) to best double with at least one clinical antigen (blue circle) to best triplet with at least one clinical antigen.
(C) Pie chart showing the composition of different gate types (high:high:high, high:high:low, and high:low:low) of triplets in the top 100 per tumor type.
(D) Each gray dot represents the precision (left) or recall (right) for one of the top antigens (single, double, and triples) for a single tumor type. Gray lines show the median illustrating the global increase in precision when including more antigens at the expense of recall. Precision has a significant increase and recall a significant decrease when going from one to two (precision: Wilcoxon rank sum p = 2.45 × 10−120, n = 2,979; recall: Wilcoxon rank sum p = 3.55 × 10−9, n = 2,979) and two to three antigens (precision: Wilcoxon rank sum p = 5.69 × 10−84, n = 1,578; recall: Wilcoxon rank sum p = 5.94 × 10−4; n = 1,578).
(E) Example 3D triplet antigen gates showing expression level of all antigens in normal tissue samples (gray) versus specific cancer-type samples (red). Tissue centroids are in dark blue. Triplets were scored by clustering as well as by F1 score.
Looking across the top 100 gates per cancer we find that the majority of triples (92%) have at least one NOT element, with over half (56%) having two antigens that have low expression in the target (AND-NOT-NOTs, Figure 5C). Such a high percentage of NOTs further highlights the importance of synthetic NOT gates and our in silico approach, as more naive approaches such as combining cancer specific markers in AND gates would miss many of the highest discriminatory combinations.
Perhaps the greatest benefit of adding a third antigen is the improvement we observe in recognizing challenging cancers. Cholangiocarcinoma, in particular, was the tumor type that was the hardest to discriminate, with a max F1 score of 0.26 for a pairing of two novel antigens. When adding a third antigen, we were able to reduce predicted off-target toxicity and increase sensitivity, by combining a lower-scoring clinical-novel pair with an additional novel antigen, increasing the max F1 score by 0.31. Encouragingly, we also see substantial increases in the maximum discrimination performance for several other cancer types as well (Figure 5B).
While we see increases in overall performance for many cancers, we also noticed that for some tumor types we do not see a big gain in recognition going from two to three antigens. This is because overall the gains in precision come at a cost of reduced recall. Looking at both the precision and recall of the top combinations per each gate per tumor in Figure 5D, we see as we go from one to two to three antigens we achieve nearly perfect average precision (μ precision single = 0.07, μtop10 precision double = 0.44, and μtop10 precision triple = 0.90), and this increase is significant from one to two (Wilcoxon rank sum p = 2.45 × 10−120, n = 2,979) and two to three (Wilcoxon rank sum p = 5.69 × 10−84, n = 1,578). However, with each additional antigen there is a significant reduction in the average recall (μ recall single = 0.66, μtop10 recall double = 0.52, μtop10 recall triple = 0.47; 1 to 2: Wilcoxon rank sum p = 3.55 × 10−9, n = 2,979; 2 to 3: Wilcoxon rank sum p = 5.94 × 10−4; n = 1,578). Taken together, we can conclude that 2–3 antigens are likely to be sufficient for precise recognition of most tumor types.
Like our examples in Figure 3, we also highlight a few examples of high-performing antigen triplets in Figure 5E. As in the 2D scatterplots, red dots are samples are those of a particular cancer type (e.g., mesothelioma [left] and melanoma [right]), light gray are normal tissues, and dark blue are highlighting the normal tissue centroids. In both of these example triplets, the additional antigen gives a big boost to the overall separation of tumor and normal tissue samples, yielding fairly precise triplets that suffer slightly from reduced recall. We show more examples along with their corresponding 2D plots in Figure S5.
DISCUSSION
Targeting Antigen Combinations Could Significantly Improve Tumor Recognition
This analysis, based on available gene expression datasets, predicts that using Boolean antigen combinations can significantly improve the selectivity of tumor recognition and avoidance of normal tissue cross-reactivity. Thus, using Boolean multi-antigen detecting engineered T cells has the potential to have a major impact on cancer recognition and the development of next generation cellular therapies.
We find that adding new antigens to current clinically actionable CAR targets via AND or AND-NOT Boolean recognition is predicted to significantly increase cancer versus normal tissue discrimination. Moreover, we can find many novel antigen pairs that show even stronger and near ideal discrimination. All cancers show at least several (> 25) antigen pairs above a clustering score cutoff of 0.85, with many having thousands of strong pairs, suggesting a potential therapeutic avenue for all tumor types when using a pair of antigens (Figure S2C). Furthermore, with the addition of triples, every cancer type examined here has a promising clustering-based score and an F1 score above 0.5 (out of ideal 1.0). Thus, there are likely to be many options of multi-antigen signatures that could be used to recognize any one type of tumor.
2–3 Antigen Combinatorial Circuits May Be Sufficient to Achieve Strong Cancer versus Normal Tissue Discrimination
Notably, when we examine the precision and recall of detection, we see for top performing gates, that as the number of antigens used for detection is increased from two to three, mean precision approaches perfection while the recall declines (Figure 5D). This suggests that further improvement in therapeutic cell discriminatory potential will require more narrow sub-classifications of tumor type; either by pathologic or molecular subtypes (e.g., triple-negative versus HER2-positive versus hormone receptorpositive breast cancer) of cancers defined in TCGA. This number of antigens also matches well with current synthetic biology tools, as integrating 2–3 receptor circuits is possible with current gene transfer methods (e.g., lentiviral transduction), while four or more receptor circuits would require significant improvements in vector payload capacity.
Limitations of This Analysis and Future Challenges
This study shows that combinatorial antigen recognition is predicted to yield much higher tumor discrimination than single antigen recognition. Nonetheless, the exact degree of this improvement, as well as the exact combinations that could be most clinically useful remain less clear, as there remain caveats in interpreting these data. First, this analysis is based on gene expression data, while T cell recognition is mediated by protein expression. At this time, there is far less extensive protein expression data covering both cancer and normal cells, so we leveraged the vast amount of gene expression data as a proxy. Second, while GTEx covers most major tissues, we still have an incomplete set of normal tissue expression. Recent developments in single-cell sequencing will help to bridge the gap, providing cleaner and more detailed snapshots of expression in healthy cells, while having the additional benefit of helping us account for normal cells that may be present in bulk cancer samples. Once more widely available, integration of single-cell RNA-seq data of normal and tumor cells would improve the specificity of this analysis even further.
Finally, optimal discrimination also involves setting antigen detection thresholds—exactly where the cutoff lines of high and low expression is important for discrimination (Figure 1B). In cases where we observe large distances between tumor and normal samples, separation is extremely robust and consequently, shifting threshold cutoffs makes little difference in F1. In the other cases, however, F1 scores are highly sensitive to shifts in cutoffs. This is why we chose to first rank potential antigen combinations by clustering scores and then use a classifier to evaluate performance. Focusing primarily on maximizing separation distance ensures that more of our top pairs are robust to thresholding. Experimentally, optimizing cutoff thresholds is challenging, since we currently have poor control in determining what antigen densities are detected by CARs. However, new efforts are ongoing to develop methods for tuning antigen detection cutoffs and sharpness, which may help significantly in taking advantage of expression level differences, providing an additional untapped source of discrimination information.
It is interesting that the list of current clinical antigens under investigation as CAR targets in solid tumors performs relatively poorly compared with the large set of novel antigens identified here. It may be that many of the computationally predicted antigens do not account for detailed biological and physiological factors. The discrepancy could also simply be observed because of poor correlation of gene expression with protein expression, or perhaps, because many of the clinically distinguishing markers have not been directly compared with all other healthy cells throughout the body. Clearly, disease-specific expert knowledge will be critical to filter the antigens identified here. In some cases, however, there might be some historical expert bias, where focus has been unequally placed on antigens identified early in the study of a disease. We suspect that several of the novel antigens that are identified to form high functioning pairs with many current clinical antigens (Figure 2D) should be further investigated in future work.
Combining Bioinformatics with Antigen Recognition Circuit Design to Optimize Solid Cancer T Cell Therapies
Despite the above caveats, this analysis indicates that combinatorial antigen recognition is likely to be able to make a significant contribution to the treatment of at least some, if not all, solid cancers by engineered therapeutic cells. This analysis drives home the point that recognition of tumors has to take into account a broad set of parameters, such as how much overlap there is in expression in normal tissues, and how shared the antigens are among patient populations. Gratifyingly, we can see the potential combinatorial antigen circuits have to carve this complex antigen recognition space into smaller defined sectors with better tumor-versus-normal tissue discrimination. In principle with AND, OR, and NOT functions, we can break up the antigen space in many flexible ways.
This analysis provides a roadmap for a new vision of precision medicine that more deeply integrates in silico data analysis with capabilities emerging from synthetic biology and cell design. In this case, large-scale genomic data are not used to stratify patients based on likelihood of response to a drug, but rather the data become the guide for how to best design a smart cellular drug. Here, we search for opportunities to discover, within the multidimensional space of antigens, the signatures that can offer us the optimal recognition discrimination. Given the very large size of this database, as well as the thousands of potential antigen combinations that could be created, we have provided these data in a webserver for the community to interrogate potential antigen pairs or triplets for all cancers within the TCGA (Figure 6B).
Figure 6. In Silico T Cell Circuit Design: Expansive Search and Provided Resources.

(A) In silico analysis of tumor-versus-normal expression data can be used to identify discriminatory antigen patterns. These potential antigen signatures can then be used as the basis for synthetic biology engineering of precision therapeutic T cells.
(B) We have generated an interactive webserver that allows public access to the datasets used in this paper; allowing users to identify potential discriminatory singlets, doublets, and triplets for cancer detection in the future (antigen.princeton.edu).
The range of recognition functions that we can achieve will likely have a major impact on how engineered cell therapies can detect cancer and other diseases. The variety of recognition modalities means that we have great potential to sector multidimensional antigen space in a diversity of ways to find those ways that best segregate disease tissues from normal tissues. Thus, harnessing the computational capabilities of living cells, and using in silico analysis to guide their deployment, provides a broad new frontier for recognizing and attacking complex diseases such as cancer.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, (Wendell.lim@ucsf.edu). To ensure a fast response, please copy Noleine.Blizzard (noleine.blizzard@ucsf.edu) and Michael Broeker (Michael.Broeker@ucsf.edu) in any requests related to the paper.
Materials Availability
Plasmids generated in this study are in the process of being deposited to AddGene.
Data and Code Availability
The R and python code used to score antigen combinations along with methods for regenerating the major figures in this paper freely available for non-commercial use and is deposited in GitHub at https://github.com/ruthanium/antigen-combos-scripts.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Source of Primary Human T cells
Blood was obtained from Blood Centers of the Pacific (San Francisco, CA) as approved by the University Institutional Review Board. Primary CD4+ and CD8+ T cells were isolated from anonymous donor blood after apheresis (described in methods).
METHOD DETAILS
Defining the Space of Candidate Antigens
We defined potential candidate antigens as genes with known or predicted cell surface expression, restricting our search space to current clinical targets and genes coding for transmembrane proteins. More specifically, we assembled a set of 29 unique clinical antigens along with their indications that have shown promise in the literature or are targets in currently active CAR or TCR trials and mapped them to their corresponding genes. To assemble the list of transmembrane proteins we started with a list of putative transmembrane genes then filtered by localization to the plasma membrane as annotated in the COMPARTMENTS database (Binder et al., 2014) with high confidence (level 3 or higher), yielding a list of 2,358 genes. The COMPARTMENTS database uses a combination of manually curated literature, text mining, high-throughput screens, and sequence prediction methods to make subcellular location predictions.
Gene Expression Data Processing
We gathered gene level RSEM processed TPM counts for healthy human tissue samples from the Genotype Tissue Expression (GTEx) project version 7 and gene level RSEM processed tumor samples from The Cancer Genome Atlas (TCGA) firehose. All together there were 21,486 samples covering 34 tissues and 33 cancer types (see Table S2 for individual breakdowns per tissue and tumor type). To remove differences due to technical variation and thus combine these data from these two different sources we applied batch correction using a parametric empirical Bayes framework using the COMBAT function in the SVA R package (Johnson et al., 2007).
Intelligent Subsampling and Data Partitioning
To increase the speed of our clustering score calculations as well as partition our data into training and test sets we used geometric sketching (Hie et al., 2019). Geometric sketching allows us to subsample the space of samples maintaining the overall structure of the data by fitting a plaid covering and sampling points from within each region of the covering. In simulations across 8 different sketch sizes for 5 iterations across 100 gene pairs (10 fixed genes paired with 10 random genes) we observed no loss of performance (see Figure S2B) when calculating Davies-Bouldin and Manhattan distance but substantial gains in runtime. Based on these simulations we chose to use a sketch size of 20% of all data for calculating clustering-based scores as well as the training data for classification and the remaining 80% of the data was held out for testing our classification models.
Clustering-Based Scores
We chose to adapt a method used to evaluate clustering, Davies-Bouldin (DB), to measure the ratio of within cluster spread to cluster distance. We considered the case where there are 2 clusters: a tumor cluster (given set of tumor samples) and a tissue cluster (all normal tissues samples). Lower DB scores are better as they indicate less within cluster distance (more tightly packed samples) and more distance between the cluster centers (more distance between tumor and normals). More formally, we use the following equations to calculate DB:
which measures the ratio of scatter between the target tumor type (Si) and the cluster of normal tissues (Sj) to the distance between the two clusters. Scatter for each cluster is calculated using:
where Ti is the number of samples in a given cluster and Xj is the location of a given sample and its distance from its cluster centroid (Ai).
The distance between the clusters, Mi, is calculated by subtracting the distance of the two cluster centers.
Where Ai is the centroid of the cancer cluster, and Aj is the centroid of the normal tissue cluster.
To give extra weight to the distance between clusters, we also calculated the Manhattan distance (d) between the normal and the tumor clusters and used this in the final clustering score. To compute a more interpretable clustering-based score to use throughout our search, we rescaled log DB and log distance values across all gene pairs and tumor samples to be between 0 and 1, where 1 represents the best (smallest) DB score and the largest scaled distance. The minimum of these two scores is the final clustscore, thus a clustscore of 1 has the smallest DB and largest distance. Formally,
where t is a tumor type and pi,j is a pair of genes made up of gene i and gene j and the min and max scores are calculated over all pairs.
Search Space Reduction for Triples
To reduce the number of transmembrane and clinical antigens for triple antigen search we looked at the performance of single antigens to create a smaller set of potential antigens per tumor type. The intuition being that each antigen must contribute at least a small amount of improvement to be a high scoring triple. To be included in the set of putative antigens per cancer, we required a single antigen to have a Davies-Bouldin score <= 5 and a Manhattan distance > 2. This filtering reduced the potential antigens to the following: Acute Myeloid Leukemia: 525, Adrenocortical Carcinoma: 169, Bladder Urothelial Carcinoma: 68, Brain Lower Grade Glioma: 361, Breast Invasive Carcinoma: 48, Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma: 118, Cholan-giocarcinoma: 69, Colon Adenocarcinoma: 131, Esophageal Carcinoma: 40, Glioblastoma Multiforme: 274, Head and Neck Squamous Cell Carcinoma: 102, Kidney Chromophobe: 140, Kidney Renal Clear Cell Carcinoma: 30, Kidney Renal Papillary Cell Carcinoma: 115, Liver Hepatocellular Carcinoma: 110, Lung Adenocarcinoma: 27, Lung Squamous Cell Carcinoma: 59, Lymphoid Neoplasm Diffuse Large B-cell Lymphoma: 416, Mesothelioma: 93, Ovarian Serous Cystadenocarcinoma: 102, Pancreatic Adenocarcinoma: 36, Pheochromocytoma and Paraganglioma: 233, Prostate Adenocarcinoma: 60, Rectum Adenocarcinoma: 118, Sarcoma: 64, Skin Cutaneous Melanoma: 194, Stomach Adenocarcinoma: 35, Testicular Germ Cell Tumors: 125, Thymoma: 205, Thyroid Carcinoma: 72, Uterine Carcinosarcoma: 90, Uterine Corpus Endometrial Carcinoma: 86, and Uveal Melanoma: 299. The clustering scores were then calculated as described in the above section.
Evaluation of Top Clustering-Based Scores
We chose the top 10 antigen pairs per antigen class (C:C, C:N, and N:N) for each tumor based on their clustering scores for a total of ~330 pairs per tumor type. Within the top 10 per class per tumor we only allowed a particular gene in a pair to appear a maximum of two times, preventing potential pairs from being dominated by a single gene with high separation. We further restricted our analysis to single antigens that are high, and pairs of antigens that have at least one antigen predicted to have high expression (high:high, high:-low, and low:high) pairs.
To calculate the discriminatory ability of any particular antigen or antigen combination we constructed decision tree (DT) models on the 20% training partition using antigen expression as features and evaluated performance on the held out 80% of the data. More explicitly, for antigen pairs we used the rpart R package to construct two single feature decision trees with c=−1 and a max depth=1 forcing each tree to have a single split. We then used these splits to draw a classification boundary and calculated precision (the proportion of predicted positives that are correct), recall (the proportion of real positives that are predicted positive), and F1 scores (the harmonic mean of precision and recall), as shown in the following:
Construct Design
All synNotch receptors used in this study were built using the mouse Notch1 (NM_008714) minimal regulatory region (Ile1427 to Arg 1752). The following binding domains were engineered into synNotch receptors: α-AXL scFv (Grada et al., 2013; Hegde et al., 2013) (https://patents.google.com/patent/WO2012175691A1/en), and the α-CDH6 scFv clone V10 (https://patents.google.com/patent/WO2016024195A1/en). synNotch receptors were designed to include either Gal4 DNA-binding domain (DBD) VP64 fusion proteins as a synthetic transcription factor. All synNotch receptors contain an N-terminal CD8α signal peptide (MALPVTALLLPLALLLHAARP) for membrane targeting. Following the CD8α signal peptide, Gal4 synNotch receptors contain a myc tag (EQKLISEEDL) for easy and orthogonal surface detection with α-myc AF647 (Cell Signaling #2233) AF488 (R&D Systems #IC8529G), respectively.
All CARs used in this study were designed by fusing scFvs to the human CD8α chain hinge and transmembrane domains and the cytoplasmic regions of the human 4-1BB and CD3ζ signaling proteins. The following binding domains were engineered into CARs: α-AXL scFv (https://patents.google.com/patent/WO2012175691A1/en), and the CD27 extracellular domain. All CARs included an N-terminal V5 tag for easy detection with α-V5 PE (Thermo Fisher #12-6796-42). All CARs contain an N-terminal CD8α signal peptide.
For experiments with T cells expressing a synNotch receptor the Gal4 system was utilized, and the receptors were cloned into a modified pHR’SIN:CSW vector containing a constitutive PGK promoter. For these experiments, the pHR’SIN:CSW vector was also modified to make the response element plasmids. Five copies of the Gal4 DNA binding domain target sequence (GGAG-CACTGTCCTCCGAACG) were cloned 5’ to a minimal CMV promoter. Also included in the response element plasmids is a PGK promoter that constitutively drives mCherry or BFP expression to easily identify transduced T cells. For all synNotch response element vectors, the inducible transgene (e.x. CAR or TCR) was cloned via a BamHI site in the multiple cloning site 3’ to the Gal4 response elements. All constructs were cloned via InFusion Cloning (Takara Bio #638910).
Primary Human T Cell Isolation and Culture
Primary CD4+ and CD8+ T cells were isolated from anonymous donor blood after apheresis by negative selection (STEMCELL Technologies #15062 and 15023). T cells were cryopreserved in RPMI-1640 (Corning #10-040-CV) with 20% human AB serum (Valley Biomedical, #HP1022) and 5% DMSO (Sigma-Aldrich #472301). After thawing, T cells were cultured in human T cell medium consisting of X-VIVO 15 (Lonza #04-418Q), 5% Human AB serum and 10 mM neutralized N-acetyl L-Cysteine (Sigma-Aldrich #A9165) supplemented with 30 units/ mL IL-2 (NCI BRB Preclinical Repository) for all experiments.
Lentiviral Transduction of Human T Cells and Target
Lenti-X293T packaging cells (Clontech #11131D) were cultured in medium consisting of Dulbecco’s Modified Eagle Medium (DMEM) (Gibco #10569-010), 10% fetal bovine serum (FBS) (University of California, San Francisco [UCSF] Cell Culture Facility), and gentamicin (UCSF Cell Culture Facility). Fresh packaging cells were thawed after cultured cells reached passage 30.
Pantropic VSV-G pseudotyped lentivirus was produced via transfection of Lenti-X 293T cells with a pHR’SIN:CSW transgene expression vector and the viral packaging plasmids pCMVdR8.91 and pMD2.G using Fugene HD (Promega #E2312). Primary T cells were thawed the same day, and after 24 hr in culture, were stimulated with Dynabeads Human T-Activator CD3/CD28 (Thermo Scientific #11131D) at a 1:3 cell:bead ratio. At 48 hr, viral supernatant was harvested, and the primary T cells were exposed to the virus for 24 hr. At day 5 post T cell stimulation, Dynabeads were removed and the T cells expanded until day 12 when they were rested and could be used in assays. T cells were sorted for assays with a FACs ARIA II on day 5 or 6 post T cell stimulation.
Cancer and Target Cell Lines
The cancer cell lines used were Raji B cell Burkitt lymphoma cells (ATCC #CCL-86), and 769-P renal cell carcinoma cells (ATCC #CRL-1933). Rajis and 769-Ps were cultured in RPMI 1640 with L-glutamine (Corning #10-040-CV) supplemented with 10% FBS. The immortalized healthy tissue cell lines or primary human cells were Beas2B lung epithelial cells (ATCC #CRL-9609). Beas2B cells were cultured in BEBM (Lonza #CC3171) supplemented with the BEGM kit (Lonza #CC-3170).
Levels of various antigens were determined via flow cytometry after staining cells with the following antibodies: α-CD70 APC (Biolegend #355109), α-AXL APC (R&D systems #FAB154A), and α-CDH6 AF647 (R&D systems #FAB2715R).
Antibody Staining and Flow Cytometry Analysis
All antibody staining for flow cytometry was carried out in wells of round-bottom 96-well tissue culture plates. Cells were pelleted by centrifugation of plates for 4 min at 400 x g. Supernatant was removed and cells were resuspended in 50 μL PBS containing the fluorescent antibody of interest. Cells stained 25 minutes at 4°C in the dark. Stained cells were then washed two times with PBS and resuspended in fresh PBS supplemented with 1% FBS and EDTA for flow cytometry with a BD LSR II. All flow cytometry data analysis was performed in FlowJo software (TreeStar).
In Vitro Stimulation of synNotch T cells
For all in vitro synNotch T cell assays (including both reporter and killing assays), T cells were co-cultured with target cells at a 1:1 ratio, with anywhere from 1e4-1e5 each/well. The Countess II Cell Counter (ThermoFisher) was used to determine cell counts for all assay set up. T cells and target cells were mixed in 96-well tissue culture plates in 200 μL T cell media, and then plates were centrifuged for 1 min at 400 x g to initiate interaction of the cells. For assays with Raji, round-bottom 96-well plates were used. For assays with all other target cells, flat-bottom 96-well plates were used. Cells were co-cultured for anywhere from 18 to 96 hours before analysis via flow cytometry with a BD LSR II.
Flow Cytometry-Based T Cell Cytotoxicity Assay
For all cytotoxicity assays, synNotch T cells, constitutive CAR or untransduced T cells were co-cultured with target cells at a 1:1 ratio as described above. After intended periods of incubation, samples were centrifuged for 4 min at 400 x g, after first being transferred to a round-bottom 96-well plate if necessary. Supernatant was then removed and cells were resuspended in PBS supplemented with 1% FBS and EDTA for flow cytometry with a BD LSR II. Control samples containing only the target cells were used to set up flow cytometry gates for live target cells based on forward and side scatter patterns. For assays with all other target cells, target cells were gated on low CellTrace Far Red dye (Thermo Fisher #C34564) or low CD3 staining, as T cells used in these assays were either labeled with CellTrace Far Red or the samples were stained with α-CD3 APC/Cy7 (Biolegend #317342) to specifically label T cells. The level of target cell survival was determined by comparing the fraction of target cells alive in the culture compared to treatment with untransduced T cell controls. Three technical replicates are performed for each experiment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa647 mouse anti-myc tag clone 9B11 | Cell Signaling Technology | Cat#2233S; RRID:AB_823474 |
| PE mouse anti-V5 tag clone TCM5 | Thermo Fisher | Cat#12-6796-42; RRID:AB_2784630 |
| APC mouse anti-human CD70 clone 113-16 | Biolegend | Cat#355109; RRID:AB_2562480 |
| APC mouse anti-human AXL clone 108724 | R&D Systems | Cat#FAB154A; RRID:AB_2062558 |
| Alexa647 mouse anti-human CDH6 clone 427909 | R&D Systems | Cat#FAB2715R; RRID:AB_2078122 |
| APC/Cy7 mouse anti-human CD3 clone OKT3 | Biolegend | Cat#317342; RRID:AB_2563410 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| In-Fusion HD Cloning Plus | Takara Bio | Cat#638910 |
| N-acetyl L-Cysteine | Sigma Aldrich | Cat#A9165 |
| Recombinant human IL-2 protein | NCI BRB Preclinical Repository | https://ncifrederick.cancer.gov/research/brb/ |
| Gentamicin | UCSF Cell Culture Core | N/A |
| Penicillin-streptomycin | UCSF Cell Culture Core | N/A |
| Fugene HD | Promega | Cat#E2312 |
| DMSO | Sigma Aldrich | Cat#472301 |
| EDTA | Thermo Fisher | Cat#AM9260G |
| CellTrace Violet | Thermo Fisher | Cat#C34557 |
| CellTrace Far Red | Thermo Fisher | Cat#C34564 |
| CellTrace CFSE | Thermo Fisher | Cat#C34554 |
| Critical Commercial Assays | ||
| RosetteSep Human CD8+ T cell Enrichment Cocktail | STEMCELL Technologies | Cat#15023 |
| RosetteSep Human CD4+ T cell Enrichment Cocktail | STEMCELL Technologies | Cat#15022 |
| AllPrep DNA/RNA Mini Kit | Qiagen | Cat#80204 |
| Cell Culture Reagents | ||
| PBS | UCSF Cell Culture Core | N/A |
| TrypLE Express | Thermo Fisher | Cat#12504013 |
| RPMI-1640 w/ L-Glutamine | Corning | Cat#10-040-CV |
| Human Ab Serum | Valley Medical | Cat#HP1022 |
| X-VIVO15 | Lonza | Cat#04-418Q |
| DMEM | Gibco | Cat#10569-010 |
| FBS | UCSF Cell Culture Core | N/A |
| GlutaMAX | Thermo Scientific | Cat#10569-044 |
| BEBM (Bronchial Epithelial Cell Growth Basal Medium) | Lonza | Cat#CC-3171 |
| BEGM Bronchial Epithelial Cell Growth Medium BulletKit | Lonza | Cat#CC-3170 |
| Dynabead Human T cell Activator anti-CD3/CD28 | Thermo Fisher | Cat#11131D |
| Experimental Models: Cell Lines | ||
| LentiX 293T | Clontech | Cat#11131D |
| Raji cells | ATCC | Cat#CCL-86 |
| 769-P | ATCC | Cat#CRL-1933 |
| Beas2B | ATCC | Cat#CRL-9609 |
| Recombinant DNA | ||
| pHR_SFFV | Addgene | ID#79121 |
| pHR_PGK | Addgene | ID#79120 |
| pHR_Gal4UAS_PGK_mCherry | Addgene | ID#79124 |
| pHR_Gal4UAS_tBFP_PGK_mCherry | Addgene | ID#79130 |
| pHR_SFFV_antiCD70_BBZ | This paper | CD27 sequence from UniPortKB – P26842 (CD27_Human) |
| pHR_SFFV_antiAXL_BBZ | This paper | scFv sequence in WO2012175691A1 |
| pHR_pGK_antiAXL_synN_G4VP64 | This paper | scFv sequence in WO2012175691A1 |
| pHR_pGK_antiCDH6_synN_G4VP64 | This paper | scFv sequence in W02016024195A1 |
| pHR_Gal4UAS_antiCD70_BBZ_PGK_BFP | This paper | CD27 sequence from UniPortKB – P26842 (CD27_Human) |
| pHR_Gal4UAS_antiAXL_BBZ_PGK_BFP | This paper | scFv sequence in WO2012175691A1 |
| Software and Algorithm | ||
| Prism Version 7a | Graphpad | N/A |
| FlowJo V10.4.0 | TreeStar | N/A |
| rpart package | R | https://cran.r-project.org/web/packages/rpart/ |
| R version 3.5.3 | R | N/A |
| ComBat in SVA package | R | https://www.bioconductor.org/packages/release/bioc/html/sva.html |
| clustscore and analysis | This paper | https://github.com/ruthanium/antigen-combos-scripts |
| Deposited Data | ||
| COMPARTMENTS (cellular location data) | Binder etal. 2014 | https://compartments.jensenlab.org |
| Other | ||
| Webserver for exploring the data in this publication | This paper | antigen.princeton.edu |
Highlights.
2- and 3-antigen AND or NOT logic gates improve tumor discrimination of CAR T cells
All transmembrane antigen combination pairs and triples are computationally screened
Combinatorial antigens that outperform current clinical CAR T cells are predicted
Adding antigens improves precision at the cost of recall; 2–3 is optimal
ACKNOWLEDGMENTS
We thank J. Bluestone, M. Broeker, K. Chang, J. Garbarino, C. Ghosh, X. Huang, P. Lopez Pazmino, N. Blizzard, W. Yu, and the W.A.L. and Troyanskaya labs for technical assistance, advice, and helpful discussion. This work was supported by a Jane Coffin Childs Memorial Fund Postdoctoral Fellowship (to G.M.A.); NIH grants U54CA244438 (to W.A.L. and O.G.T.), T32HG003284 (to R.D.), R01 CA196277 (to W.A.L.), and R01 GM071966 (to O.G.T.); and the Howard Hughes Medical Institute (to W.A.L.).
Footnotes
DECLARATION OF INTERESTS
W.A.L. is on the Scientific Advisory Board for Allogene Therapeutics and O.G.T. is on the Scientific Advisory Board for Caris Life Sciences. W.A.L. and O.G.T. have filed patents related to this work.
WEB RESOURCES
Antigen explorer, https://antigen.princeton.edu/
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2020.08.002.
REFERENCES
- Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O’Donoghue SI, Schneider R, and Jensen LJ (2014). COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database (Oxford) 2014, bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cristofaro T, Di Palma T, Soriano AA, Monticelli A, Affinito O, Cocozza S, and Zannini M (2016). Candidate genes and pathways downstream of PAX8 involved in ovarian high-grade serous carcinoma. Oncotarget 7, 41929–41947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VD, Themeli M, and Sadelain M (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med 5, 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson HJM, Wragg JW, Ward S, Heath VL, Ismail T, and Bicknell R (2016). Glutamate dependent NMDA receptor 2D is a novel angiogenic tumour endothelial marker in colorectal cancer. Oncotarget 7, 20440–20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schonfeld K, Koch J, Dotti G, et al. (2013). TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde M, Corder A, Chow KKH, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, et al. (2013). Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther 21, 2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B, Cho H, DeMeo B, Bryson B, and Berger B (2019). Geometric sketching compactly summarizes the single-cell transcriptomic landscape. Cell Syst. 8, 483–493.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzen RQ, Lens SM, Koopman G, Pals ST, Spits H, and van Lier RA (1994). CD70 represents the human ligand for CD27. Int. Immunol 6, 477–480. [DOI] [PubMed] [Google Scholar]
- Jilaveanu LB, Sznol J, Aziz SA, Duchen D, Kluger HM, and Camp RL (2012). CD70 expression patterns in renal cell carcinoma. Hum. Pathol 43, 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, and Sadelain M (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, and Gratama JW (2013). Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of ontarget toxicity. Mol. Ther 21, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. (2018). Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbalaviele G, Nishimura R, Myoi A, Niewolna M, Reddy SV, Chen D, Feng J, Roodman D, Mundy GR, and Yoneda T (1998). Cadherin-6 mediates the heterotypic interactions between the hemopoietic osteoclast cell lineage and stromal cells in a murine model of osteoclast differentiation. J. Cell Biol 141, 1467–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, and Lim WA (2016). Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. (2017). Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. (2011). T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther 19, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Ewing CM, Robinson JC, Marshall FF, Johnson KR, Wheelock MJ, and Isaacs WB (1997). Cadherin-6, a cell adhesion molecule specifically expressed in the proximal renal tubule and renal cell carcinoma. Cancer Res. 57, 2741–2748. [PubMed] [Google Scholar]
- Qu X, Liu J, Zhong X, Li X, and Zhang Q (2016). Role of AXL expression in non-small cell lung cancer. Oncol. Lett 12, 5085–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016b). Precision tumor recognition by T cells With combinatorial antigen-sensing circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016a). Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, and Riddell SR (2019). Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumia I, Pierani A, and Causeret F (2019). Kremen1-induced cell death is regulated by homo- and heterodimerization. Cell Death Discov. 5, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesselaar K, Xiao Y, Arens R, van Schijndel GMW, Schuurhuis DH, Mebius RE, Borst J, and van Lier RAW (2003). Expression of the murine CD27 ligand CD70 in vitro and in vivo. J. Immunol 170, 33–40. [DOI] [PubMed] [Google Scholar]
- Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, et al. (2017). The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioningdependent respiratory toxicity. Cancer Immunol. Immunother 66, 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Yu Z, Hanada KI, Patel K, Kleiner D, Restifo NP, and Yang JC (2017). Preclinical evaluation of chimeric antigen receptors targeting CD70-expressing cancers. Clin. Cancer Res 23, 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, Burbridge SE, Box C, Eccles SA, and Maher J (2012). Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol 32, 1059–1070. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dang Y, Wu H, He R, Ma J, Peng Z, Rong M, Li Z, Yang J, Jiang Y, et al. (2020). Effect of CELSR3 on the cell cycle and apoptosis of hepatocellular carcinoma cells. J. Cancer 11, 2830–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Liu R, Ma B, Li X, Yen HY, Zhou Y, Krasnoperov V, Xia Z, Zhang X, Bove AM, et al. (2015). AXL receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br. J. Cancer 113, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wei Y, and Wei X (2019). AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol. Cancer 18, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R and python code used to score antigen combinations along with methods for regenerating the major figures in this paper freely available for non-commercial use and is deposited in GitHub at https://github.com/ruthanium/antigen-combos-scripts.