

Abstract
This follow-up study of a subgroup of the patients seen in a natural history study of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A) addressed the adaptive and medical characteristics of their advanced disease manifestations. Of the original 24 patients, specific data was collected on only 58% primarily due to difficulty in locating families and coordinating time for interviews two to four years after the original study. At the last contact with the patient, age range was 8 to 24 years of age. Data were collected from telephone interviews from the Vineland Adaptive Behavior Scales II and medical and treatment history. We report the case data from rapid progressing and slow progressing patients separately. By the end of our data collection, 5 patients had died; 4 rapid progressing patients between 8 and 12 years of age and 1 slow progressing patient at age 21. Two patients were in out-of-home placements in the year before they died. We found that the incidence of surgeries and epilepsy was relatively low and that behavior problems largely subsided. Adaptive levels were very low with children functioning at below a two-year age equivalent level in all adaptive functions, but motor skills were slightly more intact. Only one slow progressing patient was functioning above a three-year level. Parent burden had shifted from behavioral control to physical management. Although their quality of life was clearly negatively impacted by physical management and palliative care, parents were more able to cope and adapt to such demands than in the initial stages of the disease.
Keywords: Mucopolysacharidosis type IIIA, Advanced stage disease, Survival, Adaptive Behavior
1. Introduction:
MPS III is a neurodegenerative disease of childhood that is relentless in its course and at present has no approved treatments. Most children with the rapid onset disease in both MPS A and MPS B have a rapid degenerative course in early childhood that is characterized by dementia of childhood with loss of cognitive ability, language, and motor function and is accompanied by severe behavioral abnormalities [1-3]. Three stages were proposed by Cleary and Wraith [1] and then based on our findings in a prospective natural history study [4] ages could be related to these stages. The first stage of developmental delays and slowing in cognitive development most frequently occurs between 1 to 4 years of age; a second stage from 3 to 4 years with behavioral difficulties, sleep disturbance, and cognitive decline, and finally a third stage ensues at a later but variable age with feeding and aspiration problems and loss of ambulation. Seizures are reported in this last stage [1-3]. The average age of death is reported to be approximately 18 years of age [3]. This study will further clarify the final stage.
While we have characterized the early and mid-stage disease process in a natural history study [4] [NCT01047306], this study attempted to follow these children as they decline, to delineate the course of the end of the second and the third and most advanced disease stage. Treatment of children with advanced disease is palliative and focuses on trying to prevent medical complications and attempting to ease everyday tasks so that the quality of life of the child and the family is as good as possible. Symptoms of the disease that occur more frequently late in the disease course have been reported to be epilepsy, sleep disturbance, decreased ability to be ambulatory and to feed and toilet oneself [1]. Palliative care and psychological support for families will be better served if we can better understand the end of life characteristics of these children.
2. Methods:
2.1. Subjects
Twenty-four of 25 children recruited for the original NH study completed the two year study. We attempted to find and follow-up these 24 patients by telephone beginning two years after the termination of the natural history study. The original study had 19 rapid progressing (RP) patients and 5 slowly progressing [SP] patients. Due to expected differences in rate of decline, these patients are reported separately.
2.2. Procedures
Two separate calls were made to each family. The first was an interview regarding their current psychological status that also included the Vineland Adaptive Behavior Scales-II Survey Interview form (VABS-II) administered over the phone. The VABS-II is a parent observation of adaptive behavior administered by interview that includes Communication, Daily Living Skills, Socialization, and Motor Skills [5]. Patients had either three or four previous VABS-II interviews during the natural history study. Because their functional capacity is so low, we are able to only report age equivalent scores in months [6]. We collected overall age equivalents for Communication, Daily Living Skills, and Socialization and an Adaptive Composite for comparison with the last value reported from the initial study. We also collected Motor Skills data which is separate from the Adaptive Composite.
The second telephone interview was a medical and treatment history, also using the case report form that was used for the initial natural history study with some modifications. This included medical problems since last visit, current medications, new behavioral problems, surgeries, hospitalization, and observations.
For the 10 patients who did not participate, we were able via direct telephone contact, internet, and social media to determine the vital status of all but two patients who were lost to follow-up.
Patients signed a consent form approved by the IRB at the University of Minnesota.
Because we had a limited sample, we present these data in a summarized case-by-case format.
3. Results:
3.1. Survival:
Of the 24 patients, 3 RP patients had already died when we began. During the time period that we were collecting data, two patients on whom we collected data on (one RP and one SP patient] died making a total of five deceased patients by the time of this report. The SP patient had a rapid and sudden decline over one year, was placed in a special facility because of her difficult behavior, and died shortly thereafter. The RP patient had a steady decline. Of those who died, two, one RP and one SP, were placed outside the home in a facility during the last year of their life. A Kaplan Meier survival curve for 22 of the 24 patients to the end of this study was graphed (see Figure 1).
Figure 1.

Kaplan Meier curve for 22 patients with known vital status. Probability of survival by age.
3.2. Recruitment:
It was very difficult to get parents/caretakers to commit the time to spend on the telephone to carry out this study. Apart from the three patients who had already died from the disease, we succeeded in getting data on 11 patients, four slow progressing (one who subsequently died) and 7 rapid progressing patients (one of whom subsequently died). For the other 10 patients that we were unable to collect data on, two were willing but were unable to commit to the time. We were unable to locate two patients. However, through social media, we have learned that all of these patients are alive; one patient has developed seizures, one patient has had pneumonia and a tracheostomy, and one hospitalized for feeding problems. Of the patients with detailed data, the age range at the close of the study was 8 to 24 years.
3.3. Differences in recruited and unrecruited patients:
Comparing the 10 unrecruited patients with 11 on whom we report data on (excluding the 3 patients deceased before this study), we first compared the last VABS age equivalent score of those RP patients interviewed and those not included. The mean VABS-II age equivalent score was 20 compared to 18 months, not a meaningful difference. However, age differences may be a factor in the results. Of those interviewed RP patients had a mean chronological age of 82.6 months and those not interviewed were 66.6 months at last natural history visit, about a year and a half younger.
The four SP patients at their last natural history visit had VABS-II age equivalent scores of 8.9, 11.6, 24, and 64 months at the chronological ages of 237, 244, 158 and 181 months (19.8, 20.3, 13, and 15 years). For comparison, the single SP patient lost to follow-up had an age equivalent score of 40 months at 110 months (9.2 years).
3.4. Deceased patients:
Using the last VABS-II score prior to death for the five patients who are deceased, the scores in months were 5.6 (just prior to death), 8.5, 8.9 (SP patient), 10, and 15 months age equivalent score. The 15 month score on the VABS was collected about one year prior to the patient’s death. Age at death for the four rapidly progressive patients was 9, 11, 11, and 12 years of age, and for the slow progressing patient, 21 years of age. For all patients, death was associated with MPS III disease.
3.5. Medical History data
3.5.1. Rapid progressing patients:
In the RP patients, the following were noted, MRSA infections in two, chronic cough in three, swallowing problems in two, spells of crying out in one, and sleep problems, one. Although one patient had two episodes of seizures, none of the parents were diagnosed with epilepsy. Two male patients had increasing hyperactivity and behavioral problems and two are on risperidone. Surgeries for G-tube placement were reported for two patients, inguinal hernia repair in one, and dental work under anesthesia for three. Gabapentin was being prescribed for four patients for pain.
3.5.2. Slow progressing patients:
For the three SP patients (one male and two female), one patient had hearing loss and a myringotomy and accompanying decline in communication skill. Behaviorally this male patient had increasing aggressive behavior and short attention span; he was on risperidone and melatonin for sleep. In the two SP female patients, stability was noted medically. One patient, 24 years old, had a supplementary G-tube but was mobile and able to eat and drink. She was on miglustat 300 mg per day and high dose genistein and supplements. The other patient, the highest functioning patient in the group at 17 years, was on no active medication and has had no further medical complications for the past two years. The fourth patient died during the period of interviewing. She had a rapid downhill course at the termination of the natural history study, and because of out-of-control behavior she was placed in a supervised living situation because she was unmanageable at home. She died shortly thereafter at the age of 21.
3.6. VABS-II Data
3.6.1. Rapid progressing patients:
A 13.5 year-old boy is largely bedridden, tube fed, and only marginally responsive functioning at a 1 month level having declined from 11 months two years ago. A 11 year-old girl had similarly lost her mobility and died shortly after the interview. She was functioning at a 5 to 6 month-old level, and had declined from a 13.5 month level two years previously. Her 8-year-old brother was functioning at an 8-month old age equivalent level but with slightly higher functioning in personal self-care but socially unresponsive. His motor skills were higher at 19 months. In contrast, a younger 7.5-year-old male declined from 27-months age equivalent in two years to an 11.4-month age equivalent. His motor skills remained at a 24-month age equivalent level. Another 8-year-old female is in a wheelchair but not talking, and her older 11-year-old male sibling can still say some words, is highly mobile, and still goes to school. A 9-year-old male declined from a 33-month-old age equivalent when he was 5 years of age to an 8 month level at 9 years of age. He is not talking any more, but can walk unsteadily.
3.6.2. Slow progressing patients:
In the SP group, a 17-year-old female has showed stability over the two-year period since the end of the study. Her current age equivalent score is 63 months; previous age equivalent score was 64. She continues to verbally communicate, is toilet trained with other personal self-care skills at about a 55-month age equivalent level. She has simple domestic skills and no behavioral issues. She can play games and seeks companionship but does not have friends and has some difficulties with social reciprocity. Her lowest skills are in gross motor abilities at 42 months but her fine motor abilities are at a 67-month age equivalent.
In contrast, the brother of the patient previously described, 15 years of age, has declined in the two-year interval from a 24-month age equivalent to 15.8 months. Primarily while he retains some personal care skills (partially toilet trained), he has few other daily living skills. His socialization skills are at 13 months. In contrast, his motor skills are significantly better. Gross motor skills are at 29 months and fine motor at 33-month age equivalent scores.
Another young woman of 23 years is stable over the two years; although she does not talk, she eats and drinks on her own, is mobile, and engages in a lot of therapy activities. She has declined very little in the past 4 years. Her stability has been attributed to her medication regimen by her caretaker; she has been on miglustat 300 mg per day and high dose genistein. While she is functioning at a 10-month level overall, her motor skills are better. Her gross motor skills are 15 months and fine motor at 11 months.
3.6.3. Summarized VABS-II findings.
For the RP patients, VABS-II losses ranged from 4 to 8 months per year with the youngest patient showing the 8 month per year decline. There did not seem to be differences overall in communication, daily living skills or socialization. However, motor skills remain the most preserved with the youngest patients who were 8 and under remaining at a two-year-old mobility level.
The SP patients were highly variable. The female patient who died had a sudden decline in functionality with unmanageable behavior before her death. Two females, chronological ages 17 and 21, were remarkably stable, although the 21-year-old is stable at a very low level except for motor function. The 17-year-old was the highest functioning patient that we interviewed and she has not lost function since she was seen last, 3 years prior to the interview. In contrast, her sibling, the 15-year-old SP male patient has shown an 8 month decline in age equivalent over two years.
Figure 2 is a graphic portrayal of these results.
Figure 2.
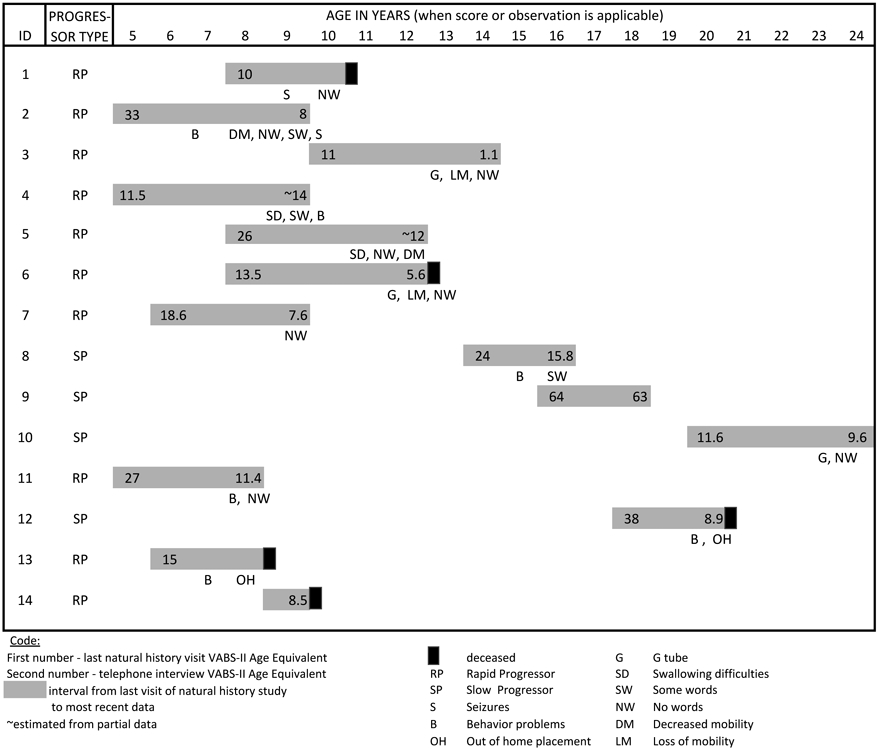
A graphic representation of late stage disease progression in MPS IIIA
4. Discussion:
While the inexorable progression of the disease makes daily care a burden on the family and caregiver, these families are providing appropriate medical care and palliative care. Examples of such care are airway clearance therapy, G-tube feedings, and for those who still are able, physical and educational interventions.
There was a low incidence of surgeries and hospitalizations as well as a low incidence of newly emerging behavioral difficulties. This is consistent with previous findings that behavioral difficulties subside in the late stages of disease [1,2,7]. Risperidone has been used successfully for behavioral difficulties, gabapentin for pain management, and melatonin for sleep in our patients. Only one of our patients was on high dose genistein and miglustat, both non-approved experimental therapies with unproven clinical efficacy. Epilepsy preceded death of one of our patients, and one, not interviewed, also has seizures. Thus, in this sample the incidence of epilepsy in this group was not as high as reported in some reports [3].
On the other hand, adaptive levels were very low with children functioning at below a two-year level in communication, daily living skills and socialization and the composite score. However, for a few children, motor skills were more intact at the 2 to 3-year level. One exception was a 17-year-old SP patient who showed stable function for the past two years in all areas with some reading and writing skills still at a five to six-year level as well as personal and social skills. Another noteworthy finding is that the rate of decline between the siblings is not congruent. This variability has been described by several authors [1-3]. However, the RP patients had consistently low functional level. Delineating the prognosis in SP from RP patients can help parents and caretakers prepare for future challenges.
For these patients, prevention of medical complications and the treatment of problem areas such as behavior, pain, and disturbed sleep contribute to quality of life. Risperidone, gabapentin, and melatonin have been used for these problems, respectively. Behavior in MPS III, was investigated in another study using a diary and interview of parents, has been reported to cause distress due to management difficulties, even more than the burden of physical symptoms [8]. As behavioral problems diminished in some of our patients, some burdensome aspects of caretaking have been attenuated.
The conclusions from this study are tempered by the knowledge that we were only able to report details on 11 patients. Three patients had already died by the time we began. We were unable to provide details on 10 patients. Comparing the two groups, the patients we had data on were somewhat older, but not significantly different in level of impairment on the VABS-II. Importantly, decrease in VABS-II age equivalent scores below one year, in the year before death, occurred in 4 patients; the fifth did not have a measurement within the year.
We can conclude that these patients with the exception of one slow progressing patient were all functioning below a 2-year-old level and below 3 years age equivalent on motor skills. Epilepsy was not as prevalent as we had anticipated from the literature. Furthermore, in the year prior to death, age equivalent scores in most patients declined to below one year. Only two RP patients continued to have behavioral issues; in contrast, shortly before her death one SP patient had major behavioral difficulties. However, for the other patients, the burden for the family shifted from behavioral control to physical management.
Although these parents did not complain about stresses directly, the burden on the family due to the loss of mobility and self-care skills, the need for tube feeding and airway clearance, and other supports continue to take a toll on the quality of life of families. However, the acute distress that was observed during the original natural history study appears to have evolved into habituation to the stress and accustomed to the situation. Understanding these end of life characteristics, can help to improve palliative care for the patient and psychological support for the family by having a better sense of what the future holds for them.
Acknowledgments:
We are grateful for the cooperation of the parents and the patients who participated in this study.
Funding Sources
Shire supported this study through an investigator initiated research grant “Observing the disease course in the natural history of Sanfilippo Syndrome Type A: A follow-up of SAN-053 participants” P.I. Elsa Shapiro
Additional support was provided by the Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NCATS. This consortium is funded through a collaboration between NCATS, NINDS, and NIDDK.
References:
- 1.Cleary MA, Wraith JE. Management of mucopolysaccharidosis type III. Arch Dis Child 1993;69:403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valstar MJ, Ruijter GJ, Van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. Journal of inherited metabolic disease. 2008. April 1;31(2):240–52. [DOI] [PubMed] [Google Scholar]
- 3.Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype– phenotype correlations. Ann Neurol 2010;68:876–87. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro E, Nestrasil I, Delaney K, Rudser K, Kovac V, Nair N, Richard C, Haslett P, Whitley C. (2016) A prospective natural history study of Mucopolysaccharidosis Type IIIA. Journal of Pediatrics. 170, 278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sparrow SS, Cicchetti DV. Diagnostic uses of the Vineland Adaptive Behavior Scales. J Pediatr Psychol 1985;10:215–25. [DOI] [PubMed] [Google Scholar]
- 6.Delaney KA, Rudser KR, Yund BD, Whitley CB, Haslett PA, Shapiro EG. Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome. JIMD Rep 2014;13:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grant S, Cross E, Wraith JE, Jones S, Mahon L, Lomax M, Bigger B, Hare D. Parental social support, coping strategies, resilience factors, stress, anxiety and depression levels in parents of children with MPS III (Sanfilippo syndrome) or children with intellectual disabilities (ID). Journal of inherited metabolic disease. 2013. March 1;36(2):281–91. [DOI] [PubMed] [Google Scholar]
- 8.Malcolm C, Hain R, Gibson F, Adams S, Anderson G, Forbat L. Challenging symptoms in children with rare life-limiting conditions: findings from a prospective diary and interview study with families. Acta Paediatrica. 2012. September 1;101(9):985–92. [DOI] [PubMed] [Google Scholar]