

Conspectus.
Light has been instrumental in the study of living cells since its use helped in their discovery in the late C17th. Further, combining chemical technology with light microscopy was an essential part of the Nobel Prize for Physiology in 1906. Such landmark scientific findings involved passive observation of cells. However, over the past 50 years a “second use” of light has emerged in cell physiology, namely one of rational control. The seminal method for this emerged in late 1970s with the invention of caged compounds. This was the point when “caged compounds” were defined as optical probes in which the active functionality of a physiological signaling molecule was blocked with a photochemical protecting group. Caged compounds are analogous to prodrugs, in both the activity of the effector is latent. However, caged compounds, unlike prodrugs, use a trigger that confers the power of full temporal and spatial manipulation of the effects of release of its latent biological cargo. Light is distinct because it is bio-orthogonal, passes through living tissue (even into the cell interior) and initiates rapid release of the “caged” biomolecule. Further, because light can be directed to broad areas, or focused to small points, caged compounds offer an array of timing scenarios for physiologists to dissect virtually any type of cellular process.
The collaborative interaction between chemists and physiologists plays a fundamental role in the development of caged compounds. First, the physiologists must define the problem to be addressed, then, with the help of chemists, decide if a caged compound would be useful. For this structure-activity relationships of the potential optical probe and receptor must be determined. If rational targets seem feasible, synthetic organic chemistry is used to make the caged compound. The crucial property of prephotolysis bio-inertness relies on physiological or biochemical assays. Second, detailed optical characterization of the caged compound requires the skill of photochemists, because the rate and efficiency of uncaging are also crucial properties for a “useful caged compound”. Often, these studies reveal limitations in the caged compound which has been developed, thus chemist and physiologist use their abilities for iterative development of even more powerful optical probes. A similar dynamic will be familiar to scientists in the pharmaceutical industry. Therefore, caged compound development provides an excellent training framework for (young) chemists both intellectually and professionally. In this Account I draw on my long experience in the field of making “useful caged compounds for cell physiology” by showing how each probe I have developed has been defined by an important physiological problem. Fundamental to this process has been my initial training by the pioneers in aromatic photochemistry, Derek Bryce-Smith and Andrew Gilbert. I discuss making a range “caged calcium” probes, ones which went on to be the most widely used of all caged compounds. Then I describe the development of caged neurotransmitters for two-photon uncaging microscopy. Finally, I survey recent work on making new photochemical protecting groups for wavelength orthogonal, two-color and ultra-efficient two-photon uncaging.
Graphical Abstract
Caged compounds are physiological signaling molecules that have been rendered biologically inert by covalent attachment of a photochemical protecting group1. They were invented in 19782, in order to study the Na+ pump. This is the integral membrane enzyme that is ubiquitous in mammalian cells which is responsible for the movement across the plasma membrane of both Na+ and K+ against their concentration gradients. Such active transport uses the energy provided by the hydrolysis of the gamma phosphate of ATP. Because the binding site for ATP is intracellular, rapid control of its concentration was technically challenging at that time. The simple idea for “caged ATP” was to prevent ATP hydrolysis by blocking ATPase access to the beta-gamma phosphate bond with the ortho-nitrobenzyl photochemical protecting group3. With such a thermally stable, photochemical precursor of ATP, a latent energy source could be loaded into red blood cells; irradiation would produce a concentration jump of ATP of sufficient quantity to initiate Na+ pump activity (e.g. Na+ efflux from the intracellular compartment). These experiments established many of the key chemical properties for such optical probes. Namely, it was (1) stable at physiological pH, (2) biologically inert before irradiation, and (3) photochemically active enough to liberate ATP in useful quantities to drive active transport. Soon after this, the probe was used for a “real biological study” in 19804. Genuine kinetic analysis of ATP uncaging was not addressed in 1978, however, in 19805 it was shown using laser flash photolysis that the rate of release of ATP was very fast6 (ca. 80 s−1), providing insight into the final property so crucial for many caged compounds. Subsequently, laser flash-photolysis of caged ATP was used to study the rate of relaxation of single skeletal muscle fibers on the millisecond time scale in 19827. These four papers2,4,6,7 firmly established the potential of caged compounds for cell physiology. Moreover, they form a template for us to understand how all useful caged compounds must be developed. This Account provides an overview of some of my work in this field (Figure 1); each probe uses photochemistry to address a fundamental biological problem, and is illustrated by an important biological application.
Figure 1.
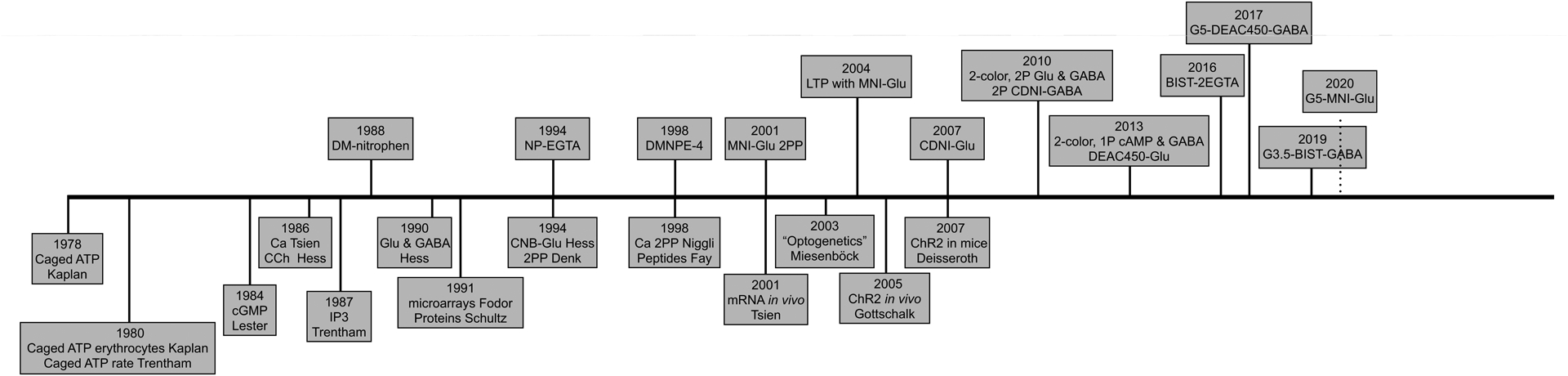
Time line of the development of “useful caged compounds for cell physiology”.
Caged calcium.
In the late 1970s, very little was known about the precise nature of Ca2+ signals inside cells. Light-based methods offered the obvious solution to this problem, as light passes through the plasma membrane, and therefore is non-invasive. Starting in 1980, Roger Tsien introduced several Ca2+-specific dyes that enabled physiologists study the kinetics of Ca2+ signaling with excellent temporal and spatial fidelity using fluorescence microscopy8. However, Jack Kaplan told me he envisaged “caged calcium” before such important, quantitative methods, as early 1976. He realized that because one cannot form covalent bonds with the divalent cation, caged Ca2+ would require an entirely different method from caged ATP. The concept was to take the widely used Ca2+-selective chelator EGTA (Ca2+ affinity 150 nM at pH 7.2, Mg2+ affinity 12 mM), or the very high affinity Ca2+ chelator ETDA (Ca2+ affinity 32 nM at pH 7.2, Mg2+ affinity 2.5 μM), and make them photosensitive. The idea was to cut the high affinity chelator in half, yielding low-affinity, di-acid molecules.
Biological context.
“Ja Kalium, das ist alles” is a famous quip from Otto Loewi9, the Nobel Laureate who discovered neurotransmission. Prescient when made in 1957, we now know that increases in intracellular Ca2+ control physiological processes such as muscle contraction and hormone secretion. Ca2+ is called “second messenger” as its concentration inside cells increases in response to some initial stimulus, thus it acts as a relay to communicate the initial impulse to some target down stream. In the case of neurotransmitter release, the pre-synaptic cell has vesicles containing a transmitter (e.g. glutamate) that are ready to release their cargo. An action potential stimulus (the “first message”) causes Ca2+ entry from outside, via voltage-gated Ca2+ channels. This rapid increase in Ca2+ in the nerve terminal (the “second message”) triggers vesicle fusion and transmitter release, which, in turn, open ligand-gated ion channels on the postsynaptic cell. This causes membrane depolarization, allowing the signal to be “passed on” through the electrical circuit (Figure 2). In the case of muscle contraction, the major source of the trigger Ca2+ is from intracellular stores. The three muscle types (skeletal, heart, and smooth) have subtle differences in their Ca2+-driven signal cascades, but in all cases it is Ca2+that functions as the crucial “second messenger”. In nerves and muscles resting Ca2+ is low, ca. 100 nM, with the stimulus increasing it into the 0.01–0.20 mM range. Note intracellular Mg2+ is at least 10,000-fold higher in concentration, and always competes with Ca2+ for divalent cations chelators. This is true of synthetic molecules such as EDTA and EGTA, and well as enzymes like calmodulin, troponin C and synaptotagmin. Readers may refer to excellent physiology textbooks for more details10. These fields have been the subjects of many Nobel Prizes. As will become clear, many of the caged compounds I have made have been used to dissect these signaling mechanisms (Figure 2).
Figure 2.
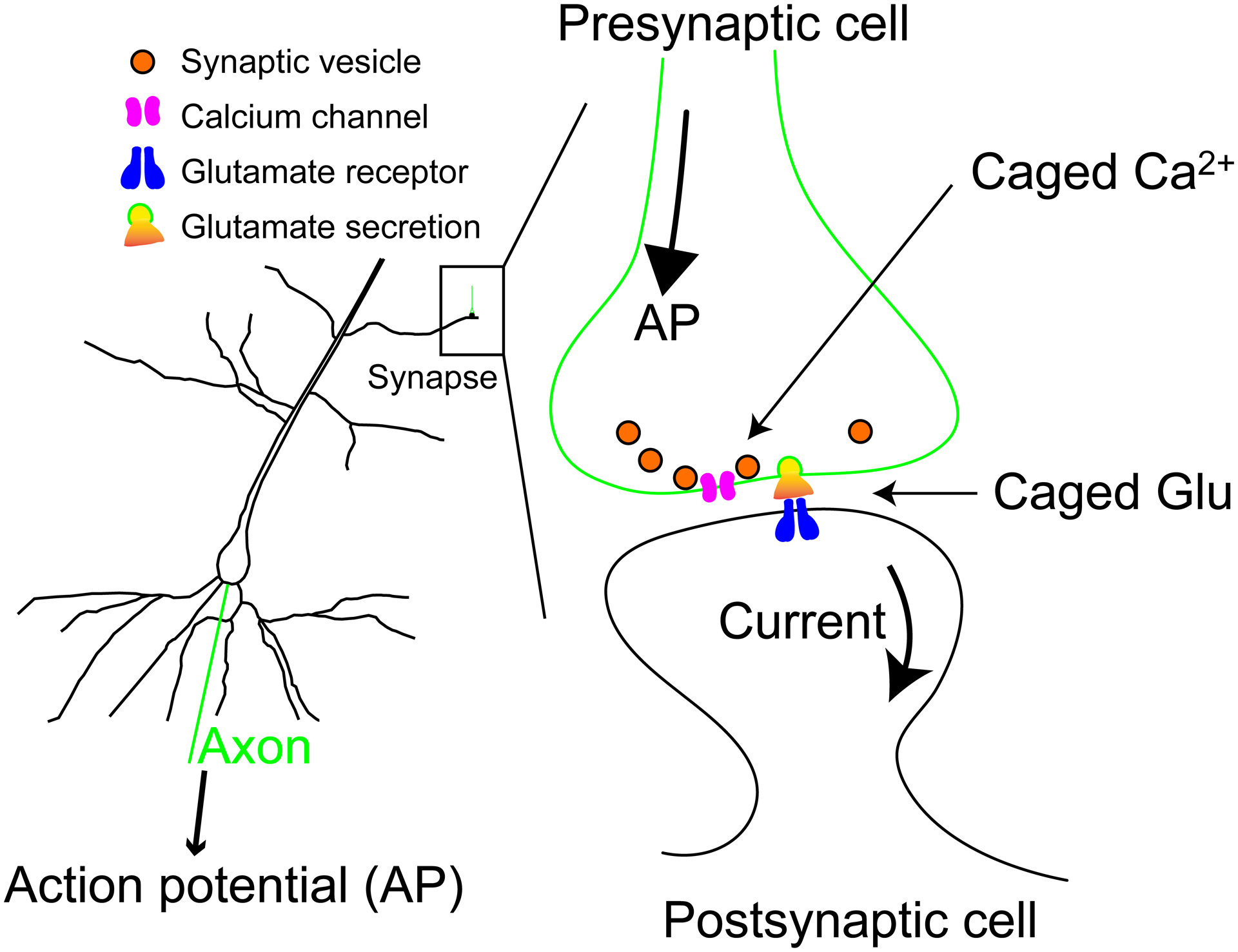
Outline of the basic physiology of neurotransmission. Left: cartoon of pyramidal neuron showing the postsynaptic dendritic tree and cell body. Right: Cartoon of a single synapse indicating where caged compounds “replace” physiological stimuli.
Highly efficient Ca2+ uncaging: DM-nitrophen.
The first useful caged Ca2+ I made was a photolabile EDTA derivative11, later called “DM-nitrophen” in 198812 (Figure 3). The key step in the synthesis was a high-pressure amination of a dibromide (Figure 3A – I want to thank Regina Zibuck for suggesting this method). We measured the rate of Ca2+ release from the DM-nitrophen:Ca2+ complex13 (Figure 3B). The rate of 38,000 s−1 is sufficient to make DM-nitrophen useful for the study of even the fastest Ca2+-triggered physiological processes (e.g. neurotransmission). If this were not the case, uncaging itself would be rate limiting and make kinetic analysis meaningless. Together with the good quantum yield (0.18), the chemical properties of DM-nitrophen are near-perfect for a caged Ca2+ probe. The only real issue is its high affinity for Mg2+. As noted above, the resting Mg2+/Ca2+ ratio is 10,000, meaning, with physiological values of these cations, DM-nitrophen becomes “caged Mg2+”. To our surprise much physiology with Mg2+-free solutions was feasible, and so DM-nitrophen proved enormously successful, being used in more than 500 studies14.
Figure 3.
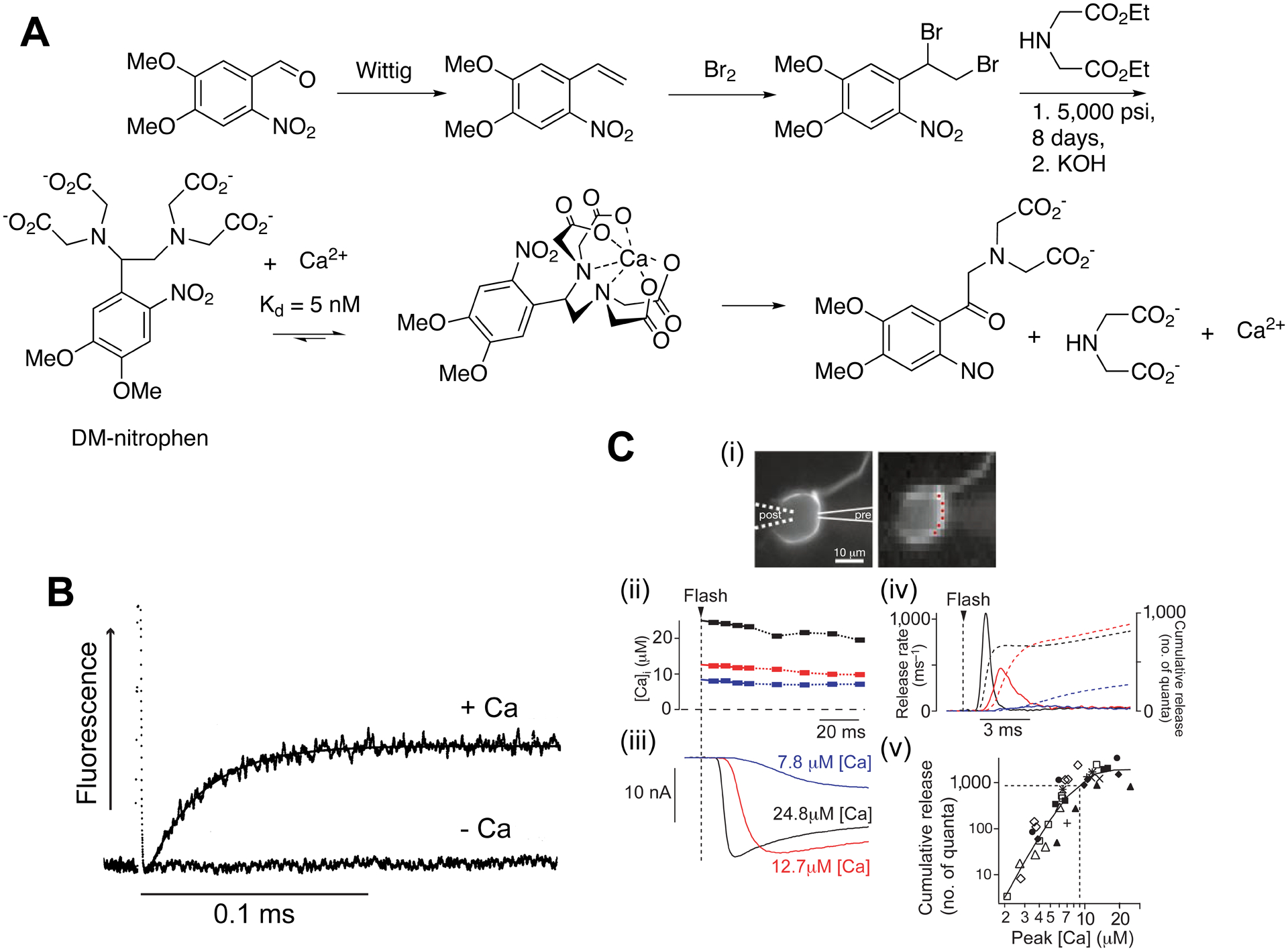
DM-nitrophen - the first highly effective caged calcium.
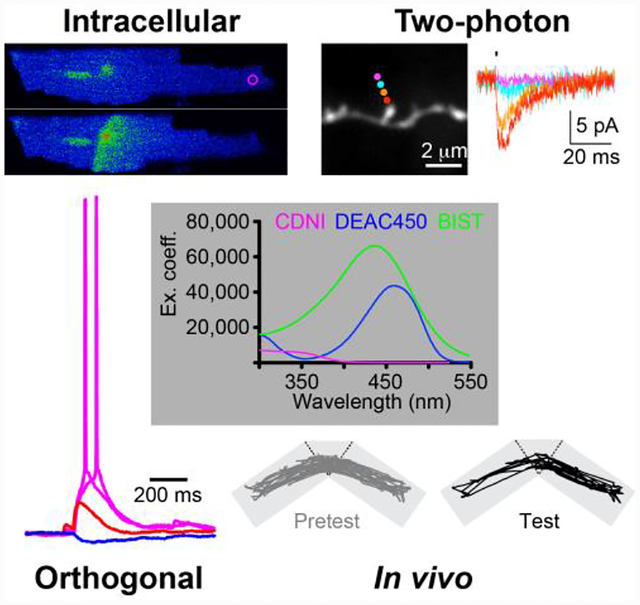
A. Synthesis and photolysis of DM-nitrophen11. B. Rate of Ca2+ release.13 C. Uncaging Ca2+ in a synaptic connection15. (i) Images of a calyx of Held synapse, left - simultaneous pre- and postsynaptic patch-clamp. Imaging the presynapse, containing DM-nitrophen and furaptra, allowed measurement of uncaged [Ca2+] (red dots). (ii) flash photolysis caused corresponding postsynaptic currents (iii), giving estimates of glutamate release rates (iv). (v) Quantitative description of peak [Ca2+] and the number of quanta released by Ca2+-driven exocytosis.
B: Reproduced with permission from ref. 13. Copyright (1996) Biophysical Society. C: Adapted with permission from ref. 15. Copyright (2000) AAAS.
One of these is highlighted in Figure 3C. Here Erwin Neher used15 the rapid, highly efficient photorelease of Ca2+ from DM-nitrophen to determine the relationship between pre-synaptic [Ca2+] and the size and rate of the postsynaptic current, thus providing a quantitative description of Sir Bernard Katz’s “Ca2+ hypothesis for neurotransmitter release.”16 Before DM-nitrophen, I did make, in 1986, a photolabile EGTA derivative11. Unfortunately this chelator had a very low affinity before photolysis (ca. 25 μM), making it useless as “caged calcium”. At the same time Roger Tsien made his first caged Ca2+ (nitr-2)17, and this was Ca2+-selective. Because EDTA has a quite high affinity for Mg2+, I made DM-nitrophen almost as an “afterthought”. Later, Kaplan would say to me: “Never let your own lack of imagination limit other peoples’ creativity”.
Cation-selective ucaging: NP-EGTA.
I solved the “Mg2+ problem” of DM-nitrophen18 by making NP-EGTA19 (Figure 4). The key steps in the synthesis are transformation of the ester to a ketal, followed by trans acetalization of this material to an intermediate incorporating the di-ether backbone of EGTA. This was reduced to the key advanced intermediate using chloroborane-dimethylsulphide complex (Figure 4A). A sequence for efficient installation of the amino acid termini, using standard thermal chemistry, allowed NP-EGTA to be made in 10 synthetic steps, in an overall yield of 24% (Figure 4A). The basicities of the amino functionalities of EDTA and EGTA are a major factor19,20 that determines the equilibrium constant of these molecules for Ca2+. The presence of the electron withdrawing nitrophenyl substituent reduces the pKa of the adjacent N when compared with EGTA, thus the Kd was decreased to 80 nM. This allows NP-EGTA to be almost fully loaded with Ca2+, whilst maintaining low [Ca2+]free (Figure 4B, blue curve). Adding 1.5 equivalents of Mg2+ had no effect on the [Ca2+]free (Figure 4B, orange dashes). Irradiation with UV laser light fully saturated the Ca2+ indicator fluo-3 (Figure 4B, pink curve). These data were gathered in one morning, and are still the most exciting in my scientific life, as I knew at that moment I had finally made a “real” caged Ca2+! The efficiency of the synthesis allowed me to make good quantities of NP-EGTA, and so initiate many collaborations with distinguished physiologists. One is highlighted21 in Figure 4C.
Figure 4.
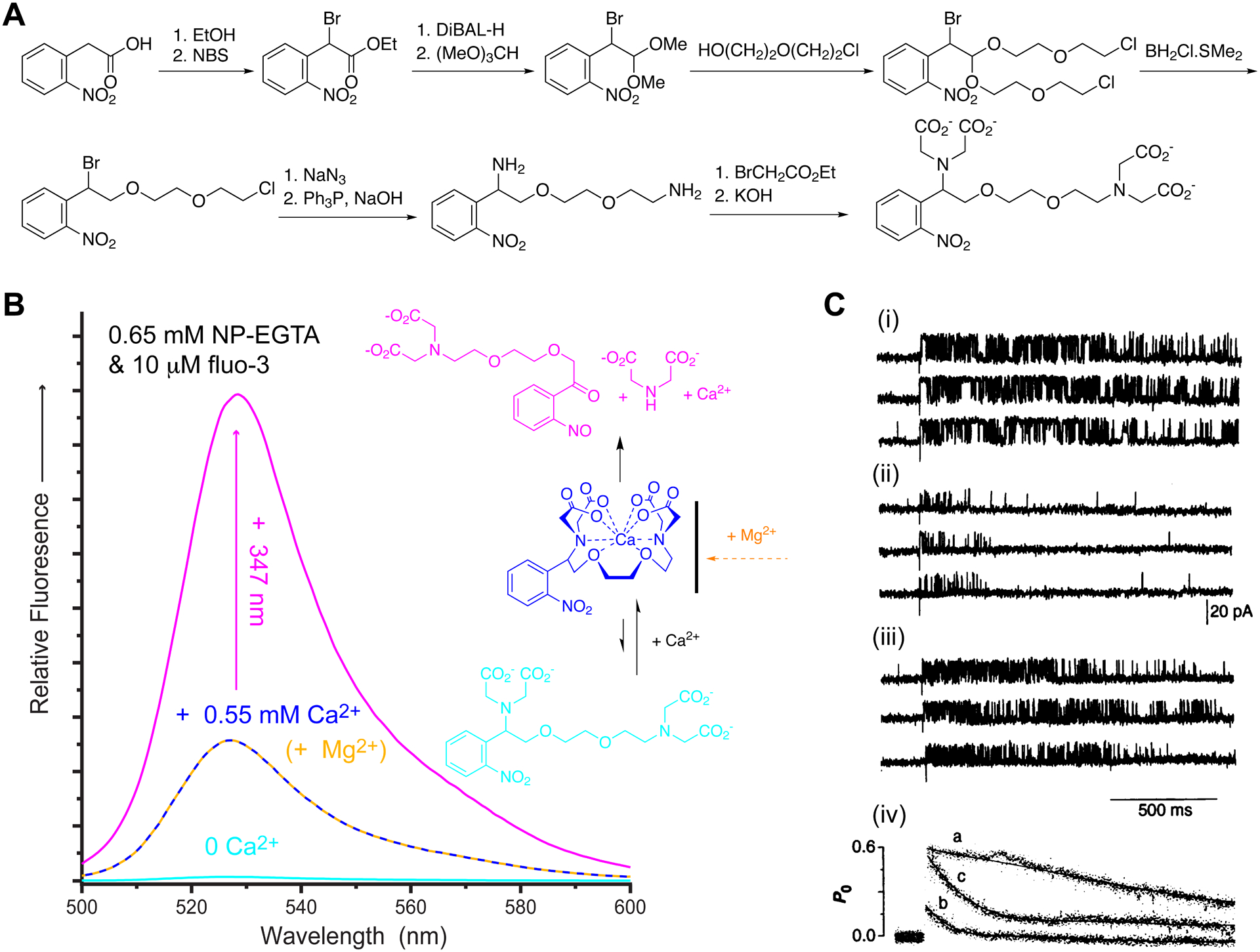
NP-EGTA - the first highly effective, cation-selective caged calcium.
A. Synthesis of NP-EGTA19. B. Fluorescence from the Ca2+ indicator fluo-3 with NP-EGTA, but before adding any Ca2+ (cyan). NP-EGTA is a high affinity Ca2+ cage (blue) that is Ca2+-specific (mustard dashes), and releases large quantities of Ca2+ upon photolysis (pink). The chelation and uncaging of Ca2+ by NP-EGTA is illustrated on the right. C. Single channel currents from RyR of black lipid membranes. Uncaging NP-EGTA in absence (i) or presence of 1 mM Mg2+ (ii & iii), step increases in [Ca2+]free to 1 μM (i) or 10 μM (ii & iii) caused rapid opening of the Ca2+ channels. The presence of Mg2+ caused a rapid adaption of the open channel probability (PO) as shown in (iv). C: Reproduced with permission from ref. 21. Copyright (1995) AAAS.
Cardiac muscles use a mechanism called Ca2+-induced Ca2+ release (CICR) to cause contraction10. When the muscle cells depolarizes, a voltage-gated Ca2+ channel in the plasma membrane opens, admitting a small quantity of Ca2+ from the extracellular environment. This triggers a large efflux of Ca2+ from the muscle intracellular store through another Ca2+ channel called the ryanodine receptor (RyR). The single channel properties of RyRs are difficult to study with patch-clamp methods, but isolated membrane fragments containing RyRs can be fused to lipid membranes at low density such that single channels can be observed (Figure 4C). Flash photolysis of NP-EGTA:Ca2+ complex allowed us to mimic Ca2+ entry into cells, and the Ca2+ selectivity of NP-EGTA enabled us to define the rapid modulatory effect of physiological Mg2+ on RyR gating21.
Summary.
The development of DM-nitrophen and NP-EGTA was followed by two other useful caged Ca2+ probes22,23, which we also used in several studies24–37. Taken together, these studies illustrate how useful the caged Ca2+ probes I have developed are for cell physiology. Here the biological problems define the required properties of the caged compound, and the way probes must often be improved iteratively. Before I developed this set of probes relatively little was known about the quantitative nature of Ca2+-controlled secretion of neurotransmitters and hormones from mammalian cells. Ca2+ uncaging allowed such fundamental biological processes to be studied38.
Two-photon uncaging of neurotransmitters.
Two-photon microscopy (2PM) was invented by Denk et al., in 199039. Chromophore excitation occurs by the simultaneous absorption of two photons of twice the wavelength normally used to produce the S1 state. This method was designed for imaging live tissue at greater depths than conventional laser-scanning confocal microscopy. In the original abstract they observed that, “This technique also provides unprecedented capabilities for three-dimensional, spatially resolved photochemistry, particularly photolytic release of caged effector molecules.” In 1994 Denk realized the first example of 2P uncaging in living tissue40. However, he used the simple ortho-nitrobenzyl probe requiring 2P uncaging using 640-nm excitation. By 2000, laser technology had advanced enabling facile excitation at > 700 nm, thus caged neurotransmitters remained the limiting factor. To address this problem I developed 4-methoxy-7-nitroindolinyl-glutamate(MNI)-Glu, a new probe designed to be photolyzed at 720 nm.
Photochemical glutamate quanta: MNI-Glu.
Glutamate is the major neurotransmitter in the brain, and its release by axons at synapses in fundamental for life. The first example of glutamate uncaging within intact tissue appeared in 199341. This study used UV photolysis on an inverted microscope giving low lateral resolution (50 microns). Following Denk’s pioneering study40, work by Niggli in 199842 inspired me to think that 2P uncaging would work with ortho-nitrobenzyl chromophores bearing electron donating substituents. Therefore, in parallel with the synthesis and 2P photolysis of DMNPE-422,24, I made DMCNB-Glu, the electron-rich analogue of CNB-Glu (Figure 5A). Laser flash-photolysis revealed that the rate of release was 200,000 s−1 (Figure 5B). A fast rate is absolutely crucial for effective 2P uncaging of glutamate, because we know that (a) the residence time of an uncaging molecule in the 2P voxel is 0.3 ms and (b) glutamatergic transmission is on the millisecond time domain (Figure 3D). In collaboration with Masanori Matsuzaki and Haruo Kasai, I showed that 2P uncaging of DMCNB-Glu yielded very high spatial resolution in the axial dimension, as would be predicted by the geometry of the laser beam. In contrast, we found that the axial resolution of CNB-Glu from UV uncaging was very low (Figure 5C). I presented these data in 1999 at the Society of General Physiologists annual conference43. Unfortunately, DMCNB-Glu was very sensitive to hydrolysis at pH 7.4, making this probe very difficult to use.
Figure 5.
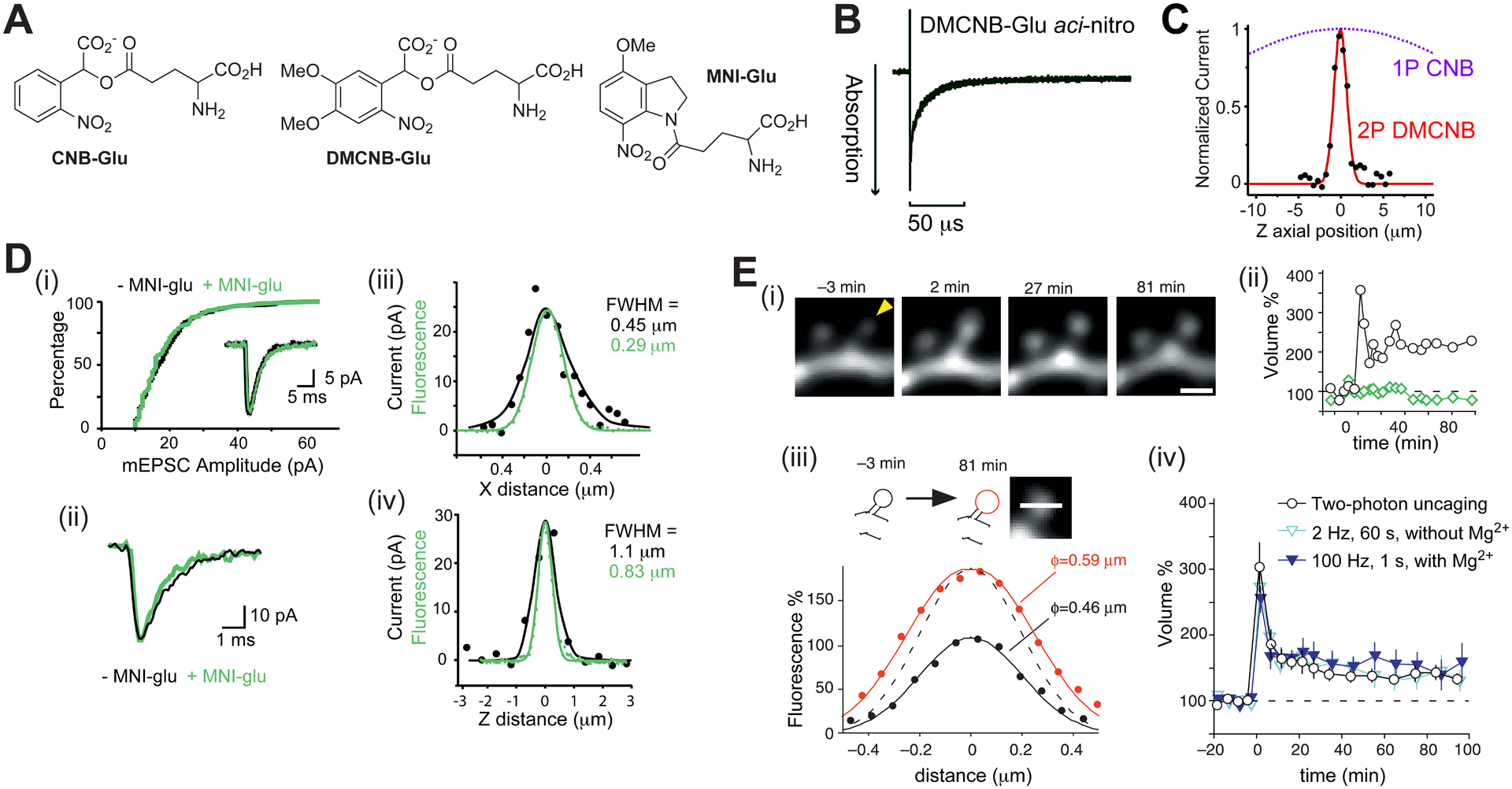
MNI-Glu – two-photon uncaging mimics quantal release of glutamate.
A. Structures of caged glutamate probes. B. Aci-nitro decay of DMCNB-Glu had a half-time of 5 μs. C. Comparison of the axial resolution of currents from CNB-Glu and DMCNB-Glu. CNB-Glu is not 2P sensitive. D. Properties of MNI-Glu44: (i) it does not block AMPA receptors; (ii) 2P uncaging mimics quantal release; lateral (iii) and axial (iv) resolution of 2P uncaging E. Structural LTP at single spines45. (i & ii) 2P uncaging (60 quanta at 1 Hz) induces input-specific, structural LTP. (iii) Example of volume increase. (iv) Structural LTP from 2P uncaging is similar to electrical LTP. D: Reproduced from ref. 47. Copyright (2001) GCRE-D. E: Reproduced from ref. 45. Copyright (2004) GCRE-D.
Nitro-indolinyl photochemical protecting groups were invented by Patchornik in 197646. I made a more electron rich, mononitro-indolinyl-caged glutamate in 1999, and presented44 the usefulness of this probe with Matsuzaki and Kasai at the Society for Neuroscience annual meeting in 2000. In comparison to DMCNB-Glu, MNI-Glu is highly stable (half-life 120 mins vs days). The other key aspects of the effectiveness of MNI-Glu are shown in Figure 5D. The probe does not block its target receptor, 2P uncaging matches optical resolution, and the glutamate dosage can be tuned to mimic the postsynaptic current evoked by single quanta47. It is this latter property which revolutionized optical neurobiology, as in the brain glutamate is always released from fusion of a single vesicle in presynaptic cells, glutamate floods the synaptic cleft, binds immediately to its postsynaptic receptor, opening its ligand-gated ion channel, causing depolarization (Figure 3C). This biological process is the fundamental “coin of communication” in the brain. 2P uncaging of MNI-Glu mimics this biological process faithfully (Figure 5Dii).
The brain is the only organ that is plastic. This ability to change in fundamental ways is the basis of learning and memory, and, more broadly, all human conscious experience. The first clues as to how the brain changes at the synaptic level were provided by a Classic experiment by Bliss and Lumo in 197348. They found that brief high-frequency electrical stimulation (HFS) of an axon tract in a live rabbit caused a strengthening of the electrical signal recorded in the brain region targeted by those axons. This increase in synaptic strength was found to last for hours, hence the term “long-term potentiation”, or LTP, was coined. Literally thousands of LTP studies have been published by physiologists since then49. In 2004 we used45 2P photolysis of MNI-Glu at visually identified, single synapses in living brain slices to show that LTP had a structural correlate (Figure 5D). Specifically, at dendritic spines, HFS caused long lasting volumetric enlargement that could be mimicked by uncaging (Figure 5D). Further experiments showed that the structural LTP was accompanied by a corresponding increase in postsynaptic current45. Importantly, LTP was synapse specific, with adjacent spines not showing any changes. Using our method and caged compound Yasuda has dissected the biochemical details of single spine LTP50. Furthermore, many laboratories have used our approach to examine the details of synaptic plasticity in many brain regions. I reviewed this field in 201951.
Dinitroindolinyl caged glutamate – photolysis with high quantum yield.
Two photochemical properties define the effectiveness of any chromophore: (1) its ability to absorb light effectively; and (2) conversion of excited-state molecules into effect. Fluorophores such as fluorescein and rhodamine are important because they have large extinction coefficients (property 1) and high quantum yield of emission (property 2). The quantum yields of caged ATP and IP3 are very high (>0.5), whereas MNI-Glu is a modest (<0.1). Following Tsien52, we made53 the “dinitro version” of MNI-Glu, called “MDNI-Glu” in 2005 (Figure 6A, this probe was “re-invented” in 2014 by Femtonics), and found the quantum yield of photolysis was about 0.5. The 2P absorptivity was similar to MNI. Dinitrobenzenes suffer from solubility problems, therefore, I made54 more buffer-amenable version bearing a negative charge, called CDNI-Glu (Figure 6A).
Figure 6.
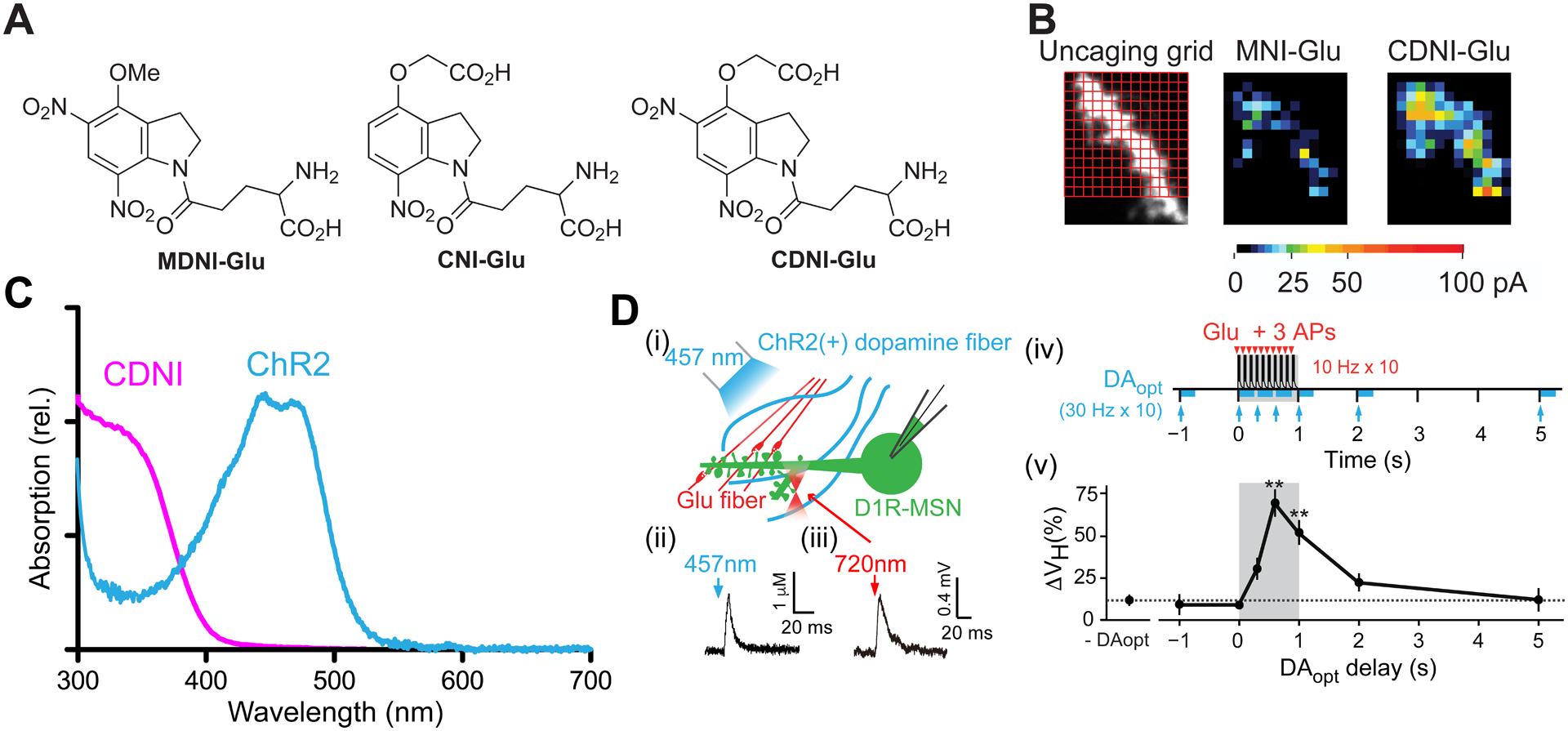
Dinitroindolinyl caged glutamates - probes with exceptional quantum yields.
A. Structures of MDNI-Glu53, CNI-Glu54 and CDNI-Glu54. B. Comparison functional 2P-mapping on the grid (left) using MNI- and CDNI-Glu on the same dendrite54. C. Spectra of the CDNI and ChR2 (normalized). D. (i) Scheme for structural LTP using 2-color actuation55. (ii) Blue-light evoked dopamine concentration measured by amperometry. (iii) 2P-evoked glutamate current at a single spine. (iv) Outline of the timing combination of 2P uncaging and optogenetic release of dopamine. (v) Summary of the relationship between dopamine timing and structural LTP. Optimum effects of dopamine input were between 0.25–1.0 s. No LTP was seen at −1 s or > +2 s. B: Adapted with permission from ref. 54. Creative Commons License. D: Adapted with permission from ref. 55. Copyright (2014) AAAS.
We compared54 the biological efficacy of MNI-Glu and CDNI-Glu by 2P uncaging on the same neuron in 2007. We found that the evoked current was more than 4x higher with CDNI (Figure 6B). This improvement of photochemical property enabled us to use less energy for 2P uncaging, for certain types of physiological experiments this is crucial. One example is shown in Figure 6C,D. For principal neurons in the cortex, LTP is known to be dependent on Ca2+ entry through NMDA receptors45, allowing a simple HFS protocol to induce LTP48,49. In many other types of neurons plasticity is not so well defined, an example of this is medium spiny neurons in nucleus accumbens. We found that structural and electrical LTP was dependent not only on glutamate, but also on the simultaneous release of dopamine onto these cells55. To photostimulate precisely the release of two neurotransmitters we used two-color actuation by combining CDNI-Glu and ChR2, as they have chromophores that absorb at different wavelengths (Figure 6C). Importantly, ChR2 is not very sensitive to 2P excitation at 720 nm, but is very easily excited by blue light, a wavelength not absorbed by CDNI. In a complex set of physiological experiments we found that, in essence, the structural LTP protocol used with hippocampal neurons (Figure 6D) needed to be “supplemented” with a timed dopamine dosage. This was evoked by blue light causing dopamine release from axons infected with ChR2, originating from the ventral tegmental area. Furthermore, there was a “timing window” for this postsynaptic coincidence detection by neurons in the nucleus accumbens (Figure 6D). Our results may explain why simple electrical stimulation of medium spiny neurons does not always induce LTP.
High-resolution, 2P uncaging of the major inhibitory transmitter: CDNI-GABA.
γ-Aminobutyric acid (GABA) is the major inhibitory transmitter in the brain. While CNB-GABA proved useful for low-resolution mapping of postsynaptic GABA receptors56, the success51 of MNI-Glu suggested a comparably effective caged GABA might open new avenues for study of this transmitter. Thus, following the development of CDNI-Glu54, I made57 CDNI-GABA (Figure 7A). Photolysis of this probe on hippocampal neurons evoked GABA-A receptor-specific currents (Figure 7B), one that could be tuned to match quantal release of GABA (Figure 7C). Detailed 2P functional mapping of GABA-A receptors was found to be reproducible, and dependent on 2P excitation (Figure 7D), enabling excellent resolution in the axial dimension (Figure 7E). These data were the first example of high-resolution, 2P uncaging of GABA appeared in 201057.
Figure 7.
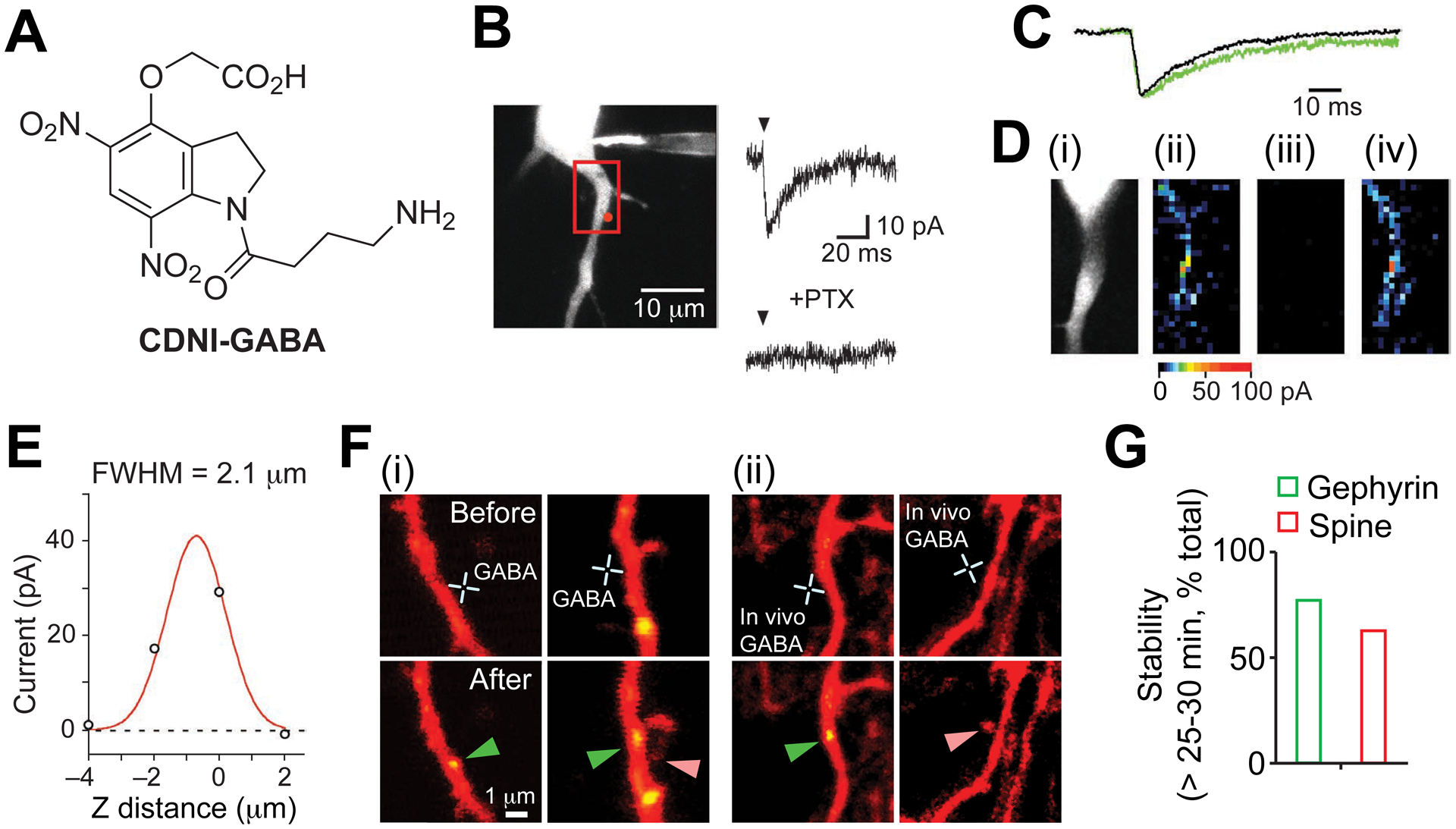
CDNI-GABA – two-photon uncaging of the major inhibitory transmitter.
A. Structure of CDNI-Glu57. B. Example of rapid, GABA-A selective 2P-evoked current57. C. 2P photolysis of CDNI-GABA mimics quantal release57. D. Functional mapping57 of neuronal GABA-A receptors (i) required a mode-locked laser (ii) vs (iii), and was reproducible (ii) & (iv). E. Axial resolution of 2P uncaging of CDNI-GABA was diffraction-limited57. F. 2P photolysis of CDNI-GABA on young neurons induces GABA synaptogenesis and spinogenesis in vitro (i) and in vivo (ii). G. Most photo-induced synapses were stable for longer than 25–30 min60. B-E Reproduced from ref. 57. Copyright (2010) GCRE-D. E,F: Reproduced with permission from ref. 60. Copyright (2016) AAAS.
Several biological studies have appeared58–60 using CDNI-GABA. Spinogenesis has been one of the most keenly debated phenomenon in neurophysiology61. Do axons seek out new postsynaptic partners, or do dendrites initiate synapse formation? In 2013 it was shown62 that glutamate caused spinogenesis on neurons between postnatal days 8–12 by using 2P photolysis of MNI-Glu. In slightly younger neurons, postnatal days 6–8, GABA is, like glutamate, excitatory60, this because the chloride ion gradient is the opposite of adult neurons. High-resolution, 2P uncaging of CDNI-GABA induced spinogenesis in vitro and in vivo. On other parts of a dendrite the same input induced the appearance of GABA-A receptor synapses, and seen by a fluorescently tagged gephyrin at these postsynaptic densities (Figure 7F). The majority of both types of synapses were found to be stable for at least 30 mins (Figure 7G). These data suggest that GABA sets the balance between inhibitory and excitatory synapses in early postnatal stages, laying the foundation for later circuit development, and were revealed by the ability of high-resolution 2P uncaging of GABA using CDNI-GABA.
Summary:
Before the development of probes for 2P uncaging of neurotransmitters51 little information was available on single synapse activation and the immediate, localized down stream biochemical events. Probes such as MNI-Glu allowed definitive, direct cause and effect to be defined in many studies, in particular, the localized biochemical events involved in LTP50.
Wavelength orthogonal, two-color uncaging: DEAC450.
In 1977 Bruce Merrifield stated63, “An orthogonal system is defined as a set of completely independent classes of protecting groups. In a system of this kind, each class of groups can be removed in any order and in the presence of all other classes.” Organic chemists take for granted such selectively using thermal reactions. However, when only one reagent is available, namely light, independence is very challenging. Because simple nitro-aromatic caged compounds work very well at short wavelengths (ca. 350 nm for 1P, and 720 nm for 2P, see above), my idea was to develop a new photochemical protecting group that was not sensitive at these wavelengths. The clue for this was found in the work of Hara, et al., who made a series of substituted aminocoumarins with extended π-electron substituents at the 3-position64. Addition of an acrylate to the 3-position 7-diethylaminocourmarin(DEAC) produced a blue-absorbing chromophore with the desired minimum at 350 nm which we called “DEAC450”.
The first DEAC450-caged compound we made was DEAC450-Glu69 (Figure 8A). DEAC450-Glu is shows no hydrolysis at RT at pH 7.4 during a working day, and such solutions show no hydrolysis when frozen for at least 3 months. However, because this chromophore absorbs room light very effectively (Figure 8B) great care must be taken when handling it in solution. Photochemically it is 44x more efficient than MNI-Glu for 1P uncaging67. More importantly, DEAC450 is almost photochemically inactive towards 2P excitation at 720 nm, while undergoing clean 2P uncaging at 900 nm (Figure 8C). When we compared uncaging at these wavelengths at the same spine head, we found we had to use extended irradiation times (6 ms) to detect any significant currents at 720 nm68. The same energy at 900 ns produced about 30x more current (Figure 8D). (In contrast, another blue light absorbing probe, RuBi-Glu, has no such minimum69 (spectrum Figure 8B), with 720 nm being only 50% less active than its maximum at 800 nm.) In practice, we could find powers at 900 nm (e.g. 1 ms irradiation) that evoked quantal responses such that when this energy was used at 720 nm no currents could be detected67. Because the latter wavelength is optimal for CDNI-GABA, we co-applied this probe with DEAC450-Glu to hippocampal brain slices, and could use 2-color, 2P uncaging to fire and block action potentials in a reproducible way68 (Figure 8E). DEAC450-caged compounds have also proved highly effective for wavelength orthogonal 1P uncaging69,70. Using DEAC450-cAMP (Figure 8A) inside cells and CDNI-GABA outside cells we were able to control the firing rate of neurons69 using 355 nm for GABA uncaging and 473 nm for cAMP uncaging (Figure 8F,G). Importantly, we established conditions quite easily under which 355-nm light did not evoke an effect from DEAC450-cAMP (Figure 8F). It is important to emphasize that 355-nm light must photolyze some DEAC450-cAMP, but the amount released is so small that no biological effect was detected. Similar results were also obtained with caged GABA in another study70.
Figure 8.
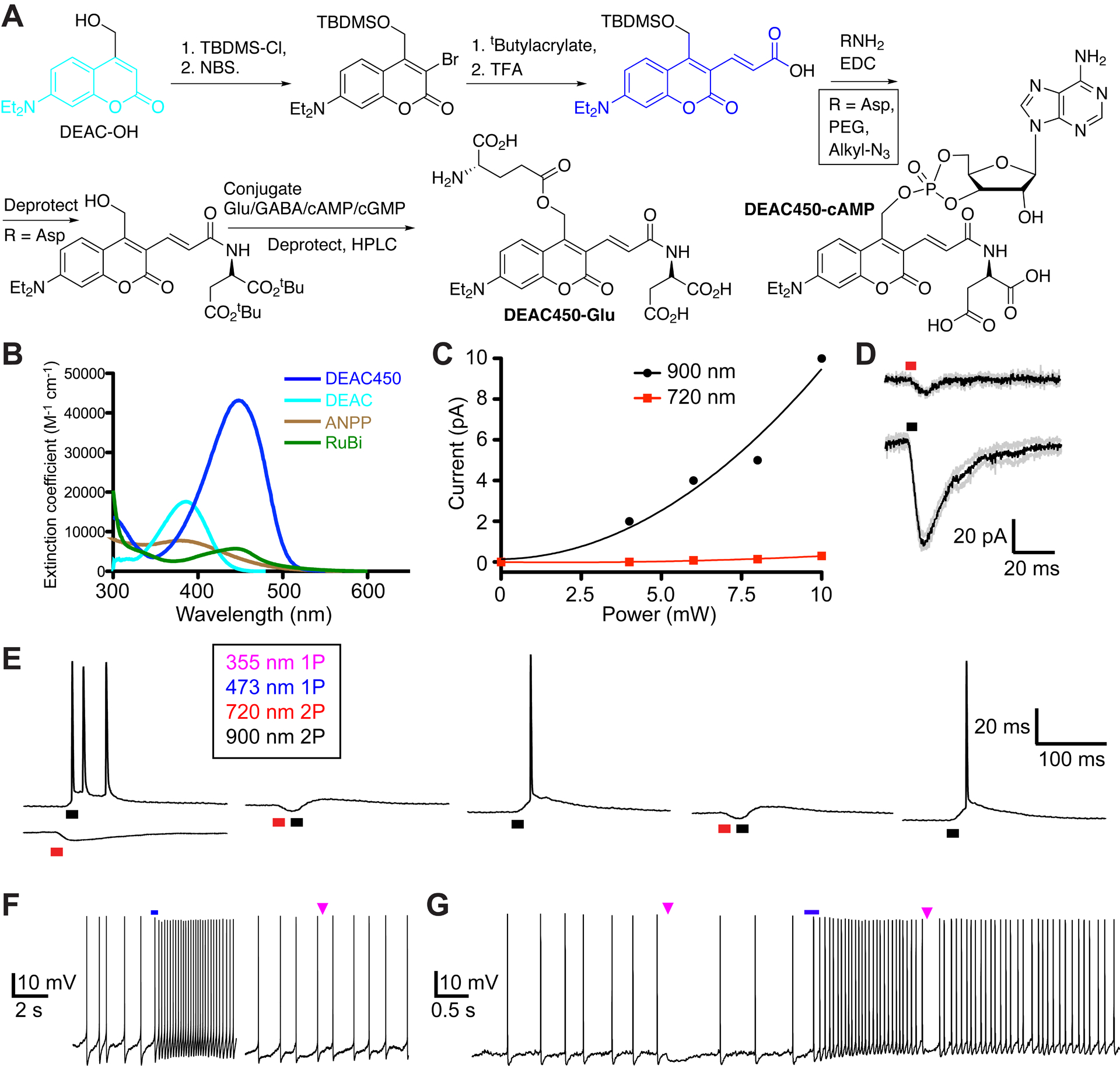
DEAC450 – enables wavelength selective, orthogonal photo-actuation with living cells.
A. Synthetic outline for the conversion of DEAC into DEAC450. Intermediate 3 is suitably bifunctional for coupling of a variety of “solubilization units” on its acid functionality, followed by coupling of the biomolecule to the subsequently deprotected methylalcohol. B. Absorption spectra of DEAC and DEAC450, with two other blue absorbing nitrobenzyl chromophores65,66. Only DEAC450 has a real minimum at 350 nm, the wavelength where traditional caged compounds are very sensitive. C. Comparison of 2P uncaging of DEAC450Glu with increasing power at 900 nm versus 720 nm67. D. Uncaging at 720 nm high powers were required to evoke detectable responses, such powers at 900 nm generated very large currents68. E. Co-application of DEAC450-Glu and CDNI-GABA allowed 2-color, 2P uncaging on CA1 neurons. Action potentials could be generated and blocked in a wavelength-selective manner. F. DEAC450-cAMP uncaged inside neurons with blue light enhances the firing rate, whereas UV light evokes no response. G. Wavelength-selective 1P UV uncaging of CDNI-GABA blocks spikes, followed by 1P-blue uncaging of cAMP enhances tonic firing, which is blocked by UV. D,E: Reproduced with permission from ref. 68. Copyright (2015) Wiley. F: Adapted with permission from ref. 69. Copyright (2013) ACS.
Summary:
Taken together, our work with DEAC450 has firmly established this photochemical protecting group can be used for all modes of wavelength selective 2-color actuation using 2P/2P, 2P/1P and 1P/1P uncaging68,71. DEAC450, when partnered with chromophores like CDNI, allows us to apply photostimulation to two receptors any number of times, in any order, with any magnitude and duration. In other words, truly arbitrary two-color uncaging is now facile71.
BIST: an extended π-electron caging chromophore with an exceptional 2P absorption cross-section.
In 1998 two seminal studies appeared72,73 defining the relationship between molecular structure of large chromophores and their 2P cross-section (2PCS). Rigidity, double bonds connected to aromatics, decoration with electron donors and acceptors were all properties found to contribute to the 2PCS. Importantly, ab initio calculations agreed with measured 2PCSs. Subsequently numerous studies substantiated these works, and have been reviewed many times74,75. In 2006, we attempted to implement these principles with the NDBF chromophore23. In same year Jullien explored the idea systematically with a library of substituted ortho-nitrobenzyl chromophores76, similar work by Goeldner appeared in 201265. Most of these new chromophores were found to have relatively modest 2PCS compared to the molecules reported in 199872,73.
In 2016 we revealed that it was possible to apply such molecular design rules successfully to caging chromophores for the first time with the development of BIST-2EGTA77. Following this we applied BIST to caged neurotransmitters, with G3.5-BIST-GABA78. Both synthetic routes have identical steps for making the core chromophore (Figure 9A, note the crucial double bond is added using the same Heck reaction). However, given the particulars each route is also quite distinct in terms of the details. The 1P absorption spectrum of BIST is also strikingly different from older nitroaromatic caging chromophores, with a much larger extinction coefficient in the blue and even some absorption in the green (Figure 9B). Joe Perry’s lab, an originator of one of the 1998 studies72, measured the absolute 2PCS of the basic BIST chromophore using the z-scan technique, and found a 2PCS of 740 GM77 (cf. MNI 0.9 GM47), with the G3.5 dendrimer leaving this essentially unaffected78. Flash photolysis using 2P excitation revealed that Ca2+ uncaging was much more effective than DM-nitrophen, as would be expected from the large 2PCS (Figure 9C). Recently, we have established that using low concentrations of BIST-2EGTA (50 μM) loaded into permeabilized cells could be used to induce local CICR transients using 2P uncaging with brief pulses of 810 nm light (data not shown). Only 1 ms and 20 mW were required, and this should be contrasted with the 50 ms and 40–80 mW required for DM-nitrophen42.
Figure 9.
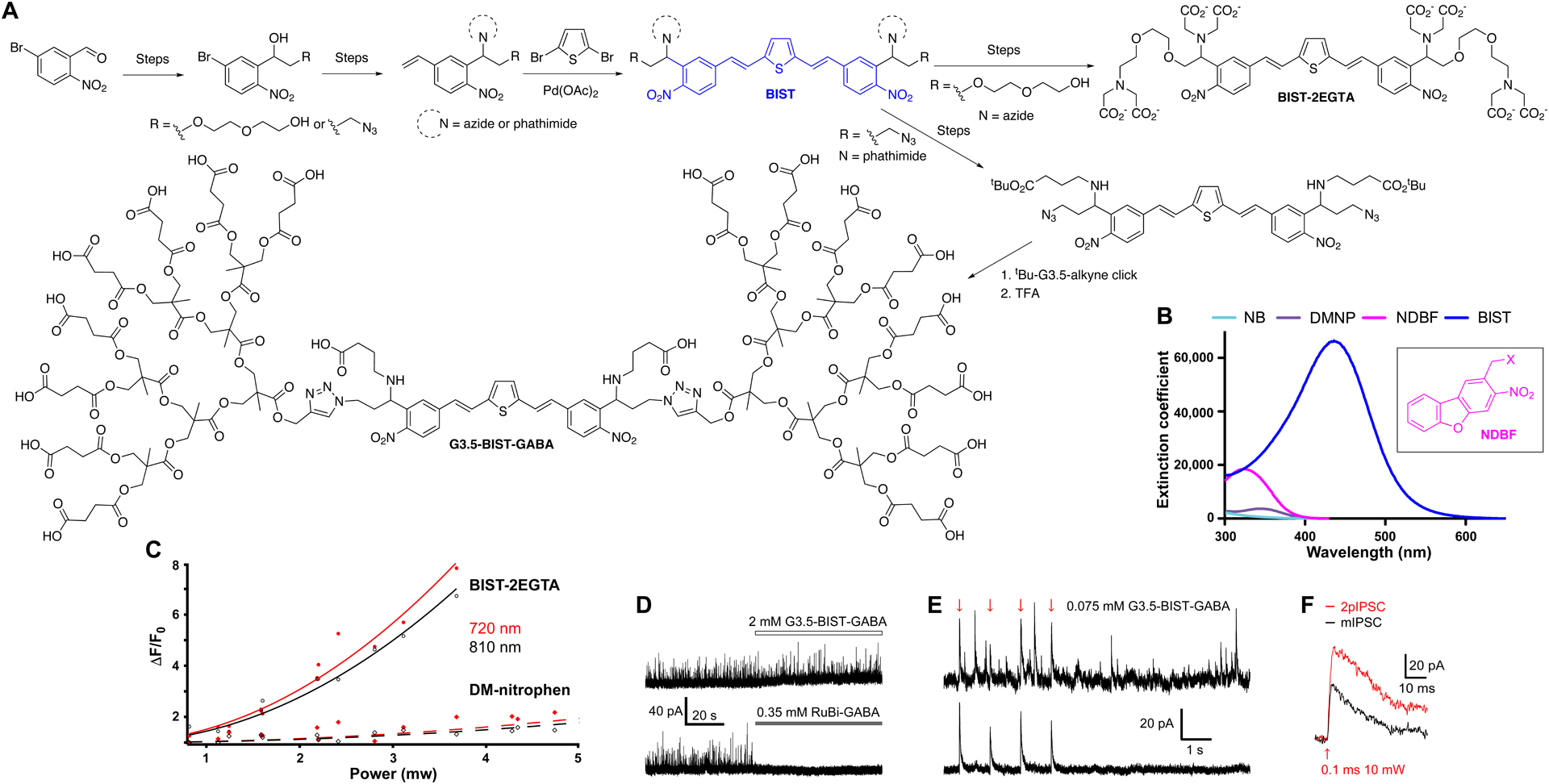
BIST-caged Ca2+ and GABA probes – chromophore construction and photochemical properties.
A. Construction of the BIST chromophore and elaboration into BIST-2EGTA and G3.5-BIST-GABA. B. Extinction coefficients of a range of o-nitrobenzyl caging chromophores. C. Comparison of the efficacy of 2P uncaging of Ca2+ from BIST-2EGTA and DM-nitrophen in a droplet. D. Structure of BIST-caged GABA. D. G3.5-BIST-GABA is inert up to 2 mM, but RuBi-GABA is highly antagonistic at 0.35 mM. E. GABA-A receptor currents (inhibitory postsynaptic currents, IPSCs) recorded from a CA1 neuron. Upper: Single sweep showing 2pIPSCs are indistinguishable from spontaneous currents. Bottom: average of 24 sweeps showing only the 2pIPSCs. F. Brief light flashes can evoke rapid IPSCs similar to spontaneous currents. D-F: Reproduced with permission from ref. 82. Copyright (2019) Wiley.
The decoration of the hydrophobic BIST chromophore with two anionic dendrimers produced a caged GABA that was inert up to 2 mM (Figure 9D). Because of the exceptionally high 2PCS of BIST, we could bath apply G3.5-BIST-GABA at a very low concentration of 0.075 mM and evoke GABA-A currents (2pIPSCs) which were indistinguishable from spontaneous mIPSCs (Figure 9E)78. Finally, very brief shutter times of 0.1 ms could be used to evoke large inhibitory currents using 2P excitation (Figure 9F). Normally, one would have to use 1–5 ms with probes like CDNI-GABA58,60.
Summary:
BIST is a new nitro-aromatic photochemical protecting group that is exceptionally sensitive to 2P uncaging.
Cloaked caged compounds.
One of the basic criteria of a “useful caged compound” is that it is biologically inert1,2. Fortunately attachment of the caging chromophore to most signaling molecules delivers a probe that satisfies this requirement. However, in the case of all the simple caged Glu and GABA probes made this is not the case. MNI-Glu does not block glutamate receptors47, but, to our surprise, we found it did have off-target antagonism towards GABA-A receptors79. CNB-GABA was reported80 to block GABA-A receptors with an IC50 = 32 μM in 2001, however this report was overlooked by those working in this field for many years. A clue to the solution of this problem was uncovered by determining that the BOC-protected precursor of PEG-DEAC450-GABA81 was almost inert, suggesting that if we could envelop GABA completely, whilst also caging it, we could make a much less antagonistic probe. Thus, attaching a neutral fifth generation (G5) dendrimer, a method we call “cloaking”, to the carboxylate side chain of DEAC450 yielded a caged GABA that was 90-fold less antagonistic than the PEG analogue82. Note that G3.5-BIST-GABA, shown above, is another example of a cloaked caged compound which is biologically inert towards GABA-A receptors (Figure 9E).
Initially we actually discovered MNI-Glu GABA-A antagonism by work in living mice, they became epileptic! Thus, any application in vivo required the co-application of TTX to control this side effect84. To solve this problem we made G5-MNI-Glu (Figure 10A) following a similar route to G5-DEAC450-GABA83. Bath application of up to 1 mM G5-MNI-Glu produced no detectable effect on the size or frequency of miniature excitatory postsynaptic currents (mEPSCs), in contrast, this concentration of MNI-Glu fully blocks these currents (Figure 10B). Cloaking had no significant effect on the photochemical properties of MNI-Glu, so local perfusion of 1 mM on neurons could be photolyzed very effectively with a violet laser (Figure 10C). These data suggested that G5-MNI-Glu could be used in vivo without any of the side effects seen with MNI-Glu. Using a newly developed method called “optofluidics”85–87, we delivered G5-MNI-Glu into ventral tegmental area (VTA) of the mouse brain, along with violet light during a conditioned place preference test. Uncaging in this brain region stimulated dopamine release which mediated Pavlovian conditioning in a way that was indistinguishable from traditional methods (Figure 10D). This work forms a useful paradigm for experiments with future caged probes that will allow phasic manipulation of the behavior of higher order animals.
Figure 10.
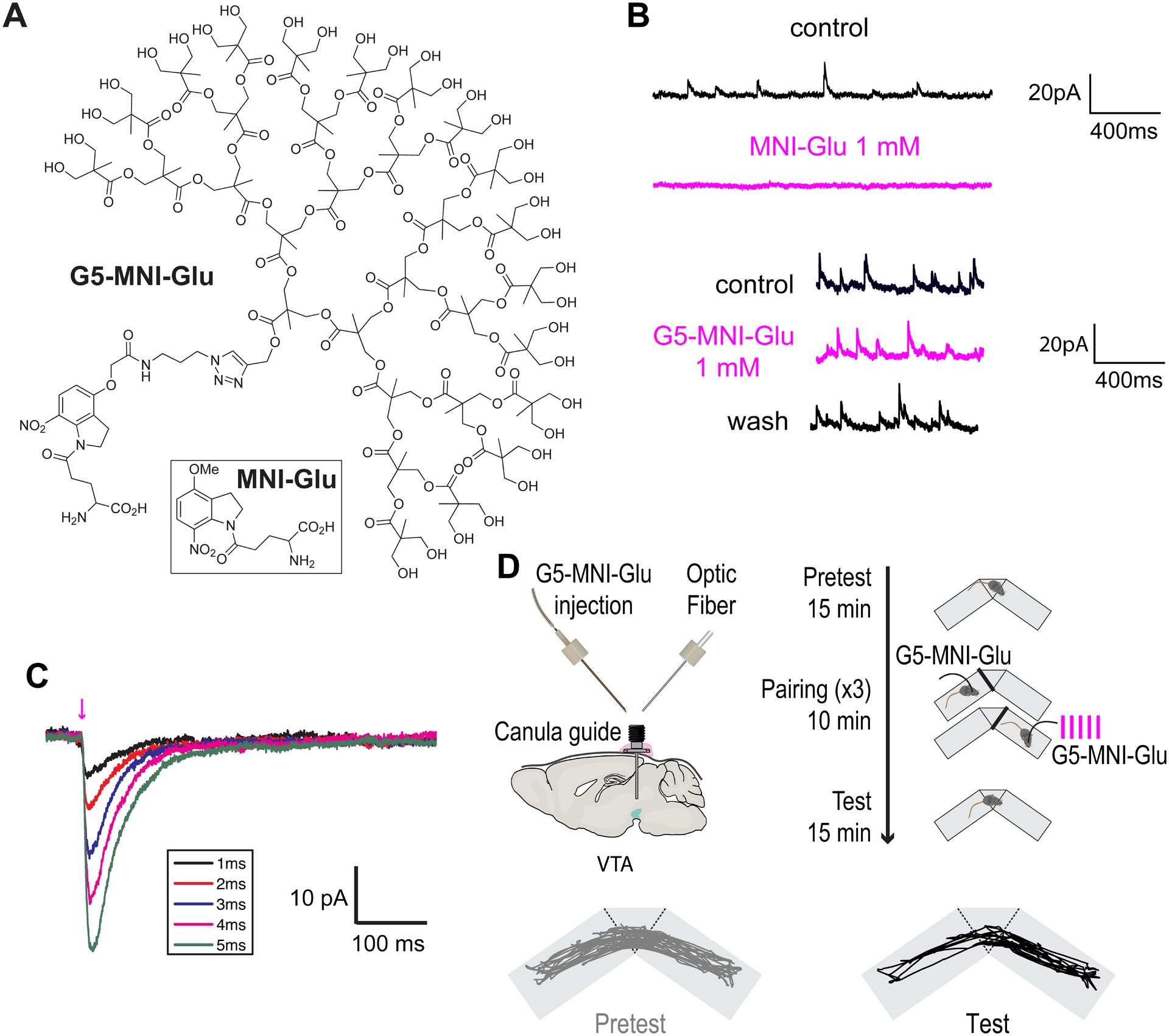
Cloaked caged glutamate.
A. Structures of G5-MNI-Glu and MNI-Glu. B. Effects of bath application of MNI-Glu and G5-MNI-Glu of GABA-A receptor minis recorded in CA1 neurons. C. Currents from a CA1 neuron evoked from uncaging G5-MNI-Glu (0.5 mM) with 405 nm laser (1.3 mW). D. Outline of optofluidic uncaging experiment in a freely moving mouse83. Uncaging in the VTA mediates Pavlovian conditioning. C,D: Adapted from ref. 83. Copyright (2020) GCRE-D.
Summary:
“cloaking” provides a general solution to the GABA-A receptor antagonism problem of caged neurotransmitters.
Summary.
In 1956, CP Snow wrote a famous article in The New Statesman titled “The Two Cultures”88. In it he lamented that “science” and “the humanities” seemed incommensurable. By analogy chemists and physiologists often find it difficult to appreciate the needs and abilities of each other’s fields. With many chemists now making photolabile compounds89 I hope this Account illustrates how important it is for them to listen to the needs of biologists in terms of optical probe development. On the other hand, it is vital that biologists appreciate the huge effort chemists must exert to make a “useful caged compound for cell physiology”. I hope symbiotic dialogue between these “two cultures” is catalyzed is some small ways by this Account.
Acknowledgements.
My lab has been supported by the NIH since 1995. I want to thank the many collaborators and postdocs who have helped me develop the “useful caged compounds” described in this Account.
Graham Ellis-Davies. I was born in 29 Feb 1957 (Liverpool, UK). I obtained my Ph.D. in Chemistry in 1982 from the University of Reading. This was followed by postdoctoral work at King’s College London (Chemistry, 1982-3); Rijksuniversiteit Leiden (Chemistry, 1983-4); unemployment (1984-5); and postdoctoral work at the University of Pennsylvania (Physiology, 1985-1995). I have held faculty positions in: Physiology at Drexel University (1996-2009) and Neuroscience at Mount Sinai, New York City (2010-now).
References.
- (1).Ellis-Davies GCR Nat Methods 2007, 4, 619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kaplan JH; Forbush B; Hoffman JF Biochemistry 1978, 17, 1929–35. [DOI] [PubMed] [Google Scholar]
- (3).Barltrop JA; Plant PJ; Schofield P Chem. Commun 1966, 822–3. [Google Scholar]
- (4).Kaplan JH; Hollis RJ Nature 1980, 288, 587–9. [DOI] [PubMed] [Google Scholar]
- (5).Herbette LG; Kaplan JH; Kihara T; McCray JA; Trentham DR Biophys. J 1979, 25, 244a. [Google Scholar]
- (6).McCray JA; Herbette L; Kihara T; Trentham DR Proc Natl Acad Sci U S A 1980, 77, 7237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Goldman YE; Hibberd MG; McCray JA; Trentham DR Nature 1982, 300, 701–5. [DOI] [PubMed] [Google Scholar]
- (8).Tsien RY Annu Rev Neurosci 1989, 12, 227–53. [DOI] [PubMed] [Google Scholar]
- (9).Carafoli E Proc Natl Acad Sci USA 2002, 99, 1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Boron W; Boulpaep E Medical Physiology; 2nd ed.; Elsivier: New York. [Google Scholar]
- (11).Ellis-Davies GCR; Kaplan JH Journal of Organic Chemistry 1988, 53, 1966–1969. [Google Scholar]
- (12).Kaplan JH; Ellis-Davies GCR Proc Natl Acad Sci U S A 1988, 85, 6571–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ellis-Davies GCR; Kaplan JH; Barsotti RJ Biophys J 1996, 70, 1006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ellis-Davies GCR Chem Rev 2008, 108, 1603–1613. [DOI] [PubMed] [Google Scholar]
- (15).Schneggenburger R; Neher E Nature 2000, 406, 889–93. [DOI] [PubMed] [Google Scholar]
- (16).Katz B; Miledi R J Physiol (Lond) 1967, 192, 407–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tsien RY; Zucker RS Biophys J 1986, 50, 843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ellis-Davies GCR Cell Calcium 2006, 39, 471–473. [DOI] [PubMed] [Google Scholar]
- (19).Ellis-Davies GCR; Kaplan JH Proc Natl Acad Sci U S A 1994, 91, 187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Grell E; Lewitzki E; Ruf H; Bamberg E; Ellis-Davies GC; Kaplan JH; de Weer P Cell Mol Biol 1989, 35, 515–22. [PubMed] [Google Scholar]
- (21).Valdivia HH; Kaplan JH; Ellis-Davies GCR; Lederer WJ Science 1995, 267, 1997–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ellis-Davies GCR Tetrahedron Letters 1998, 39, 4. [Google Scholar]
- (23).Momotake A; Lindegger N; Niggli E; Barsotti RJ; Ellis-Davies GCR Nat Methods 2006, 3, 35–40. [DOI] [PubMed] [Google Scholar]
- (24).DelPrincipe F; Egger M; Ellis-Davies GCR; Niggli E Cell Calcium 1999, 25, 85–91. [DOI] [PubMed] [Google Scholar]
- (25).Maeda H; Ellis-Davies GCR; Ito K; Miyashita Y; Kasai H Neuron 1999, 24, 989–1002. [DOI] [PubMed] [Google Scholar]
- (26).Takahashi N; Kadowaki T; Yazaki Y; Ellis-Davies GCR; Miyashita Y; Kasai H Proc Natl Acad Sci U S A 1999, 96, 760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kishimoto T; Liu TT; Ninomiya Y; Takagi H; Yoshioka T; Ellis-Davies GCR; Miyashita Y; Kasai H J Physiol 2001, 533, 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ng YK; Lu X; Watkins SC; Ellis-Davies GCR; Levitan ES J Neurosci 2002, 22, 3890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xu J; Xu Y; Ellis-Davies GCR; Augustine GJ; Tse FW J Neurosci 2002, 22, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lu X; Ellis-Davies GCR; Levitan ES Cell Calcium 2003, 33, 267–71. [DOI] [PubMed] [Google Scholar]
- (31).Soeller C; Jacobs MD; Jones KT; Ellis-Davies GCR; Donaldson PJ; Cannell MB J Biomed Opt 2003, 8, 418–27. [DOI] [PubMed] [Google Scholar]
- (32).Millar AG; Zucker RS; Ellis-Davies GCR; Charlton MP; Atwood HL J Neurosci 2005, 25, 3113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tanaka K; Khiroug L; Santamaria F; Doi T; Ogasawara H; Ellis-Davies GCR; Kawato M; Augustine GJ Neuron 2007, 54, 787–800. [DOI] [PubMed] [Google Scholar]
- (34).Gordon GR; Choi HB; Rungta RL; Ellis-Davies GCR; MacVicar BA Nature 2008, 456, 745–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tritsch NX; Zhang YX; Ellis-Davies GCR; Bergles DE Purinergic Signal 2010, 6, 155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang HC; Lin CC; Cheung R; Zhang-Hooks Y; Agarwal A; Ellis-Davies GCR; Rock J; Bergles DE Cell 2015, 163, 1348–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Watkins S; Robel S; Kimbrough IF; Robert SM; Ellis-Davies GCR; Sontheimer H Nat Commun 2014, 5, 4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Neher E Neuron 1998, 20, 389–99. [DOI] [PubMed] [Google Scholar]
- (39).Denk W; Strickler JH; Webb WW Science 1990, 248, 73–6. [DOI] [PubMed] [Google Scholar]
- (40).Denk W Proc Natl Acad Sci U S A 1994, 91, 6629–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Callaway EM; Katz LC Proc Natl Acad Sci U S A 1993, 90, 7661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lipp P; Niggli E J Physiol 1998, 508, 801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ellis-Davies GCR J. Gen. Physiol 1999, 114, 1a.10398688 [Google Scholar]
- (44).Matsuzaki M; Ellis-Davies GCR; Kasai H Society for Neuroscience Annual Conference 2000, 426.12. [Google Scholar]
- (45).Matsuzaki M; Honkura N; Ellis-Davies GCR; Kasai H Nature 2004, 429, 761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Amit B; Ben-Efraim DA; Patchornik AJ Chem. Soc. Perkin Trans. I 1976, 57–63. [Google Scholar]
- (47).Matsuzaki M; Ellis-Davies GCR; Nemoto T; Miyashita Y; Iino M; Kasai H Nat Neurosci 2001, 4, 1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bliss TV; Lomo T J Physiol 1973, 232, 331–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sjöström PJ; Rancz EA; Roth A; Häusser M Physiol Rev 2008, 88, 769–840. [DOI] [PubMed] [Google Scholar]
- (50).Nakahata Y; Yasuda R Front Synaptic Neurosci 2018, 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ellis-Davies GCR Front Synaptic Neurosci 2019, 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Adams SR; Kao JPY; Tsien RY J. Am. Chem. Soc 1989, 111, 7957–68. [Google Scholar]
- (53).Fedoryak OD; Sul JY; Haydon PG; Ellis-Davies GCR Chem Commun 2005, 3664–6. [DOI] [PubMed] [Google Scholar]
- (54).Ellis-Davies GCR; Matsuzaki M; Paukert M; Kasai H; Bergles DE J Neurosci 2007, 27, 6601–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yagishita S; Hayashi-Takagi A; Ellis-Davies GCR; Urakubo H; Ishii S; Kasai H Science 2014, 345, 1616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Eder M; Zieglgansberger W; Dodt HU Rev Neurosci 2004, 15, 167–83. [DOI] [PubMed] [Google Scholar]
- (57).Matsuzaki M; Hayama T; Kasai H; Ellis-Davies GCR Nat Chem Biol 2010, 6, 255–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gross GG; Junge JA; Mora RJ; Kwon HB; Olson CA; Takahashi TT; Liman ER; Ellis-Davies GCR; McGee AW; Sabatini BL; Roberts RW; Arnold DB Neuron 2013, 78, 971–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Villa KL; Berry KP; Subramanian J; Cha JW; Oh WC; Kwon HB; Kubota Y; So PT; Nedivi E Neuron 2016, 89, 756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Oh WC; Lutzu S; Castillo PE; Kwon HB Science 2016, 353, 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Matus A Curr Op Neurobiol 2005, 15, 67–72. [DOI] [PubMed] [Google Scholar]
- (62).Kwon H-B; Sabatini BL Nature 2011, 474, 100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Barany G; Merrifield RB J Am Chem Soc 1977, 99, 7363–5. [DOI] [PubMed] [Google Scholar]
- (64).Hara K; Sato T; Katoh R; Furube A; Ohga Y; Shinpo A; Suga S; Sayama K; Sugihara H; Arakawa HJ Phys. Chem. B 2003, 107, 597–606. [Google Scholar]
- (65).Specht A; Bolze F; Donato L; Herbivo C; Charon S; Warther D; Gug S; Nicoud J-F; Goeldner M Photochem Photobiol Sci 2012, 11, 578–86. [DOI] [PubMed] [Google Scholar]
- (66).Zayat L; Calero C; Alborés P; Baraldo L; Etchenique R J Am Chem Soc 2003, 125, 882–3. [DOI] [PubMed] [Google Scholar]
- (67).Olson JP; Kwon HB; Takasaki KT; Chiu CQ; Higley MJ; Sabatini BL; Ellis-Davies GCR J Am Chem Soc 2013, 135, 5954–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Amatrudo JM; Olson JP; Agarwal HK; Ellis-Davies GCR Eur J Neurosci 2015, 41, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Olson JP; Banghart MR; Sabatini BL; Ellis-Davies GCR J Am Chem Soc 2013, 135, 15948–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Passlick S; Kramer PF; Richers MT; Williams JT; Ellis-Davies GCR PLoS One 2017, 12, e0187732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Passlick S; Ellis-Davies GCR In Optochemical Biology; Deiters A, Ed.; Elsevier: New York, 2019; Vol. 624. [Google Scholar]
- (72).Albota M; Beljonne D; Bredas J; Ehrlich J; Fu J; Heikal A; Hess S; Kogej T; Levin M; Marder S; McCord-Maughon D; Perry J; Rockel H; Rumi M; Subramaniam C; Webb W; Wu X; Xu C Science 1998, 281, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (73).Reinhardt B; Brott L; Clarson S; Dillard A; Bhatt J; Kannan R; Yuan L; He G; Prasad P Chem Mater 1998, 10, 1863–1874. [Google Scholar]
- (74).Rumi M; Barlow S; Wang J; Perry JW; Marder SR Adv Polym Sci 2008, 213, 1–95. [Google Scholar]
- (75).He GS; Tan LS; Zheng Q; Prasad PN Chem. Rev 2008, 108, 1245–1330. [DOI] [PubMed] [Google Scholar]
- (76).Aujard I; Benbrahim C; Gouget M; Ruel O; Baudin JB; Neveu P; Jullien L Chemistry- Eur. J 2006, 12, 6865–6879. [DOI] [PubMed] [Google Scholar]
- (77).Agarwal HK; Janicek R; Chi SH; Perry JW; Niggli E; Ellis-Davies GCR J Am Chem Soc 2016, 138, 3687–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Richers MT; Passlick S; Agarwal H; Ellis-Davies GCR Angew Chem Int Ed 2019, 58, 12086–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ellis-Davies GCR; Meucci O; Shimizu S Society for Neuroscience Annual Conference 2007, 480.6/S14. [Google Scholar]
- (80).Molnár P; Nadler JV Eur J Pharmacol 2000, 391, 255–62. [DOI] [PubMed] [Google Scholar]
- (81).Amatrudo JM; Olson JP; Lur G; Chiu CQ; Higley MJ; Ellis-Davies GCR ACS Chem Neurosci 2014, 5, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Richers MT; Amatrudo JM; Olson JP; Ellis-Davies GCR Angew Chem Int Ed 2017, 56, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Durand-de Cuttoli R; Chauhan PS; Petriz Reyes A; Faure P; Mourot A; Ellis-Davies GCR Proc Natl Acad Sci U S A 2020, 117, 6831–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Noguchi J; Nagaoka A; Watanabe S; Ellis-Davies GCR; Kitamura K; Kano M; Matsuzaki M; Kasai H J Physiol 2011, 589, 2447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Jeong JW; McCall JG; Shin G; Zhang Y; Al-Hasani R; Kim M; Li S; Sim JY; Jang KI; Shi Y; Hong DY; Liu Y; Schmitz GP; Xia L; He Z; Gamble P; Ray WZ; Huang Y; Bruchas MR; Rogers JA Cell 2015, 162, 662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Park S; Guo Y; Jia X; Choe HK; Grena B; Kang J; Park J; Lu C; Canales A; Chen R; Yim YS; Choi GB; Fink Y; Anikeeva P Nat Neurosci 2017, 20, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Durand-de Cuttoli R; Mondoloni S; Marti F; Lemoine D; Nguyen C; Naude J; d’Izarny-Gargas T; Pons S; Maskos U; Trauner D; Kramer RH; Faure P; Mourot A Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Snow CP In The New Statesman; Fabian Society: London. [Google Scholar]
- (89).Klán P; Solomek T; Bochet CG; Blanc A; Givens R; Rubina M; Popik V; Kostikov A; Wirz J Chem Rev 2013, 113, 119–91. [DOI] [PMC free article] [PubMed] [Google Scholar]